

Abstract
Conditional gene silencing by RNA interference in Trypanosoma brucei can be inconclusive if knockdowns are inefficient or have off-target effects. To enable efficient, specific silencing of single-copy genes in mammalian-infective, bloodstream form trypanosomes, we developed a system that targets the heterologous and functional Trypanosoma cruzi U2AF35 3′ untranslated region (UTR) (Tc3) or, alternatively, the sequence of the PTP tag, which can be fused to any mRNA of interest. Two cell lines were created, single-marker Tc3 (smTc3) and smPTP, which conditionally express Tc3 and PTP double-stranded RNA (dsRNA), respectively. The system depends on manipulating both alleles of the gene of interest so that cells exclusively express the target mRNA as a fusion to one of these heterologous sequences. We generated allele integration vectors in which the C-terminal part of a gene's coding sequence can be fused to either heterologous sequence in a single cloning step. We first tested this system with CITFA7, which encodes a well-characterized subunit of the class I transcription factor A (CITFA), an essential factor for transcription initiation by RNA polymerase I. Targeting either Tc3 or PTP fused to the CITFA7 mRNA resulted in gene knockdowns that were as efficient and specific as targeting the endogenous CITFA7 mRNA. Moreover, application of this system to CITFA1, which could not be silenced by established methods, demonstrated that the gene encodes an essential CITFA subunit that mediates binding of the transcription factor complex to RNA polymerase I promoters.
INTRODUCTION
Among kinetoplastid organisms, the tsetse-borne lethal human parasite Trypanosoma brucei allows specific gene silencing through the RNA interference (RNAi) pathway (1, 2). This system is based on strong, conditional expression of an ∼500-bp-long double-stranded RNA (dsRNA) that targets a gene's mRNA and is available for both the insect-stage procyclic form (PF) and the mammalian-infective bloodstream form (BF) of the parasite (3). Given that direct transfection of dsRNA into cells is transient in nature and does not reach all cells in a sample (1), conditional dsRNA expression from genome-integrated vectors is the system of choice for gene-silencing experiments in trypanosomes. The first step in developing such a system was the generation of trypanosomes expressing the bacterial tetracycline (Tet) repressor (TetR) that controlled tetracycline-inducible promoters (4). Reproducible, tightly regulated Tet-inducible expression was originally established in trypanosomes that express the heterologous T7 RNA polymerase (pol), as well as TetR, by a mutated T7 promoter (3). Two T. brucei brucei 427 cell lines were generated that have been widely used for RNAi experiments: the PF cell line 29-13, harboring the selectable NEO and HYG genes, which encode neomycin and hygromycin phosphotransferase, respectively, and the “single-marker” (sm) (NEO) BF cell line (3).
Several genome integration vectors were generated for Tet-inducible expression of gene-specific dsRNA in T. brucei. The simplest vector type has opposing T7 promoters. Since gene fragments can be cloned between the promoters in a single step, this type of vector, termed p2T7 (5) or pZJM (6), enabled the generation of RNAi libraries and the first successful forward genetic RNAi screen (7). However, the T7 promoters of these vectors, which are each regulated by a single TetR binding site, the Tet operator, were partially active even in the absence of tetracycline (3, 6), which could be lethal to transformed trypanosomes in some cases. A more tightly regulated vector was the stem-loop vector, in which a target sequence is cloned in sense and antisense directions around a stuffer fragment (8, 9). In the original vector, the stem-loop cassette is expressed from the strong EP1 procyclin gene promoter under the control of two Tet operators (3). Although it takes two or three cloning steps to produce a gene-specific stem-loop construct, the tight regulation of its expression proved to be very useful in PFs (10). Procyclin is the major cell surface antigen of PFs, and procyclin genes are highly expressed by virtue of a multifunctional RNA pol I, which is recruited to the procyclin gene promoter (11). However, since procyclin is not expressed in BFs and the procyclin promoter is severalfold less active in that life cycle stage (12), replacement of the procyclin promoter by a T7 promoter made the pT7-stl stem-loop construct more suitable for gene knockdowns in BFs; two Tet operators appear to be sufficient to minimize leakiness from the strong T7 promoter (13). Recently established vectors that made conditional transgene expression in trypanosomes independent of T7 RNA pol revealed effective gene knockdowns in PFs (14). Other vector improvements simplified the cloning procedure. The pQuadra system is based on a four-component ligation assay and allows the generation of a stem-loop vector in a single cloning step (15), whereas other strategies made both dual T7 promoter and stem-loop constructions compatible with the recombination-based Gateway cloning system (16, 17).
In addition to different vector designs, the conditional-gene-silencing system in T. brucei has benefitted from several modifications. Since high T7 RNA polymerase levels appear to be toxic for trypanosomes, putting the expression of the enzyme under tetracycline control enhanced the success rate in obtaining PF cell lines that exhibited an inducible phenotype (18). However, this modification did not generally improve the functionality of p2T7 constructs. Historically, the preferred genome integration site of inducible vectors has been the transcriptionally silent spacer of rRNA gene (RRNA) arrays. It appears that not all RRNA loci provide equal conditions for regulated dsRNA expression, and thus, a standard procedure has been to test several clonal cell lines to find the cells with the best knockdown efficiency. To avoid this position effect, Alsford et al. marked a specific RRNA locus so that vectors could be specifically and reproducibly targeted to this particular RRNA spacer, thereby reducing the variability of gene-silencing experiments (19). In an independent approach, Wickstead et al. found that targeting the p2T7 vector p2T7-177 to a 177-bp-long, transcriptionally silent repeat region of trypanosome minichromosomes improved regulation of the T7 promoters (20). Nevertheless, despite these modifications and improvements, the RNAi-mediated gene knockdowns have remained inefficient in some cases. Furthermore, due to the need to express rather long dsRNA molecules, it is possible that small interfering RNAs (siRNAs) are produced that affect trypanosome proliferation by targeting the wrong RNA, causing a so-called off-target effect.
We have been using conditional gene silencing to characterize the T. brucei class I transcription factor A (CITFA) complex, which is indispensable for the multifunctional RNA pol I system in T. brucei. The parasite uses RNA pol I to transcribe the RRNA array, as in all other eukaryotes, yet also employs it to express its major cell surface proteins, procyclins in PFs and variant surface glycoprotein (VSG) in BFs (13, 21, 22). The latter is expressed from a single VSG gene, drawn from a large repertoire, in one of 15 40- to 60-kb-long BF telomeric expression sites (BESs) (23). While trypanosome RRNA transcription is localized to the nucleolus, as in other eukaryotes, the active BES is transcribed outside this compartment (24) in the DNase I-resistant expression site body (ESB) (25). CITFA consists of the subunits CITFA1 to -7, which are conserved only among kinetoplastid organisms, and the dynein light chain DYNLL1 (also known as LC8). Data obtained thus far strongly indicate that CITFA is a promoter-binding transcription initiation factor. CITFA stably bound the BES promoter in gel shift assays and required both promoter elements for efficient binding, depletion or inhibition of CITFA resulted in loss of transcription within 121 to 146 bp of the transcription initiation site, CITFA7 silencing strongly reduced promoter-proximal RNA pol I occupancy at RRNA repeats, and a genome-wide chromatin immunoprecipitation-sequencing (ChIP-seq) analysis found CITFA7 occupancy within a BES to be restricted to the promoter region (13, 21, 22). To analyze specific functions of individual subunits, we attempted to silence the expression of each subunit gene. Our previous results demonstrated that CITFA2 and CITFA7 are essential for RNA pol I transcription and trypanosome viability (13, 21). We also found that three of the eight subunits are required to maintain the integrity of the complex (T. N. Nguyen and A. Günzl, unpublished results). However, we have been unable to generate an unambiguous CITFA1 knockdown. Although we targeted two different regions of the CITFA1 mRNA, the RNA levels were not significantly affected (data not shown). To circumvent this setback, we set out to develop a generally applicable system in BFs for efficient and specific gene knockdowns that targets a heterologous sequence (HS) fused to the mRNA of interest. Here, we demonstrate that the 3′ gene flank of the Trypanosoma cruzi U2AF35 gene (TcU2AF35) (here abbreviated as Tc3) is functional in T. brucei and that Tc3 can be specifically targeted when fused to a T. brucei mRNA. In addition, we show that the sequence of the composite PTP tag, consisting of a protein C epitope (ProtC), a TEV protease cleavage site, and tandem protein A domains (ProtA), is an equally good target for gene silencing. Employing this system, we were able to demonstrate that CITFA1 is an indispensable component of the CITFA complex and that it is required for CITFA to bind to the RRNA and BES promoters.
MATERIALS AND METHODS
DNAs.
pT7-CITFA7-stl (21) and pCITFA7-PTP-BLA (22) were described previously. For the generation of pT7-Tc3-stl, the entire 679-bp-long 3′ intergenic region of the T. cruzi U2AF35 gene (accession number TcCLB.510943.60 [www.genedb.org or www.tritrypdb.org]; note that this gene has been annotated as U2AF26 instead of U2AF35), from position 703 to position 1381 relative to the translation initiation codon, was amplified from T. cruzi genomic DNA and inserted into the pT7-stl vector (13) in a sense-stuffer-antisense arrangement according to a published protocol (8). A T924C point mutation was introduced to remove an XbaI restriction site that would have interfered with the stem-loop cloning strategy. pT7-PTP-stl was generated analogously using the entire PTP coding sequence (498 bp) (26). The cloning strategy required the removal of two MluI restriction sites, which was achieved by introducing T153C and T327C point mutations.
The tagging vector pCITFA7-HA-Tc3-BLA (see Fig. S1 in the supplemental material) has two cassettes, a C-terminal tagging module and a selectable marker cassette. It is a direct derivative of pCITFA7-HA-BLA (21) and was obtained by replacing the T. brucei RPA1 3′ gene flank with Tc3 using the vector's XhoI and ClaI restriction sites. Furthermore, our tagging vectors are derived from pBluescript II SK(+), which has a T7 promoter that, after integration into an endogenous allele, could lead to overexpression of downstream genes in T7 RNA polymerase-expressing sm BF and 29-13 PF cells. We therefore removed 27 bp from pCITFA7-HA-Tc3-BLA, beginning precisely at the T7 promoter and ending in the downstream KpnI restriction site. In the same way, we removed the T7 promoter from pCITFA7-PTP-BLA and named the corresponding plasmid pCITFA7-PTP-BLAv2 (see Fig. S2 in the supplemental material). pCITFA1-HA-Tc3-BLA was obtained from pCITFA7-HA-Tc3-BLA by replacing the CITFA7 sequence with 696 bp of the C-terminal CITFA1 coding region (position 700 to position 1395) using the ApaI and NotI restriction sites.
The following DNA oligonucleotides were used in semiquantitative RT-PCR: 5′-CCTACGGTGCAGCCATGCCGTTGG-3′/5′-TTCGGCACTGCCATATGCGAC-3′ (CITFA7 coding sequence), 5′-CAATAACAGGAACAGCTGCACCAAG-3′/5′-GAGGAAACTCAAGTGCATTG-3′ (Tc3), and 5′-GTAGACAACAAATTCAACAAAG-3′/5′-ATTTAGCTTTTTAGCTTCTGC-3′ (PTP). Oligonucleotides for VSG221, TFIIB, and CITFA2 amplification were previously described (21). Quantitative RT-PCR of CITFA1 mRNA was carried out either with the oligonucleotide pair 5′-ATCGGATGTTGAGTCGCTGCGTTG G-3′/5′-AAAGTCATTCCATGCCACTGGAACC-3′ (CITFA1 coding sequence) or the pair 5′-AATACGCCAG GCAGATTGATGC-3′/5′-TTAAGCGTAGTCAGGTACGTCGTAAGG-3′ (CITFA7 coding region/HS). BES and RRNA promoter consensus oligonucleotides and oligonucleotides specific to the TFIIB gene and the β-/α-tubulin intergenic region, used in quantitative PCR (qPCR), were previously specified (21, 27).
Cells.
BFs were cultured in HMI-9 medium as specified previously (27). Transfections were done with 1 × 107 to 2 × 107 BF trypanosomes using the Amaxa Basic Parasite Nucleofector kit (Lonza). Specifically, trypanosomes were pelleted at room temperature at 1,500 × g for 5 min, and the cell pellets were resuspended in 100 μl of solution 1 containing 18% supplement 1, both part of the kit. After mixing DNA (5 μg of the PCR product or 10 μg of plasmid DNA) into the resuspension, trypanosomes were electroporated using program X-001 on the Nucleofector 2b unit (Lonza). Prewarmed HMI-9 medium (500 μl) was added immediately to the transfected cells, which were then transferred to 50 ml of ∼37°C warm medium. After allowing the cells to recover for 15 min at 37°C, the cell culture was distributed into two 24-well plates. Trypanosomes were cultivated without antibiotic selection overnight, after which additional medium containing selecting antibiotics was added to each well. Transfectants typically reached a transferrable cell density 6 to 9 days later. Cells were cultured in 2.5 μg/ml G418, 1 μg/ml phleomycin, and/or 2 μg/ml blasticidin.
Cell lines C7HA-Tb3 and C7HA-Tc3 were obtained by transfecting wild-type BF 427 cells with PshAI-linearized plasmids CITFA7-HA-BLA and CITFA7-HA-Tc3-BLA, respectively. The basal BF cell lines for conditional expression of Tc3 or PTP dsRNA, smTc3 and smPTP, were obtained by transfecting sm cells with the EcoRV-linearized vectors pT7-Tc3-stl and pT7-PTP-stl, respectively. These vectors were targeted to the transcriptionally silent RRNA spacer. smC7HA-Tc3 and smC7-PTP cell lines were generated by targeted integration of PshAI-linearized plasmids CITFA7-HA-Tc3-BLA and CITFA7-PTP-BLAv2, respectively, into the CITFA7 locus in the first step and by replacing in the second step the remaining CITFA7 wild-type allele with a PCR product in which 100 bp of CITFA7 gene flanks surrounded the hygromycin phosphotransferase coding sequence. The cell line smC1HA-Tc3 was obtained analogously using the MfeI-linearized plasmid CITFA1-HA-Tc3-BLA. Correct DNA integrations were analyzed by PCR of genomic DNA with at least one oligonucleotide placed outside the cloned or amplified sequence (data not shown).
For gene-silencing experiments, dsRNA synthesis was induced with doxycycline, a more stable derivative of tetracycline, at 2 μg/ml. Cells were counted and diluted to 2 × 105 cells/ml daily.
Antibodies and protein analysis.
For the sedimentation analysis, extract was prepared from noninduced BFs and from CITFA1-silenced BFs as specified previously (27); 100 μl of extract was then loaded onto 4-ml 10 to 40% linear sucrose gradients, ultracentrifuged, and fractionated exactly as has been described previously (13). Immunoblots of hemagglutinin (HA)- and PTP-tagged proteins were probed with a rat monoclonal anti-HA antibody (Roche) and the mouse monoclonal anti-ProtC antibody HPC4 (Roche), respectively. Immune sera against CITFA7, TFIIB, and U2A/ (also known as U2-40K) were described previously (21, 28, 29). To obtain recombinant CITFA3 for antibody production, the entire CITFA3 coding region was placed downstream of the glutathione S-transferase (GST) sequence in the pGEX-4T-2 vector (GE Healthcare). Recombinant GST-CITFA3 was expressed in Escherichia coli strain BL21Star(DE3) and purified by glutathione affinity chromatography (GE Healthcare) following the manufacturer's recommendations. Generation of anti-CITFA3 immune serum was achieved by immunization of female Sprague-Dawley rats with purified GST-CITFA3, as detailed previously (29). Polyclonal anti-GST-CITFA3 antibodies were purified from rat immune serum by first preclearing the serum of antibodies that nonspecifically interact with trypanosome proteins. This was done by separating whole-cell lysates of a total of 4.5 × 108 wild-type PF trypanosomes in nine lanes on a 12% SDS-polyacrylamide gel. The gel was transferred to a polyvinylidene difluoride (PVDF) membrane and blocked for 2 h in Tris-buffered saline (TBS), pH 7.5, containing 5% milk and 0.1% Tween 20. After washing twice in TBS, the 50- to 55-kDa range of the membrane that contained CITFA3 was removed, and the remaining membrane was incubated with 1 ml of antiserum and 9 ml of blocking solution at 4°C for 16 h. Recombinant GST-CITFA3 (100 μg) was then run on four different SDS-PAGE gels, which were then transferred and blocked as described above. The 70- to 80-kDa range of these four membranes, corresponding to the size of GST-CITFA3, was excised, cut into small strips, and incubated with the precleared antiserum and blocking solution mixture for 16 h at 4°C with rotation. These membrane strips were washed three times with TBS and then rinsed briefly one time with water. Antibodies were eluted by incubating the strips in 1 ml of 0.2 M glycine, pH 2.8, for 5 min. The eluted antibodies were then immediately quenched by adding 1 M Tris-HCl, pH 8.0, until the solution pH reached 7.5, after which bovine serum albumin was added to a final concentration of 1%. The purified antibody was aliquoted and stored at −20°C until use.
RNA analysis.
Total RNA was prepared by the hot-phenol method as described previously (30). For the analysis of rRNA, total RNA was separated in Reliant precast 1.25% SeaKem Gold agarose RNA gels (Lonza), and rRNA was detected by ethidium bromide staining. Relative amounts of spliced leader (SL) RNA and U2 snRNA were determined by primer extension of 10 μg of total RNA using the 5′-32P-end-labeled oligonucleotides SL-1394 and U2f (31) and Superscript reverse transcriptase II (Invitrogen) according to the manufacturer's specifications. The primer extension products were separated on denaturing 8% polyacrylamide-50% urea gels and detected by autoradiography. For PCR analyses, total RNA was reverse transcribed with Superscript reverse transcriptase II and either oligo(dT) or random hexanucleotides (Roche). Semiquantitative PCR was performed using cycle numbers that were empirically determined to be within the linear amplification range for each oligonucleotide pair. Oligonucleotide pairs used in qPCR were verified for their specificity and suitability by standard agarose gel electrophoresis. Additionally, for every round of qPCRs, a melt curve analysis was included to ensure the amplification of only a single product, and linear regression analysis of a serial dilution of input material confirmed that the coefficient of determination (r2) was within the 0.98 to 1.0 range.
Immunofluorescence microscopy.
BF microscopy was carried out as described previously (21), except that coverslips with settled BF cells were incubated with a 1:100 dilution of the affinity-purified polyclonal rat anti-CITFA3 antibody and were then, after washing, incubated with the 1:500-diluted anti-rat IgG antibody Alexa 488 (Invitrogen) and 4,6-diamidino-2-phenylindol (DAPI) at a final concentration of 2 ng/μl. Imaging was performed using a Zeiss AxioVert 200 microscope and Zeiss Axiovision 4.6.3.0 software. CITFA3 localizations were captured using a fluorescein isothiocyanate (FITC) filter and a fixed exposure time of 6 s, while DAPI images were captured using a DAPI filter and a variable exposure time, which averaged 0.5 s.
ChIP.
Anti-CITFA3 ChIP assays, using the purified polyclonal anti-CITFA3 antibody, were performed with smC1HA-Tc3 cells that were either not induced or induced by doxycycline for 42 h, as recently described (22). In negative controls, chromatin was immunoprecipitated with a nonspecific rat immune serum. The precipitated DNA was analyzed by qPCR using consensus oligonucleotides for the slightly varying copies of RRNA and BES promoters and an oligonucleotide pair specific for the β-/α-tubulin intergenic region. The percent immunoprecipitation (IP) was calculated relative to the input material and corrected by subtracting the percent IP of the negative-control assays.
RESULTS
System outline.
Our aim was to generate a system in which the same heterologous sequence can be functionally fused to any mRNA and provide a target for efficient and reproducible gene knockdown. In the first step, we would generate an sm BF line that conditionally expresses dsRNA of an HS without consequences on cell proliferation (Fig. 1). This smHS cell line would carry NEO used to generate sm cells (3) and the bleomycin resistance marker (BLE) as part of the stem-loop vector for conditional HS dsRNA expression. For a specific gene knockdown, the smHS cell line would be used in two consecutive transfections to enable specific knockdown of any single-copy gene of interest, here denoted gene X. In the first transfection, site-specific integration of a PCR amplification product of the hygromycin phosphotransferase coding region (HYG) surrounded by 5′ and 3′ flanks of gene X would lead to the knockout of one wild-type X allele, while in the consecutive transfection, targeted integration of plasmid X-HS-BLA (BLA stands for the blasticidin-S deaminase selectable marker gene) into the X locus would fuse the HS to gene X. The corresponding smX-HS cell line would exclusively express the X-HS fusion that can be targeted by doxycycline-mediated induction of HS dsRNA synthesis (Fig. 1).
FIG 1.
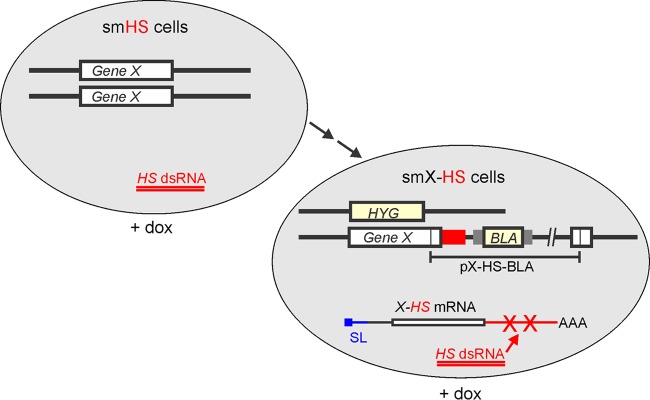
Gene-silencing system targeting an HS. The system is based on a single-marker BF cell line that has been stably transfected with a stem-loop construct (smHS cell line) containing a doxycycline (dox)-inducible promoter for the expression of a heterologous dsRNA. In the absence of further genetic manipulations, induction of this dsRNA expression has no consequence for trypanosome proliferation. To enable a specific knockdown of any single-copy gene X, two further consecutive transfections of smHS cells are necessary: one to eliminate an X allele by the hygromycin resistance marker (HYG) and one to integrate a plasmid into the remaining X allele, which will fuse the HS to the X mRNA sequence. Doxycycline-induced (+ dox) expression of HS dsRNA then targets and destroys the X-HS mRNA hybrid.
The T. cruzi U2AF35 3′ gene flank is functional in T. brucei.
We speculated that a T. cruzi 3′ gene flank could provide an HS in the form of a functional 3′ untranslated region (UTR), because all trypanosomatids process their mRNAs by SL trans splicing and polyadenylation (32), making it likely that a T. cruzi gene flank is able to direct these RNA-processing steps in T. brucei. At the same time, the T. cruzi intergenic sequences are divergent from their T. brucei counterparts and are unlikely to give rise to siRNAs that target T. brucei mRNAs. After comparing known T. cruzi 3′ UTRs (33), we chose to analyze the suitability of the 3′ UTR of the TcU2AF35 mRNA because its complete cDNA had been characterized and the 3′ UTR length of 390 nucleotides (nt) appeared to be sufficiently large for efficient gene knockdown (34). Moreover, the complete intergenic region between TcU2AF35 (accession number TcCLB.510943.60) and its downstream neighbor TcCLB.510943.50, encoding putative delta-1-pyrroline-5-carboxylate dehydrogenase, is only 679 bp long and should harbor all necessary RNA-processing signals. Finally, U2AF35 encodes an essential RNA-splicing factor that in trypanosomes is involved in the initial steps of the ubiquitous SL trans splicing process (35), which suggested that the TcU2AF35 3′ UTR could support a sufficient level of constitutive gene expression.
We first tested the functionality of the TcU2AF35 3′ gene flank (Tc3) in T. brucei with the well-characterized CITFA7 gene. CITFA7 is an essential subunit of the CITFA complex, and CITFA7 silencing in BFs led to clear defects in RNA pol I transcription, e.g., a decrease of the RNA pol I transcripts rRNA and VSG221 mRNA of the active VSG gene (21). Moreover, CITFA7 can be functionally tagged at the C terminus, and there is no haploinsufficiency effect after deleting one CITFA7 allele either in BFs or PFs (21). We generated two cell lines by targeting the integration of a plasmid to the endogenous CITFA7 gene (Fig. 2A). In both cases, the HA tag sequence was fused 3′ to the CITFA7 coding region, followed by the T. brucei RPA1 (TbRPA1) 3′ gene flank in the C7HA-Tb3 cell line or by the Tc3 gene flank in the C7HA-Tc3 cell line. RPA1 is the largest subunit of RNA pol I. The TbRPA1 3′ gene flank is present in all our C-terminal tagging constructs and supported the expression of a variety of factors, so that the knockout of the remaining wild-type allele did not cause haploinsufficiency phenotypes (26). Accordingly, CITFA7-HA was easily detectable in C7HA-Tb3 cell lysates in multiple bands (Fig. 2B) that represent the various (un)phosphorylated forms of CITFA7 (21). In C7HA-Tc3 cells, Tc3 supported 78% of the CITFA7-HA expression observed in C7HA-Tb3 cells (Fig. 2B). These results showed that in T. brucei, the heterologous Tc3 sequence directed the processing of functional CITFA7-HA mRNA, and the corresponding 3′ UTR supported an adequate level of CITFA7 expression.
FIG 2.
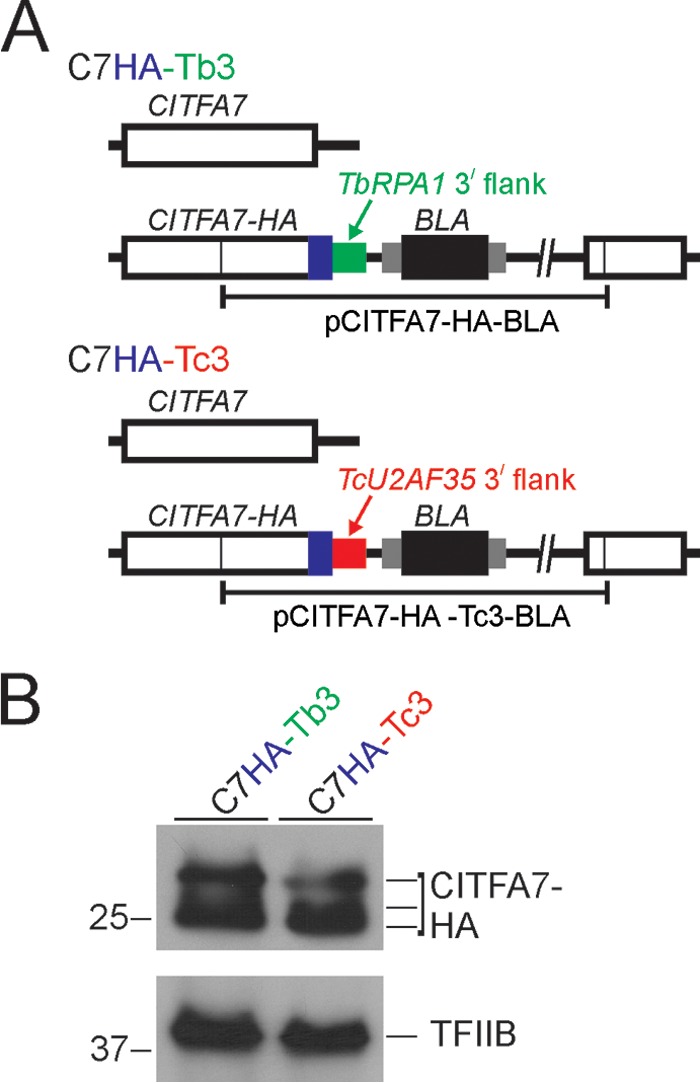
The 3′ gene flank and UTR of T. cruzi TcU2AF35 are functional in T. brucei. (A) Schematic outline (not to scale) of the CITFA7 locus in the control cell line C7HA-Tb3 and in the cell line C7HA-Tc3. In both cell lines, the HA tag sequence was fused to the CITFA7 coding region by targeted integration of the specified linearized plasmid into one CITFA7 allele. The difference is that in C7HA-Tb3 and C7HA-Tc3 cells, the manipulated CITFA7 allele is under the control of the T. brucei RPA1 (green) and the heterologous TcU2AF35 (red) 3′ gene flanks, respectively. The CITFA7 coding region, the HA tag, and the BLA sequences are indicated by open, blue, and black boxes, respectively. The smaller gray boxes surrounding BLA represent T. brucei gene flanks providing RNA-processing signals. (B) Immunoblot of C7HA-Tb3 and C7HA-Tc3 whole-cell lysates detecting CITFA7-HA with a monoclonal anti-HA antibody and, as a loading control, the transcription factor TFIIB with an anti-TFIIB polyclonal immune serum. Note that, due to phosphorylation, CITFA7 separates into multiple bands.
Expression of Tc3 dsRNA does not affect BF trypanosome proliferation.
Comparing the Tc3 sequence to the T. brucei brucei Lister 427 genome returned a single, siRNA-size stretch of identical sequence (27 bp) downstream of the nonsyntenic gene Tb427.10.1000. Since this sequence motif was found in only a fraction of the heterogenous Tb427.10.1000 3′ UTRs (www.tritrypdb.org), we anticipated Tc3 dsRNA would not give rise to deleterious siRNAs. To test this, we inserted a stem-loop construct of the Tc3 sequence into the pT7-stl vector, which has a tetracycline-inducible T7 promoter (13). The vector, targeted to the ribosomal spacer, was transfected into sm BF cells, which express both the tetracycline repressor and T7 RNA polymerase (3). Immediately after transfection, the cells were cloned by limiting dilution. Three of the resulting cell lines were evaluated for their knockdown competence by transfecting them with the CITFA7-HA-Tc3-BLA construct and monitoring knockdown efficiencies on the RNA and protein levels (see below) (data not shown). Based on these results, one of the parent cell lines, termed smTc3, was chosen for all subsequent experiments. As shown in Fig. 3A, induction of Tc3 dsRNA synthesis by doxycycline did not affect the proliferation of smTc3 cells in culture, suggesting that no deleterious off-target effects occurred.
FIG 3.
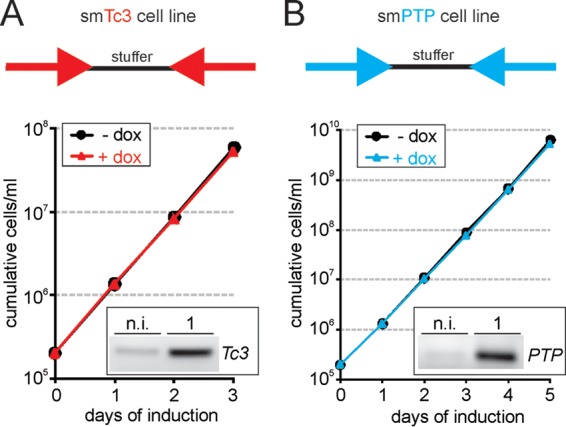
Proliferation of smTc3 and smPTP cells is not affected by heterologous dsRNA expression. The smTc3 (A) and smPTP (B) cell lines are single-marker BF cell lines in which stem-loop constructs were integrated into the RRNA spacer for inducible expression of Tc3 and PTP dsRNA, respectively. (Insets) Addition of doxycycline to the medium did not inhibit cell proliferation in either cell line while it induced strong expression of Tc3 or PTP RNA, as analyzed by semiquantitative PCR of random-hexamer-derived cDNA. n.i., noninduced; 1, 1 day induced.
Generation of an alternative smPTP cell line.
At this point, we considered testing a second heterologous sequence in parallel. We decided on the large PTP tag because we have utilized it repeatedly for tandem-affinity purification (26), indirect fluorescence microscopy (27), and chromatin immunoprecipitation (27, 36) experiments. The composite, heterologous PTP tag, designed for a modified tandem-affinity purification approach (26), comprises 166 amino acids and consists of human-derived ProtC (37), a tobacco etch virus-derived protease cleavage site, and a tandem ProtA domain of Staphylococcus aureus (38). Importantly, the PTP tagging strategy was based on integration of a PTP plasmid into an endogenous allele, as shown in Fig. 1. Hence, we wanted to know whether the PTP tag sequence could be used as a target for efficient gene knockdowns, as well. A bioinformatic analysis could not detect any sequence match longer than 18 bp between the PTP sequence and the T. brucei brucei 427 genome (data not shown). Analogously to cell line smTc3, we generated clonal smPTP cell lines that conditionally express PTP dsRNA. To pick the most efficient line for subsequent experiments, we determined PTP dsRNA levels before and after doxycycline induction by reverse transcription (RT) of total RNA using random hexamers and semiquantitative PCR of the PTP sequence (Fig. 3B and data not shown). Induction of PTP dsRNA by doxycycline in the chosen smPTP cell line did not affect trypanosome proliferation, again indicating that this heterologous dsRNA does not target vitally important endogenous T. brucei RNAs (Fig. 3B).
The Tc3 3′ UTR and the PTP sequence are efficient knockdown targets.
To test whether targeting either of the two HSs by RNAi leads to efficient gene knockdowns, we fused both of these sequences to the CITFA7 gene. As depicted in Fig. 1, this required two consecutive transfections of smTc3 and of smPTP cells so that they exclusively expressed CITFA7-HA mRNA with the Tc3 3′ UTR and CITFA7-PTP mRNA, respectively. In the first transfection of smTc3 cells, we integrated plasmid CITFA7-HA-Tc3-BLA into one of the two CITFA7 alleles, and in the second step, we eliminated the remaining CITFA7 allele by transfecting a PCR product (39) that comprised the hygromycin phosphotransferase coding region surrounded by 100-bp-long CITFA7 gene flanks (Fig. 4A). Three independently derived clonal smC7HA-Tc3-RNAi cell lines exhibited nearly identical growth defects upon induction of Tc3 dsRNA synthesis. As shown for one representative line in Fig. 4B, addition of doxycycline stopped culture growth after 24 h and reduced the number of surviving trypanosomes within the next 48 h. Since a very similar growth curve was obtained previously when CITFA7 mRNA was targeted directly in smC7 cells (21), this result suggested that targeting of the Tc3 3′ UTR resulted in effective depletion of CITFA7.
FIG 4.

Effective and specific CITFA7 silencing by targeting heterologous sequences. (A) Schematic outline (not to scale) of the CITFA7 locus in smC7HA-Tc3 cells. One CITFA7 allele was replaced by the hygromycin resistance marker (HYG), and the second CITFA7 allele was modified by targeted integration of plasmid CITFA7-HA-Tc3-BLA. The colors of the boxes correspond to the description in the legend to Fig. 2A. (B) Culture growth of a representative smC7HA-Tc3 cell line in the presence (+) and absence (−) of doxycycline. (C) Schematic outline (not to scale) of smC7-PTP cells in which integration of plasmid CITFA7-PTP-BLA fused the PTP sequence (cyan box) to the 3′ end of the CITFA7 coding region. (D) Corresponding growth curve of a representative smC7-PTP cell line. (E) Semiquantitative PCR analysis of oligo(dT)-primed and reverse transcribed CITFA7-HA, CITFA7-PTP, and CITFA7 mRNAs in noninduced (n.i.) and 1-day-induced (1) trypanosomes of the cell lines smC7HA-Tc3, smC7-PTP, and smC7, respectively. TFIIB mRNA was analyzed in parallel as a control. (F) Relative abundances of rRNA and of VSG221 and TFIIB mRNAs were determined by ethidium bromide staining and semiquantitative RT-PCR, respectively, in smC7HA-Tc3 and smC7-PTP cells that were noninduced or induced for 1 and 2 days. (G) Immunoblot detecting CITFA7-HA with an anti-HA antibody in smC7HA-Tc3 cell lysates, CITFA7-PTP with an anti-ProtC antibody in smC7-PTP cell lysates, and wild-type CITFA7 with a polyclonal immune serum in smC7 cell lysates. Detection of TFIIB on the same blots served as a loading control. Cells were analyzed in their noninduced state or when they were grown in the presence of doxycycline for 1 and 2 days. Marker sizes in kDa are indicated on the left of each panel.
Analogously to smC7HA-Tc3 cells, we generated the cell line smC7-PTP, in which integration of plasmid pCITFA7-PTP-BLA fused the PTP sequence to the CITFA7 coding region in one CITFA7 allele and the hygromycin resistance marker replaced the remaining wild-type allele (Fig. 4C). Again, inducing the synthesis of PTP dsRNA stopped proliferation of smC7-PTP cells after 1 day and led to cell death thereafter (Fig. 4D). Semiquantitative RT-PCR analyses showed that CITFA7-HA, CITFA7-PTP, and CITFA7 mRNAs were strongly reduced after 1 day of induction in smC7HA-Tc3, smC7-PTP, and smC7 cells, respectively (Fig. 4E). This reduction was observed despite the fact that the level of the control TFIIB mRNA increased in each of these experiments. As we have shown previously, silencing of CITFA subunit genes rapidly decreases the levels of RNA pol I transcripts in induced cells, including rRNA, the most abundant component in total RNA preparations, which in turn leads to relative increases of RNA pol II and III transcripts (13, 21). As anticipated from these previous results, targeting the HSs that were fused to the CITFA7 mRNA led to a decrease of rRNA and of VSG221 mRNA from the active VSG gene (Fig. 4F). In addition, immunoblotting showed that the gene knockdowns led to a rapid loss of CITFA7 protein (Fig. 4G). Together, these results clearly demonstrated that the HS-targeted CITFA7 gene knockdowns were specific and efficient, affecting the abundance of RNA pol I transcripts in the same way as did silencing of CITFA7 by dsRNA of the endogenous sequence, as previously shown (21).
Tc3 3′-UTR-mediated efficient CITFA1 silencing.
Next, we applied this gene knockdown system to the CITFA1 gene, which we had not been able to silence efficiently thus far. Again, two consecutive transfections of smTc3 cells generated the cell line smC1HA-Tc3, in which, after the knockout of one CITFA1 allele, targeted integration of the plasmid CITFA1-HA-Tc3-BLA fused the HA sequence and the Tc3 gene flank to the remaining CITFA1 allele (Fig. 5A). In the absence of doxycycline, smC1HA-Tc3 cells proliferated as fast as smTc3 cells, indicating that the Tc3 3′ UTR supported sufficient CITFA1 expression from a single allele and that the C-terminal HA tag did not impair the functionality of CITFA1 (Fig. 5B). Adding doxycycline to the medium was similarly deleterious to trypanosome proliferation and viability, as in the CITFA7 knockdown. Trypanosome proliferation was affected 1 day after induction, and trypanosome numbers started to decline after 2 days of induction (Fig. 5B). RT-qPCR analysis revealed that the Tc3-targeted knockdown reduced CITFA1 mRNA abundance relative to that of TFIIB mRNA by ∼80% (Fig. 5C). Interestingly, we obtained slightly different results depending on which part of the cDNA was amplified. With oligonucleotides specific for the CITFA1 coding region, the reduction of CITFA1 mRNA was, on average, 77%, whereas the reduction with an HS-specific oligonucleotide was 83%. Although this difference does not seem that dramatic, we point out that we have made similar observations with other gene knockdowns. Amplification of different parts of the reverse-transcribed mRNA revealed different knockdown efficiencies, with the targeted region typically resulting in the greatest reduction of mRNA abundance (Sung Hee Park, Bao N. Nguyen, Daniela L. Ambrósio, and Arthur Günzl, unpublished results). Further RNA analysis showed that, as expected, CITFA1 silencing decreased the abundance of the major RNA pol I transcripts rRNA and VSG221 mRNA and, consequently, elevated the relative abundances of RNA pol II-synthesized TFIIB mRNA, CITFA2 mRNA, and SL RNA, as well as that of the RNA pol III transcript U2 snRNA (Fig. 5D). Together, these results demonstrated that CITFA1 was efficiently and specifically silenced by targeting the Tc3 3′ UTR, identified CITFA1 as the third CITFA subunit that is essential for trypanosome viability in culture, and indicated that CITFA1 has an essential function in trypanosome RNA pol I transcription.
FIG 5.
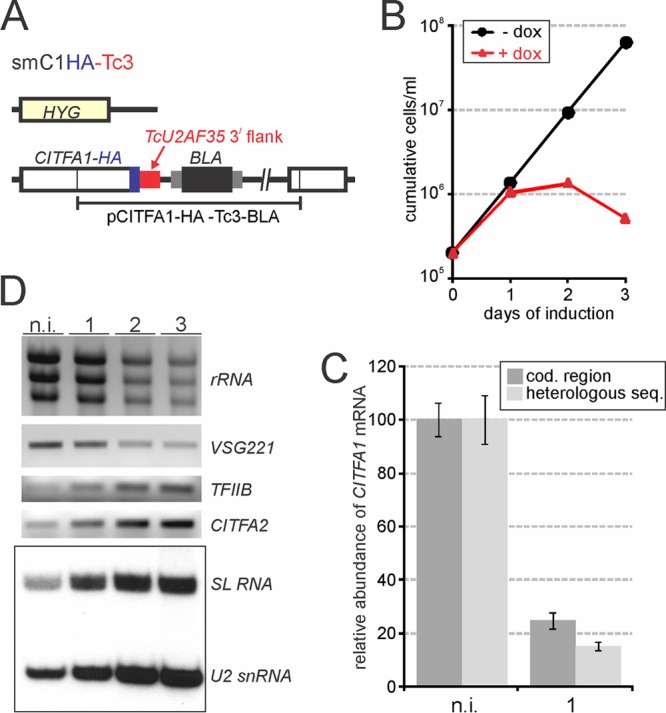
Effective and specific CITFA1 silencing by targeting the TcU2AF35 3′ UTR. (A) Schematic outline (not to scale) of the CITFA1 locus in smC1HA-Tc3 cells. (B) Culture growth of a representative smC1HA-Tc3 cell line in the presence and absence of doxycycline. (C) RT-qPCR analysis of CITFA1 mRNA in noninduced (n.i.) and 1-day-induced trypanosomes. The oligo(dT)-derived cDNA was amplified either in the CITFA1 coding (cod.) region or in the heterologous TcU2AF35 3′ UTR (heterologous seq.). CITFA1 mRNA abundance was normalized with that of TFIIB, and its level in noninduced cells was set to 100 in each of three independent experiments. The error bars indicate standard deviations. (D) Relative RNA abundances in total RNA preparations of noninduced cells and of cells induced for 1, 2, or 3 days were analyzed by ethidium bromide staining (rRNA), semiquantitative RT-PCR (VSG221, TFIIB, and CITFA2 mRNAs), or a primer extension assay (SL RNA and U2 snRNA).
CITFA1 is required for binding of the transcription factor complex to RNA pol I promoters.
The analysis of specific functions of individual CITFA subunits has been hampered by the fact that amino acid sequences of CITFA subunits have not revealed functional motifs, such as DNA binding domains. In addition, procedures intended to break up the complex into smaller functional units have failed thus far (data not shown). However, we recently found that depletion of CITFA7 from cells resulted in the concomitant rapid loss of other CITFA subunits within 2 days of CITFA7 silencing, indicating that CITFA7 has a scaffold function and that the stability of CITFA subunits in trypanosomes depends on the integrity of the transcription factor complex (Nguyen and Günzl, unpublished). An immunoblot analysis of whole-cell lysates showed that CITFA1-HA was strongly depleted after 1 and 2 days of CITFA1 silencing (Fig. 6A). To assess the abundances of other CITFA subunits in these samples, we used previously established immune sera against CITFA2, CITFA6, and CITFA7 and a newly generated polyclonal anti-CITFA3 antibody (see below). The results demonstrate that, besides a minor reduction of CITFA2 on day 2, the CITFA subunits analyzed exhibited robust expression during the 2-day experiment, suggesting that CITFA1, in contrast to CITFA7, is not required for overall complex integrity. To substantiate this notion, we analyzed the CITFA complex upon CITFA1 depletion by sucrose gradient sedimentation, in which fractions were taken from top to bottom of the gradient (Fig. 6B). In noninduced cells, the sedimentation profile was exactly as determined previously (13, 21): CITFA1, -3, -6, and -7 exhibited sedimentation peaks in fractions 13 and 14, while the less abundant CITFA2 peaked in fractions 14 and 15, shifting part of the complex one fraction down the gradient. Loss of CITFA1 decreased the sedimentation of CITFA2, -3, -6, and -7 by ∼2 fractions (note that the sedimentation peak of the minor amount of detectable CITFA1 remained in fraction 13). However, the CITFA subunits still cosedimented between the 6.6S IgG marker (150-kDa) and the 230-kDa large trypanosome TRF4-SNAPc-TFIIA transcription factor complex (40). Since the CITFA complex without CITFA1 and without CITFA1 and -2 has calculated masses of 221 kDa and 173 kDa, respectively, this result strongly indicates that CITFA1 depletion did not affect the integrity of the CITFA complex. Accordingly, a pulldown of CITFA3 in extract efficiently coprecipitated CITFA2, -6, and -7 in both noninduced and CITFA1-depleted cells (see Fig. S3 in the supplemental material).
FIG 6.

CITFA1 is required for CITFA promoter binding in vivo. (A) Immunoblot detecting the indicated CITFA subunits in whole-cell lysates of noninduced cells (n.i.) and in cells in which CITFA1-HA was silenced for 1 or 2 days (boxed) on the same blot. Detection of the spliceosomal U2A/ protein served as a loading control. (B) Sedimentation of extracts by ultracentrifugation in a 10 to 40% linear sucrose gradient. Fractions 5 to 20, taken from top to bottom, were analyzed by immunoblotting. Note that the two CITFA1 blots were codeveloped. For comparison, sedimentations of TEV protease (29 kDa), Taq DNA polymerase (95 kDa), IgG (150 kDa; 6.6S), the TRF4-SNAPc-TFIIA transcription factor complex (TST) (230 kDa), apoferritin (AP) (444 kDa; 17S), and thyroglobin (TG) (660 kDa; 19S) were analyzed in parallel gradients (arrowheads). Fractions with CITFA3, -6, and -7 cosedimentation peaks are indicated by red lettering and the shifted CITFA2 peaks by asterisks. (C) Immunoblot using the purified polyclonal anti-CITFA3 antibody for detection of PTP-CITFA3 in PF PTPC3ee cells and untagged CITFA3 in PF and BF wild-type (WT) whole-cell lysates. Loading was controlled by the detection of the RNA pol II transcription factor TFIIB. (D) Indirect immunofluorescence microscopy of CITFA3 in noninduced cells and cells in which CITFA1 was silenced for 42 h. Nucleolar areas are indicated by arrows. For the induced cells, an example was chosen in which the putative ESB was detected as an additional spot outside the nucleolus. Bars, 5 μm. (E) Anti-CITFA3 ChIP experiments with noninduced cells or with cells in which CITFA1 was silenced for 1 or 2 days. Occupancy by CITFA3 was determined by qPCR at RRNA and BES promoters and, as a control, at the β-/α-tubulin intergenic region (Tub). The percent precipitation was corrected by subtracting the percent precipitation from negative-control precipitations using a comparable nonspecific immune serum. Each experiment including the negative control was carried out three times independently, and differences in occupancy were statistically analyzed by a two-tailed Student t test assuming equal variance. **, P < 0.01. The error bars indicate standard deviations.
The polyclonal anti-CITFA3 antibody was obtained by raising an immune serum in rats against a recombinant GST-CITFA3 fusion protein that was expressed in E. coli and purified by glutathione affinity chromatography (data not shown) and by affinity purifying the antibody from serum with immobilized antigen. While the calculated molecular mass of CITFA3 is 47 kDa, the antibody recognized a single band of ∼55 kDa in both BF and PF whole-cell lysates (Fig. 6C). This is the correct band, because in cell lysates of the PF line, PTPC3ee, exclusively expressing CITFA3 with an N-terminally fused PTP tag, the band shifted up by ∼20 kDa, the size of the tag. Hence, the polyclonal antibody detected CITFA3 with high specificity. We therefore used this antibody for two further assays. First, we analyzed CITFA3 localization in noninduced cells and in cells in which CITFA1 was silenced for 42 h, because there was a possibility that CITFA1 directs the CITFA complex to the nucleus or, within the nucleus, to the nucleolus and the ESB. We analyzed 70 randomly selected noninduced cells and 83 CITFA1-silenced cells in detail. In all the cells, CITFA3 exhibited subnuclear localization. In DAPI staining, the nucleolus becomes clearly visible as a spherical area of low DNA density resulting in a fainter DAPI stain (41). In 93% of noninduced cells and in 86% of CITFA1-silenced cells, the CITFA3 signal was found predominantly in the nucleolus (Fig. 6D). We also detected an additional, smaller extranucleolar spot in 11% and 6% of noninduced and induced cells, respectively (Fig. 6D). This spot is likely the ESB, since we have shown previously that CITFA7 reliably colocalized with RNA pol I in an extranucleolar compartment of similar size and signal intensity (21, 22). Since the vast majority of cells in this analysis exhibited CITFA3 localization in the nucleolus independently of the CITFA1 knockdown, it appears that CITFA1 is not required for localizing the complex to the sites of RNA pol I transcription.
Second, we conducted an anti-CITFA3 ChIP assay in noninduced and CITFA1-silenced cells (Fig. 6E). For the PCR analysis of precipitated DNA, we used consensus oligonucleotide pairs that recognize either all copies of the RRNA promoter or all BES promoters (27). The antibody effectively precipitated RRNA promoter DNA, whereas BES promoter DNA was enriched 7.8-fold less than the RRNA promoter. This was expected, because we had recently obtained a similar result in anti-CITFA7 ChIPs due to the fact that CITFA predominantly binds the promoter of the active BES and occupies promoters of silent BESs to a much lesser extent. This is in contrast to RRNA promoters, which, according to a ChIP-seq analysis, appear to be generally occupied by CITFA (22). Hence, in this assay, most if not all RRNA promoters were precipitated while BES promoter enrichment was mainly restricted to the active BES. Independent of the enrichment efficiency, CITFA1 silencing reduced CITFA3 occupancy of both promoter types. For efficient RRNA precipitation, this reduction was highly significant (Fig. 6E). Since we showed that CITFA1 has no role in the formation of a stable CITFA complex or in localizing CITFA to the nucleolus, these results strongly indicate that CITFA1 has a specific function in binding of the complex to RRNA and BES promoters.
DISCUSSION
We have established a system in BF T. brucei for specific and efficient gene silencing that is based on targeting an HS that is fused to the mRNA of a gene of interest. We have established two such HSs, namely, the 3′ UTR of the T. cruzi U2AF35 gene and the coding sequence of the large PTP tag. The system is based on the BF cell lines smTc3 and smPTP, which inducibly express Tc3 and PTP hairpin RNAs, respectively. Two consecutive transfections are required to fuse the HS to the gene of interest in one allele and to eliminate the remaining allele. Although these transfections are time-consuming, the system has distinct benefits. Since doxycycline did not alter the rate of proliferation of the smTc3 and smPTP cell lines (Fig. 3) or affect trypanosome morphology, as observed by light microscopy, it can be inferred that Tc3 and PTP dsRNA-derived siRNAs do not target genes that are important for trypanosome culture growth. Furthermore, since both of these cell lines have the regulatable stem-loop vector already integrated, an RRNA-specific position effect is highly unlikely. Moreover, the vectors pCITFA7-HA-Tc3-BLA (see Fig. S1 in the supplemental material) and pCITFA7-PTP-BLAv2 (see Fig. S2 in the supplemental material) offer a straightforward cloning strategy for fusing the HS to the gene of interest. C-terminal coding sequences can be amplified and inserted into the ApaI and NotI restriction sites in a single step. The only requirement for successful targeting of the plasmid to an endogenous allele is a restriction site within the gene coding sequence that is surrounded by at least 100 bp of coding sequence on either side and that can be used to linearize the plasmid. Since this system is based on targeting the same sequence independently of the gene of interest, we anticipate that it can provide unambiguous gene-silencing data in those cases where targeting endogenous sequences was not successful or was inefficient. Finally, it is likely that the system can be implemented in PF 29-13 cells, as well. However, the knockout of wild-type alleles in 29-13 cells would have to be accomplished with the PURO marker instead of the HYG marker, as shown in Fig. 1, because 29-13 cells already harbor HYG.
The system also has its limitations, however. First, it can be applied only to single-copy genes, because it is obligatory to produce a cell line that exclusively expresses the mRNA of interest as an HS fusion and no wild-type mRNA. Second, the system, as presented here, will not function if the expression of both alleles is required for trypanosome viability and proliferation. It should be noted, though, that we have so far generated many viable BF and PF cell lines that expressed various essential nuclear proteins exclusively as PTP fusions from a single allele. This has included RNA pol subunits, various transcription and RNA-splicing factors, and kinases, suggesting that haploinsufficiency rarely affects trypanosome viability in culture (30, 36, 42–44). If haploinsufficiency is a concern, it may be possible to integrate one HS plasmid into each endogenous allele instead of knocking one allele out. For this, the blasticidin marker gene has to be replaced with the puromycin marker, which can be achieved by a single PCR amplification and cloning step, since either the selectable marker cassettes or the coding region can be excised by restriction digests (see Fig. S1 and S2 in the supplemental material). Third, the HS may negatively affect the expression of the mRNA or the functionality of the resulting protein. If the tag interferes with the function of the protein, there are two options. The PTP tag can be fused to the N terminus with our published pN-PURO-PTP vector (26). Although in the N-terminal PTP tag the ProtA and ProtC domains have different N- to C-terminal sequence orders, the PTP dsRNA produced in smPTP cells effectively silenced a CITFA2 gene in which the PTP tag sequence was inserted after the initiation codon (J. K. Kirkham and A. Günzl, unpublished data). Alternatively, Tc3 can be employed without the HA tag, which was used here to enable specific detection of the protein translated from the fusion mRNA. In this case the 27-bp-long HA coding sequence needs to be removed from pCITFA7-HA-Tc3-BLA. Furthermore, if the gene of interest produces an mRNA whose regulation through its 3′ UTR is critical for cell viability, then replacing it either with Tc3 or with the T. brucei RPA1 3′ UTR in pC-PTP-BLA may be deleterious. In this case, tagging the protein N terminally with the PTP tag or replacing the RPA1 3′ gene flank with that of the gene of interest in pC-PTP-BLA may be a solution.
Recently, a conditional gene knockout system has been established in T. brucei that employs Cre recombinase and loxP sites (45, 46). As with our approach, this system requires the manipulation of both gene alleles, e.g., it also depends on two consecutive transfection steps. The system offers instantaneous removal of the gene of interest in the genome and an unambiguous assessment of gene essentiality. However, in contrast to our system, this approach is irreversible, preventing the analysis of temporary gene knockdowns or the titration of the gene-silencing level.
Finally, we have successfully employed this new gene-silencing approach to evaluate CITFA1's role in the CITFA complex. We could unambiguously show that CITFA1 is essential for BF viability in culture and, as expected, has a vital role in rRNA and VSG mRNA expression. After CITFA2 and CITFA7, this is the third CITFA subunit whose knockdown led to rapid trypanosome death in culture, underscoring the indispensability of this transcription initiation factor for trypanosome viability. Furthermore, we showed that CITFA1 depletion strongly reduced CITFA occupancy at the RRNA and BES promoters. Since we could not detect mislocalization of CITFA3 or the loss of CITFA subunits, which are indicative of complex disruption as a consequence of CITFA1 silencing, it appears that CITFA1 has a direct role in binding to promoter DNA. Most interestingly, CITFA3 remained localized to the nucleolus/ESB after CITFA1 knockdown, which suggests that subnuclear CITFA localization is not mediated by its binding to DNA. This finding further supports a recently described model in which DNA-independent concentration and confinement of CITFA to the nucleolus and ESB restrict maximal transcription initiation by RNA pol I to these compartments (22).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Barbara Burleigh (Harvard University) for providing T. cruzi genomic DNA.
This work was supported by grants R01 AI059377 and R01 AI AI073300 to A.G. from the National Institute of Allergy and Infectious Diseases (NIAID) of the U.S. National Institutes of Health (NIH).
Footnotes
Published ahead of print 11 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00014-14.
REFERENCES
- 1.Ngo H, Tschudi C, Gull K, Ullu E. 1998. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc. Natl. Acad. Sci. U. S. A. 95:14687–14692. 10.1073/pnas.95.25.14687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kolev NG, Tschudi C, Ullu E. 2011. RNA interference in protozoan parasites: achievements and challenges. Eukaryot. Cell 10:1156–1163. 10.1128/EC.05114-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wirtz E, Leal S, Ochatt C, Cross GAM. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 99:89–101. 10.1016/S0166-6851(99)00002-X [DOI] [PubMed] [Google Scholar]
- 4.Wirtz E, Clayton C. 1995. Inducible gene expression in trypanosomes mediated by a prokaryotic repressor. Science 268:1179–1183. 10.1126/science.7761835 [DOI] [PubMed] [Google Scholar]
- 5.LaCount DJ, Bruse S, Hill KL, Donelson JE. 2000. Double-stranded RNA interference in Trypanosoma brucei using head-to-head promoters. Mol. Biochem. Parasitol. 111:67–76. 10.1016/S0166-6851(00)00300-5 [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Morris JC, Drew ME, Englund PT. 2000. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 275:40174–40179. 10.1074/jbc.M008405200 [DOI] [PubMed] [Google Scholar]
- 7.Morris JC, Wang Z, Drew ME, Englund PT. 2002. Glycolysis modulates trypanosome glycoprotein expression as revealed by an RNAi library. EMBO J. 21:4429–4438. 10.1093/emboj/cdf474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi H, Djikeng A, Mark T, Wirtz E, Tschudi C, Ullu E. 2000. Genetic interference in Trypanosoma brucei by heritable and inducible double-stranded RNA. RNA 6:1069–1076. 10.1017/S1355838200000297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bastin P, Ellis K, Kohl L, Gull K. 2000. Flagellum ontogeny in trypanosomes studied via an inherited and regulated RNA interference system. J. Cell Sci. 113:3321–3328 [DOI] [PubMed] [Google Scholar]
- 10.Tschudi C, Djikeng A, Shi H, Ullu E. 2003. In vivo analysis of the RNA interference mechanism in Trypanosoma brucei. Methods 30:304–312. 10.1016/S1046-2023(03)00047-1 [DOI] [PubMed] [Google Scholar]
- 11.Günzl A, Bruderer T, Laufer G, Schimanski B, Tu LC, Chung HM, Lee PT, Lee MG. 2003. RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei. Eukaryot. Cell 2:542–551. 10.1128/EC.2.3.542-551.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biebinger S, Rettenmaier S, Flaspohler J, Hartmann C, Pena Diaz J, Wirtz LE, Hotz HR, Barry JD, Clayton C. 1996. The PARP promoter of Trypanosoma brucei is developmentally regulated in a chromosomal context. Nucleic Acids Res. 24:1202–1211. 10.1093/nar/24.7.1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandenburg J, Schimanski B, Nogoceke E, Nguyen TN, Padovan JC, Chait BT, Cross GA, Günzl A. 2007. Multifunctional class I transcription in Trypanosoma brucei depends on a novel protein complex. EMBO J. 26:4856–4866. 10.1038/sj.emboj.7601905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sunter J, Wickstead B, Gull K, Carrington M. 2012. A new generation of T7 RNA polymerase-independent inducible expression plasmids for Trypanosoma brucei. PLoS One 7:e35167. 10.1371/journal.pone.0035167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inoue M, Nakamura Y, Yasuda K, Yasaka N, Hara T, Schnaufer A, Stuart K, Fukuma T. 2005. The 14-3-3 proteins of Trypanosoma brucei function in motility, cytokinesis, and cell cycle. J. Biol. Chem. 280:14085–14096. 10.1074/jbc.M412336200 [DOI] [PubMed] [Google Scholar]
- 16.Lacomble S, Portman N, Gull K. 2009. A protein-protein interaction map of the Trypanosoma brucei paraflagellar rod. PLoS One 4:e7685. 10.1371/journal.pone.0007685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalidas S, Li Q, Phillips MA. 2011. A Gateway(R) compatible vector for gene silencing in bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 178:51–55. 10.1016/j.molbiopara.2011.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alibu VP, Storm L, Haile S, Clayton C, Horn D. 2005. A doubly inducible system for RNA interference and rapid RNAi plasmid construction in Trypanosoma brucei. Mol. Biochem. Parasitol. 139:75–82. 10.1016/j.molbiopara.2004.10.002 [DOI] [PubMed] [Google Scholar]
- 19.Alsford S, Kawahara T, Glover L, Horn D. 2005. Tagging a T. brucei RRNA locus improves stable transfection efficiency and circumvents inducible expression position effects. Mol. Biochem. Parasitol. 144:142–148. 10.1016/j.molbiopara.2005.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wickstead B, Ersfeld K, Gull K. 2002. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol. Biochem. Parasitol. 125:211–216. 10.1016/S0166-6851(02)00238-4 [DOI] [PubMed] [Google Scholar]
- 21.Nguyen TN, Nguyen BN, Lee JH, Panigrahi AK, Günzl A. 2012. Characterization of a novel class I transcription factor A (CITFA) subunit that is indispensable for transcription by the multifunctional RNA polymerase I of Trypanosoma brucei. Eukaryot. Cell 11:1573–1581. 10.1128/EC.00250-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen TN, Müller LS, Park SH, Siegel TN, Günzl A. 2014. Promoter occupancy of the basal class I transcription factor A differs strongly between active and silent VSG expression sites in Trypanosoma brucei. Nucleic Acids Res. 42:3164–3176. 10.1093/nar/gkt1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, Brooks K, Churcher C, Fahkro S, Goodhead I, Heath P, Kartvelishvili M, Mungall K, Harris D, Hauser H, Sanders M, Saunders D, Seeger K, Sharp S, Taylor JE, Walker D, White B, Young R, Cross GA, Rudenko G, Barry JD, Louis EJ, Berriman M. 2008. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One 3:e3527. 10.1371/journal.pone.0003527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaves I, Zomerdijk J, Dirks MA, Dirks RW, Raap AK, Borst P. 1998. Subnuclear localization of the active variant surface glycoprotein gene expression site in Trypanosoma brucei. Proc. Natl. Acad. Sci. U. S. A. 95:12328–12333. 10.1073/pnas.95.21.12328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Navarro M, Gull K. 2001. A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414:759–763. 10.1038/414759a [DOI] [PubMed] [Google Scholar]
- 26.Schimanski B, Nguyen TN, Günzl A. 2005. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot. Cell 4:1942–1950. 10.1128/EC.4.11.1942-1950.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park SH, Nguyen TN, Kirkham JK, Lee JH, Günzl A. 2011. Transcription by the multifunctional RNA polymerase I in Trypanosoma brucei functions independently of RPB7. Mol. Biochem. Parasitol. 180:35–42. 10.1016/j.molbiopara.2011.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cross M, Wieland B, Palfi Z, Günzl A, Röthlisberger U, Lahm HW, Bindereif A. 1993. The trans-spliceosomal U2 snRNP protein 40K of Trypanosoma brucei: cloning and analysis of functional domains reveals homology to a mammalian snRNP protein. EMBO J. 12:1239–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schimanski B, Brandenburg J, Nguyen TN, Caimano MJ, Günzl A. 2006. A TFIIB-like protein is indispensable for spliced leader RNA gene transcription in Trypanosoma brucei. Nucleic Acids Res. 34:1676–1684. 10.1093/nar/gkl090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen TN, Schimanski B, Günzl A. 2007. Active RNA polymerase I of Trypanosoma brucei harbors a novel subunit essential for transcription. Mol. Cell. Biol. 27:6254–6263. 10.1128/MCB.00382-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Günzl A, Cross M, Bindereif A. 1992. Domain structure of U2 and U4/U6 small nuclear ribonucleoprotein particles from Trypanosoma brucei: identification of trans-spliceosomal specific RNA-protein interactions. Mol. Cell. Biol. 12:468–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Günzl A. 2010. The pre-mRNA splicing machinery of trypanosomes: complex or simplified? Eukaryot. Cell 9:1159–1170. 10.1128/EC.00113-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brandao A, Jiang T. 2009. The composition of untranslated regions in Trypanosoma cruzi genes. Parasitol. Int. 58:215–219. 10.1016/j.parint.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 34.Vazquez M, Atorrasagasti C, Bercovich N, Volcovich R, Levin MJ. 2003. Unique features of the Trypanosoma cruzi U2AF35 splicing factor. Mol. Biochem. Parasitol. 128:77–81. 10.1016/S0166-6851(03)00007-0 [DOI] [PubMed] [Google Scholar]
- 35.Vazquez MP, Mualem D, Bercovich N, Stern MZ, Nyambega B, Barda O, Nasiga D, Gupta SK, Michaeli S, Levin MJ. 2009. Functional characterization and protein-protein interactions of trypanosome splicing factors U2AF35, U2AF65 and SF1. Mol. Biochem. Parasitol. 164:137–146. 10.1016/j.molbiopara.2008.12.009 [DOI] [PubMed] [Google Scholar]
- 36.Lee JH, Cai G, Panigrahi AK, Dunham-Ems S, Nguyen TN, Radolf JD, Asturias FJ, Günzl A. 2010. A TFIIH-associated mediator head is a basal factor of small nuclear spliced leader RNA gene transcription in early-diverged trypanosomes. Mol. Cell. Biol. 30:5502–5513. 10.1128/MCB.00966-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stearns DJ, Kurosawa S, Sims PJ, Esmon NL, Esmon CT. 1988. The interaction of a Ca2+-dependent monoclonal antibody with the protein C activation peptide region. Evidence for obligatory Ca2+ binding to both antigen and antibody. J. Biol. Chem. 263:826–832 [PubMed] [Google Scholar]
- 38.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030–1032. 10.1038/13732 [DOI] [PubMed] [Google Scholar]
- 39.Arhin GK, Shen S, Ullu E, Tschudi C. 2004. A PCR-based method for gene deletion and protein tagging in Trypanosoma brucei. Methods Mol. Biol. 270:277–286. 10.1385/1-59259-793-9:277 [DOI] [PubMed] [Google Scholar]
- 40.Schimanski B, Nguyen TN, Günzl A. 2005. Characterization of a multisubunit transcription factor complex essential for spliced-leader RNA gene transcription in Trypanosoma brucei. Mol. Cell. Biol. 25:7303–7313. 10.1128/MCB.25.16.7303-7313.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daniels JP, Gull K, Wickstead B. 2012. The trypanosomatid-specific N terminus of RPA2 is required for RNA polymerase I assembly, localization, and function. Eukaryot. Cell 11:662–672. 10.1128/EC.00036-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Lee JH, Chu F, Burlingame AL, Günzl A, Wang CC. 2008. Identification of a novel chromosomal passenger complex and its unique localization during cytokinesis in Trypanosoma brucei. PLoS One 3:e2354. 10.1371/journal.pone.0002354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luz Ambrósio D, Lee JH, Panigrahi AK, Nguyen TN, Cicarelli RM, Günzl A. 2009. Spliceosomal proteomics in Trypanosoma brucei reveal new RNA splicing factors. Eukaryot. Cell 8:990–1000. 10.1128/EC.00075-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Badjatia N, Ambrósio DL, Lee JH, Günzl A. 2013. Trypanosome cdc2-related kinase 9 controls spliced leader RNA cap4 methylation and phosphorylation of RNA polymerase II subunit RPB1. Mol. Cell. Biol. 33:1965–1975. 10.1128/MCB.00156-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim HS, Park SH, Günzl A, Cross GA. 2013. MCM-BP is required for repression of life-cycle specific genes transcribed by RNA polymerase I in the mammalian infectious form of Trypanosoma brucei. PLoS One 8:e57001. 10.1371/journal.pone.0057001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim HS, Li Z, Boothroyd C, Cross GA. 2013. Strategies to construct null and conditional null Trypanosoma brucei mutants using Cre-recombinase and loxP. Mol. Biochem. Parasitol. 191:16–19. 10.1016/j.molbiopara.2013.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.