

Abstract
Here, we report the genome of one gammaproteobacterial member of the gut microbiota, for which we propose the name “Candidatus Schmidhempelia bombi,” that was inadvertently sequenced alongside the genome of its host, the bumble bee, Bombus impatiens. This symbiont is a member of the recently described bacterial order Orbales, which has been collected from the guts of diverse insect species; however, “Ca. Schmidhempelia” has been identified exclusively with bumble bees. Metabolic reconstruction reveals that “Ca. Schmidhempelia” lacks many genes for a functioning NADH dehydrogenase I, all genes for the high-oxygen cytochrome o, and most genes in the tricarboxylic acid (TCA) cycle. “Ca. Schmidhempelia” has retained NADH dehydrogenase II, the low-oxygen specific cytochrome bd, anaerobic nitrate respiration, mixed-acid fermentation pathways, and citrate fermentation, which may be important for survival in low-oxygen or anaerobic environments found in the bee hindgut. Additionally, a type 6 secretion system, a Flp pilus, and many antibiotic/multidrug transporters suggest complex interactions with its host and other gut commensals or pathogens. This genome has signatures of reduction (2.0 megabase pairs) and rearrangement, as previously observed for genomes of host-associated bacteria. A survey of wild and laboratory B. impatiens revealed that “Ca. Schmidhempelia” is present in 90% of individuals and, therefore, may provide benefits to its host.
INTRODUCTION
Autochthonous gut microorganisms greatly influence animal health by providing a range of nutritional, developmental, and protective benefits (e.g., energy, vitamins, immune priming, detoxification, and pathogen exclusion) to their hosts (1–3). Highly consistent gut-associated microbes are common among eusocial insect species (4–8). These microbes can be actively passed between generations through trophallaxis (mouth-to-mouth or anus-to-mouth food sharing) or passively transmitted via a fecal-oral route due to communal living (9, 10). Beneficial gut bacteria are often selectively transmitted between generations and form well-established interactions with their hosts (11); however, many of the eusocial-insect-associated microbes have unknown relationships with their hosts. Genome sequencing can provide insight into the metabolic capabilities and biological significance of these host-associated microbes.
Guts of bumble bee (Bombus sp.) are commonly inhabited by two bacterial species that are closely related to the honey bee (Apis mellifera)-associated lineages of Gilliamella apicola (previously called the “Gamma-1” phylotype, Gammaproteobacteria) and Snodgrassella alvi (previously called the “Beta” phylotype, Betaproteobacteria) (6, 12–14). Metagenomic sequencing of the A. mellifera gut bacteria revealed that Gilliamella and Snodgrassella contain genes that may contribute to pollen digestion, detoxification of mannose, and defense against pathogens (15). The closely related gut microbiota of Bombus terrestris was experimentally determined to provide an extended-immune phenotype against the trypanosomatid gut parasite Crithidia bombi, yet the mechanism of this defense was not identified (16).
The Bombus impatiens genome sequencing project recovered the genome sequence of a gammaproteobacterium related to the Gilliamella apicola clade. This genome sequence provides insights into the phylogenetic relationships and lifestyle of Gilliamella-related bumble bee symbionts, as well as clues about the role of this gammaproteobacterium in B. impatiens biology. Here, we describe the first genome sequence from the gammaproteobacterial order Orbales and compare it to the metagenome recovered from the A. mellifera microbiota to identify basic metabolic and ecological attributes and potential effects that this symbiont may have on its host.
MATERIALS AND METHODS
Source DNA, sequencing, identification and assembly of bacterial reads.
A single adult male Bombus impatiens approximately at 24 h posteclosion was collected from a colony purchased from Biobest Biological Systems (Leamington, Ontario, Canada). DNA was extracted from the entire specimen using a standard phenol-chloroform preparation.
Three paired-end libraries were constructed with fragment sizes of 400, 4,000, and 8,000 bp. DNA was sequenced using Illumina GAIIx sequencing technology to generate eight lanes of 125-bp raw sequences. After error correction, the average read length was 105 bp, and the total number of reads used in assembled contigs was 150,442,748. Based on an estimated genome size of 250 megabases (Mb), this yields approximately 65-fold coverage of the B. impatiens genome. The reads were assembled using the CABOG assembler (17) modified to handle short Illumina reads. The draft assembly contained 69,944 contigs (including symbiont contigs) with a length greater than 100 bp, of which 6,658 contigs were longer than 10,000 bp. In addition, the assembly contained 1,086,650 degenerate contigs, primarily small repetitive sequences or contigs with very low quality. Another 60,355,858 reads remained as unassembled singletons.
To scan the 69,944 contigs for sequences of possible bacterial symbionts, we initially aligned them to the genomes of four strains of Wolbachia, which we considered the first candidates because Wolbachia bacteria are the most ubiquitous endosymbionts of invertebrates. We aligned all assembled contigs against genomes of each of these four strains using the promer program from the MUMmer package (18), which translates both the reference and the query sequences to amino acids in all six reading frames. This allowed a more sensitive alignment than a DNA-based approach. We identified multiple contigs that had strong homology to at least one of the Wolbachia species, indicating that bacterial sequences might be present in the whole-genome data. We extracted these contigs and used the BLASTX program to map them against the complete nonredundant protein database at NCBI in order to find bacteria that were closer to the symbiont in B. impatiens. The best hits from this mapping were to three Gammaproteobacteria species: Photorhabdus asymbiotica, Yersinia enterocolitica, and Proteus mirabilis, indicating that the bacterial sequences were from Enterobacteriales (Gammaproteobacteria) and not Wolbachia (Alphaproteobacteria).
We then aligned all assembled contigs and all singleton reads to the complete genomes of each of these three bacteria. We ran DNA alignments and translated protein alignments using the nucmer and promer programs from the MUMmer package and mapped all contigs against both the DNA and the protein sequences of the three bacterial genomes. The more sensitive protein-based alignments were compared to the annotated coordinates of the proteins in each of the bacterial genomes. Each contig that contained at least one complete protein was considered possibly bacterial. In addition, contigs not longer than 500 bp that contained at least a partial match at greater than 60% identity to any bacterial protein were also considered bacterial. This analysis identified 367 regular contigs, 1,129 degenerate contigs, and 255,589 singleton reads as possibly bacterial in origin. Next, we used BLASTX to search each of these contigs and reads against the entire NCBI protein database, and we eliminated any contig with a better match to a eukaryotic species than to a bacterial genome. This left 343 regular contigs, 941 degenerate contigs, and 255,589 singleton reads as likely bacterial sequences.
We used the CABOG assembler's raw output files to locate all reads used to build these bacterial contigs and extracted these reads from the original sequence files with their paired-end mates. This resulted in 615,185 mate pairs from the 400-bp insert size library, 20,716 mate pairs from the 4-lb insert size library, 8,164 mate pairs from the 9-kb insert size library, and 121,568 unpaired reads. These reads were assembled de novo with the CABOG assembler. The final bacterial assembly contained 1,998,543 bp in just 79 contigs. The largest contig contained 110,984 bp, and the assembly had an N50 size of 39,885 bp. The 79 contigs were combined into 33 scaffolds spanning 2,004,741 bp, with a scaffold N50 size of 98,624 bp and a maximum scaffold of 204,248 bp. The approximate average coverage of the genome is 37-fold.
Annotation of the bacterial genes.
Gene annotation was completed in the automated Integrated Microbial Genomes Expert Review (IMG/ER; Joint Genome Institute) pipeline (19). Protein-coding sequences and RNA-coding genes were predicted within its framework using Prodigal and tRNAS-can-1.23 (19). Functional annotations were assigned to genes based on protein domain characterization according to clusters of orthologous groups of proteins (COG) terms, Pfam, TIGRfam, InterPro domains, Gene Ontology (GO) terms, and KEGG Orthology (KO) terms with metabolic pathway maps. Additional manual assessment with KEGG (20), EcoCyc (21), and the MetaCyc Pathological program (22) was performed to check pathway completeness. Genes identified as missing from main pathways (e.g., tricarboxylic acid [TCA] cycle or NADH dehydrogenase I) were manually investigated using BLASTP searches against the B. impatiens symbiont genome with corresponding genes from Escherichia coli. A metabolic map was manually created for the B. impatiens gammaproteobacterium (BiG). The annotated contigs for this genome are available at the IMG/ER website (https://img.jgi.doe.gov/cgi-bin/er/main.cgi) (proposed name, “Candidatus Schmidhempelia bombi Bimp”).
Core gene phylogeny and metagenome gene phylogenies.
A set of 89 single-copy orthologous (SiCO) genes was selected from an original set of 203 consistently present, single copy, non-horizontally transferred core genes (23); this set was used to reconstruct the phylogenetic placement of BiG. SiCO genes were selected from 28 gammaproteobacterial genomes using SiCO gene lists in MaGE (24) or using a cutoff of a bit-score ratio of >0.30 in a BLASTP search with the 203 SiCO genes from E. coli (23). Inferred protein sequences of the 89 genes were individually aligned in MUSCLE (25) and concatenated together. The Gblocks (26) program was used to remove poorly aligned regions, and the resulting alignment consisted of 27,452 amino acid sites. Maximum-likelihood reconstruction was performed with RAxML (27) on 100 bootstrap replicates using the PROTGAMMA algorithm and the WAG substitution matrix, which were selected with ProtTest, version 2.4 (28). Individual SiCO gene trees were built with the same methods as the multiprotein data set and subsequently sorted with PhyloSort (29).
Gene content of the BiG genome was compared to the gene content of the metagenome of the A. mellifera gut microbiota, which was determined in a previous study (15). The comparison was made to the taxonomic bins from that study including the all-bacteria bin and the gammaproteobacteria (Gamma) bin using the COG (clusters of orthologous genes) annotations from IMG/ER (Joint Genome Institute). Of the 195 SiCO genes present in BiG, a set of 193 was identified as being shared between BiG and the Gamma bin using BLASTP to identify pairwise protein sequence identities (SiCOs identified with a bit-score ratio of >0.30 to the set found in Lerat et al. [23]). The subset of 89 SiCO genes used to construct the multiprotein phylogeny was amended with corresponding SiCO genes from the Gamma bin, and individual gene trees were constructed using previously described methods. The phylogenetic relationships between the Gamma bin sequences, which contained genes from both G. apicola and Frischella perrara, and the BiG were collected for each tree.
16S rRNA gene phylogeny.
The 16S rRNA gene sequence was used to reconstruct and refine the phylogenetic relationships of the BiG among a representative set of G. apicola, F. perrara (collected from the genera Apis and Bombus), and closely related sequences from the NCBI database. One of the four 16S rRNA gene copies in the BiG genome was selected and aligned with the 16S rRNA data set using Infernal (30). A maximum-likelihood phylogeny was constructed with RAxML using the GTRCAT parameter and 100 bootstrap replicates (27).
Putative HGT.
The IMG annotation pipeline identified putative horizontally transferred genes. Further analysis of potential horizontal gene transfer (HGT) was assayed with a phylogenetic pipeline modified from Moustafa and Bhattacharya (29). Briefly, the pipeline identifies closely related genes in the NCBI database, aligns the amino acid sequences, and constructs phylogenies for each. PhyloSort (29) was used to find trees which indicated horizontal transfer from Firmicutes to BiG. The vast majority of genes from the BiG genome have best hits to Gammaproteobacteria. The potential HGT genes are the outstanding examples having best hits to and clustering in a phylogeny with Firmicutes. These potential HGT genes come from many different contigs, most of which are fairly long and are otherwise composed of genes with top hits to Gammaproteobacteria. Because the phylogenetically near neighbors to BiG have not been densely sampled, these computational findings may reflect sampling bias rather than true horizontal gene transfer, and further work will be needed to validate these findings.
PCR screen for BiG among B. impatiens individuals.
Specific primers BombusG2f (5′-CTGGTCGTCTGGAGTATTGT-3′) and BombusG2r1 (5′-AGGTCCGCTCACCATCGCTG-3′) were used to search for BiG within B. impatiens individuals or gut organs from wild and laboratory individuals. Cycling was performed with an annealing temperature of 54°C for 35 cycles and a 1-min extension.
Nucleotide sequence accession number.
The bacterial symbiont genome assembly was deposited in the GenBank database under accession number AWGA00000000.
RESULTS AND DISCUSSION
Retrieval of a nearly complete genome of a B. impatiens symbiont.
Sequencing of the B. impatiens genome resulted in the by-product sequencing of a gammaproteobacterial genome corresponding to an organism present in the bee from which DNA was extracted. From the ∼250 Mb of assembled sequence from the project, 79 contigs representing 2 Mb were assigned to the bacterial genome (see Materials and Methods). Contigs that represent the gammaproteobacterial genome harbor at least one open reading frame (ORF) per contig and have an N50 length of 39.9 kb. To determine the completeness of the genome and the number of distinct bacterial genomes present, coverage of a preselected set of 203 single-copy, near-universal bacterial genes was assessed using BLASTP. We identified 195 of these 203 genes (96%), each with exactly one copy indicating the complete or nearly complete coverage of a single bacterial genome. Calculation of G+C content of the contigs produced a unimodal distribution with a mean of 36.6%, and the average depth of coverage (37-fold) was consistent across the contigs, providing further evidence for the retrieval of a single bacterial genome. Here, we refer to this organism as the B. impatiens gammaproteobacterium, or BiG.
The BiG genome is at least 1.99 Mb in size (Table 1). A total of 50 tRNA genes and 23 tRNA synthetase genes were identified, corresponding to all 20 amino acids. Altogether, 1,694 protein-coding genes were identified from the assembly, with 14% (236) of them having unknown functions. Roughly 72% had functions predicted as clusters of orthologous groups (COGs) (31). The largest COG categories represented were translation (9.7%), general function only (9.3%), amino acid transport and metabolism (8.6%), cell wall biogenesis (7.6%), replication (6.8%), coenzyme transport and metabolism (5.8%), and carbohydrate transport and metabolism (5.3%).
TABLE 1.
General features of the BiG genome
| Parametera | Value for the parameter |
|---|---|
| Chromosome length (bp) | >1,999,325 |
| Extrachromosomal elements | Presence unknown |
| GC content (%) | 36.6 |
| Total no. of predicted CDSs | 1,770 |
| Coding density (%) | 82 |
| Average CDS length (bp) | 954 |
| No. of rRNA operons | |
| 5S rRNA | 7 |
| 16S rRNA | 4 |
| 23S rRNA | 5 |
| No. of tRNAs | 50 |
CDS, coding sequence.
The small size and low G+C content resemble genomes of previously sequenced host-dependent commensals and pathogens (e.g., “Candidatus Hamiltonella defensa” [32] or Histophilus somnus [33]). Further, gut bacteria with strict host associations (e.g., Helicobacter sp., Lactobacillus reuteri, and Pasteurellales species) often have small genomes (34, 35), suggesting that BiG may have a restricted host distribution. Overall, the genome lacks large regions of chromosomal synteny with other genomes, even for regions conserved among many species of Enterobacteriales and Pasteurellales (see Fig. S1 and S2 in the supplemental material for examples). However, contigs from the A. mellifera Gamma bin harbor regions with strikingly similar gene orders, even with interrupted operons for TCA cycle enzymes (sucABCD; only sucCD are retained) and the NADH dehydrogenase I complex (nuoA-nuoN; only nuoJ-nuoN are retained) (see Fig. S1 and S2). The conserved synteny between BiG and sequences from the gut microbiota of A. mellifera confirms the presence of similar bacteria in both bee species and substantiates the robust assembly of the BiG contigs.
The majority of BiG protein-coding genes (98%, 1,659/1,694) were shared with the Gamma bin of the A. mellifera gut microbiota metagenome. Notably, genes for the four subunits of the respiratory nitrate reductase A are present in BiG but absent from the Gamma bin (see Table S1 in the supplemental material).
Consistent association of BiG with laboratory-raised and wild Bombus impatiens individuals.
The BiG genome sequence was retrieved from a single male individual from a laboratory-raised B. impatiens colony. To understand the consistency of the association between BiG and B. impatiens, we undertook a PCR survey of workers, males, and queens from natural and laboratory environments. Specific primers that differentiated between BiG and the closely related Gilliamella revealed that BiG is nearly ubiquitous (90%, 18/20) among laboratory adults and larvae and wild adults (see Fig. S3 in the supplemental material). The bacterium was detected in all gut organs (crop, midgut, ileum, and rectum), as well as from the leg DNA extraction. The leg sample may have been surface contaminated with feces because the source colonies lacked locations to remove excrement, possibly increasing fecal contamination within the hive relative to normal conditions.
The presence of this bacterium in both wild and laboratory-raised B. impatiens bees implies that BiG is passed from one colony to the next, as documented for members of the closely related genus Gilliamella in honey bees and bumble bees (14, 16). Queens were shown to harbor this bacterium, thus affording a transmission link between the annual disintegration of Bombus colonies in the fall and the founding of new colonies in the spring. Overall, BiG is a common associate of B. impatiens and potentially many other bumble bee species.
BiG is a relative of Gilliamella apicola in the newly described order Orbales.
Our 16S rRNA gene tree placed BiG as a member of Orbales, a bacterial order previously recovered from numerous honey bee and bumble bee species (13). BiG clusters among sequences collected from native and commercially reared bumble bee species from around the world (Fig. 1b; see also Fig. S4 in the supplemental material). A survey of the bacterial associates of bumble bees (36) shows that BiG clusters within a separate clade from the genus Gilliamella (found in honey bees and bumble bees [13]), Frischella perrara (Gamma-2 of honey bees [37]), and other Orbales species, with strong bootstrap support (95%) (Fig. 1b; see also Fig. S4). The BiG sequences were identified in geographically and phylogenetically diverse bumble bees, but their 16S rRNA sequences are very similar (>98% identical). Therefore, BiG is a member of a geographically widespread bacterial clade that is strictly associated with bumble bees, based on surveys conducted to date.
FIG 1.

(a) Phylogenetic placement of BiG as a singleton clade among five orders of Gammaproteobacteria. Maximum-likelihood reconstruction inferred from 89 concatenated SiCO genes (27,452 aligned amino acid sites). (b) Location of BiG among members of the insect gut-associated Orbales. The tree is based on maximum likelihood with the 16S rRNA gene. (c) Proportions of the 89 individual SiCO gene trees that returned each phylogenetic pattern with their average (Avg) bootstrap support at the indicated node and average gene length. (d) Proportions of individual SiCO gene trees that united the BiG and the Apis mellifera Gamma bin (Gbin) gene copies as a monophyletic clade and average bootstrap support. Asterisks represent bootstrap support values of 100, and values below 50 are not shown. Str, strain; SS, secondary symbiont.
Our concatenated, multiprotein phylogeny retrieved high bootstrap support for previously established evolutionary relationships between the bacterial orders of Gammaproteobacteria included in the analysis (Enterobacteriales, Pasteurellales, Vibrionales, Alteromonadales, and Pseudomonadales) (23, 38, 39) (Fig. 1a). In this tree, BiG is sister to Enterobacteriales, which supports the previous placement of Orbales (Fig. 1a) (13). Trees created with individual SiCO genes varied in their support, mainly due to their differing sequence complexities (46 to 1,407 amino acids in length) (Fig. 1c). The majority (61/89) of individual SiCO genes resulted in a tree topology uniting BiG, Enterobacteriales, and Pasteurellales. Nearly three-fourths of those trees (44/61) placed BiG sister to, or within, the Enterobacteriales, with an average bootstrap support of >80% (Fig. 1c).
To identify the relationship between BiG and the sequences from the Gamma bin of the A. mellifera metagenome, which included sequences from the related genera Gilliamella and Frischella, the corresponding Gamma bin SiCO sequences were analyzed with each of the 89 SiCO genes. Many strains of Gilliamella and Frischellaare present within the metagenomic data set for the A. mellifera microbiota, and the number of copies corresponding to the 89 BiG SiCOs varies among the loci. The majority (92%, 81/89) of SiCOs retrieves the Gamma bin sequences and the BiG sequence as a clade (Fig. 1d). The BiG gene copy usually branches basal to the Gamma bin sequences (69%, 61/81), with a high average bootstrap support (90%) for this placement (Fig. 1d), which adds further support that BiG represents a new genus of Orbales and not a member of Gilliamella or Frischella. This data set of protein-coding genes supports the sister relationship between the Orbales (i.e., BiG and the Gamma bin clade) and Enterobacteriales, with high support (71% average bootstrap value; 36/57 trees) for this pattern, rather than Pasteurellales (50% average bootstrap value; 21/57 trees).
Pairwise protein identities of homologs in the A. mellifera metagenome reveal considerable strain variation within the Gamma bin. By collecting only the top hit in the Gamma bin of the A. mellifera metagenome for each of the 193 BiG SiCO genes, we find that BiG has a lower average protein identity to copies within the Gamma bin than these copies have to one another (Fig. 2) (15). Thus, the BiG genome is considerably more divergent than the entire community of gammaproteobacterial strains within the A. mellifera microbiota, suggesting that BiG be considered a separate genus of Orbales.
FIG 2.
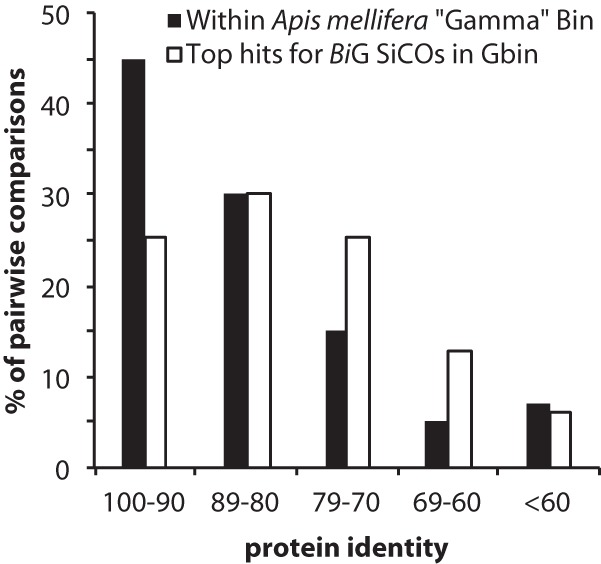
Pairwise comparisons between copies of SiCO genes separated by protein identity determined with BLASTP. Comparisons within the Apis mellifera Gamma bin show protein identities between 80 and 100% (15), whereas identities between the BiG genome SiCOs and the Gamma bin (Gbin) are between 70 and 90%.
All phylogenies and the shared synteny of BiG with contigs in the Gamma bin support placement of BiG within the newly described order Orbales, close to Gilliamella (13). The 16S rRNA of the BiG group sequences have an average distance of 5% or greater to Gilliamella, Frischella, and other Orbales, indicating sufficient divergence to be ranked as a separate genus.
Metabolic reconstruction of BiG and comparisons to the A. mellifera gut microbiota.
The BiG genome contains the majority of genes in predicted metabolic pathways for glycolysis, gluconeogenesis, the full pentose phosphate pathway, nucleotide metabolism, lipopolysaccharide production, peptidoglycan fabrication, heme and siroheme, and ubiquinone assembly (Fig. 3). Biosynthetic capabilities remain intact for the majority of the 20 protein amino acids and many cofactors. However, individual genes are missing for several pathways, including the final step in biosynthesis of the branched-chain amino acids (Ile, Val, and Leu), conversion of Ser to Gly, and synthesis of Pro (Fig. 3). Insects, including B. impatiens, encode enzymes for these reactions, and BiG contains transporters for these amino acids, suggesting that these products may be imported from the host.
FIG 3.

Metabolic reconstruction of BiG. Dots next to each connecting line (arrows) represent genes involved with that pathway; filled dots represent intact genes that are present, and open dots represent missing genes. Amino acids are in solid boxes, and vitamins and cofactors are in dashed boxes; ovals represent membrane transporters, with their putative targets listed next to them outside the cell. T1SS, type 1 secretion system; T5SS, type 5 secretion system; T6SS, type 6 secretion system; TAT, twin-arginine translocation pathway; SEC, SEC system; NDHI, NADH dehydrogenase 1; ATPase, F-type ATPase; NDHII, NADH dehydrogenase 2; NAR, respiratory nitrate reductase A; FDN, formate dehydrogenase N; cyt bd, cytochrome bd complex; cyt o, cytochrome o complex; FRD, fumarate reductase.
Numerous genes underlying the pathways in aerobic energy metabolism that are conserved in many Enterobacteriales and Pasteurellales are not encoded in the BiG genome (Fig. 3). Incomplete pathways include NADH dehydrogenase I (missing nuoABCDEFGHI) and cytochrome o (missing cyoABCDE) (for synteny evidence, see Fig. S2 in the supplemental material). Of the 16 core genes that normally encode enzymes underlying the TCA cycle, BiG is missing 14: gltA, acnAB, icd, sucAB, sdhABCD, fumABC, and mdh. BiG has retained only the succinyl-coenzyme A (CoA) synthetase genes sucC and sucD (for synteny evidence, see Fig. S1).
In contrast, the BiG genome possesses several low-oxygen and anaerobic mechanisms for energy production. It has kept the NADH dehydrogenase II (ndh) and cytochrome bd (cydAB), which have optimal conditions at low oxygen levels and have been shown to be critical for colonization of the hypoxic mouse gut by Escherichia coli (40, 41). In E. coli this cytochrome has its maximal level of expression and optimal conditions at low oxygen concentrations; additionally cytochrome bd reinforces the hypoxic environment by scavenging the remaining oxygen.
Additionally, BiG encodes genes for anaerobic respiration. Together the periplasmic respiratory nitrate reductase A (narGHI) and the nitrate-induced formate dehydrogenase N (fdnGHI) can produce a respiratory chain, resulting in a proton motive force and cytoplasmic nitrite (42, 43). The NADH-dependent nitrite reductase (nirBD) detoxifies resulting nitrite to ammonia (44). Notably, the narGHI genes are not present in the A. mellifera gut microbiota metagenome Gamma bin, which further suggests that this Gilliamella-like bacterium is a divergent lineage among this group of common bee associates (see Table S1 in the supplemental material). However, the absence of the narGHI genes in the A. mellifera metagenome could reflect incomplete sampling of the gammaproteobacterial genomes.
BiG has retained genes for citrate fermentation and for several branches of mixed acid fermentation. A Na/citrate symporter is adjacent to the complete citrate fermentation gene locus (citABCDEFGX), encoding capabilities for transporting citrate into the cell, sensing citrate in the environment, and degrading citrate to oxaloacetate and acetate via citrate lyase. Mixed-acid fermentation genes are present for lactate fermentation (ldhA), pyruvate cleavage to formate and acetyl-CoA (pflB), ATP generation via acetate formation (pta and ackA), and NAD+ generation through ethanol production (adhE and adhP). Fermentation results in the excreted metabolites of ethanol, formate, and the short-chain fatty acids acetate and lactate. Short-chain fatty acids are the bulk of carbon and energy sources of ruminant animals (45). This raises the possibility that the bacterium is providing its host a nutritional benefit through biosynthesis of needed compounds (46). However, biosynthetic contributions (e.g., amino acids or vitamins) may not be very significant because the bee diet is composed of both easily accessible mono- and disaccharides and protein-rich pollen (47).
A primary constituent of the bee diet is pollen, which presents plant macromolecules that are difficult to degrade and that form barriers surrounding the pollen germ, the primary source of protein for bees. Some strains of G. apicola from A. mellifera encode pectate lyase and are able to degrade pectin, a function that may aid their host in nutrient acquisition by releasing pollen contents (15). This capability is not encoded within the BiG genome; however, BiG has transporters (bglF) and beta-glucosidases (bglA, glucoside hydrolase family 4) (48) that may confer the ability to import and metabolize some of the products of cellulase activity (e.g., cellobiose and cellotriose) potentially produced by other bacteria present in the gut or present in nectar or pollen. Metabolic scavenging of these compounds could provide the majority of energy for this organism since glucose and fructose (the major sugars in honey) are absorbed rapidly in the midgut and are not abundant in the hindgut (49, 50).
The pattern of missing genes implies that BiG may inhabit a low-oxygen niche within the bumble bee gut, which is consistent with the cultivation conditions for related members of Orbales, both microaerophilic (Gilliamella apicola) and anaerobic (Frischella perrara) (13, 37). Recent in situ analysis of the A. mellifera microbiota showed that the majority of the bacteria (including G. apicola) reside within the hindgut (14). Oxygen levels could govern colonization of the gut organs (i.e., anaerobic hindgut and aerobic foregut), restricting members of the Orbales to low-oxygen or anaerobic environments. Fluorescent in situ microscopy of the Bombus microbiota and microsensor surveys (i.e., O2) of the Apis and Bombus gut are needed to test this hypothesis and determine the breadth of this pattern in corbiculate bees (51).
Candidate horizontal gene transfers from Firmicutes to BiG.
A total of 54 genes were identified by IMG as putatively horizontally transferred from Firmicutes to BiG. Of these genes, PhyloSort supported horizontal transfer from Firmicutes for 39 genes (Table 2). Because BiG is a novel genus, phylogenetic placement of its genes may not be as reliable as with a more thoroughly sequenced clade, and these 39 genes should be considered candidates worthy of further scrutiny rather than confirmed horizontal gene transfers. Closer analysis of potentially transferred genes identified several sugar uptake and degradation genes, including the previously mentioned bglA (Table 2). These genes may enable BiG to utilize the numerous sugars found in nectar that cannot be metabolized by B. impatiens or that are abundant in the gut. An intact operon for the uptake of mannose (phosphotransferase system [PTS]) may have been transferred from a species related to Bacillus (Table 2). The mannose PTS has been shown to have an extensive history of horizontal transfer in bacteria and is mostly found in bacteria associated with animal guts (52). Mannose is toxic to honey bees and bumble bees; therefore, microbial assimilation of this sugar could protect the host from small amounts of mannose, which is often present in nectar (53, 54). Alternatively, these transport systems can often act on a broad range of substrates (52). Evidence that mannose PTSs may be linked to the bee gut environment also comes from the finding that they are overrepresented in the A. mellifera gut metagenome relative to other gut metagenomes (15); however, this overrepresentation may merely reflect the taxonomic composition of the bacterial community.
TABLE 2.
Genes putatively horizontally transferred into the BiG genome from species of Firmicutes
| IMG gene object IDa | IMG locus tag | Gene product name | Length (aa) | Scaffold IMG accession no. | Scaffold length (bp) | Contig GC% | COG | Genome source |
|---|---|---|---|---|---|---|---|---|
| 2505924216 | GBi_0012.00000200 | Arabinose efflux permease | 405 | GBi_ctg820001289386 | 38219 | 0.39 | COG2814 | Staphylococcus carnosus subsp. carnosus TM300 |
| 2505924217 | GBi_0012.00000210 | Uridine phosphorylase (EC 2.4.2.3) | 251 | GBi_ctg820001289386 | 38219 | 0.39 | COG2820 | Lactobacillus rhamnosus Lc 705 |
| 2505924690 | GBi_0029.00000900 | Peptide methionine sulfoxide reductase (EC 1.8.4.11, EC 1.8.4.12, EC 1.8.4.11) | 177 | GBi_ctg820001289403 | 102,764 | 0.35 | COG0225 | Listeria grayi DSM 20601 |
| 2505924734 | GBi_0036.00000050 | Nitrous oxide-stimulated promoter. | 112 | GBi_ctg820001289410 | 14,474 | 0.35 | Bacillus coagulans 2-6 | |
| 2505924830 | GBi_0041.00000130 | Uncharacterized protein conserved in bacteria | 124 | GBi_ctg820001289416 | 38,071 | 0.35 | COG4687 | Enterococcus faecium E980 |
| 2505924831 | GBi_0041.00000140 | PTS, mannose/fructose/N-acetylgalactosamine-specific component IID | 302 | GBi_ctg820001289416 | 38,071 | 0.35 | COG3716 | Enterococcus sp. strain 7L76 |
| 2505924832 | GBi_0041.00000150 | PTS, mannose/fructose/N-acetylgalactosamine-specific component IIC | 267 | GBi_ctg820001289416 | 38,071 | 0.35 | COG3715 | Bacillus thuringiensis subsp. israelensis ATCC 35646 |
| 2505924833 | GBi_0041.00000160 | PTS, mannose/fructose/N-acetylgalactosamine-specific component IIB (EC 2.7.1.69) | 323 | GBi_ctg820001289416 | 38,071 | 0.35 | COG3444 | Bacillus thuringiensis IBL200 |
| 2505924834 | GBi_0041.00000170 | Uncharacterized protein conserved in bacteria | 244 | GBi_ctg820001289416 | 38,071 | 0.35 | COG1636 | Enterococcus italicus DSM 15952 |
| 2505924868 | GBi_0043.00000060 | Predicted hydrolases of the HAD superfamily | 257 | GBi_ctg820001289418 | 10,510 | 0.37 | COG0561 | Listeria welshimeri serovar 6b, SLCC5334 |
| 2505924928 | GBi_0048.00000120 | Adenine-specific DNA methylase Mod (EC 2.1.1.72) | 628 | GBi_ctg820001289423 | 59,508 | 0.38 | COG2189 | Clostridium cf. saccharolyticum K10 |
| 2505924929 | GBi_0048.00000130 | Restriction endonuclease (EC 3.1.21.5) | 1,035 | GBi_ctg820001289423 | 59,508 | 0.38 | COG3587 | Clostridium cf. saccharolyticum K10 |
| 2505924938 | GBi_0048.00000220 | Pyrimidine-nucleoside phosphorylase (EC 2.4.2.2) | 433 | GBi_ctg820001289423 | 59,508 | 0.38 | COG0213 | Bacillus atrophaeus 1942 |
| 2505924940 | GBi_0048.00000240 | Nucleoside permease | 398 | GBi_ctg820001289423 | 59,508 | 0.38 | COG1972 | Bacillus pumilus SAFR-032 |
| 2505925004 | GBi_0051.00000020 | Predicted membrane protein | 173 | GBi_ctg820001289427 | 39,885 | 0.37 | COG4905 | Eubacterium cylindroides T2–87 |
| 2505925044 | GBi_0052.00000060 | Putative glucose uptake permease | 291 | GBi_ctg820001289428 | 34,555 | 0.36 | COG4975 | Listeria welshimeri serovar 6b, SLCC5334 |
| 2505925059 | GBi_0052.00000200 | Type I site-specific restriction-modification system, R (restriction) subunit and related helicases (EC 3.1.21.3) | 1,026 | GBi_ctg820001289428 | 34,555 | 0.36 | COG0610 | Bacillus coagulans 36D1 |
| 2505925060 | GBi_0052.00000210 | Restriction endonuclease S subunits (EC 3.1.21.3) | 417 | GBi_ctg820001289428 | 34,555 | 0.36 | COG0732 | Enterococcus italicus DSM 15952 |
| 2505925061 | GBi_0052.00000220 | Type I restriction-modification system methyltransferase subunit (EC 2.1.1.72) | 859 | GBi_ctg820001289428 | 34,555 | 0.36 | COG0286 | Bacillus coagulans 36D1 |
| 2505925076 | GBi_0054.00000020 | Chorismate mutase (EC 5.4.99.5) | 88 | GBi_ctg820001289430 | 20,279 | 0.36 | COG1605 | Anaerofustis stercorihominis DSM 17244 |
| 2505925077 | GBi_0054.00000030 | Hypothetical protein | 77 | GBi_ctg820001289430 | 20,279 | 0.36 | Desulfitobacterium hafniense DCB-2 | |
| 2505925164 | GBi_0059.00000040 | Transposase | 44 | GBi_ctg820001289435 | 37,254 | 0.37 | Staphylococcus aureus subsp. aureus ST398 | |
| 2505925221 | GBi_0060.00000290 | Predicted branched-chain amino acid permease (azaleucine resistance) | 234 | GBi_ctg820001289436 | 77,226 | 0.37 | COG1296 | Listeria welshimeri serovar 6b, SLCC5334 |
| 2505925222 | GBi_0060.00000300 | Predicted membrane protein | 107 | GBi_ctg820001289436 | 77,226 | 0.37 | COG4392 | Listeria welshimeri serovar 6b, SLCC5334 |
| 2505925262 | GBi_0061.00000100 | PTS beta-glucoside-specific IIA component, Glc family (TC 4.A.1.2.5) | 631 | GBi_ctg820001289437 | 27,350 | 0.37 | COG1263 | Bacillus subtilis subsp. spizizenii W23 |
| 2505925263 | GBi_0061.00000110 | Beta-glucosidase/6-phospho-beta-glucosidase/beta-galactosidase (EC 3.2.1.86) | 478 | GBi_ctg820001289437 | 27,350 | 0.37 | COG2723 | Clostridium bartlettii DSM 16795 |
| 2505925412 | GBi_0068.00000040 | Transcriptional regulator | 307 | GBi_ctg820001289444 | 65,738 | 0.37 | COG0583 | Syntrophomonas wolfei subsp. wolfei strain Goettingen, DSM 2245B |
| 2505925413 | GBi_0068.00000050 | Methionine synthase (B12-independent) (EC 2.1.1.14) | 761 | GBi_ctg820001289444 | 65,738 | 0.37 | COG0620 | Paenibacillus curdlanolyticus YK9 |
| 2505925414 | GBi_0068.00000060 | Tagatose-1,6-bisphosphate aldolase (EC 4.1.2.40) | 345 | GBi_ctg820001289444 | 65,738 | 0.37 | COG3684 | Enterococcus gallinarum EG2 |
| 2505925484 | GBi_0069.00000240 | Fructose-2,6-bisphosphatase (EC 5.4.2.1) | 253 | GBi_ctg820001289445 | 47,248 | 0.37 | COG0406 | Listeriaceae bacterium TTU M1-001 |
| 2505925526 | GBi_0070.00000200 | Amidases related to nicotinamidase | 182 | GBi_ctg820001289446 | 64,926 | 0.34 | COG1335 | Geobacillus sp. strain WCH70 |
| 2505925638 | GBi_0072.00000210 | Serine/threonine exchange transporter, LAT family (TC 2.A.3.8.12) | 485 | GBi_ctg820001289448 | 31,379 | 0.36 | COG0531 | Bacillus cereus subsp. cytotoxis NVH 391-98 |
| 2505925734 | GBi_0076.00000160 | Predicted membrane protein | 221 | GBi_ctg820001289453 | 45,947 | 0.35 | COG2323 | Leptotrichia goodfellowii F024 |
| 2505925735 | GBi_0076.00000170 | Acetyltransferases (EC 2.3.1.-) | 150 | GBi_ctg820001289453 | 45,947 | 0.35 | COG0456 | Desulfitobacterium dehalogenans JW/IU-DC1, ATCC 51507 |
| 2505925751 | GBi_0076.00000330 | Predicted hydrolases of the HAD superfamily | 279 | GBi_ctg820001289453 | 45,947 | 0.35 | COG0561 | Listeria seeligeri serovar 1/2b, SLCC3954 |
| 2505925796 | GBi_0078.00000400 | Predicted HD superfamily hydrolase | 222 | GBi_ctg820001289456 | 51,284 | 0.37 | COG1418 | Pediococcus pentosaceus ATCC 25745 |
| 2505925870 | GBi_0082.00000030 | Predicted glutamine amidotransferase involved in pyridoxine biosynthesis | 197 | GBi_ctg820001289460 | 6,516 | 0.39 | COG0311 | Bacillus subtilis subsp. subtilis strain 168 |
| 2505925871 | GBi_0082.00000040 | Pyridoxine biosynthesis enzyme | 295 | GBi_ctg820001289460 | 6,516 | 0.39 | COG0214 | Bacillus subtilis subsp. subtilis strain 168 |
| 2505925872 | GBi_0082.00000050 | Transcriptional regulator, GntR family | 467 | GBi_ctg820001289460 | 6,516 | 0.39 | COG1167 | Listeria monocytogenes 08-5923 |
ID, identifier.
Potential interactions with the host and other gut microorganisms.
Several putative host interaction factors were present in the BiG genome, including Sel1 repeat proteins, bacterial Ig-like domains, and bacterial α2-macroglobulins; these could be critical for recognition of this bacterial strain by the host epithelium (55). A full Flp pilus gene set was present; this apparatus is known to be critical for adhesion and biofilm formation (56). Strains of Gilliamella associated with the honey bee have been shown to form thick biofilms within the ileum (14), and genes involved in biofilm formation/adhesion were overrepresented in the A. mellifera Gamma bin (15). More generally, adhesion to the gut wall may play a critical role for insect gut associates because, unlike mammals, insects do not secrete a mucus layer that facilitates microbial residence (57).
Similar to the A. mellifera Gamma bin of the metagenome, BiG has a marked abundance of antibiotic/multidrug resistance transporters, including several ABC, drug metabolic transporter (DMT), multidrug/oligosaccharidyl-lipid/polysaccharide (MOP), membrane fusion protein (MFP), Eam/Emr, and arabinose efflux pumps. As mentioned in Engel et al. (15), the bee gut is exposed to plant defense compounds during pollen/nectar foraging and ingestion. These compounds could be a selective force for mechanisms enabling efficient elimination of these toxins in bee gut bacteria.
Complex interactions with the host epithelium are additionally supported by the presence of multiple secretion systems (types I, V, and VI) (Fig. 3). The recently described type VI secretion system (T6SS) evolved from viral tail fibers into a syringe-like effector delivery mechanism that is present in many bacteria and is becoming understood as pivotal to interactions among bacteria and between bacteria and eukaryotes (58–61). The cell-puncturing device (valine-glycine repeat protein G [VgrG] gene) of the T6SS, is critical for discriminating and attaching to target cells (62). The BiG genome contains three intact VgrGs and 13 additional VgrG fragments (see Table S2 in the supplemental material), which may correspond to between 12 and 16 full VgrGs, a relatively large number for a genome of this size (63). These VgrG elements cannot be annotated with target specificity or function, but some have genes adjacent to the C terminus that may be T6SS effectors that interact with the host and other microbes present in the gut (64, 65) (see Table S2). For example, asymptomatic colonization of the mouse gut by Helicobacter hepaticus was disrupted when its T6SS was knocked out, leading to overcolonization by H. hepaticus and an inflammatory reaction by the host (66). This example suggests that the H. hepaticus T6SS facilitates signaling between the bacterium and the host or that the bacterium inoculates the gut epithelium with anti-inflammatory compounds. T6SS effectors also have lytic effects (67) that can be delivered to specific unicellular eukaryotic or bacterial targets, depending upon the VgrG utilized (62, 68). Thus, further investigation of the BiG T6SS and its biological effects on the host bumble bee, gut pathogens, and members of the indigenous microbiota may demonstrate context-dependent interactions.
The bumble bee hive is a resource-rich environment that has a number of autochthonous bacterial species but also attracts a variety of pathogens, many of which are specific to the gut environment (69). Koch et al. (16) showed that protection from pathogens is provided by the resident microbiota in Bombus terrestris. The BiG genome provides candidate mechanisms for protection against antagonistic microorganisms invading the gut, and future efforts to cultivate BiG would facilitate direct tests of its effect on bumble bee biology.
Conclusions.
As a member of the Orbales, the BiG genome represents a recently described order of Gammaproteobacteria that is found in honey bees and bumble bees and that has been repeatedly collected from other insects (6, 12, 13, 36, 70). Our analyses show that the BiG genome, sequenced concurrently with the genome of B. impatiens, is nearly complete, as indicated by the unimodal G+C content, the presence of a complete tRNA synthetase complement, exactly one copy of 96% of a defined set of single-copy genes, and a consistent coverage of 37-fold across all contigs. Previous genome sequencing projects have been shown to produce data sets corresponding to symbiotic microorganisms (71); however, most of these symbionts do not assemble as well as the BiG genome retrieved in our study (79 contigs), indicating the presence of a clonal or near-clonal bacterial population in the source DNA sample. The loss of central metabolism components (i.e., TCA cycle) and the lack of synteny with other bacterial genomes suggest that this genome and genomes of closely related organisms (Orbales) underwent rearrangement, reduction, and specialization to the host environment similar to the processes observed in other symbiotic genomes (72, 73).
“Candidatus Schmidhempelia bombi.”
We propose the name “Candidatus Schmidhempelia bombi” for the bacterium identified within the bumble bee Bombus impatiens and several other bumble bee species (36). Phylogenetic reconstruction places this bacterium within the Orbales, a recently described family of Gammaproteobacteria that has been identified nearly exclusively within insect guts. “Ca. Schmidhempelia” is a distinct clade (95% bootstrap support) of Orbales that is separate from the named genera Gilliamella and Frischellathat are symbiotic in honey bees and bumble bees, as well as from other non-bee-associated members of the Orbales (Fig. 1b; see also Fig. S4 in the supplemental material). This bacterium has an average of 5% 16S rRNA gene sequence divergence from Gilliamella apicola sequences and has been identified in several bumble bee species from around the world, yet it has not been found in the thoroughly surveyed honey bee microbiota (36). The proposed genus for “Ca. Schmidhempelia bombi” refers to the Swiss evolutionary parasitologist Paul Schmid-Hempel, who has studied the evolutionary ecology of bumble bee species and associated organisms, while the specific epithet reflects that this bacterium resides within bumble bees. Unique features of this organism include its apparent restriction to bumble bee hosts and the 16S rRNA gene sequence 5′-TTTAAAACTGGTCGTCTGGAGTATTGT-3′ (positions 636 to 662 of the 16S rRNA gene, with E. coli numbering).
Supplementary Material
ACKNOWLEDGMENTS
We thank Ellen O. Martinson for her helpful discussion and comments on the manuscript and Jennifer H. Wisecaver for bioinformatics assistance.
V.G.M. was supported by the Center for Insect Science (University of Arizona) and a National Science Foundation award to N.A.M. (NSF 1046153). Sequencing and analysis were supported by NIH Director's Pioneer Award 1DP1OD006416-01 (G. E. Robinson) and by NIH grant R01-HG006677 (S.L.S.). H.K. was supported by Swiss National Science Foundation grants 140157 and 147881.
Footnotes
Published ahead of print 18 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00322-14.
REFERENCES
- 1.Dillon RJ, Dillon VM. 2004. The gut bacteria of insects: nonpathogenic interactions. Annu. Rev. Entomol. 49:71–92. 10.1146/annurev.ento.49.061802.123416 [DOI] [PubMed] [Google Scholar]
- 2.Shin SC, Kim SH, You H, Kim B, Kim AC, Lee KA, Yoon JH, Ryu JH, Lee WJ. 2011. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334:670–674. 10.1126/science.1212782 [DOI] [PubMed] [Google Scholar]
- 3.Stecher B, Hardt WD. 2011. Mechanisms controlling pathogen colonization of the gut. Curr. Opin. Microbiol. 14:82–91. 10.1016/j.mib.2010.10.003 [DOI] [PubMed] [Google Scholar]
- 4.Dietrich C, Köhler T, Brune A. 2014. The cockroach origin of the termite gut microbiota: patterns in bacterial community structure reflect major evolutionary events. Appl. Environ. Microbiol. 80:2261–2269. 10.1128/AEM.04206-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funaro CF, Kronauer DJC, Moreau CS, Goldman-Huertas B, Pierce NE, Russell JA. 2011. Army ants harbor a host-specific clade of Entomoplasmatales bacteria. Appl. Environ. Microbiol. 77:346–350. 10.1128/AEM.01896-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinson VG, Danforth BN, Minckley RL, Rueppell O, Tingek S, Moran NA. 2011. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol. Ecol. 20:619–628. 10.1111/j.1365-294X.2010.04959.x [DOI] [PubMed] [Google Scholar]
- 7.Noda S, Kitade O, Inoue T, Kawai M, Kanuka M, Hiroshima K, Hongoh Y, Constantino R, Uys V, Zhong J, Kudo T, Ohkuma M. 2007. Cospeciation in the triplex symbiosis of termite gut protists (Pseudotrichonympha spp.), their hosts, and their bacterial endosymbionts. Mol. Ecol. 16:1257–1266. 10.1111/j.1365-294X.2006.03219.x [DOI] [PubMed] [Google Scholar]
- 8.Russell JA, Moreau CS, Goldman-Huertas B, Fujiwara M, Lohman DJ, Pierce NE. 2009. Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc. Natl. Acad. Sci. U. S. A. 106:21236–21241. 10.1073/pnas.0907926106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lombardo MP. 2008. Access to mutualistic endosymbiotic microbes: an underappreciated benefit of group living. Behav. Ecol. Sociobiol. 62:479–497. 10.1007/s00265-007-0428-9 [DOI] [Google Scholar]
- 10.Weiss MR. 2006. Defecation behavior and ecology of insects. Annu. Rev. Entomol. 51:635–661. 10.1146/annurev.ento.49.061802.123212 [DOI] [PubMed] [Google Scholar]
- 11.Bright M, Bulgheresi S. 2010. A complex journey: transmission of microbial symbionts. Nat. Rev. Microbiol. 8:218–230. 10.1038/nrmicro2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koch H, Schmid-Hempel P. 2011. Bacterial communities in central European bumblebees: low diversity and high specificity. Microb. Ecol. 62:121–133. 10.1007/s00248-011-9854-3 [DOI] [PubMed] [Google Scholar]
- 13.Kwong WK, Moran NA. 2012. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: Snodgrassella alvi gen. nov., sp. nov., a member of the Neisseriaceae family of the Betaproteobacteria; and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the Enterobacteriales order of the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 63:2008–2018. 10.1099/ijs.0.044875-0 [DOI] [PubMed] [Google Scholar]
- 14.Martinson VG, Moy J, Moran NA. 2012. Establishment of characteristic gut bacteria during development of the honeybee worker. Appl. Environ. Microbiol. 78:2830–2840. 10.1128/AEM.07810-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engel P, Martinson VG, Moran NA. 2012. Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. U. S. A. 109:11002–11007. 10.1073/pnas.1202970109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koch H, Schmid-Hempel P. 2011. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc. Natl. Acad. Sci. U. S. A. 108:19288–19292. 10.1073/pnas.1110474108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G. 2008. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24:2818–2824. 10.1093/bioinformatics/btn548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5:R12. 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40:D115–D122. 10.1093/nar/gkr1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. 2008. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36:D480–D484. 10.1093/nar/gkm882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keseler IM, Bonavides-Martinez C, Collado-Vides J, Gama-Castro S, Gunsalus RP, Johnson DA, Krummenacker M, Nolan LM, Paley S, Paulsen IT, Peralta-Gil M, Santos-Zavaleta A, Shearer AG, Karp PD. 2009. EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Res. 37:D464–D470. 10.1093/nar/gkn751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F, Kaipa P, Karthikeyan AS, Kothari A, Krummenacker M, Latendresse M, Mueller LA, Paley S, Popescu L, Pujar A, Shearer AG, Zhang P, Karp PD. 2010. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 38:D473–D479. 10.1093/nar/gkp875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lerat E, Daubin V, Moran NA. 2003. From gene trees to organismal phylogeny in prokaryotes: the case of the gamma-proteobacteria. PLoS Biol. 7:e19. 10.1371/journal.pbio.1000019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, Lajus A, Pascal G, Scarpelli C, Medigue C. 2006. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res. 34:53–65. 10.1093/nar/gkj406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56:564–577. 10.1080/10635150701472164 [DOI] [PubMed] [Google Scholar]
- 27.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. 10.1093/bioinformatics/btl446 [DOI] [PubMed] [Google Scholar]
- 28.Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105. 10.1093/bioinformatics/bti263 [DOI] [PubMed] [Google Scholar]
- 29.Moustafa A, Bhattacharya D. 2008. PhyloSort: a user-friendly phylogenetic sorting tool and its application to estimating the cyanobacterial contribution to the nuclear genome of Chlamydomonas. BMC Evol. Biol. 8:6. 10.1186/1471-2148-8-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nawrocki EP, Kolbe DL, Eddy SR. 2009. Infernal 1.0: inference of RNA alignments. Bioinformatics 25:1335–1337. 10.1093/bioinformatics/btp157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. 10.1186/1471-2105-4-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Degnan PH, Yu Y, Sisneros N, Wing RA, Moran NA. 2009. Hamiltonella defensa, genome evolution of protective bacterial endosymbiont from pathogenic ancestors. Proc. Natl. Acad. Sci. U. S. A. 106:9063–9068. 10.1073/pnas.0900194106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siddaramappa S, Challacombe JF, Duncan AJ, Gillaspy AF, Carson M, Gipson J, Orvis J, Zaitshick J, Barnes G, Bruce D, Chertkov O, Detter JC, Han CS, Tapia R, Thompson LS, Dyer DW, Inzama TJ. 2011. Horizontal gene transfer in Histophilus somni and its role in the evolution of pathogenic strain 2336, as determined by comparative genomic analyses. BMC Genomics 12:570. 10.1186/1471-2164-12-570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walter J, Britton RA, Roos S. 2011. Host-microbial symbiosis in the vertebrate gastrointestinal tract and the Lactobacillus reuteri paradigm. Proc. Natl. Acad. Sci. U. S. A. 108:4645–4652. 10.1073/pnas.1000099107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walter J, Ley RE. 2011. The human gut microbiome: ecology and recent evolutionary changes. Annu. Rev. Microbiol. 65:411–429. 10.1146/annurev-micro-090110-102830 [DOI] [PubMed] [Google Scholar]
- 36.Koch H, Abrol DP, Li J, Schmid-Hempel P. 2013. Diversity and evolutionary patterns of bacterial gut associates of corbiculate bees. Mol. Ecol. 22:2028–2044. 10.1111/mec.12209 [DOI] [PubMed] [Google Scholar]
- 37.Engel P, Kwong WK, Moran NA. 2013. Frischella perrara gen. nov., sp. nov., a gammaproteobacterium isolated from the gut of the honey bee, Apis mellifera. Int. J. Syst. Evol. Microbiol. 63:3646–3651. 10.1099/ijs.0.049569-0 [DOI] [PubMed] [Google Scholar]
- 38.Gao B, Mohan R, Gupta RS. 2009. Phylogenomics and protein signatures elucidating the evolutionary relationships among the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 59:234–247. 10.1099/ijs.0.002741-0 [DOI] [PubMed] [Google Scholar]
- 39.Williams KP, Gillespie JJ, Sobral BWS, Nordberg EK, Snyder EE, Shallom JM, Dickerman AW. 2010. Phylogeny of Gammaproteobacteria. J. Bacteriol. 192:2305–2314. 10.1128/JB.01480-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones SA, Chowdhury FZ, Fabich AJ, Anderson A, Schreiner DM, House AL, Autieri SM, Leatham MP, Lins JJ, Jorgensen M, Cohen PS, Conway T. 2007. Respiration of Escherichia coli in the mouse intestine. Infect. Immun. 75:4891–4899. 10.1128/IAI.00484-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones SA, Gibson T, Maltby RC, Chowdhury FZ, Stewart V, Cohen PS, Conway T. 2011. Anaerobic respiration of Escherichia coli in the mouse intestine. Infect. Immun. 79:4218–4226. 10.1128/IAI.05395-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones RW. 1980. Proton translocation by the membrane-bound fomate dehydrogenase of Escherichia coli. FEMS Microbiol. Lett. 8:167–171. 10.1111/j.1574-6968.1980.tb05072.x [DOI] [Google Scholar]
- 43.Jormakka M, Tornroth S, Byrne B, Iwata S. 2002. Molecular basis of proton motive force generation: structure of formate dehydrogenase-N. Science 295:1863–1868. 10.1126/science.1068186 [DOI] [PubMed] [Google Scholar]
- 44.Cole JA, Brown CM. 1980. Nitrite reduction to ammonia by fermentative bacteria: short circuit in the biological nitrogen cycle. FEMS Microbiol. Lett. 7:65–72. 10.1111/j.1574-6941.1980.tb01578.x [DOI] [Google Scholar]
- 45.Dehority BA. 1997. Foregut fermentation, p 39–83 In Mackie RI, White BA, Isaacson RE. (ed), Gastrointestinal microbiology, vol 1 Chapman & Hall, New York, NY [Google Scholar]
- 46.Breznak JA, Kane MD. 1990. Microbial H2/CO2 acetogenesis in animal guts—nature and nutritional significance. FEMS Microbiol. Rev. 7:309–313. 10.1111/j.1574-6941.1990.tb01698.x [DOI] [PubMed] [Google Scholar]
- 47.Kane MD. 1997. Microbial fermentation in insect guts, p 231–268 In Mackie RI, White BA, Isaacson RE. (ed), Gastrointestinal microbiology, vol 2 Chapman & Hall, New York, NY [Google Scholar]
- 48.Grabnitz F, Seiss M, Rucknagel KP, Staudenbauer WL. 1991. Structure of the beta-glucosidase gene bglA of Clostridium thermocellum. Eur. J. Biochem. 200:301–309. 10.1111/j.1432-1033.1991.tb16186.x [DOI] [PubMed] [Google Scholar]
- 49.Crailsheim K. 1988. Intestinal transport of sugars in the honeybee (Apis mellifera). J. Insect Physiol. 34:839–845. 10.1016/0022-1910(88)90117-5 [DOI] [Google Scholar]
- 50.Crailsheim K. 1988. Regulation of food passage in the intestine of the honeybee (Apis mellifera). J. Insect Physiol. 34:85–90. 10.1016/0022-1910(88)90158-8 [DOI] [Google Scholar]
- 51.Schramm A. 2006. Microsensors for the study of microenvironments and processes in the intestine of invertebrates, p 463–473 In Konig H, Varma A. (ed), Intestinal microorganisms of soil invertebrates, vol 6 Springer-Verlag, Berlin, Germany [Google Scholar]
- 52.Zúñiga M, Comas I, Linaje R, Monedero V, Yebra MJ, Esteban CD, Deutscher J, Pérez-Martinez G, González-Candelas F. 2005. Horizontal gene transfer in the molecular evolution of mannose PTS transporters. Mol. Biol. Evol. 22:1673–1685. 10.1093/molbev/msi163 [DOI] [PubMed] [Google Scholar]
- 53.Barker RJ, Lehner Y. 1974. Influence of diet on sugars found by thin-layer chromatography in thoraces of honey bees, Apis mellifera L. J. Exp. Zool. 188:157–164. 10.1002/jez.1401880204 [DOI] [PubMed] [Google Scholar]
- 54.Pawlikowski T. 2010. Pollination activity of bees (Apoidea: Apiformes) visiting the flowers of Tilia cordata Mill. and Tilia tomentosa Moench in an urban environment. J. Apic. Sci. 54:73–79 [Google Scholar]
- 55.Budd A, Blandin S, Levashina EA, Gibson TJ. 2004. Bacterial α2-macroglobulins: colonization factors acquired by horizontal gene transfer from the metazoan genome? Genome Biol. 5:R38. 10.1186/gb-2004-5-6-r38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomich M, Planet PJ, Figurski DH. 2007. The tad locus: postcards from the widespread colonization island. Nat. Rev. Microbiol. 5:363–375. 10.1038/nrmicro1636 [DOI] [PubMed] [Google Scholar]
- 57.Bignell DE, Oskarsson H, Anderson JM. 1980. Specialization of the hindgut wall for the attachment of symbiotic microorganisms in a termite Procubitermes aburiensis (Isoptera, Termitidae, Termitinae). Zoomorphology 96:103–112. 10.1007/BF00310080 [DOI] [Google Scholar]
- 58.Leiman PG, Basler M, Ramagopal UA, Bonanno JB, Sauder JM, Pukatzki S, Burley SK, Almo SC, Mekalanos JJ. 2009. Type VI secretion apparatus and phage tail-associated protein complexes share a common evolutionary origin. Proc. Natl. Acad. Sci. U. S. A. 106:4154–4159. 10.1073/pnas.0813360106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, Goodman AL, Joachimiak G, Ordonez CL, Lory S, Walz T, Joachimiak A, Mekalanos JJ. 2006. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 312:1526–1530. 10.1126/science.1128393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc. Natl. Acad. Sci. U. S. A. 103:1528–1533. 10.1073/pnas.0510322103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwarz S, Hood RD, Mougous JD. 2010. What is type VI secretion doing in all those bugs? Trends Microbiol. 18:531-537. 10.1016/j.tim.2010.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zheng J, Ho B, Mekalanos JJ. 2011. Genetic analysis of anti-amoebae and anti-bacterial activities of the type VI secretion system in Vibrio cholerae. PLoS One 6:e23876. 10.1371/journal.pone.0023876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pukatzki S, McAuley SB, Miyata ST. 2009. The type VI secretion system: translocation of effectors and effector-domains. Curr. Opin. Microbiol. 12:11–17. 10.1016/j.mib.2008.11.010 [DOI] [PubMed] [Google Scholar]
- 64.Jani AJ, Cotter PA. 2010. Type VI secretion: not just for pathogenesis anymore. Cell Host Microbe 8:2–6. 10.1016/j.chom.2010.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murdoch SL, Trunk K, English G, Fritsch MJ, Pourkarimi E, Coulthurst SJ. 2011. The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J. Bacteriol. 193:6057–6069. 10.1128/JB.05671-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chow J, Lee SM, Shen Y, Khosravi A, Mazmanian SK. 2010. Host-bacterial symbiosis in health and disease. Adv. Immunol. 107:243–274. 10.1016/B978-0-12-381300-8.00008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD. 2011. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475:343–347. 10.1038/nature10244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hood RD, Singh P, Hsu FS, Guvener T, Carl MA, Trinidad RRS, Silverman JM, Ohlson BB, Hicks KG, Plemel RL, Li M, Schwarz S, Wang WY, Merz AJ, Goodlett DR, Mougous JD. 2010. A type VI secretion system of Pseudomonas aeruginosa targets, a toxin to bacteria. Cell Host Microbe 7:25–37. 10.1016/j.chom.2009.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schmid-Hempel P. 1998. Parasites in social insects. Princeton University Press, Princeton, NJ [Google Scholar]
- 70.Chandler JA, Lang JM, Bhatnagar S, Eisen JA, Kopp A. 2011. Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLoS Genet. 7:e1002272. 10.1371/journal.pgen.1002272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salzberg SL, Hotopp JCD, Delcher AL, Pop M, Smith DR, Eisen MB, Nelson WC. 2005. Serendipitous discovery of Wolbachia genomes in multiple Drosophila species. Genome Biol. 6:402. 10.1186/gb-2005-6-7-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burke GR, Moran NA. 2011. Massive genomic decay in Serratia symbiotica, a recently evolved symbiont of aphids. Genome Biol. Evol. 3:195–208. 10.1093/gbe/evr002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nikoh N, Hosokawa T, Oshima K, Hattori M, Fukatsu T. 2011. Reductive evolution of bacterial genome in insect gut environment. Genome Biol. Evol. 3:702–714. 10.1093/gbe/evr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.