

Abstract
Inhibition of enzyme activity by high concentrations of substrate and/or cofactor is a general phenomenon demonstrated in many enzymes, including aldehyde dehydrogenases. Here we show that the uncharacterized protein BetB (SA2613) from Staphylococcus aureus is a highly specific betaine aldehyde dehydrogenase, which exhibits substrate inhibition at concentrations of betaine aldehyde as low as 0.15 mM. In contrast, the aldehyde dehydrogenase YdcW from Escherichia coli, which is also active against betaine aldehyde, shows no inhibition by this substrate. Using the crystal structures of BetB and YdcW, we performed a structure-based mutational analysis of BetB and introduced the YdcW residues into the BetB active site. From a total of 32 mutations, those in five residues located in the substrate binding pocket (Val288, Ser290, His448, Tyr450, and Trp456) greatly reduced the substrate inhibition of BetB, whereas the double mutant protein H448F/Y450L demonstrated a complete loss of substrate inhibition. Substrate inhibition was also reduced by mutations of the semiconserved Gly234 (to Ser, Thr, or Ala) located in the BetB NAD+ binding site, suggesting some cooperativity between the cofactor and substrate binding sites. Substrate docking analysis of the BetB and YdcW active sites revealed that the wild-type BetB can bind betaine aldehyde in both productive and nonproductive conformations, whereas only the productive binding mode can be modeled in the active sites of YdcW and the BetB mutant proteins with reduced substrate inhibition. Thus, our results suggest that the molecular mechanism of substrate inhibition of BetB is associated with the nonproductive binding of betaine aldehyde.
INTRODUCTION
Inhibition of enzyme activity at high concentrations of substrate and/or cofactor is a general phenomenon observed in over 20% of known enzymes, including dehydrogenases, kinases, methyltransferases, and hydroxylases (1–3). Presently, it is considered to be a biologically relevant regulatory mechanism with important biological functions in several metabolic pathways (2). For example, substrate inhibition of tyrosine hydroxylase stabilizes the level of dopamine despite large changes in the tyrosine concentration, whereas the inhibition of acetylcholinesterase enhances the neural signal (2). However, substrate inhibition has a negative effect on biotechnological application of enzymes, because it reduces the reaction rate and product yield in industrial processes, which are usually performed at high substrate concentrations (4). For example, the inhibition of β-galactosidases by glucose and galactose limits the production of galacto-oligosaccharides (5).
For dehydrogenases, several molecular mechanisms of substrate inhibition have been proposed, including the formation of a covalent adduct between the oxidized forms of substrate and cofactor, allosteric inhibition (which occurs away from the active site, e.g., in d-3-phosphoglycerol dehydrogenase from Mycobacterium tuberculosis), and the formation of a nonproductive enzyme complex with cofactor and/or substrate (6–13). The latter mechanism can be associated with the dehydrogenase residues located both in the substrate and in cofactor binding sites. Recent biochemical studies have demonstrated that substrate inhibition of several dehydrogenases can be significantly reduced or eliminated by single mutations in the enzyme active site, which can be accompanied by a reduction or increase in the reaction rate and usually correlates with a reduction of substrate affinity (increasing Km) (4, 7, 10, 13). For example, the mutation S163L in human lactate dehydrogenase eliminates substrate inhibition with a minor effect on its turnover rate, whereas in Bacillus stearothermophilus lactate dehydrogenase the D38R mutation reduces substrate inhibition 3-fold (4, 14). In addition, natural dehydrogenases resistant to substrate inhibition have been discovered, e.g., lactate dehydrogenase from Plasmodium falciparum (15). Understanding of the molecular mechanisms of substrate inhibition of enzymes is important for our fundamental knowledge, as well as for the optimization of the enzyme-catalyzed processes in biocatalysis and for engineering of new biosynthetic pathways for production of biofuels and chemicals (16).
Aldehyde dehydrogenases (ALDHs; PF00171) are a large group of enzymes that oxidize a wide range of aliphatic and aromatic aldehydes to corresponding carboxylic acids using NAD(P)+ as an electron acceptor. ALDHs are ubiquitous in nature, being present in archaea, bacteria, and eukaryotes, indicating that they play an essential role in the cell (17). In addition to the detoxification of aldehydes, ALDHs are believed to modulate cellular homeostasis and organismal functions, and mutations in the human ALDH genes are associated with several diseases (18). Presently, the ALDH superfamily is organized into more than 14 families active against a broad range of substrates, including betaine aldehyde (BA), succinate semialdehyde, and glyceraldehyde 3-phosphate (19).
Betaine aldehyde dehydrogenases (BADHs, EC 1.2.1.8) catalyze the irreversible NAD(P)+-dependent oxidation of betaine aldehyde (or glycine betaine aldehyde) to glycine betaine (GB) (Fig. 1A), which functions as an osmoprotectant in both prokaryotic and eukaryotic organisms (20–25). The physiological role of BADHs is associated with the protection of cells from osmotic stress, as well as with the catabolism of choline or choline precursors (26–29). In bacteria, betaine synthesis is determined by two enzymes: choline dehydrogenase (which oxidizes choline to betaine aldehyde) and betaine aldehyde dehydrogenase (which converts betaine aldehyde into betaine) (26). In Brucella abortus, the betaine aldehyde dehydrogenase BetB is an essential virulence factor required for osmotic-stress resistance and replication in mammalian cells (30). BADHs are related to the aldehyde dehydrogenase families ALDH9, ALDH10, ALDH25, and ALDH26 (19). To date, several BADHs have been characterized biochemically and structurally, including the enzymes from cod liver (23), Spinacia oleracea (20), Pisum sativum (31), Pseudomonas aeruginosa (32), and Escherichia coli (YdcW in ALDH26) (22). These enzymes share similar three-dimensional structures containing three domains: the nucleotide binding domain with the Rossmann fold, the substrate binding domain, and the oligomerization domain. The proposed chemical mechanism of BADHs involves four steps, including the formation of a tetrahedral intermediate by a nucleophilc cysteine (Cys280 in YdcW) attacking BA, the transfer of the hydride from the hemithioacetal to the NAD(P)+ resulting in a thioester, the formation of a second tetrahedral intermediate from the thioester via a nucleophilic water molecule activated by a glutamate side chain (Glu246 in YdcW), and the release of glycine betaine and NAD(P)H (33). BADHs have a complex kinetic mechanism compared to other ALDHs, most of which follow an ordered binding mechanism, whereby NAD(P)+ binds prior to an aldehyde, and then the resulting carboxylic acid is released before NAD(P)H (34). However, the BADHs from the plant Amaranthus hypochondriacus (ALDH10) and porcine kidney (ALDH9) have been proposed to follow an iso-ordered Bi Bi mechanism, since the rate-limiting step occurs in the isomerization of free enzyme after the release of the last product and before the binding of the first substrate (21, 24). In contrast, the P. aeruginosa BADH (PaBADH, ALDH9) follows a random Bi Bi mechanism, although the mechanism is predominantly ordered in the case of the NADP+-dependent reaction (35). A kinetic study revealed that the activity of PaBADH is inhibited by the cofactor and partially by the aldehyde (25, 35), and the structure of this enzyme in complex with the cofactor suggested that this inhibition is caused by a novel NADPH-cysteine covalent adduct (8).
FIG 1.

Reaction catalyzed by S. aureus BetB and proposed mechanism. (A) NAD+-dependent oxidation of betaine aldehyde to glycine betaine. (B) Ordered Bi Bi mechanism proposed for BetB.
Recently, we have determined the crystal structures of the putative betaine aldehyde dehydrogenase BetB from Staphylococcus aureus (SA2613) in the apo form and in the complex with NAD+ (Protein Data Bank accession codes 4MPB and 4MPY) (unpublished data). In the present work, we have performed detailed biochemical, mutational, and substrate docking studies of this enzyme and its inhibition by BA. We have identified several residues of BetB essential for substrate inhibition, and our substrate docking experiments based on the BetB structure suggested that substrate inhibition of BetB is associated mainly with the nonproductive binding of BA in its active site.
MATERIALS AND METHODS
Expression and purification of BetB and YdcW.
The genes encoding S. aureus BetB (GenBank 3237065, Uniprot ID Q5HCU0), and E. coli YdcW (GenBank 945876, Uniprot ID P77674) were PCR amplified using S. aureus and E. coli genomic DNA and cloned into the IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible protein expression vector p15TV-L as described previously (36). The resulting plasmids were transformed into the E. coli BL21(DE3) Gold strain (Stratagene, USA), and the recombinant proteins were purified using affinity chromatography on Ni-nitrilotriacetic acid (Ni-NTA) resin (Qiagen, Germany). The purity of proteins (usually higher than 95%) was confirmed by SDS-PAGE, and the protein concentration was measured using the Bradford method (Bio-Rad) using bovine serum albumin (BSA) as a standard.
Site-directed mutagenesis of BetB.
Site-directed mutagenesis was performed based on the QuikChange site-directed mutagenesis kit (Stratagene, USA). The standard PCR mixture (50 μl) contains 100 ng of template DNA and 100 to 250 ng of each mutagenic primer. The PCR cycling conditions were as follows: 95°C (30 s); 16 cycles of 95°C (30 s), 55°C (1 min), and 68°C (2 min/kb). After the PCRs, DpnI (10 U) was added, and the mixture was incubated at 37°C for 1 h. An additional 10 U of DpnI was added and incubated for another hour to completely digest parental DNA. The PCR mixtures were transformed into E. coli competent DH5α cells and plated on LB medium supplemented with 0.1 g/liter ampicillin. All mutations were verified by sequencing, and the mutant proteins were purified as described for the wild-type enzyme.
Enzymatic assays.
Aldehyde dehydrogenase activity against a range of substrates was determined using a continuous assay following the increase in absorbance at 340 nm [ε340 (NADH) = 6.22 mM−1 · cm−1, and ε340 (NADPH) = 6.2 mM−1 · cm−1]. Substrate screens were performed at 30°C in a reaction mixture (200 μl) containing 100 mM potassium phosphate buffer (pH 7.5), 1 mM NAD+, 1 mM EDTA, 1 mM substrate, and 10 μg of purified BetB. Initial velocity patterns of the S. aureus BetB reaction were obtained by varying the concentrations of NAD+ at fixed concentrations of betaine aldehyde. Kinetic parameters of the wild-type and mutant enzymes were determined with varying concentrations of one substrate in the presence of the saturating or subinhibitory concentration of the second substrate in 100 mM potassium phosphate buffer at pH 8.0 and 30°C and were calculated from the initial reaction rates using equation 1 or 2 and the Prism program (version 5.02; GraphPad Software, San Diego, CA):
| (1) |
| (2) |
where [S] is the concentration of the varied substrate, Km is the Michaelis-Menten constant of the enzyme for the varied substrate, and Ki is the dissociation constant for the enzyme of the varied substrate. BA concentrations were varied between 0.01 and 200 mM in the presence of 5 mM NAD+, and NAD+ concentrations were between 0.05 and 10 mM in the presence of a constant BA concentration (see Table 2). One unit (U) of enzyme activity corresponds to the release of 1 μmol of NADH per 1 min.
TABLE 2.
Product and dead-end inhibition patterns and inhibition constants for the NAD+-dependent oxidation of BA by BetB
| Variable substrate | Fixed substrate | Product or inhibitor | Ki (μM) | αKi (μM) | Inhibition pattern |
|---|---|---|---|---|---|
| NAD+ | BA | NADH | 37 | Competitive | |
| BA | NAD+ | NADH | 8.0 × 102 | Noncompetitive | |
| NAD+ | BA | GB | 8.4 × 105 | Mixed | |
| BA | NAD+ | GB | 2.2 × 106 | Uncompetitive | |
| NAD+ | BA | Choline | 3.4 × 104 | Uncompetitive | |
| BA | NAD+ | Choline | 2.6 × 104 | Competitive | |
| NAD+ | BA | Benzaldehyde | 5.2 × 102 | Uncompetitive | |
| BA | NAD+ | Benzaldehyde | 2.2 × 102 | Competitive |
Product and dead-end inhibition studies.
Inhibition studies were conducted by measuring initial velocities of the reaction, in which the concentration of one substrate was varied in the presence of a constant (and subsaturating) concentration of the second substrate and several concentrations of product (NADH or glycine betaine [GB]) or dead-end inhibitors (choline and benzaldehyde). The data of initial velocities were fitted using equation 3, 4, 5, or 6, corresponding to competitive inhibition, noncompetitive inhibition, mixed inhibition, or uncompetitive inhibition, respectively (GraphPad Prism, version 5.02, GraphPad Software, San Diego, CA):
| (3) |
| (4) |
| (5) |
| (6) |
where [I] is the concentration of the inhibitor and αKi and Ki are estimated inhibition constants.
Homology modeling and substrate docking.
The NAD+-bound structures of S. aureus BetB (PDB code 4MPY) and E. coli YdcW (PDB code 1WNB) were used in docking simulations. The structure of the S. aureus BetB H448F/Y450L mutant protein was constructed using SCWRL4 software (37) and the wild-type BetB structure (4MPY) as a template. For docking simulations, the structures of protein and ligand were formatted by AutoDockTools 4.2 (38). The enzyme side chains were set as rigid, while two bonds of C-1 to C-2 and C-2 to N in BA were set as rotatable. A grid box was set to 50 by 50 by 50 points with a grid point spacing of 0.375 Å. Molecular docking simulations of the wild-type BetB, the double mutant H448F/Y450L, and YdcW were performed using AutoDock 4.2 (38). The number of genetic algorithm runs and the maximum number of evaluations were set to 100 and 2,500,000, respectively. All other parameters were set to default values. The resulting binding modes with comparable binding energies calculated by Lamarckian genetic algorithm were further classified into productive and nonproductive modes based on the distances between the catalytic residue (Cys289 for BetB and Cys280 for YcdW) and BA, and the conformation with the lowest energy in each mode was referred to as a substrate binding mode. All structure figures were generated using PyMOL software (PyMOL molecular graphics system, version 1.4.1; Schrödinger, LLC).
RESULTS AND DISCUSSION
Substrate selectivity and inhibition of BetB.
The putative BADH BetB from S. aureus was overexpressed in E. coli, and the purified BetB exhibited significant NAD+-dependent dehydrogenase activity against BA (>1 μmol/min per mg protein). Since ALDHs, including BADHs, can react with a wide range of substrates (19), we also examined BetB for dehydrogenase activity toward various aldehydes and carbohydrates. This screening demonstrated that BetB is highly selective toward BA with low (but detectable) activity against 2-pyridinecarboxaldehyde, 3-pyridinecarboxaldehyde, and 4-pyridinecarboxaldehyde (<5% activity of maximal activity with BA) (see Fig. S1 in the supplemental material). Thus, the BetB enzyme is a NAD+-dependent betaine aldehyde dehydrogenase. In contrast to BetB, E. coli YdcW has been reported to have a broad substrate profile with high activity toward phenyl acetaldehyde, undecanal, nitrobenzaldehyde, and BA (22, 39). The optimal pH for dehydrogenase activity of BetB was 8.0, which is similar to other characterized BADHs (25, 40, 41). Both BetB and YdcW can use both NAD+ and NADP+ as cofactors, but the NAD+-dependent activity with BA was at least 10 times higher for both enzymes (BetB, 3.5 U/mg versus 0.2 U/mg) (22). Compared to YdcW, BetB exhibited a higher activity and affinity to BA (kcat, 0.55 and 11.0 s−1; Km, 32.8 and 0.17 mM, respectively), but YdcW had a lower Km for NAD+ (Table 1 and Fig. 2A and B). Analysis of the wild-type BetB kinetics also revealed strong inhibition by BA at substrate concentrations higher than 0.15 mM with an inhibition constant (Ki) value of 0.35 mM (Fig. 2A). Substrate saturation experiments using variable concentrations of NAD+ and a fixed concentration of BA (0.15 mM) showed no inhibition of the BetB activity at high concentrations of NAD+ (data not shown).
TABLE 1.
Apparent kinetic parameters of the wild-type and mutant BetB proteins and YdcW
| Protein(s) (BA concn in mM)a | Location of residues | Km (mM) | Ki (mM) | kcat (s−1) | kcat/Km (s−1 mM−1) | Km (NAD+, mM) |
|---|---|---|---|---|---|---|
| YdcW (200) | 32.8 ± 0.5 | 0.55 ± 0.003 | 0.017 | 0.11 ± 0.004 | ||
| BetB-WT (0.2–0.3) | 0.17 ± 0.03 | 0.34 ± 0.06 | 11.0 ± 1.4 | 64.7 | 0.26 ± 0.03 | |
| Q162 M (1–5) | Nicotinamide and BA | 0.91 ± 0.08 | 8.82 ± 0.71 | 9.9 ± 0.5 | 10.9 | 0.68 ± 0.05 |
| G234S (1–5) | Pyrophosphate | 0.17 ± 0.01 | 15.2 ± 1.9 | 2.92 ± 0.07 | 17.2 | 0.12 ± 0.01 |
| G234T (1–5) | Pyrophosphate | 0.11 ± 0.01 | 108 ± 21 | 0.10 ± 0.004 | 0.91 | 0.30 ± 0.05 |
| G234A (1–5) | Pyrophosphate | 0.12 ± 0.01 | 36.9 ± 5.4 | 1.14 ± 0.026 | 9.50 | 1.12 ± 0.10 |
| V288D (15–50) | BA | 4.76 ± 0.58 | 86.4 ± 18.0 | 0.25 ± 0.02 | 0.053 | 0.27 ± 0.01 |
| S290T (15–50) | BA | 13.8 ± 1.4 | 458 ± 37 | 1.63 ± 0.07 | 0.12 | 0.41 ± 0.05 |
| H448F (1–5) | BA | 0.26 ± 0.05 | 51.8 ± 5.2 | 0.94 ± 0.06 | 3.62 | 0.42 ± 0.04 |
| Y450L (15–50) | BA | 1.92 ± 0.14 | 71.1 ± 9.7 | 1.35 ± 0.11 | 0.70 | 1.01 ± 0.12 |
| W456H (15–50) | BA | 14.8 ± 2.3 | 83.0 ± 9.1 | 3.20 ± 0.38 | 0.22 | 0.38 ± 0.03 |
| L161 M/Q162 M (1–5) | Nicotinamide and BA | 2.16 ± 0.13 | 7.81 ± 0.54 | 4.46 ± 0.46 | 2.06 | 0.71 ± 0.02 |
| H448F/P449 M (1–5) | BA | 0.57 ± 0.06 | 7.00 ± 1.34 | 4.06 ± 0.34 | 7.12 | 1.12 ± 0.15 |
| H448F/Y450L (15–50) | BA | 1.18 ± 0.06 | 1.09 ± 0.01 | 0.92 | 0.34 ± 0.04 | |
| P449 M/Y450L (15–50) | BA | 1.94 ± 0.12 | 137 ± 15 | 1.87 ± 0.12 | 0.96 | 0.43 ± 0.10 |
| H448F/P449 M/Y450L (15–50) | BA | 4.61 ± 0.41 | 7,600 ± 790 | 1.80 ± 0.11 | 0.39 | 3.42 ± 0.40 |
All reactions were performed at pH 8.0 and 30°C, using varied BA at 5 mM NAD+ or using varied NAD+ at different concentrations of BA as indicated in parentheses.
FIG 2.
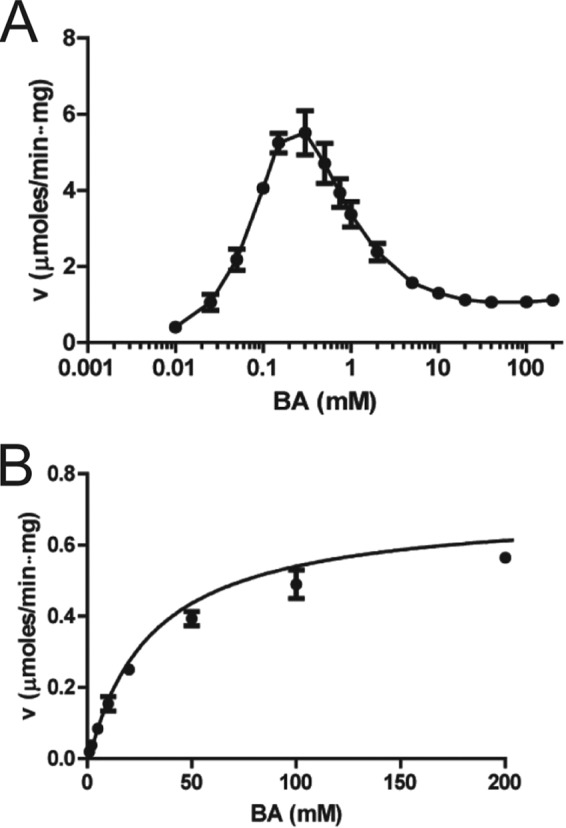
Dehydrogenase activity of BetB (A) and YdcW (B) as a function of BA concentration. Velocity (v) versus substrate concentration curves for S. aureus BetB (A) and E. coli YdcW (B). The dehydrogenase activity was determined using varied concentrations of BA, 5 mM NAD+, and 1 μg of BetB (10 μg for YdcW). Results are means ± standard deviations (SD) from at least two independent experiments.
Kinetic mechanism of BetB.
Known BADHs follow the ternary-complex kinetic mechanism, which can be either ordered (when substrates bind in a particular order) or random (33). Initial velocity studies of S. aureus BetB revealed linear double-reciprocal plots in the presence of 0.16 to 10 mM BA and 0.04 to 1.25 mM NAD+ (Fig. 3). As shown in Fig. 3, the series of lines intersecting on the left side of the y (1/v) axis suggests that BetB also follows the ternary-complex kinetic mechanism. The enzyme kinetic mechanism can be determined by the investigation of product inhibition patterns and/or dead-end inhibition patterns (3). In the product inhibition experiment, the inhibition of the wild-type BetB by NADH can be compensated for by increasing NAD+ concentrations (at a fixed concentration of BA), indicating that this inhibition is competitive (Fig. 3B). With BA as the variable substrate (within the noninhibiting concentration range, <0.15 mM; in the presence of 5 mM NAD+), the double-reciprocal plots of the NADH effect suggested a noncompetitive inhibition by NADH (Fig. 3C and Table 2; see Fig. S2 in the supplemental material). These results are consistent with an ordered Bi Bi mechanism for irreversible dehydrogenase reactions, where NAD+ is bound first to the enzyme and NADH is released last. In contrast to NADH, GB behaved as a mixed inhibitor with respect to NAD+ and as an uncompetitive inhibitor with respect to BA. The estimated inhibition constants for GB were larger than that for the reduced nucleotide by almost 3 orders of magnitude (Table 2), which is similar to that reported for PaBADH (35). GB showed significant inhibition of enzyme activity only at concentrations higher than 50 mM, suggesting that BetB has low affinity to GB. This is in line with high osmotolerance of S. aureus and its ability to accumulate high concentrations of intracellular betaine (200 mM to 2 M) (42, 43). Since the inhibition of BetB by GB can also be a result of the unspecific binding at the active site, we performed a dead-end inhibitor analysis to confirm the kinetic mechanism. Both BA analogues (choline and benzaldehyde) demonstrated competitive inhibition with regard to BA and uncompetitive inhibition with regard to NAD+, suggesting that BetB binds NAD+ first and then BA (see Fig. S3 in the supplemental material). As indicated in Table 2, benzaldehyde is an effective inhibitor with a Ki 2 orders of magnitude lower than that for choline. In contrast to BetB, several BADHs have been proposed to use the isomerization mechanism (iso-mechanism) with the noncompetitive product inhibition patterns, in which the first substrate and the last product bind to different forms of enzyme (33). Therefore, our results suggest that BetB follows an ordered Bi Bi mechanism, whereby NAD+ binds to the enzyme first, followed by BA, and GB is released first, followed by NADH (Fig. 1B).
FIG 3.
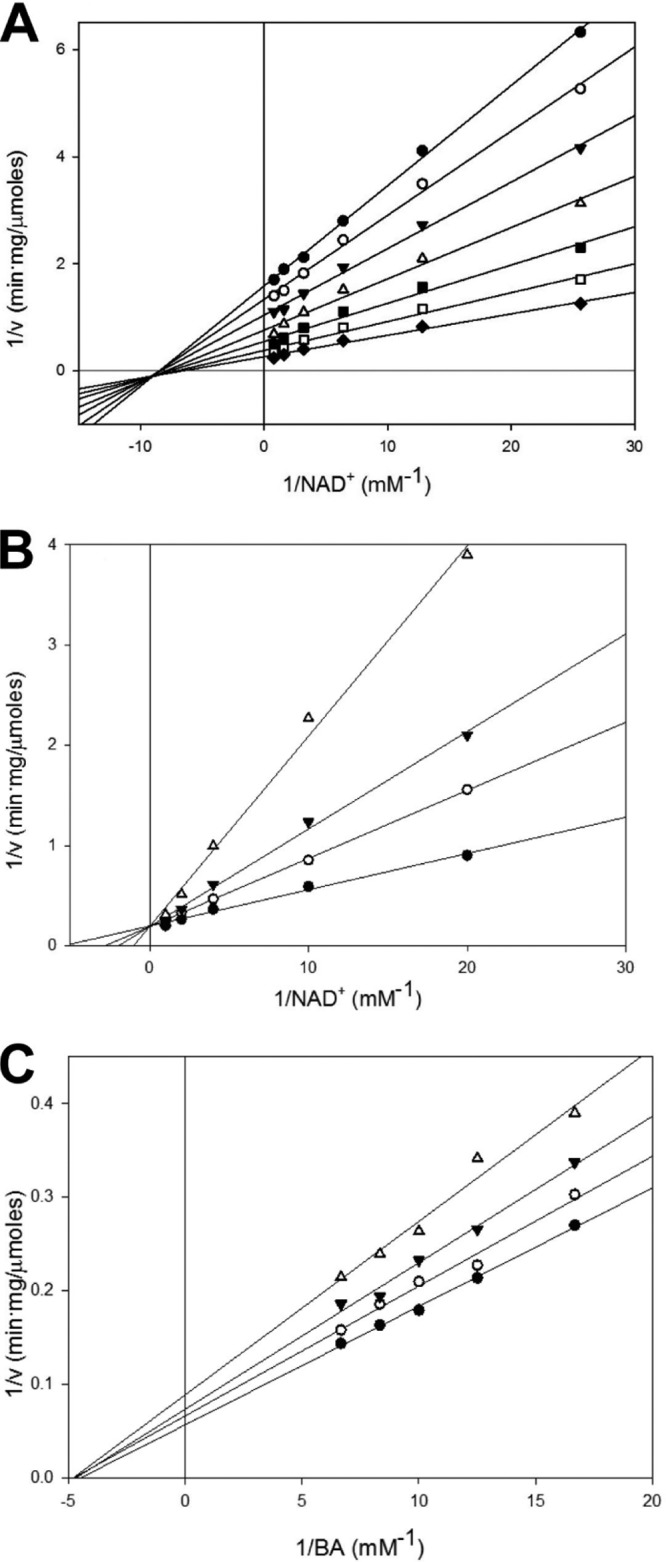
Kinetic studies of BetB. (A) Initial velocity patterns of the BetB reaction. Dehydrogenase activity of BetB was determined with varied NAD+ (0.04 to 1.25 mM) in the presence of fixed concentrations of BA: 0.16 mM (diamonds), 0.31 mM (open squares), 0.62 mM (closed squares), 1.25 mM (open triangles), 2.5 mM (closed triangles), 5 mM (open circles), and 10 mM (closed circles). (B, C) Inhibition patterns of BetB activity by NADH. (B) Dehydrogenase activity of BetB as a function of NAD+ concentration in the presence of various concentrations of NADH: 2.5 μM (closed circles), 25 μM (open circles), 50 μM (closed triangles), and 100 μM (open triangles). BA concentration was 0.15 mM. The control (0 μM NADH) has the same line as 2.5 μM NADH. (C) Dehydrogenase activity of BetB as a function of BA concentration in the presence of various concentrations of NADH: 100 μM (closed circles), 250 μM (open circles), 375 μM (closed triangles), and 500 μM (open triangles). The control (0 μM NADH) has the same line as 100 μM NADH.
Site-directed mutagenesis of BetB.
The crystal structures of both BetB and YdcW have been determined (PDB codes 4MPY and 1WNB, respectively) revealing highly similar structures (Dali Z-score, 55.2; root mean square deviation, 1.3 Å). Although BetB and YdcW share low sequence similarity (37% overall sequence identity), they have identical catalytic residues, including the nucleophile (Cys289 in BetB and Cys280 in YdcW) and the general base in the deacylation step (Glu255 in BetB and Glu246 in YdcW) (22). Additional conserved residues are located in the binding sites for BA (Asn157, Trp165, Lys166, and Leu418 in BetB; Asn149, Trp157, Lys158, and Leu406 in YdcW) and cofactor (Trp156, Lys180, Glu390, and Phe392 in BetB; Trp148, Lys172, Glu378, and Phe380 in YdcW) (see Fig. S4 in the supplemental material) (32).
Since YdcW shows no substrate inhibition at BA concentrations of up to 200 mM (Fig. 2B), we used its structure for the mutational analysis of substrate inhibition of BetB with the aim of determining if the introduction of the YdcW residues into BetB would reduce or eliminate substrate inhibition of the latter enzyme. A structural comparison of the BetB and YdcW substrate and cofactor binding sites revealed 23 different amino acids (Fig. 4). The BetB residues were then replaced by those present in YdcW or other residues using site-directed mutagenesis. We also created several double or triple mutant BetB proteins to analyze the potential synergistic effect of the neighbor residues on substrate inhibition. The mutated BetB proteins were purified, and their dehydrogenase activity was compared with that of the wild-type enzyme. The activity profile shown in Fig. 5 revealed that two mutant proteins (Q162M and P449M) were more active than the wild-type BetB immediately, suggesting that these mutant proteins have a reduced substrate inhibition. At least 10 mutant proteins showed wild-type level activity, whereas 11 mutant proteins had 20% to 70% lower activity, and 6 mutant proteins exhibited a greatly reduced activity within less than 1 order of magnitude of wild-type activity (D111A, G234T, V288D, S290T, I332S, and Y343A). Kinetic analysis revealed that 12 mutant proteins (A107N, T154A, L210F, S214K, M220L, S221T, E236A, F283Y, E335A, K339R, and M220L/S221T) retained strong inhibition by BA, whereas F231L, I332S, D111A, and P449M showed a moderate reduction in substrate inhibition, with Ki values in the range from 0.74 to 2.97 mM (see Table S1 in the supplemental material).
FIG 4.
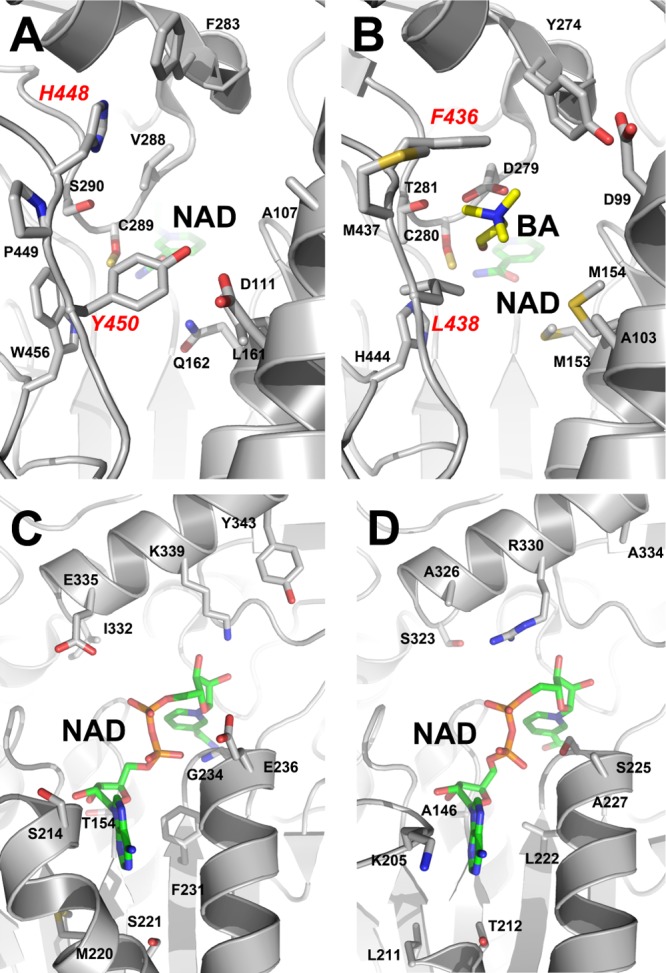
Active sites of BetB and YdcW. (A, B) Substrate binding pockets of BetB (A) and YdcW (B): a close-up view. The amino acid side chains and ligands are shown as sticks and labeled along with the protein ribbon. The residues of the double mutant protein H448F/Y450L are labeled in red. (C, D) Cofactor binding sites of BetB (C) and YdcW (D): a close-up view. The amino acid side chains and cofactors (NAD+) are shown as sticks and labeled along with the protein ribbon.
FIG 5.

Activity profiles of the wild-type and mutant BetB proteins. Dehydrogenase activity was determined in the presence of 2 mM BA, 5 mM NAD+, and 1 μg protein. Results are means ± SD from at least two independent experiments. v, velocity.
BetB variants with greatly reduced or removed substrate inhibition.
Our kinetic studies also identified several BetB mutant proteins with greatly suppressed substrate inhibition (Table 1). Both Leu161 and Gln162 are located between the substrate and cofactor binding sites, and the L161M and Q162M mutant proteins retained the wild-type activity (Table 1 and Fig. 6; see Table S1 in the supplemental material). However, whereas the L161M mutation reduced substrate inhibition only 3-fold (Ki = 0.96 mM), Q162M exhibited a greatly reduced substrate inhibition (23-fold). Interestingly, the double mutant protein L161M/Q162M also showed a greatly reduced substrate inhibition, but with lower activity (Table 1).
FIG 6.

Dehydrogenase activity of the wild-type and mutant BetB proteins as a function of BA concentration. (A) Wild-type BetB; (B) S290T; (C) Q162M; (D) G234T; (E) H448F/Y450L; (F) W456H. Reaction mixtures contained 5 mM NAD+ and 1 to 5 μg of enzyme. Results are means ± SD from at least two independent experiments. v, velocity.
The BetB residues Val288, Ser290, and Trp456 are located near the catalytic nucleophile Cys289, and the V288D, S290T, and W456H mutant proteins exhibited reduced substrate inhibition with high Ki values (86.4, 458, and 83.0 mM, respectively) (Table 1 and Fig. 6). However, these mutations dramatically lowered the catalytic activity and affinity to BA (Km = 4.76, 13.8, and 14.8 mM; kcat = 0.25, 1.63, and 3.20 s−1, respectively), retaining only 0.1%, 0.2%, and 0.3% of the wild-type catalytic efficiency (kcat/Km). In addition, Val288 and Ser290 in BetB and the homologous residues in other dehydrogenases have been proposed to be the determinants of substrate specificity for ALDHs (19). However, the replacement of these BetB residues by aspartate or threonine (as in YdcW), respectively, did not broaden its substrate specificity, and the V288D and S290T mutant proteins showed no activity against valeradehyde and 4-nitrobenzaldehyde, which are good substrates for YdcW (22).
In the active sites of BetB and YdcW, there is a short loop located close to the bound BA in YdcW and containing several nonconserved residues (Phe436, Met437, and Leu438 in YdcW and His448, Pro449, and Tyr450 in BetB) (Fig. 7). In the YdcW structure (PDB code 3FG0), the side chains of Phe436 and Leu438 are positioned close to the bound BA (2.9 to 3.4 Å) and likely contribute to substrate binding (Fig. 7C). Since this loop might also contribute to substrate binding and inhibition in BetB, we mutated the three BetB residues to the corresponding residues of YdcW. The triple BetB mutant protein H448F/P449M/Y450L showed a 27-fold decrease in kcat but exhibited a greatly reduced inhibition by BA, with the Ki value higher than 7 M (Table 1). To investigate the role of each residue in the triple mutant protein, we prepared the corresponding single mutant proteins. Both H448F and Y450L displayed a greatly reduced substrate inhibition compared to P449M, with Ki values more than 2 orders of magnitude higher, indicating that both His448 and Tyr450 play an important role in substrate inhibition of BetB (Table 1). Accordingly, the purified double mutant protein H448F/Y450L exhibited even higher resistance to BA with complete loss of substrate inhibition (up to 50 mM BA) (Table 1 and Fig. 6). In contrast, the double mutant proteins P449M/Y450L and H448F/P449M were still sensitive to inhibition by BA, with inhibition constants of 137 and 7 mM, respectively. The Y450L mutation also significantly decreased the substrate affinity of BetB, which was indicated by high Km values for BA in the BetB mutant proteins containing this mutation. Surprisingly, this mutation also lowered the affinity to NAD+, but its effect on the double and triple mutant protein appears to be complex (Table 1). The succinate semialdehyde dehydrogenase YneI from Salmonella enterica serovar Typhimurium contains Ser245 at the position of Tyr450 in BetB, and the YneI residue has been proposed to be important for substrate coordination (44). Thus, the BetB residues His448 and Tyr450 make a major contribution to substrate inhibition in this enzyme, probably through the interaction with the BA nitrogen atom.
FIG 7.

Structural basis of BetB inhibition by BA: productive and nonproductive binding of BA. Docking simulation of BA binding in the active sites of wild-type BetB (A, B), YdcW (C), and the homology model of the double mutant protein H448F/Y450L (D). Panels A, C, and D represent the productive binding mode of BA, with a hydrogen bond formed between the carbonyl group of BA and the backbone nitrogen atom of Cys289 (in BetB). In contrast, panel B presents BA in a nonproductive binding mode with similar binding affinity and a hydrogen bond formed between the carbonyl group of BA and the amide group of Asn157. The amino acid side chains and ligands are shown as sticks and labeled. The residues of the double mutant protein H448F/Y450L are labeled in a larger font.
Although the BetB residue Gly234 is located close to the pyrophosphate group of the bound NAD+, its mutation to threonine also greatly reduced the inhibition by BA (Ki = 108 mM) with a strong negative effect on activity (less than 1% of the wild-type activity) (Table 1 and Fig. 6). The replacement of Gly234 by serine (which is present in YdcW and several other ALDHs [44]) also reduced the inhibition by BA and further increased its affinity to NAD+, suggesting the formation of a new hydrogen bond between the cofactor pyrophosphate and Ser234 (Table 1). In contrast, the replacement of Gly234 by a nonpolar Ala residue significantly decreased its affinity for the nucleotide but also reduced substrate inhibition (Table 1). Interestingly, these substitutions of Gly234 did not change the BetB affinity for BA, as indicated by Km values. Thus, substrate inhibition of BetB not only is associated with the residues of the substrate binding pocket but also can be affected by the residues interacting with the cofactor, suggesting some cooperativity between these sites. It should also be noted that the BetB mutant proteins with reduced or eliminated substrate inhibition showed a significant decrease in kcat/Km. Similar effects were also demonstrated in substrate inhibition studies with other enzymes (4, 13), and in some cases they might be associated with the reduced substrate affinity of mutant proteins.
Substrate docking analysis of the BetB and YdcW active sites.
For the ordered Bi Bi reaction mechanism, substrate inhibition can be caused by the binding of excess substrate to the enzyme-NAD(P)+ complex (13, 45). Therefore, we performed a UV scanning analysis (280 to 400 nm) of BetB to detect a thio-adduct between its cysteine (Cys289 in BetB) and the pyridine ring of NAD+, which has a typical absorbance peak at 325 nm (8). However, the addition of NAD+ to BetB did not result in the increase in absorbance around 325 nm (data not shown), which is consistent with the absence of BetB inhibition by NAD+ and suggests that the BetB inhibition is not caused by the formation of a thio-adduct. The inhibition of BetB by NADH is not likely to be the result of the formation of a dead-end enzyme-BA-NADH complex, because the NAD+ saturation experiments in the presence of different BA concentrations produced no parallel plots in the 1/v versus 1/NAD+ graph (3) at high BA concentrations (data not shown).
Furthermore, our substrate docking experiments with the wild-type BetB revealed two binding modes for BA in the BetB active site with similar binding energies (Fig. 7). In the first binding mode, the carbonyl oxygen of BA is stabilized by the formation of a hydrogen bond with the main-chain NH group of the catalytic nucleophile Cys289 (1.8 Å), whereas in the second binding mode it forms a hydrogen bond with the side chain amide group of Asn157 (2.0 Å) (Fig. 7A and B). In contrast, the BA docking experiments with the YdcW structure indicated the presence of only one binding mode corresponding to the first BetB model (with an H bond to the main-chain NH of Cys280) (Fig. 7C). Since YdcW exhibits no substrate inhibition by BA, we propose calling this binding mode the productive binding mode, whereas the second type of BA coordination (with the H bond to the Asn157 side chain) is the nonproductive binding mode. The productive and nonproductive substrate binding modes have also been described for the human sulfotransferases (SULT1A, SULT1E, and SULT2A1) and for salutaridine reductase SalR from Papaver bracteatum (13, 46, 47).
In BetB, the two substrate binding modes also have different distances between the BA carbonyl carbon and the catalytic Cys289 Sγ atom, which is smaller in the productive binding mode (3.2 and 3.8 Å for wild-type BetB and YcdW, respectively) compared to the nonproductive binding (4.6 Å) (Fig. 7). It is likely that this distance is critical for the BADH catalysis because it requires a favorable position of the catalytic Cys289 Sγ atom for the nucleophilic attack of the BA carbonyl carbon. This is supported by our results with the BetB double mutant protein H448F/Y450L, which shows no substrate inhibition (Table 1) and only the productive binding mode of BA in the H448F/Y450L docking experiments with the Cys289 Sγ-to-carbonyl-C distance of 3.4 Å (Fig. 7). These results suggest that the hydrophobic interactions between the side chains of Phe448 and Leu450 in the H448F/Y450L mutant protein and methyl groups of BA might contribute to the productive substrate binding. In addition, the BA docking simulations with the BetB V288D mutant protein showed only the productive binding mode with the Cys289 Sγ-to-carbonyl-C distance of 3.4 Å and also suggested the presence of ionic interactions between the Asp288 side chain oxygens and the positively charged nitrogen group of BA (3.5 to 3.7 Å) (see Fig. S5 in the supplemental material). S290T also showed one binding mode that is the productive binding of BA (like H448F/Y450L and V288D) (see Fig. S5F in the supplemental material). Q162M had two binding modes (see Fig. S5C and D in the supplemental material) like the wild-type BetB, but the number of productive binding modes is much higher than that of nonproductive binding modes (76 versus 31; 4 modes were not in the BA binding site). All mutants with strong reduction of substrate inhibition (Q162M, V288D, S290T, and H448F/Y450L) showed consistent docking results suggesting that mutations in the BA binding site can reduce nonproductive BA binding. However, the docking simulation for G234T could not find a difference between the wild type and G234T. Rigid body docking was employed in this study, and thus, the conformations of the residues in the BA binding site of G234T were the same as those of the wild-type, resulting in the same binding modes as those of the wild type (data not shown). On the other hand, structural comparison of G234S and the wild type revealed that the G234S mutation changes the conformation of NAD in the cofactor binding site (see Fig. S6 in the supplemental material). In particular, the nicotinamide ring of G234S is moved further into the BA binding site than in the wild-type protein. This conformational change of NAD implies that substrate inhibition of BetB can also be affected by the residues interacting with the cofactor, suggesting the presence of some kind of cooperativity between the BA and NAD binding sites. Further work on the cocrystallization of the BetB mutant proteins with BA is required to prove the role of these residues in substrate inhibition of BetB.
The general concept of nonproductive substrate binding was proposed by Niemann for the explanation of the properties of chymotrypsin (48). The generality of this concept is supported by numerous studies with different enzymes, including lysozyme and carboxypeptidase (49). For lysozyme, two modes of N-acetylglucosamine binding have been distinguished using structural, binding, and kinetic approaches (49, 50). Nonproductive binding is also the mechanism used by yeast aldose reductases to distinguish between different D-aldoses resulting in a decrease in kcat for poor substrates (51). It is assumed that nonproductive complexes can form not only because of functionally important energy requirements in the productive mode but also because the flexibility of the enzyme active site allows anomalous substrate binding (49). For some enzymes with two substrates (like BetB), productive binding of one substrate may be conditional upon binding of the other substrate, resulting in an induced fit-like effect. Based on the kinetic mechanism proposed for BetB (Fig. 1B), binding of NAD+ in the cofactor-binding site may facilitate the productive binding of BA. This type of interaction between the BetB cofactor and substrate binding sites is in line with the results of our mutational studies, specifically with the reduced substrate inhibition in the G234T mutant protein (Table 1). Thus, nonproductive substrate binding can be part of the enzyme reaction mechanism, and it can have a role in enzyme inhibition or substrate specificity.
Conclusion.
Thus, our product inhibition studies have demonstrated that the betaine aldehyde dehydrogenase BetB from S. aureus follows an ordered Bi Bi mechanism, whereby NAD+ is the first substrate bound to the enzyme and NADH is the last product released by the enzyme. We have found that substrate inhibition of BetB is sensitive to mutational changes in the protein with mutations in both substrate and cofactor binding pockets contributing to the reduction in substrate inhibition. These results suggest the presence of at least two possible mechanisms underlying the reduction in substrate inhibition associated with mutations in both the BA and NAD+ binding sites. Based on the substrate docking experiments, the reduction or elimination of the inhibition by BA in the BetB mutant proteins H448F/Y450L can be explained by the reduction of the nonproductive BA binding in the active site. The second possible mechanism is based on the predicted cooperativity between the cofactor and substrate binding sites through which the mutations located in the cofactor binding site can affect the balance between the productive and unproductive modes of substrate binding. Thus, we propose that the nonproductive binding of BA in the BetB active site is the major cause of substrate inhibition of this enzyme. Glycine betaine, the product of BA dehydrogenase activity, is the most effective osmoprotectant in S. aureus and several other pathogenic bacteria, protecting them against the high-osmolarity stress prevalent in the infected tissues (52). BA dehydrogenases might be the key enzymes in the establishment and growth of the pathogen. The inhibition of BetB may be effective in counteracting infection by S. aureus, as this will lead to growth arrest due to increased osmosensitivity and accumulation of BA. Thus, the demonstration of high sensitivity of S. aureus BetB to BA in this work represents an important finding contributing to our knowledge of enzyme inhibition and to the development of potential antimicrobial therapies.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Centre for Structural Proteomics in Toronto (SPiT) for help in conducting these experiments.
This work was supported by Samsung grant GRO-2012 and by the Government of Canada through Genome Canada, the Ontario Genomics Institute, and Ontario Research Fund (2009-OGI-ABC-1405; ORF-GL2-01-004). The Center for Structural Genomics of Infectious Diseases (CSGID) project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract numbers HHSN272200700058C and HHSN272201200026C.
Footnotes
Published ahead of print 18 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00215-14.
REFERENCES
- 1.Chaplin MF, Bucke C. 1990. Enzyme technology. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 2.Reed MC, Lieb A, Nijhout HF. 2010. The biological significance of substrate inhibition: a mechanism with diverse functions. Bioessays 32:422–429. 10.1002/bies.200900167 [DOI] [PubMed] [Google Scholar]
- 3.Segel IH. 1993. Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems. John Wiley & Sons, Toronto, Ontario, Canada [Google Scholar]
- 4.Hewitt CO, Eszes CM, Sessions RB, Moreton KM, Dafforn TR, Takei J, Dempsey CE, Clarke AR, Holbrook JJ. 1999. A general method for relieving substrate inhibition in lactate dehydrogenases. Protein Eng. 12:491–496. 10.1093/protein/12.6.491 [DOI] [PubMed] [Google Scholar]
- 5.Kim CS, Ji ES, Oh DK. 2004. A new kinetic model of recombinant beta-galactosidase from Kluyveromyces lactis for both hydrolysis and transgalactosylation reactions. Biochem. Biophys. Res. Commun. 316:738–743. 10.1016/j.bbrc.2004.02.118 [DOI] [PubMed] [Google Scholar]
- 6.Bernstein LH, Grisham MB, Cole KD, Everse J. 1978. Substrate inhibition of the mitochondrial and cytoplasmic malate dehydrogenases. J. Biol. Chem. 253:8697–8701 [PubMed] [Google Scholar]
- 7.Burton RL, Chen S, Xu XL, Grant GA. 2007. A novel mechanism for substrate inhibition in Mycobacterium tuberculosis d-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 282:31517–31524. 10.1074/jbc.M704032200 [DOI] [PubMed] [Google Scholar]
- 8.Díaz-Sánchez AG, González-Segura L, Rudiño-Piñera E, Lira-Rocha A, Torres-Larios A, Muñoz-Clares RA. 2011. Novel NADPH-cysteine covalent adduct found in the active site of an aldehyde dehydrogenase. Biochem. J. 439:443–452. 10.1042/BJ20110376 [DOI] [PubMed] [Google Scholar]
- 9.Gutfreund H, Cantwell R, McMurray CH, Criddle RS, Hathaway G. 1968. Kinetics of reversible inhibition of heart lactate dehydrogenase through formation of enzyme-oxidized nicotinamide-adenine dinucleotide-pyruvate compound. Biochem. J. 106:683–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsutsumi M, Tsuse N, Fujieda N, Kano K. 2010. Site-directed mutation at residues near the catalytic site of histamine dehydrogenase from Nocardioides simplex and its effects on substrate inhibition. J. Biochem. 147:257–264. 10.1093/jb/mvp162 [DOI] [PubMed] [Google Scholar]
- 11.Vesell ES. 1965. Lactate dehydrogenase isozymes: substrate inhibition in various human tissues. Science 150:1590–1593. 10.1126/science.150.3703.1590 [DOI] [PubMed] [Google Scholar]
- 12.Winberg JO, McKinley-McKee JS. 1994. Drosophila melanogaster alcohol dehydrogenase: product-inhibition studies. Biochem. J. 301(Pt 3):901–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziegler J, Brandt W, Geissler R, Facchini PJ. 2009. Removal of substrate inhibition and increase in maximal velocity in the short chain dehydrogenase/reductase salutaridine reductase involved in morphine biosynthesis. J. Biol. Chem. 284:26758–26767. 10.1074/jbc.M109.030957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Binay B, Karaguler NG. 2007. Attempting to remove the substrate inhibition of L-lactate dehydrogenase from Bacillus stearothermophilus by site-directed mutagenesis. Appl. Biochem. Biotechnol. 141:265–272. 10.1007/BF02729067 [DOI] [PubMed] [Google Scholar]
- 15.Vander Jagt DL, Hunsaker LA, Heidrich JE. 1981. Partial purification and characterization of lactate dehydrogenase from Plasmodium falciparum. Mol. Biochem. Parasitol. 4:255–264. 10.1016/0166-6851(81)90058-X [DOI] [PubMed] [Google Scholar]
- 16.Eriksen DT, Lian J, Zhao H. 2014. Protein design for pathway engineering. J. Struct. Biol. 185:234–242. 10.1016/j.jsb.2013.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson B, Brocker C, Thompson DC, Black W, Vasiliou K, Nebert DW, Vasiliou V. 2011. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genomics 5:283–303. 10.1186/1479-7364-5-4-283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchitti SA, Brocker C, Stagos D, Vasiliou V. 2008. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 4:697–720. 10.1517/17425255.4.6.697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riveros-Rosas H, González-Segura L, Julián-Sánchez A, Díaz-Sánchez AG, Muñoz-Clares RA. 2013. Structural determinants of substrate specificity in aldehyde dehydrogenases. Chem. Biol. Interact. 202:51–61. 10.1016/j.cbi.2012.11.015 [DOI] [PubMed] [Google Scholar]
- 20.Díaz-Sánchez Á, González-Segura L, Mújica-Jiménez C, Rudiño-Piñera E, Montiel C, Martínez-Castilla LP, Muñoz-Clares RA. 2012. Amino acid residues critical for the specificity for betaine aldehyde of the plant ALDH10 isoenzyme involved in the synthesis of glycine betaine. Plant Physiol. 158:1570–1582. 10.1104/pp.112.194514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Figueroa-Soto CG, Valenzuela-Soto EM. 2000. Kinetic study of porcine kidney betaine aldehyde dehydrogenase. Biochem. Biophys. Res. Commun. 269:596–603. 10.1006/bbrc.2000.2337 [DOI] [PubMed] [Google Scholar]
- 22.Gruez A, Roig-Zamboni V, Grisel S, Salomoni A, Valencia C, Campanacci V, Tegoni M, Cambillau C. 2004. Crystal structure and kinetics identify Escherichia coli YdcW gene product as a medium-chain aldehyde dehydrogenase. J. Mol. Biol. 343:29–41. 10.1016/j.jmb.2004.08.030 [DOI] [PubMed] [Google Scholar]
- 23.Johansson K, El-Ahmad M, Ramaswamy S, Hjelmqvist L, HJörnvall Eklund H. 1998. Structure of betaine aldehyde dehydrogenase at 2.1 Å resolution. Protein Sci. 7:2106–2117. 10.1002/pro.5560071007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valenzuela-Soto EM, Muñoz-Clares RA. 1993. Betaine-aldehyde dehydrogenase from leaves of Amaranthus hypochondriacus L. exhibits an iso ordered Bi Bi steady state mechanism. J. Biol. Chem. 268:23818–23823 [PubMed] [Google Scholar]
- 25.Velasco-García R, Mújica-Jiménez C, Mendoza-Hernández G, Muñoz-Clares RA. 1999. Rapid purification and properties of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa. J. Bacteriol. 181:1292–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burg MB, Ferraris JD. 2008. Intracellular organic osmolytes: function and regulation. J. Biol. Chem. 283:7309–7313. 10.1074/jbc.R700042200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Perez A, Burg MB. 1991. Renal medullary organic osmolytes. Physiol. Rev. 71:1081–1115 [DOI] [PubMed] [Google Scholar]
- 28.Smith LT, Pocard JA, Bernard T, Le Rudulier D. 1988. Osmotic control of glycine betaine biosynthesis and degradation in Rhizobium meliloti. J. Bacteriol. 170:3142–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Velasco-García R, Villalobos MA, Ramírez-Romero MA, Mújica-Jiménez C, Iturriaga G, Muñoz-Clares RA. 2006. Betaine aldehyde dehydrogenase from Pseudomonas aeruginosa: cloning, over-expression in Escherichia coli, and regulation by choline and salt. Arch. Microbiol. 185:14–22. 10.1007/s00203-005-0054-8 [DOI] [PubMed] [Google Scholar]
- 30.Lee JJ, Kim JH, Kim DG, Kim DH, Simborio HL, Min WG, Rhee MH, Lim JH, Chang HH, Kim S. 2014. Characterization of betaine aldehyde dehydrogenase (BetB) as an essential virulence factor of Brucella abortus. Vet. Microbiol. 168:131–140. 10.1016/j.vetmic.2013.10.007 [DOI] [PubMed] [Google Scholar]
- 31.Tylichová M, Kopecný D, Moréra S, Briozzo P, Lenobel R, Snégaroff J, Sebela M. 2010. Structural and functional characterization of plant aminoaldehyde dehydrogenase from Pisum sativum with a broad specificity for natural and synthetic aminoaldehydes. J. Mol. Biol. 396:870–882. 10.1016/j.jmb.2009.12.015 [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Segura L, Rudino-Pinera E, Munoz-Clares RA, Horjales E. 2009. The crystal structure of a ternary complex of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa provides new insight into the reaction mechanism and shows a novel binding mode of the 2′-phosphate of NADP+ and a novel cation binding site. J. Mol. Biol. 385:542–557. 10.1016/j.jmb.2008.10.082 [DOI] [PubMed] [Google Scholar]
- 33.Muñoz-Clares RA, Díaz-Sánchez AG, González-Segura L, Montiel C. 2010. Kinetic and structural features of betaine aldehyde dehydrogenases: mechanistic and regulatory implications. Arch. Biochem. Biophys. 493:71–81. 10.1016/j.abb.2009.09.006 [DOI] [PubMed] [Google Scholar]
- 34.Perez-Miller SJ, Hurley TD. 2003. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry 42:7100–7109. 10.1021/bi034182w [DOI] [PubMed] [Google Scholar]
- 35.Velasco-García R, González-Segura L, Muñoz-Clares RA. 2000. Steady-state kinetic mechanism of the NADP+- and NAD+-dependent reactions catalysed by betaine aldehyde dehydrogenase from Pseudomonas aeruginosa. Biochem. J. 352(Pt 3):675–683. 10.1042/0264-6021:3520675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez CF, Proudfoot M, Brown G, Korniyenko Y, Mori H, Savchenko AV, Yakunin AF. 2006. Molecular basis of formaldehyde detoxification. Characterization of two S-formylglutathione hydrolases from Escherichia coli, FrmB and YeiG. J. Biol. Chem. 281:14514–14522. 10.1074/jbc.M600996200 [DOI] [PubMed] [Google Scholar]
- 37.Krivov GG, Shapovalov MV, Dunbrack RL. 2009. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77:778–795. 10.1002/prot.22488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. 2009. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30:2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samsonova NN, Smirnov SV, Novikova AE, Ptitsyn LR. 2005. Identification of Escherichia coli K12 YdcW protein as a γ-aminobutyraldehyde dehydrogenase. FEBS Lett. 579:4107–4112. 10.1016/j.febslet.2005.06.038 [DOI] [PubMed] [Google Scholar]
- 40.Chern MK, Pietruszko R. 1995. Human aldehyde dehydrogenase E3 isozyme is a betaine aldehyde dehydrogenase. Biochem. Biophys. Res. Commun. 213:561–568. 10.1006/bbrc.1995.2168 [DOI] [PubMed] [Google Scholar]
- 41.Oishi H, Ebina M. 2005. Isolation of cDNA and enzymatic properties of betaine aldehyde dehydrogenase from Zoysia tenuifolia. J. Plant Physiol. 162:1077–1086. 10.1016/j.jplph.2005.01.020 [DOI] [PubMed] [Google Scholar]
- 42.Peddie BA, Wong-She J, Randall K, Lever M, Chambers ST. 1998. Osmoprotective properties and accumulation of betaine analogues by Staphylococcus aureus. FEMS Microbiol. Lett. 160:25–30. 10.1111/j.1574-6968.1998.tb12885.x [DOI] [PubMed] [Google Scholar]
- 43.Pourkomailian B, Booth IR. 1992. Glycine betaine transport by Staphylococcus aureus: evidence for two transport systems and for their possible roles in osmoregulation. J. Gen. Microbiol. 138:2515–2518. 10.1099/00221287-138-12-2515 [DOI] [PubMed] [Google Scholar]
- 44.Zheng H, Beliavsky A, Tchigvintsev A, Brunzelle JS, Brown G, Flick R, Evdokimova E, Wawrzak Z, Mahadevan R, Anderson WF, Savchenko A, Yakunin AF. 2013. Structure and activity of the NAD(P)+-dependent succinate semialdehyde dehydrogenase YneI from Salmonella typhimurium. Proteins 81:1031–1041. 10.1002/prot.24227 [DOI] [PubMed] [Google Scholar]
- 45.Gamage NU, Duggleby RG, Barnett AC, Tresillian M, Latham CF, Liyou NE, McManus ME, Martin JL. 2003. Structure of a human carcinogen-converting enzyme, SULT1A1: structural and kinetic implications of substrate inhibition. J. Biol. Chem. 278:7655–7662. 10.1074/jbc.M207246200 [DOI] [PubMed] [Google Scholar]
- 46.Gamage NU, Tsvetanov S, Duggleby RG, McManus ME, Martin JL. 2005. The structure of human SULT1A1 crystallized with estradiol: an insight into active site plasticity and substrate inhibition with multi-ring substrates. J. Biol. Chem. 280:41482–41486. 10.1074/jbc.M508289200 [DOI] [PubMed] [Google Scholar]
- 47.Gulcan HO, Duffel MW. 2011. Substrate inhibition in human hydroxysteroid sulfotransferase SULT2A1: studies on the formation of catalytically non-productive enzyme complexes. Arch. Biochem. Biophys. 507:232–240. 10.1016/j.abb.2010.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niemann C. 1964. Alpha-chymotrypsin and the nature of enzyme catalysis. Science 143:1287–1296. 10.1126/science.143.3612.1287 [DOI] [PubMed] [Google Scholar]
- 49.Hess GP, Rupley JA. 1971. Structure and function of proteins. Annu. Rev. Biochem. 40:1013–1044. 10.1146/annurev.bi.40.070171.005053 [DOI] [PubMed] [Google Scholar]
- 50.Blake CC, Mair GA, North AC, Phillips DC, Sarma VR. 1967. On the conformation of the hen egg-white lysozyme molecule. Proc. R. Soc. Lond. B Biol. Sci. 167:365–377. 10.1098/rspb.1967.0034 [DOI] [PubMed] [Google Scholar]
- 51.Nidetzky B, Mayr P, Hadwiger P, Stutz AE. 1999. Binding energy and specificity in the catalytic mechanism of yeast aldose reductases. Biochem. J. 344(Pt 1):101–107. 10.1042/0264-6021:3440101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Graham JE, Wilkinson BJ. 1992. Staphylococcus aureus osmoregulation: roles for choline, glycine betaine, proline, and taurine. J. Bacteriol. 174:2711–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.