

Abstract
Mammalian DREAM is a conserved protein complex that functions in cellular quiescence. DREAM contains an E2F, a retinoblastoma (RB)-family protein, and the MuvB core (LIN9, LIN37, LIN52, LIN54, and RBBP4). In mammals, MuvB can alternatively bind to BMYB to form a complex that promotes mitotic gene expression. Because BMYB-MuvB is essential for proliferation, loss-of-function approaches to study MuvB have generated limited insight into DREAM function. Here, we report a gene-targeted mouse model that is uniquely deficient for DREAM complex assembly. We have targeted p107 (Rbl1) to prevent MuvB binding and combined it with deficiency for p130 (Rbl2). Our data demonstrate that cells from these mice preferentially assemble BMYB-MuvB complexes and fail to repress transcription. DREAM-deficient mice show defects in endochondral bone formation and die shortly after birth. Micro-computed tomography and histology demonstrate that in the absence of DREAM, chondrocytes fail to arrest proliferation. Since DREAM requires DYRK1A (dual-specificity tyrosine phosphorylation-regulated protein kinase 1A) phosphorylation of LIN52 for assembly, we utilized an embryonic bone culture system and pharmacologic inhibition of (DYRK) kinase to demonstrate a similar defect in endochondral bone growth. This reveals that assembly of mammalian DREAM is required to induce cell cycle exit in chondrocytes.
INTRODUCTION
Cellular differentiation is generally controlled by transcriptional activation or repression of specific genes. Consequently, a host of different molecular genetic events can shape the properties of cells during development. Recent evidence indicates that an evolutionarily conserved protein complex known as DREAM is capable of regulating diverse gene expression programs, thereby unifying many disparate events in development into a single molecular machine (1, 2).
The DREAM complex was isolated, and its composition was determined from a number of different model organisms. Studies of aberrant growth factor signaling in Caenorhabditis elegans lead to the discovery of complementation groups that contribute to a multivulval (Muv) phenotype (3). Mutation of any two of the synthetic multivulval (synMuv) group A, B, or C genes resulted in worms with elevated numbers of vulvae (4). Group B contains a number of genes (the Lin-9, Lin-37, Lin-52, Lin-53/RBBP4, and Lin-54 genes) whose encoded proteins form the MuvB core complex (5–7). In addition, worm homologues of retinoblastoma protein (RB), E2F, and DP are also group B members (8, 9). The MuvB core was also found to interact with MYB in transcriptional control of cell cycle progression in fruit flies (6, 7). Isolation of MYB and RB revealed that they copurify with MuvB proteins, and this has formed the basis of the DREAM complex (Drosophila RB, E2F, and MuvB). In some organisms it also contains epigenetic readers and writers such as histone deacetylases (HDACs) and L3MBT (6, 7, 10). The model that has emerged is one in which the DREAM complex can confer both positive and negative regulation of transcription, under proliferating and differentiated conditions, in a wide variety of cell and tissue types in Drosophila and C. elegans (11).
DREAM was also identified in mammalian systems from immunoprecipitation (IP) and mass spectrometry approaches (12–14). The mammalian DREAM complex contains p107 or p130, E2F4 or E2F5, and DP1 or DP2, as well as the MuvB core (12, 13, 15). In contrast, the mammalian BMYB-MuvB complex contains BMYB and FOXM1 but not the RB, E2F, and DP proteins (14, 16). These data have revealed that BMYB-MuvB and DREAM are distinct, with RB family proteins being excluded from BMYB-containing complexes and vice versa (12, 14, 17). These cell culture-based studies indicate that BMYB-MuvB in mammals is critical for expression of cell cycle-regulated genes in S phase, and its absence results in mitotic defects (15, 16, 18, 19). On the other hand, a form of DREAM containing p130 is largely present at E2F target genes in quiescence and functions to repress their expression, thus contributing to growth arrest (12). The switch that regulates the MuvB transition from BMYB-MuvB to DREAM upon cell cycle exit is dependent on the phosphorylation of LIN52 by the DYRK1A (dual-specificity tyrosine phosphorylation-regulated protein kinase 1A) kinase that stimulates DREAM assembly (17). Understanding of the role of the DREAM complex in mammalian physiology has been elusive. Elimination of the MuvB component LIN9 cripples DREAM and BMYB-MuvB simultaneously and blocks proliferation (20). Similarly, elimination of RB family proteins such as p107 or p130 not only blocks DREAM formation but may also influence E2F regulation (21–23). In short, multifunctionality of DREAM components prevents its study by classic loss of function approaches.
In a mixed 129-BL6 genetic background, deficiency of p107 or p130 alone has little effect in mice (24, 25). Combined deficiency of p107 and p130 causes defects in chondrocyte proliferation and differentiation, leading to neonatal lethality (24). Furthermore, cell culture-based assays of chondrocyte differentiation reveal that p107 and p130 are essential for arrest and the commitment to differentiate (26). In a mixed 129-BALB/c background, deficiency for either p107 or p130 alone causes overt deregulation of cell proliferation (27, 28). Notably, p130-deficient mice die at midgestation, with ectopic proliferation and apoptosis in neurogenic and myogenic precursors (27). These defects may be attributable to deficiency of DREAM function, but these distinctions are not possible at present. One challenge that has become apparent in these studies is that loss of p130 leads to elevated expression of p107 and vice versa (29, 30). Indeed, work on DREAM has demonstrated that either p130 or p107 can bind to the MuvB core and that when p130 was depleted by RNA interference (RNAi), p107 binds to the MuvB core during quiescence (12). In sum, competition between BMYB and RB family proteins in contacting the MuvB core, as well as redundancy between p107 and p130 proteins, has limited insight into DREAM function by classical loss-of-function approaches in mammals.
In order to investigate the role of DREAM in mammalian physiology, we utilized the sensitivity to viral oncoproteins such as T antigen (TAg) and E7 as a means to map the interaction between the MuvB core and the LXCXE binding cleft of p107. Using gene targeting, we created a form of p107 such that its LXCXE binding cleft is incapable of binding to MuvB. This mutant allele is called p107ΔD because it is DREAM assembly deficient, and it was combined with a p130 knockout to prevent complex formation through other RB family proteins. Fibroblast cultures from p107ΔD/ΔD; p130−/− embryos demonstrate that MuvB becomes preferentially associated with BMYB, even in quiescence, and repression of DREAM cell cycle target genes is lost. These animals have distinct bone morphogenic defects characterized by missing ossicles, as well as abnormal occipital and long bones, and most of these mice die neonatally. A detailed histological analysis of the growth plate of long bones as well as chondrocyte abundance indicates overproliferation of these cells, with a subsequent delay in endochondral ossification. Lastly, we inhibited DYRK kinases to block DREAM assembly in embryonic bone cultures which also resulted in shorter bones. These data suggest a unique connection between DREAM assembly and chondrocyte proliferative control.
MATERIALS AND METHODS
Recombinant proteins and interaction assays.
Glutathione S-transferase (GST)–p107LP (where LP is large pocket) expresses amino acids 385 to 1068 of the human p107 in Escherichia coli. GST-RBLP expressed amino acids 379 to 928. All GST fusions were purified using standard procedures. Nuclear extracts were prepared as previously described (31). Briefly, mouse embryonic fibroblasts (MEFs) were plated at a density of 2 × 106 cells/15-cm dish and allowed to attach overnight in growth medium (Dulbecco's modified Eagle's medium [DMEM] plus 10% fetal bovine serum [FBS]). For serum starvation conditions the cells were washed three times with phosphate-buffered saline (PBS) and fed with DMEM containing 0.1% FBS. The cells were incubated for 72 h, after which they were washed with PBS, scraped, collected by centrifugation, suspended in hypotonic lysis buffer (20 mM Tris-Cl, pH 7.5, 10 mM KCl, 3 mM MgCl2, 1 mM EDTA, 250 μM Na3VO4, 5 mM NaF, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 1 mM dithiothreitol [DTT]), and incubated on ice for 5 min. NP-40 was added to a final concentration of 0.05%, and cells were incubated on ice for a further 5 min. Nuclei were recovered by centrifugation at 1,700 × g for 10 min, followed by three sequential washes with hypotonic lysis buffer containing 0.05% NP-40. The purified nuclei were suspended in GSE buffer (20 mM Tris-Cl, pH 7.5, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM NaF, 0.1 mM Na3VO4, 25% glycerol, 5 mg/ml leupeptin, 5 mg/ml aprotinin, 1 mM DTT) and stored at −80°C until use. To prepare whole-cell extracts, asynchronously growing cells were washed with ice-cold PBS and collected as above. The cells were resuspended in lysis buffer (20 mM Tris-Cl, pH 7.5, 100 mM NaCl, 5% glycerol, 1% NP-40, 0.1 mM Na3VO4, 1 mM NaF, 5 μg/ml leupeptin, 5 μg/ml aprotinin, and 1 mM PMSF) and incubated on ice for 10 min, followed by centrifugation in the cold for 15 min at 20,000 × g to remove detergent-insoluble protein. Protein concentrations were determined by the Bradford method, and the extracts were used immediately for immunoprecipitations.
Purified GST fusion proteins were dialyzed against 20 mM Tris-Cl, pH 7.5, 100 mM KCl, 10% glycerol, 0.02% NP-40, and 1 mM DTT for 48 h to remove glutathione. Nuclei were thawed on ice, and nuclear envelopes and insoluble matrix proteins were removed by centrifugation at 20,800 × g at 4°C for 20 min. Protein concentrations in nuclear extracts were quantitated, and the NaCl concentration was brought to 200 mM by the addition of no-salt immunoprecipitation (IP) wash buffer (20 mM Tris-Cl, pH 7.5, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM NaF, 0.1 mM Na3VO4, 0.1% NP-40, 1 mM DTT, 5 μg/ml leupeptin, and 5 μg/ml aprotinin). The purified GST-p107LP and GST-p107ΔDLP (5 μg) were added to 500 μg of nuclear extract prepared as described above, together with 20 μl of glutathione-Sepharose beads that had been equilibrated in IP wash buffer (20 mM Tris-Cl, pH 7.5, 200 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM NaF, 0.1 mM Na3VO4, 0.1% NP-40, 1 mM DTT, 5 μg/ml aprotinin, 5 μg/ml leupeptin) and incubated with constant mixing overnight at 4°C. The beads were collected by centrifugation and washed twice with IP wash buffer, and protein complexes were eluted in SDS-PAGE sample buffer containing 25 mM DTT. Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) using standard protocols, and proteins were identified by immunoblotting using the antibodies listed below.
DREAM complex proteins were coimmunoprecipitated from nuclear extracts prepared as described above (1 mg protein) with an antibody to LIN37 (5 μg). Following the addition of 20 μl of protein G-coupled Dynabeads (30 mg/ml), the immunoprecipitations were incubated at 4°C overnight with constant mixing. The Dynabeads with bound proteins were collected, washed three times with IP wash buffer, and eluted with SDS-PAGE sample buffer containing 25 mM DTT. The immune complexes were analyzed by SDS-PAGE/Western blotting as described above.
To identify BMYB in coimmunoprecipitations, nuclear extracts from MEFs of the indicated genetic background were prepared from cells maintained under conditions of asynchronous growth or growth arrest (with or without FBS). Nuclear extracts were prepared as described above, and the MuvB core was immunoprecipitated from 1.5 mg of protein using an antibody to LIN37 (5 μg). Following the addition of 20 μl of protein G Dynabeads, immunoprecipitations were incubated overnight at 4°C with constant mixing, washed, eluted in SDS-PAGE sample buffer, and analyzed by SDS-PAGE/Western blotting using an antibody to BMYB (see below).
The interaction between p107 and CDK2 was assessed by coimmunoprecipitation from asynchronously growing MEFs using an antibody against p107. Briefly, 5 μg of an anti-p107 antibody was added to 1.5 mg of whole-cell extract protein (prepared as described above) and incubated overnight with constant mixing. The next day, 20 μl of protein G Dynabeads was added to each immunoprecipitation, and the incubation continued with mixing for a further 2 h. Protein complexes bound to the Dynabeads were collected as described above and analyzed by SDS-PAGE/Western blotting using an antibody to Cdk2.
CDK2 complexes were immunoprecipitated as above and washed twice in IP wash buffer, followed by two washes in kinase buffer (50 mM Tris pH 7.5, 10 mM MgCl2, 1 mM DTT), and then resuspended in kinase buffer. The kinase activity assay was measured by incubating CDK2 complexes with 1 μg of recombinant histone H1 (sc-221729; Santa Cruz) and 10 μCi of 32P-labeled ATP (PerkinElmer) for 20 min at 30°C. Reactions were ended by the addition of 12.5 μl of 5× SDS-PAGE sample buffer and boiling at 95°C for 5 min. The eluted material was resolved by electrophoresis on a sodium dodecyl sulfate–15% polyacrylamide gel (SDS-PAGE). The gel was then dried and exposed to film overnight at −80°C.
Electrophoretic mobility shift assays (EMSAs) were performed essentially as described by Hurford et al. (29) using nuclear extracts from proliferating cells prepared as described above.
Antibodies.
Rabbit anti-LIN9, anti-LIN37, anti-LIN52, and anti-LIN54 have been previously published (12). The specificity of antibodies for murine LIN proteins and the expected sizes of each were confirmed by comparing GST-p107LP pulldowns alongside nuclear extracts (data not shown). Antihemagglutinin (anti-HA) hybridoma culture supernatant was used as a negative control for EMSAs. Rabbit anti-E2F4 (A20; Santa Cruz), rabbit anti-p107 (C-18; Santa Cruz), mouse anti-pRB (G3-245; BD Pharmingen), rabbit anti-p130 (C-20; Santa Cruz), rabbit anti-β-actin (A2066; Sigma), rabbit anti-SP1 (H-225; Santa Cruz), rabbit anti-BMYB (H-115; Santa Cruz), mouse anti-Cdk2 (D-12; Santa Cruz), rabbit anti-HDAC1 (H-51; Santa Cruz), rabbit anti-HDAC2 (H-54; Santa Cruz), and mouse anti-PCNA (2586; Cell Signaling) were used as indicated.
Generation of mice and genotyping.
A targeting vector for the Rbl1 gene (that encodes p107) was generated from PCR-amplified fragments surrounding exon 20. Genomic DNA from bacterial artificial chromosome (BAC) clone 222M2 from the RPCI-22 library was obtained from The Centre for Applied Genomics (Toronto, Canada). The linearized plasmid was electroporated into embryonic stem (ES) cells by the Transgenic Core Facility at the Van Andel Research Institute (Grand Rapids, MI). Targeted clones were used for blastocyst injection, and chimeric animals were bred with EIIa-cre transgenic mice (32) to generate F1 progeny without the neo selectable marker. These mice were intercrossed to obtain homozygous p107ΔD/ΔD mutant mice and to remove the cre transgene. Rbl2 knockout mice were obtained from Jackson Laboratory on a mixed 129 and B6 background (24). Crosses of p107ΔD/ΔD; p130+/− mice with either p107ΔD/+; p130−/− mice or p107ΔD/ΔD; p130+/− mice were used to generate progeny for bone staining, histology, micro-computed tomography (micro-CT) experiments, and preparation of fibroblasts. Female mice were monitored for vaginal mucous plugs each morning to determine the date of conception. At the appropriate developmental time point, tail or organ tissue from embryos was processed to obtain genomic DNA. PCR genotyping was performed using primers that amplify across the site of LoxP integration in intron 20 to generate 490-bp wild-type and 580-bp mutant products. All mice were housed and handled as approved by the University of Western Ontario animal use subcommittee and in accordance with the Canadian Council on Animal Care guidelines.
Fibroblast culture and gene expression measurements.
Primary murine embryonic fibroblasts (MEFs) were generated from day 13.5 embryos (E13.5) using standard procedures and cultured in DMEM with 10% FBS (29). Where indicated, serum-starved cells were cultured in 0.1% FBS for 72 h to induce DREAM assembly and cell cycle arrest. Cell cycle phase proportions were determined by propidium iodide and bromodeoxyuridine (BrdU) staining, followed by flow cytometry (33). RNA was isolated from proliferating or serum-starved cultures using TRIzol reagent as recommended by the manufacturer (Life Technologies). Total RNA was prepared from proliferating or serum-starved cell cultures using TRIzol reagent (Life Technologies), and 2 μg was reverse transcribed using a SuperScript III first-strand synthesis system (Invitrogen) to create cDNA. Real-time PCR measurements used to evaluate the transcript levels of specific DREAM target genes were made on a CFX Connect real-time PCR system from Bio-Rad using iQ SYBR green supermix (Bio-Rad). Each gene was normalized to the expression of Rplp0 (acidic ribosomal phosphoprotein P0). The mouse primer pairs for the indicated genes are as follows: Rplp0, 5′-CAAGAACACCATGATGCGCA-3′ and 5′-GCCAACAGCATATCCCGAATC-3′; Mybl2, 5′-AACAGTGGACGCTGATAGCCA-3′ and 5′-TTCACAGCATTGTCCGTCCTC-3′; Cdc2, 5′-AGAAGGTACTTACGGTGTGGT-3′ and 5′-GAGAGATTTCCCGAATTGCAGT-3′; Ccna2, 5′-CGGAGCAAGAAAACCACTGAC-3′ and 5′-ACCAGCCAGTCCACAAGGATG-3′; Cdc6, 5′-AATCCATCCTGCTGAATTGC-3′ and 5′-ATACATCCTGCCCTTTGCTG-3′.
Chromatin immunoprecipitation.
Mouse embryonic fibroblasts of the specified genotypes were cultured in DMEM containing 0.1% FBS for 72 h to induce quiescence. Cells were fixed in 1% formaldehyde in PBS for 10 min at room temperature. Chromatin was extracted and used for immunoprecipitations as described before (34). Fifty micrograms of precleared chromatin was incubated with 2.5 μg of rabbit IgG or anti-p107 antibody overnight at 4°C with gentle rotation. Real-time PCR amplification was performed using iQSYBRGreen master mix on a Bio-Rad CFX Connect real-time system. Primer sequences are as follows: MYBl2 (transcription start site [TSS]) forward primer, 5′-ACGCACTTGGCGGGAGATAG-3′; MYBl2 (TSS) reverse primer, 5′-CTCAGGCGTCAGCGTGTCT-3′; MYBl2 (−1 kb) forward primer, 5′-GCCTGAGCCTAAAGGGCATT-3′; and MYBl2 (−1 kb) reverse primer, 5′-TCTGATGGCAAGGGTTGTCTC-3′.
Alcian blue and alizarin red whole-mount staining.
Pregnant mice were euthanized at 18.5 days postconception (E18.5). Embryos were excised from the uterus and stained using the protocol of McLeod for mineralized bone and cartilage (35). Stained embryos were stored in a 1:1 glycerol–70% ethanol solution at room temperature. Bones were imaged using a Zeiss Stemi 2000-C Stereomicroscope, a Qcam camera system, and EyeImage, version 3.0, software. Measurement of the bones in both forelimbs and both hind limbs of 8 heterozygotes and 10 mutant embryos were made using Volocity, version 5.0, software. Specific landmarks, such as the medial epicondyle and the medial anatomic neck of the humerus, were chosen for each bone to ensure that measurements were consistent between animals.
Histological analyses.
At E18.5 embryos were fixed in 10% formalin acetate for 7 days and then stored in 70% ethanol. The forelimbs and hind limbs were embedded in paraffin and sectioned into 5-μm slices. Hematoxylin and eosin (H&E) stains were used to visualize the bones and connective tissues. Safranin O/Fast Green staining was performed as described previously (36). Slides for 4′,6′-diamidino-2-phenylindole (DAPI) immunofluorescence were cleared of paraffin using xylene and then rehydrated with ethanol and H2O. Slides were sequentially washed in PBS and dried. VectaShield (100 μl) with DAPI was placed on each slide and covered with a coverslip. Specimens were viewed under a Zeiss Axioskop 40 microscope, and images were taken using a Spotflex camera. DAPI-stained nuclei in the resting zone were counted using Volocity, version 5.0, software. A total of 40 fields of view (275 μm by 275 μm) were counted.
PCNA staining was performed essentially as described by Gillespie et al. (37). Briefly, sections were rehydrated as for Safranin-O, and endogenous peroxidases were blocked with 3% hydrogen peroxide in methanol and rinsed once with water; antigens were retrieved in 0.1% Triton X-100 for 20 min, washed once with water, blocked for 2 h in 5% goat serum in PBS at room temperature, and incubated with mouse anti-PCNA diluted 1:500 in 5% goat serum at 4°C overnight. Sections were then washed three times in PBS, incubated with horseradish peroxidase-coupled goat anti-mouse antibody (sc-2005; Santa Cruz) diluted 1:200 in 5% goat serum at 4°C for 1 h, washed three times in PBS, and visualized with diaminobenzidine (DAB) substrate (Dako).
Micro-CT parameters and analysis.
Each sample contained two weight-matched, formalin-fixed embryos (one p107ΔD/+; p130+/− heterozygote and one p107ΔD/ΔD; p130−/− mutant) set in 1.5% agar in a 50-ml polypropylene centrifuge tube. The five samples were scanned for 15 h at 80 peak kV (kVp) and 80 μA on an eXplore Locus SP cone beam micro-CT (GE Healthcare) and reconstructed with isotropic 26-μm voxels as previously reported (38). Data were linearly rescaled into Houndsfield units (HU) using air, water, and SB3 (cortical bone mimic) internal calibrators. Volume data sets were rebinned 2 by 2 by 2 to further decrease noise, resulting in an effective voxel size of 52 μm for quantitative investigation. Analysis was performed using MicroView, version 2.1.2 (GE Healthcare), to determine bone volume (BV) and bone mineral content (BMC). Bone mineral density (BMD) was calculated from the BMC and BV. Subvolumes of the original, full-resolution (26 μm) reconstructions were used to perform qualitative analysis of trabeculae.
Tibia organ cultures.
Tibiae were isolated from E15.5 CD1 mice and cultured as described previously (39, 40). Cultures were performed for 6 days in the presence of dimethyl sulfoxide (DMSO) or the Dyrk inhibitor harmine (5 μM), with medium change every second day. Lengths of tibiae were determined at the beginning and end of culture using an eyepiece in a stereomicroscope, and the difference represents the growth over this period. At the end of culture, tibiae were fixed, embedded in paraffin, sectioned, and stained as described above.
RESULTS
The MuvB core binds RB family proteins using the LXCXE binding cleft.
Numerous proteins have been reported to interact with RB family members using an LXCXE-type interaction that is analogous to viral oncoproteins, such as adenovirus E1A, simian virus 40 (SV40) TAg, and human papillomavirus (HPV) E7 (41). This interaction is mediated by the LXCXE sequence on the viral protein and a well-conserved “pocket” in RB family proteins (42). While proteins such as histone deacetylases are reported to bind to RB family members in an analogous manner (43–46), evidence for these complexes with p107 or p130 at an endogenous level have been elusive (12). Figure 1A shows a GST-pulldown experiment that compares HDAC1 and -2 binding between pRB and p107 and mutant forms of these proteins that contain analogous point mutations in their LXCXE binding cleft region (I753A, N757A, and M761A for pRBΔL and I926A, N930A, and V934A in p107ΔD). This experiment demonstrates specificity between pRB and p107 in their ability to bind HDACs even though they both contain analogous binding sites for the LXCXE motif, and HDACs use it to bind to pRB. This motivated us to search for proteins that interact preferentially with the LXCXE binding cleft of p107.
FIG 1.
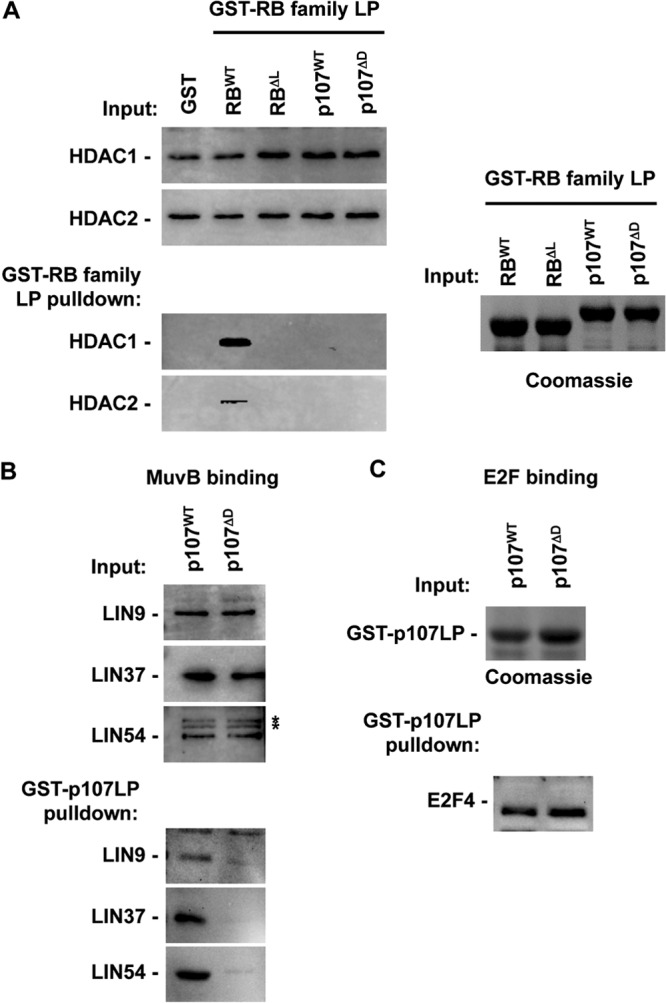
Interactions between p107 and the MuvB core complex are disrupted by LXCXE binding cleft mutations in p107. (A) The indicated GST fusion proteins were used to isolate interacting proteins from serum-starved nuclear extracts. Large pocket (LP) GST-RBΔL contains I753A, N757A, and M761A substitutions and GST-p107ΔD contains I926A, N930A, and V934A changes. Western blots demonstrate the relative input levels of HDACs and their ability to bind each GST-RB family LP protein. A Coomassie-stained gel indicates input levels of the relevant GST proteins. (B) Nuclear extracts from serum-starved MEFs were similarly used in GST-pulldown experiments with GST-p107LP proteins. Input and precipitated proteins were subjected to Western blotting with antibodies against the indicated components of the MuvB core complex. (C) Pulldown samples were also blotted for E2F4 as a control for the specificity of the p107 mutation. A Coomassie-stained gel serves as an internal control for GST-p107LP protein loading. An asterisk indicates a nonspecific band. WT, wild type.
Given previous data that DNA tumor virus proteins such as E7 and TAg can inhibit DREAM function (47, 48), we investigated whether MuvB and viral proteins bind the same region of RB family proteins. To test this possibility, we used a mutant form of p107 whose substitutions are known to inhibit interactions between viral oncoproteins and the LXCXE binding cleft in pRB (49). Figure 1B demonstrates that the MuvB proteins LIN9, LIN37, and LIN54 bind to recombinant p107 and are sensitive to substitutions in the LXCXE binding cleft region. We have named this mutant ΔDREAM, or ΔD for short, because it fails to interact with a number of components of the MuvB core, thus preventing DREAM assembly. Importantly, this mutant form of p107 retained the ability to interact with E2F4 (Fig. 1C). Taken together, these experiments suggest that the MuvB core and viral oncoproteins require similar amino acid contacts at the LXCXE binding domain on p107.
Generation of a DREAM-deficient mouse model.
We sought to generate a mutant mouse strain in which all components of the DREAM complex were present but were unable to assemble into a functional complex. Figure 2A shows the organization of the murine p107 gene (called Rbl1 but referred to as p107 for simplicity). A targeting vector was designed to introduce the murine equivalent of I926A, N930A, and V934A substitutions into exon 20 and to mark them with a new MscI restriction site. Generation of two correctly targeted ES cell clones (185 and 261) was confirmed by Southern blotting (Fig. 2B). Probes that are external to the targeting vector at the 5′ and 3′ ends were used to detect homologous recombination to generate the p107ΔD allele. Mutations in exon 20 were detected by MscI digestion and probing with the 5′ external probe (Fig. 2B). Single integration of the targeting vector was confirmed with BamHI-digested genomic DNA, and hybridization was performed with a neo selectable marker probe (Fig. 2B). Following selection of targeted clones, chimeric mice were generated and bred to cre transgenic females to remove the neomycin resistance marker. This created the targeted gene structure shown at the bottom of Fig. 2A, and mice were bred to homozygosity. Exon 20 was PCR amplified from homozygous tissue and sequenced to confirm the introduction of the desired mutations (data not shown).
FIG 2.

Generation of p107 mutant mice. (A) The wild-type p107 locus is diagramed on top. Relevant features of the targeting vector are shown below. A correctly targeted mutant locus is shown, as is the final genomic structure following cre-mediated excision of the selectable marker. (B) Southern blots of correctly targeted clones are shown to demonstrate homologous recombination at the 5′ and 3′ ends of the targeting vector as well as single integration. (C) Extracts were prepared from wild-type and p107ΔD/ΔD mutant cells. Western blotting was performed to investigate the relative expression level of p107 and its related family members pRB and p130. Pulldown experiments were performed using GST-E7 as a means to test LXCXE-dependent interactions by wild-type and mutant p107. An asterisk indicates a nonspecific band. (D) Electrophoretic mobility shift assays were performed using nuclear extracts (Ext) from wild-type and p107ΔD/ΔD mutant MEFs. Complexes that were formed are labeled to the right of the gels. E2F-dependent complexes were identified by competition with 100× wild-type cold competitor probe and 100× mutant probe. p107-containing complexes were identified using antibodies to supershift it. Anti-HA antibodies were used as a negative control. Ab, antibody; α, anti.
We generated protein extracts from p107ΔD/ΔD MEFs and performed Western blotting to confirm expression of RB family proteins and to test if the p107ΔD-encoded protein was capable of interacting with the LXCXE motif present in GST-E7 (Fig. 2C). These experiments confirmed normal expression of each pocket protein, as well as the LXCXE interaction defect in the mutant p107 protein. Lastly, we prepared nuclear extracts and subjected them to electrophoretic mobility shift assays to investigate p107 interactions with E2Fs (Fig. 2D). The identities of E2F-containing complexes are labeled on the right. p107-containing complexes were identified through antibody supershift in the rightmost lane of each gel. This analysis indicates similar p107-E2F complex formation between wild-type and mutant p107 proteins per amount of input extract. Collectively, these experiments reveal that the p107ΔD targeted allele is active and expresses normal levels of the p107 protein and that this protein is defective for LXCXE interactions but not E2F binding.
We then investigated the effects of the p107ΔD mutation on cell cycle exit and transcriptional repression of known DREAM target genes. Serum starvation of MEFs followed by BrdU and propidium iodide staining and flow cytometry revealed that wild-type and p107ΔD/ΔD MEFs withdraw from the cell cycle equivalently (Fig. 3A). Lastly, examination of known DREAM transcriptional targets Mybl2, Cdc2, and Ccna2 revealed that they were repressed similarly under these growth arrest conditions (Fig. 3B). These experiments suggest that loss-of-function effects of the p107ΔD mutation may be obscured by redundancy with other pocket proteins such as p130.
FIG 3.
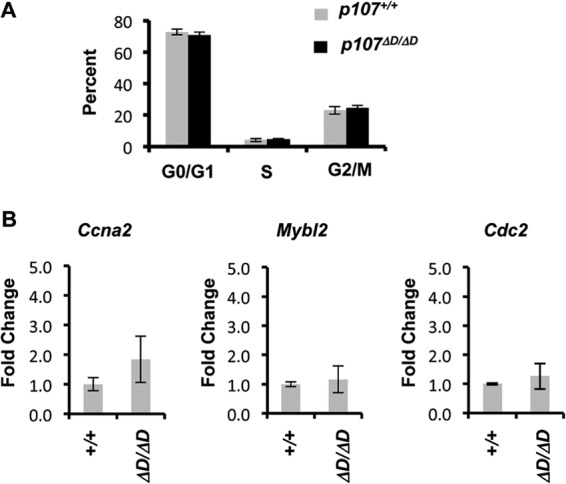
Regulation of cell cycle and gene expression in p107ΔD/ΔD cells. (A) Wild-type and p107ΔD/ΔD fibroblasts were serum starved for 3 days, followed by BrdU pulse-labeling and analysis by flow cytometry. The percentage of cells in each cell cycle phase are shown. (B) RNA was prepared from serum-starved cells, and the relative expression levels of Mybl2, Cdc2, and Ccna2 were determined. Error bars indicate 1 standard deviation from the mean (n = 3).
In order to create a mouse model system in which DREAM assembly is inhibited, we bred our p107ΔD allele into a p130−/− background and prepared MEFs from wild-type, p130−/−, and p107ΔD/ΔD; p130−/− mutants. Cells were cultured in low serum to induce quiescence and stimulate DREAM assembly as previously reported (17). Figure 4A displays Western blots of nuclear extracts obtained from each of the indicated genotypes of cells. These blots confirm that RB family proteins and MuvB components are all expressed under these conditions. Extract from each genotype was then subjected to IP-Western blot analysis with antibodies against LIN37. Precipitation of LIN37 from wild-type cells revealed that it is associated with LIN9 and LIN54 and also forms complexes with p107 or p130, but pRB is undetectable in these immunoprecipitates (Fig. 4B). In the absence of p130, LIN37 association with LIN9 and LIN54 remains unchanged, but the abundance of p107 associated with LIN37 is greatly elevated while interactions with pRB remain undetectable. Lastly, LIN37 immunoprecipitation of p107ΔD/ΔD; p130−/− nuclear extracts failed to result in detectable levels of RB family proteins. Based on these experiments, we conclude that DREAM, in mouse fibroblasts, can contain either p107 or p130, and the increased abundance of p107 in DREAM complexes found in p130−/− nuclear extracts suggests that they are interchangeable. In addition, these experiments suggest that pRB itself was unable to replace p107 and p130 in DREAM from p107ΔD/ΔD; p130−/− extracts. The failure to assemble DREAM in p107ΔD/ΔD; p130−/− MEFs suggests that these mice may be an effective way to probe where DREAM assembly is required in mammalian development.
FIG 4.

Disruption of endogenous DREAM complexes in p130−/− and p107ΔD/ΔD; p130−/− mutant cells. (A) Nuclear extracts were prepared from serum-starved cells of the indicated genotypes. Expression levels of RB family proteins, and components of the MuvB core were detected by Western blotting. SP1 levels serve as a loading control. Nonspecific bands are indicated by asterisks. (B) Anti-LIN37 immunoprecipitations were performed using serum-starved nuclear extracts, and RB family proteins, along with the indicated MuvB core proteins, were detected by Western blotting. The lysate lane was included with the pRB blot as a technical control for pRB detection. (C) Nuclear extracts were prepared from proliferating and serum-starved cells. The relative levels of LIN37 and BMYB in these extracts were demonstrated by Western blotting. SP1 serves as a loading control. (D) Anti-LIN37 immunoprecipitations were performed, and associated BMYB levels were detected by Western blotting. (E) ChIP experiments were performed for p107 at the Mybl2 promoter in the indicated genotypes of serum-starved cells. Error bars represent 1 standard deviation (n = 3). An asterisk represents a statistically significant difference from the relevant controls (t test, P < 0.05). (F) Extracts were prepared from proliferating cells of the indicated genotypes. The relative expression levels of CDK2 and p107 were determined by Western blotting. (G) p107 was immunoprecipitated, and CDK2 levels were detected by Western blotting. An asterisk marks an IgG-related band. (H) CDK2 was precipitated using the indicated antibodies from wild-type, p130−/−, and p107ΔD/ΔD; p130−/− cells. Precipitates were incubated with [32P]ATP and histone H1 and analyzed by SDS-PAGE. H1 was detected by Coomassie staining, and 32P incorporation was determined by autoradiography.
To determine the molecular consequences of defective DREAM assembly, we investigated the abundance of BMYB-MuvB complexes under serum starvation and asynchronously proliferating conditions. LIN37 was again precipitated from nuclear extracts, and associated BMYB was detected by Western blotting (Fig. 4C and D). In wild-type cells BMYB was absent from serum-starved LIN37 immunoprecipitates but was present in IPs from proliferating cells, as expected. Interestingly, BMYB appears in LIN37 immunoprecipitates from p130−/− serum-starved cells in modest levels, but in p107ΔD/ΔD; p130−/− extracts LIN37-BMYB complexes are of similar abundances between serum-starved and proliferating cells. This suggests that reduction of DREAM assembly may preferentially shift MuvB into BMYB-containing complexes.
Since BMYB and MuvB interact in p107ΔD/ΔD; p130−/− cells under growth conditions where DREAM would normally be assembled, we investigated the fate of p107ΔD that is expressed in these cells. We used chromatin immunoprecipitation (ChIP) to search for p107ΔD at the Mybl2 promoter (Fig. 4E). Not surprisingly, p107 is largely absent from this promoter in genotypes of cells that express p130, as p130 is known to form the most abundant E2F complexes in G0 (29, 50–52). In the absence of p130, both p107 and p107ΔD occupy this promoter; however, p107ΔD levels are lower, commensurate with BMYB-MuvB complexes being assembled in their place. We also investigated cyclin/Cdk2 interactions with p107 in proliferating cells. In these IP-Western blotting experiments, Cdk2 was observed in p107ΔD immunoprecipitates (Fig. 4F and G), and overall Cdk2 catalytic levels were slightly reduced in p107ΔD/ΔD; p130−/− cells compared with controls (Fig. 4H). Thus, the defect in p107ΔD function appears to be specifically at the level of MuvB recruitment as other molecular interactions are retained at an endogenous level.
We investigated the effects of impaired DREAM function on proliferation and quiescence in p107ΔD/ΔD; p130−/− MEFs using propidium iodide and BrdU staining of cells followed by flow cytometry. As shown in Fig. 5A, p107ΔD/ΔD; p130−/− cells have similar levels of G0/G1, S, and G2/M cells as p107ΔD/ΔD controls while proliferating. Furthermore, both genotypes of MEFs respond similarly to serum deprivation and display minimal S-phase content (Fig. 5B). This result is in agreement with previous finding that loss of all three RB family proteins is required to override G0/G1 arrest in MEFs (53, 54). To determine the consequences of the p107ΔD/ΔD; p130−/− genotype on known DREAM and BMYB-MuvB target genes, we assessed their expression by real-time PCR. Figure 5C shows gene expression levels of Mybl2, Cdc2, Ccna2, and Cdc6 in serum-starved cells. This demonstrates that repression of these known DREAM target genes is lost in p107ΔD/ΔD; p130−/− MEFs. We also investigated the expression of these genes in proliferating populations of cells. Asynchronously proliferating p107ΔD/ΔD; p130−/− cells had elevated levels of these target genes compared to controls (Fig. 5D). Given that proliferating p107ΔD/ΔD; p130−/− fibroblasts have similar percentages of cells in G0/G1, S, and G2/M as control genotypes (Fig. 5A), elevated levels of BMYB-MuvB complexes at earlier stages of the cell cycle, or loss of DREAM in G1 cells, may lead to premature expression of these genes and overall higher levels.
FIG 5.

Increased expression of DREAM target genes in p107ΔD/ΔD; p130−/− cells. (A) Asynchronously proliferating cells were pulse-labeled with BrdU and processed for propidium iodide and BrdU analysis by flow cytometry. The percentage of cells in each phase is shown. Error bars indicate 1 standard deviation (n = 3). (B) Serum-starved cells were BrdU labeled and analyzed as described for panel A. The percentage of cells in each phase is shown. (C) Total RNA was prepared from serum-starved cells and subjected to real-time PCR for the indicated DREAM target genes. The relative expression of each target in p107ΔD/ΔD; p130−/− cells is shown, and error bars indicate 1 standard deviation (n = 3). An asterisk indicates a statistically significant difference (t test, P < 0.05). (D) A similar analysis of the same DREAM target genes is shown from asynchronously proliferating cells.
Taken together, these experiments demonstrate that cells from p107ΔD/ΔD; p130−/− mice are impaired for assembly of DREAM even though the mutant p107 and the MuvB core are present. This may lead to a shift in molecular organization such that BMYB is associated with the MuvB core during quiescence or earlier in the cell cycle than normal. Loss of DREAM function in p107ΔD/ΔD; p130−/− cells disrupts repression of target genes in quiescence, and these cells overexpress the same target genes during proliferation.
Neonatal mortality and distinct bone defects in p107ΔD/ΔD; p130−/− mice.
In order to understand the physiological role of DREAM in mammals, we set out to generate p107ΔD/ΔD; p130−/− mice and study their associated phenotypes. We intercrossed p107ΔD/+; p130+/− mice and failed to obtain viable p107ΔD/ΔD; p130−/− mutants (data not shown). Crosses between p107ΔD/ΔD; p130+/− parents generated one runted weanling in 85 offspring (Table 1). This prompted us to search at earlier time points to determine when p107ΔD/ΔD; p130−/− mutants lose viability. Table 2 demonstrates the normal abundance of p107ΔD/ΔD; p130−/− embryos at embryonic day 18.5 of gestation (E18.5), just before birth. To determine precisely when in development p107ΔD/ΔD; p130−/− mutants were dying, newborns were observed daily for 1 week, with the number of live and dead pups of each genotype being determined. This experiment demonstrated that 30% of p107ΔD/ΔD; p130−/− pups were dead by postnatal day 0.5, and all pups of this genotype had died within 1.5 days following birth (Fig. 6A). In contrast, all other genotypes were viable during the same period (Fig. 6A).
TABLE 1.
Rare p107ΔD/ΔD; p130−/− mutants at postnatal day 14
| Genotype | No. of offspring with the genotypea |
|
|---|---|---|
| Expected | Observed | |
| p107ΔD/ΔD; p130+/+ | 42.5 | 59 |
| p107ΔD/ΔD; p130+/− | 21.25 | 25 |
| p107ΔD/ΔD; p130−/− | 21.25 | 1 |
Intercrosses were performed between p107ΔD/ΔD; p130+/− and p107ΔD/ΔD; p130+/− parents, and offspring were genotyped as early as postnatal day 14. Genotypes were determined by PCR as reported in Materials and Methods (n = 85).
TABLE 2.
Viable p107ΔD/ΔD; p130−/− mutants at E18.5
| Genotype | No. of offspring with the genotypea |
|
|---|---|---|
| Expected | Observed | |
| p107ΔD/+; p130+/− | 19.5 | 22 |
| p107ΔD/+; p130−/− | 19.5 | 16 |
| p107ΔD/ΔD; p130+/− | 19.5 | 19 |
| p107ΔD/ΔD; p130−/− | 19.5 | 21 |
Intercrosses were performed between p107ΔD/ΔD; p130+/− and p107ΔD/+; p130−/− parents, and offspring were isolated at day 18.5 of pregnancy. Viability was determined by the presence of a heartbeat upon visual inspection, and genotypes were determined by PCR (n = 78).
FIG 6.

p107ΔD/ΔD; p130−/− embryos display distinct bone defects and neonatal lethality. (A) Transheterozygous p107 and p130 mutant mice were intercrossed to produce offspring. Percent survival of newborn pups was determined daily for 1 week. For p107ΔD/ΔD; p130−/− n = 12, and for all other genotypes n = 42. (B) Alcian blue and alizarin red staining of E18.5 embryos. Arrows indicate the interparietal (red) and supraoccipital (green) bones in the skull base. Lower images show these bones at higher magnification. Scale bars, 3 mm. (C) Micro-CT analysis of E18.5 skeletons. Red arrows indicate the positions of ossicles in a control mouse. Scale bars, 1.01 mm. (D) Pairs of control and p107ΔD/ΔD; p130−/− mice were scanned together in a micro-CT to measure bone properties. Pairwise comparisons in the same scan were used to minimize variation. Bone mineral densities were determined for all long bones (humerus, ulna, radius, femur, tibia, and fibula) and were averaged within each mouse. Average bone density was compared between five mutant and control pairs. Error bars represent one standard deviation from the mean. (E) Longitudinal and sagittal images of the humerus are shown from micro-CT scans of control and p107ΔD/ΔD; p130−/− mice at E18.5. The leftmost scale bars are 0.25 mm and the rightmost are 0.1 mm.
Neonatal lethality in p107ΔD/ΔD; p130−/− mice is very similar to the previously reported phenotype of p107−/−; p130−/− mice (24). Furthermore, even though p107ΔD/ΔD; p130−/− mice were not smaller by weight at E18.5, the only survivors we have observed were severely runted, suggesting that smaller pups have the best chance to survive. Based on this line of reasoning, we investigated the skeletal structure of p107ΔD/ΔD; p130−/− mice at E18.5 to search for defects that could lead to death at, or shortly after, birth. Bones and cartilage of clarified embryos were stained using alizarin red and alcian blue. This revealed that the endocranial bones forming the base of the skull, namely, the interparietal and supraoccipital bones, were smaller and underdeveloped in mutant embryos compared to heterozygotes (Fig. 6B). This phenotype is highly reminiscent of p107−/−; p130−/− mice, and cranial bone and cartilage defects may be linked to loss of neonatal viability (24).
Micro-CT scans were used to search for and characterize other bone abnormalities in these mice. Similar to the defects in the skull, the inner ear ossicles were too poorly mineralized in p107ΔD/ΔD; p130−/− mutants to be visualized in the micro-CT images (Fig. 6C). Bone mineral density was measured for the humerus, ulna, radius, femur, tibia, and fibula, as well as for the interparietal and supraoccipital bones of the cranium, and averaged for pairs of control and p107ΔD/ΔD; p130−/− mutant mice. These measurements showed that there was no detectable difference between control and mutant mice (Fig. 6D). The structural properties of trabecular bones could not be quantified due to the low level of mineralization in embryonic bones; however, qualitative observations of high-resolution (26-μm) cross-sections of the humerus displayed little difference in morphology between heterozygotes and mutants. Taken together, these observations indicate that p107ΔD/ΔD; p130−/− mice have discrete defects in bone morphogenesis whereby later aspects of osteogenesis appear normal while particular bones are defective or absent.
While not statistically significant, micro-CT analysis revealed that some long bones in p107ΔD/ΔD; p130−/− mice were shorter than controls. We investigated this further by measuring the long bones of alcian blue- and alizarin red-stained E18.5 skeletons and compared their averages (Fig. 7A and B). This revealed a statistically significant reduction in long bone length in p107ΔD/ΔD; p130−/− mutants compared with that of controls. In order to determine if there was a defect in endochondral ossification, sections stained with hematoxylin and eosin and with Safranin O were examined. Upon observation of H&E- and Safranin O-stained samples, the growth plates appeared slightly shorter in p107ΔD/ΔD; p130−/− bones (Fig. 7C). Blinded counting of fluorescent DAPI-stained nuclei in the resting zone of epiphyses determined that there was hyperplasia of chondrocytes in this region (Fig. 7D). Furthermore, PCNA staining revealed elevated levels of proliferating cells in both the resting and proliferating zones of the tibial growth plate (Fig. 7E and F). These experiments revealed defective chondrocyte proliferative control and endochondral ossification in p107ΔD/ΔD; p130−/− mice. This phenotype is highly reminiscent of p107−/−; p130−/− knockout mice (24), suggesting that the DREAM complex is a significant contributor to p107 and p130 activity during mouse development.
FIG 7.

Defective endochondral ossification in p107ΔD/ΔD; p130−/− mice. (A) E18.5 skeletons were stained with alcian blue and alizarin red to visualize bone and cartilage. The lengths of forelimb long bones were determined for p107ΔD/+; p130+/− mice and p107ΔD/ΔD; p130−/− mice. The graph to the right shows average length measurements. The number of bones measured for each control and p107ΔD/ΔD; p130−/− comparison were as follows (in respective order): humerus, n = 16 and n = 20; radius, n = 16 and n = 20; ulna, n = 16 and n = 20; scapular spine, n = 16 and n = 17. All error bars indicate 1 standard deviation from the mean. A single asterisk indicates a statistically significant difference (t test, P < 0.01). Scale bars, 3 mm. (B) Hind limbs were similarly analyzed for length. The graph shows average measurements from control and p107ΔD/ΔD; p130−/− mutants (in respective order, femur, n = 15 and n = 19; tibia, n = 16 and n = 19; fibula, n = 16 and n = 18). Scale bars, 3 mm. (C) Hematoxylin and eosin staining of the proximal tibial epiphysis from control and p107ΔD/ΔD; p130−/− mice. Safranin O staining of cartilage from a similarly cut bone section from control and mutant mice is also shown. Scale bars, 200 μm. Arrows alongside indicate the growth plate. (D) Sections were stained with DAPI and imaged to quantitate cell density of the resting zone of long bone epiphyses. The average number of cells in a 0.75-mm2 field of view was determined and graphed. Cells were counted in 19 fields of view for control, and 20 fields of view were counted for p107ΔD/ΔD; p130−/−. Two asterisks indicate a statistically significant difference (t test, P < 0.00001). (E) Proliferation of chondrocytes in long-bone epiphyses was detected by PCNA staining. Blue bars alongside the image indicate the resting zone, and red bars indicate the proliferating zone. Scale bars, 200 μm. (F) PCNA-positive cells in the resting and proliferating zones were determined from four tibias each. Error bars indicate 1 standard deviation from the mean. Two asterisks indicate a statistically significant difference (t test, P < 0.005).
Since it is known that p107 and p130 contribute to cell cycle arrest in models of chondrocyte biology (22, 24, 26, 55), we sought further evidence that assembly of the DREAM complex is required for proliferative control in these cells. Recent work indicates that DYRK1A is required to phosphorylate LIN52 to signal the assembly of DREAM in cell culture experiments (17). For this reason we determined whether DYRK signaling affects endochondral bone growth in a relatively intact developing bone. We performed organ cultures of wild-type mouse embryonic tibiae with or without the DYRK inhibitor harmine (Fig. 8A). Safranin O staining of sections showed a shorter growth plate in harmine-treated tibiae. Tibial length was measured at the beginning and end of this 6-day culture experiment, and measurements revealed that harmine slows the growth of tibiae by 30% (Fig. 8B), a similar reduction in bone growth as that found in the p107ΔD/ΔD; p130−/− mutant mice. Importantly, treatment of quiescent cells with the same concentration of harmine reduces DREAM assembly as judged by LIN37 IP and p130 Western blotting (Fig. 8C). The similarity in phenotypes between p107ΔD/ΔD; p130−/− mice and harmine-treated bones further suggests that defective DREAM assembly impairs proliferative control in chondrocytes.
FIG 8.
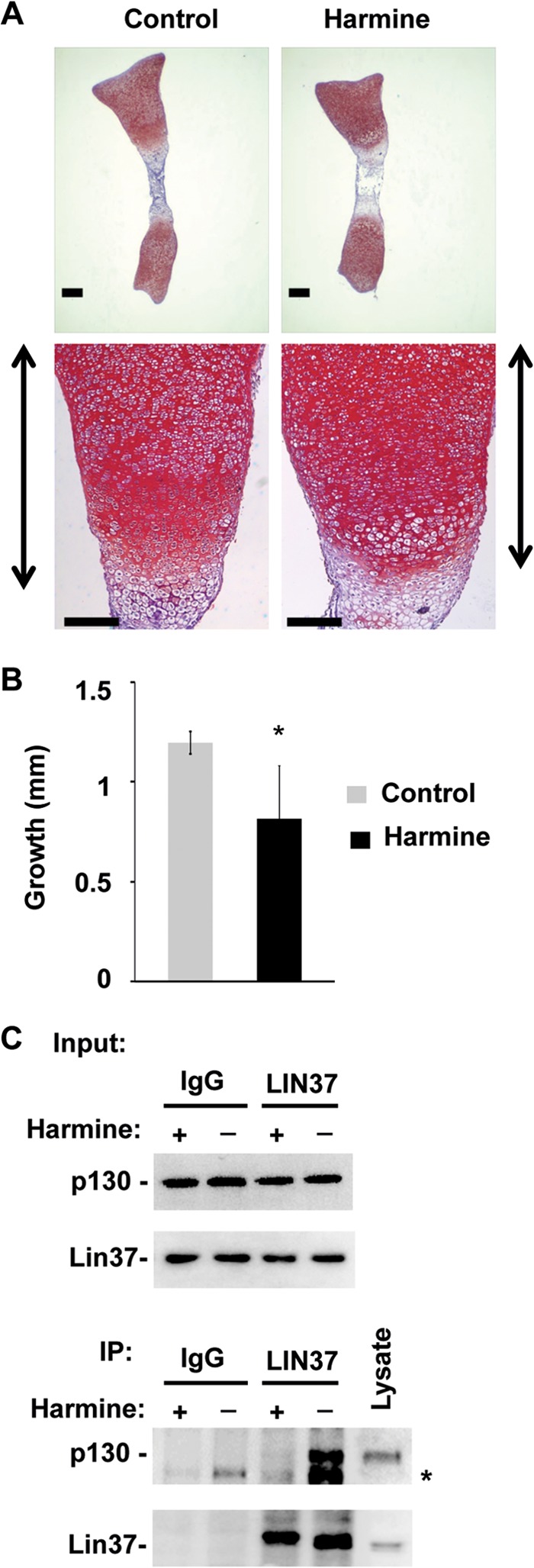
Harmine treatment leads to shortened tibiae. (A) Tibiae were excised from E15.5 embryos and cultured in the presence of DMSO vehicle as well as 5 μM harmine for 6 days. Bones were embedded in paraffin, sectioned, and stained with Safranin O. Scale bars, 200 μm. Growth plates are indicated to the sides of each lower image. (B) Bone length was measured microscopically at the beginning and end of the culture period. The average increase in length (growth) was determined (n = 4 for control and harmine-treated cells). All error bars indicate 1 standard deviation from the mean. A single asterisk indicates a statistically significant difference (t test, P < 0.01). (C) Serum-starved cells were treated with 5 μM harmine for 48 h, and nuclear extracts were isolated. Input levels of relevant proteins are shown, as are the proteins present in anti-LIN37 immunoprecipitations. An asterisk indicates a nonspecific band.
DISCUSSION
In this paper we report the generation of a targeted allele of p107 (p107ΔD) that disrupts its interaction with the MuvB core proteins. It demonstrates that MuvB interacts with the analogous region of p107 as viral oncoproteins such as human papillomavirus E7. When combined with deficiency for p130, this p107 mutation prevents the assembly of the DREAM complex. Cells from p107ΔD/ΔD; p130−/− mice have elevated expression levels of DREAM target genes, and p107ΔD/ΔD; p130−/− mice have defects in endochondral ossification and die shortly after birth. These data suggest a key role for DREAM in regulating the transition between resting and proliferating chondrocytes in the growth plate. Our structure-function approach of creating a targeted mutant mouse line with DREAM assembly-deficient properties does not rule out that other proteins can be sensitive to the same p107ΔD mutation. There are two main reasons why we do not anticipate that such a protein contributes to this phenotype. First, IP mass spectrometry approaches used in the original discovery of DREAM (12) would have been expected to reveal such a factor. In addition, since harmine treatment inhibits DYRKs and DREAM assembly and causes a similar phenotype in our bone culture system, such an unknown factor would have to be regulated similarly. We expect that this is relatively unlikely and for this reason favor the interpretation that chondrocyte proliferative control defects are most likely caused by defective DREAM assembly.
Based on prior reports of protein interactions with the LXCXE binding site on pocket proteins, our data demonstrating MuvB binding specificity for p107 and p130 are surprising. Approximately 30 proteins have been reported to interact with pRB using an LXCXE-like interaction (41), and many are also described as p107 and p130 binding proteins (56). Given the MuvB interaction with p107 or p130 is dependent on LIN52 phosphorylation (17), we expect that signals that regulate assembly likely play a key role in creating specificity for pocket protein interactions. We have recently demonstrated a similar regulatory paradigm for LXCXE-dependent interactions between PML and pRB during senescence (34).
The phenotype of p107ΔD/ΔD; p130−/− mice is highly reminiscent of that of the previously reported p107−/−; p130−/− knockout mice (24). Strong similarities between these phenotypes are evident based on the bones that are affected. Both studies highlight shorter long bones and defective interparietal and supraoccipital bones in the skull. The hyperplasia in the resting zones of bone epiphyses is also very similar on a morphological level and with regard to cell density. In contrast, there are also some subtle differences that may be meaningful, such as the survival of rare p107ΔD/ΔD; p130−/− mice beyond neonatal stages. In crosses other than those documented in this report, we have obtained a few more of these animals, and they lived into adulthood without obvious phenotypes besides being small. This may suggest that loss of DREAM assembly does not manifest in other major developmental or physiological defects. Unfortunately, we did not obtain enough of them to undertake any type of systematic analysis. Interestingly, no newborn p107−/−; p130−/− mice were reported to survive (24), and this suggests a difference between our study and the previous double knockout mice. In addition, some developmental aspects, such as the failure to form ossicles by E18.5 in p107ΔD/ΔD; p130−/− animals, are unreported for p107−/−; p130−/− mice. An important difference in these mouse models is the retention of cyclin/Cdk binding and E2F interactions in the p107ΔD mutant. Since p107 and p130 are also capable of interacting with activator E2Fs in chondrocytes, interpretation of the p107ΔD/ΔD; p130−/− phenotype as being identical to double knockout mice would suggest that these interactions lack functional consequences. However, since there are instances, such as neonatal survival, where the p107ΔD/ΔD; p130−/− phenotype is milder, it is possible that these are places where Cdk and E2F functions can contribute. Further investigation is needed to distinguish between these possibilities. Another important consideration in the interpretation of the chondrocyte phenotypes presented here, which is also true of the double knockout, is that failure of DREAM assembly seems to obligatorily lead to BMYB-MuvB complex formation, even under growth arrest conditions. It is possible that hyperplasia in the growth plates of p107 and p130 mutant mice is as much a consequence of a gain in BMYB-MuvB as a loss of DREAM. At present we are not able to make a distinction between these possibilities and note only that this phenotype begins with the loss of DREAM assembly.
This study suggests that DREAM assembly may play an evolutionarily divergent role in mammals compared to lower eukaryotes because the p107ΔD/ΔD; p130−/− mice do not have more extensive phenotypic consequences outside bone development. Studies in Drosophila highlight broad roles in transcriptional control (6, 11, 57) that are not necessarily proliferation related. However, it is possible that some aspects of DREAM function in lower organisms could not be assessed using our mouse model because they are reproduction related and only rare p107ΔD/ΔD; p130−/− animals reach adulthood, preventing the investigation of these phenotypes. In addition, some aspects of DREAM function in Drosophila, such as olfactory receptor expression patterns (57), might not be evident in E18.5 embryos and difficult to identify even in conditional DREAM assembly-deficient mice. Alternatively, it is possible that the mutually exclusive nature of MuvB interactions with RB family proteins and BMYB in mammals created a more limited function for the DREAM complex in mammals than in lower organisms. We suggest that additional work is needed to search for other circumstances where DREAM may be relevant in mammals. In addition, a better understanding of the DYRK1A regulatory pathway that induces DREAM assembly may illuminate more broadly where else this complex may function.
ACKNOWLEDGMENTS
We are indebted to many colleagues for discussions and encouragement during the course of this work.
C.F. was supported by an SRP award from the Schulich School of Medicine and Dentistry. S.A.H. and C.A.I. were supported by fellowship awards from CaRTT, and S.A.H. also acknowledges fellowship support from CBCF-Ontario. J.R.B. is the recipient of CIHR postdoctoral fellowship (Priority Announcement: Epigenetics). F.B. is the Canada Research Chair in musculoskeletal research. F.A.D. is the Wolfe Senior Fellow in Tumor Suppressor Genes at Western University. Funding for this work was provided by the Canadian Cancer Society Research Institute (2011-700720) to F.A.D. and the Canadian Institutes of Health Research (MOP43899) to F.B.
Footnotes
Published ahead of print 7 April 2014
REFERENCES
- 1.Lipsick JS. 2004. synMuv verite—Myb comes into focus. Genes Dev. 18:2837–2844. 10.1101/gad.1274804 [DOI] [PubMed] [Google Scholar]
- 2.Sadasivam S, DeCaprio JA. 2013. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer. 13:585–595. 10.1038/nrc3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferguson EL, Horvitz HR. 1989. The multivulva phenotype of certain Caenorhabditis elegans mutants results from defects in two functionally redundant pathways. Genetics 123:109–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ceol CJ, Horvitz HR. 2004. A new class of C. elegans synMuv genes implicates a Tip60/NuA4-like HAT complex as a negative regulator of Ras signaling. Dev. Cell 6:563–576. 10.1016/S1534-5807(04)00065-6 [DOI] [PubMed] [Google Scholar]
- 5.Harrison MM, Ceol CJ, Lu X, Horvitz HR. 2006. Some C. elegans class B synthetic multivulva proteins encode a conserved LIN-35 Rb-containing complex distinct from a NuRD-like complex. Proc. Natl. Acad. Sci. U. S. A. 103:16782–16787. 10.1073/pnas.0608461103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korenjak M, Taylor-Harding B, Binne UK, Satterlee JS, Stevaux O, Aasland R, White-Cooper H, Dyson N, Brehm A. 2004. Native E2F/RBF complexes contain Myb-interacting proteins and repress transcription of developmentally controlled E2F target genes. Cell 119:181–193. 10.1016/j.cell.2004.09.034 [DOI] [PubMed] [Google Scholar]
- 7.Lewis PW, Beall EL, Fleischer TC, Georlette D, Link AJ, Botchan MR. 2004. Identification of a Drosophila Myb-E2F2/RBF transcriptional repressor complex. Genes Dev. 18:2929–2940. 10.1101/gad.1255204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ceol CJ, Horvitz HR. 2001. dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol. Cell 7:461–473. 10.1016/S1097-2765(01)00194-0 [DOI] [PubMed] [Google Scholar]
- 9.Lu X, Horvitz HR. 1998. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell 95:981–991. 10.1016/S0092-8674(00)81722-5 [DOI] [PubMed] [Google Scholar]
- 10.Beall EL, Manak JR, Zhou S, Bell M, Lipsick JS, Botchan MR. 2002. Role for a Drosophila Myb-containing protein complex in site-specific DNA replication. Nature 420:833–837. 10.1038/nature01228 [DOI] [PubMed] [Google Scholar]
- 11.Lee H, Ragusano L, Martinez A, Gill J, Dimova DK. 2012. A dual role for the dREAM/MMB complex in the regulation of differentiation-specific E2F/RB target genes. Mol. Cell. Biol. 32:2110–2120. 10.1128/MCB.06314-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, Chen R, Washburn MP, Liu XS, DeCaprio JA. 2007. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 26:539–551. 10.1016/j.molcel.2007.04.015 [DOI] [PubMed] [Google Scholar]
- 13.Pilkinton M, Sandoval R, Colamonici OR. 2007. Mammalian Mip/LIN-9 interacts with either the p107, p130/E2F4 repressor complex or B-Myb in a cell cycle-phase-dependent context distinct from the Drosophila dREAM complex. Oncogene 26:7535–7543. 10.1038/sj.onc.1210562 [DOI] [PubMed] [Google Scholar]
- 14.Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, von Eyss B, Gagrica S, Hanel F, Brehm A, Gaubatz S. 2007. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle 6:1903–1913. 10.4161/cc.6.15.4512 [DOI] [PubMed] [Google Scholar]
- 15.Osterloh L, von Eyss B, Schmit F, Rein L, Hubner D, Samans B, Hauser S, Gaubatz S. 2007. The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J. 26:144–157. 10.1038/sj.emboj.7601478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadasivam S, Duan S, DeCaprio JA. 2012. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 26:474–489. 10.1101/gad.181933.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. 2011. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 25:801–813. 10.1101/gad.2034211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight AS, Notaridou M, Watson RJ. 2009. A Lin-9 complex is recruited by B-Myb to activate transcription of G2/M genes in undifferentiated embryonal carcinoma cells. Oncogene 28:1737–1747. 10.1038/onc.2009.22 [DOI] [PubMed] [Google Scholar]
- 19.Pilkinton M, Sandoval R, Song J, Ness SA, Colamonici OR. 2007. Mip/LIN-9 regulates the expression of B-Myb and the induction of cyclin A, cyclin B, and CDK1. J. Biol. Chem. 282:168–175. 10.1074/jbc.M609924200 [DOI] [PubMed] [Google Scholar]
- 20.Reichert N, Wurster S, Ulrich T, Schmitt K, Hauser S, Probst L, Gotz R, Ceteci F, Moll R, Rapp U, Gaubatz S. 2010. Lin9, a subunit of the mammalian DREAM complex, is essential for embryonic development, for survival of adult mice, and for tumor suppression. Mol. Cell. Biol. 30:2896–2908. 10.1128/MCB.00028-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calbo J, Parreno M, Sotillo E, Yong T, Mazo A, Garriga J, Grana X. 2002. G1 cyclin/cyclin-dependent kinase-coordinated phosphorylation of endogenous pocket proteins differentially regulates their interactions with E2F4 and E2F1 and gene expression. J. Biol. Chem. 277:50263–50274. 10.1074/jbc.M209181200 [DOI] [PubMed] [Google Scholar]
- 22.Kurimchak A, Haines DS, Garriga J, Wu S, De Luca F, Sweredoski MJ, Deshaies RJ, Hess S, Grana X. 2013. Activation of p107 by fibroblast growth factor, which is essential for chondrocyte cell cycle exit, is mediated by the protein phosphatase 2A/B55α holoenzyme. Mol. Cell. Biol. 33:3330–3342. 10.1128/MCB.00082-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee EY, Cam H, Ziebold U, Rayman JB, Lees JA, Dynlacht BD. 2002. E2F4 loss suppresses tumorigenesis in Rb mutant mice. Cancer Cell 2:463–472. 10.1016/S1535-6108(02)00207-6 [DOI] [PubMed] [Google Scholar]
- 24.Cobrinik D, Lee MH, Hannon G, Mulligan G, Bronson RT, Dyson N, Harlow E, Beach D, Weinberg RA, Jacks T. 1996. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 10:1633–1644. 10.1101/gad.10.13.1633 [DOI] [PubMed] [Google Scholar]
- 25.Lee M-H, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, Harlow E, Jacks T. 1996. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev. 10:1621–1632. 10.1101/gad.10.13.1621 [DOI] [PubMed] [Google Scholar]
- 26.Laplantine E, Rossi F, Sahni M, Basilico C, Cobrinik D. 2002. FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J. Cell Biol. 158:741–750. 10.1083/jcb.200205025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LeCouter JE. 1998. Strain-dependent embryonic lethality in mice lacking the retinoblastoma-related p130 gene. Development 125:4669–4679 [DOI] [PubMed] [Google Scholar]
- 28.LeCouter JE, Kablar B, Hardy WR, Ying C, Megeney LA, May LL, Rudnicki MA. 1998. Strain-dependent myeloid hyperplasia, growth deficiency, and accelerated cell cycle in mice lacking the Rb-related p107 gene. Mol. Cell. Biol. 18:7455–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hurford RK, Jr, Cobrinik D, Lee MH, Dyson N. 1997. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 11:1447–1463. 10.1101/gad.11.11.1447 [DOI] [PubMed] [Google Scholar]
- 30.Mulligan GJ, Wong J, Jacks T. 1998. p130 is dispensable in peripheral T lymphocytes: evidence for functional compensation by p107 and pRB. Mol. Cell. Biol. 18:206–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cecchini MJ, Dick FA. 2011. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem. J. 434:297–308. 10.1042/BJ20101210 [DOI] [PubMed] [Google Scholar]
- 32.Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. 1996. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. U. S. A. 93:5860–5865. 10.1073/pnas.93.12.5860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cecchini MJ, Amiri M, Dick FA. 2012. Analysis of cell cycle position in mammalian cells. J. Vis. Exp. 59:pii3491. 10.3791/3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talluri S, Dick FA. 2014. The retinoblastoma protein and PML collaborate to organize heterochromatin and silence E2F-responsive genes during senescence. Cell Cycle 13:641–651. 10.4161/cc.27527 [DOI] [PubMed] [Google Scholar]
- 35.McLeod MJ. 1980. Differential staining of cartilage and bone in whole mouse fetuses by Alcian blue and alizarin red S. Teratology 22:299–301. 10.1002/tera.1420220306 [DOI] [PubMed] [Google Scholar]
- 36.Ulici V, Hoenselaar KD, Agoston H, McErlain DD, Umoh J, Chakrabarti S, Holdsworth DW, Beier F. 2009. The role of Akt1 in terminal stages of endochondral bone formation: angiogenesis and ossification. Bone 45:1133–1145. 10.1016/j.bone.2009.08.003 [DOI] [PubMed] [Google Scholar]
- 37.Gillespie JR, Ulici V, Dupuis H, Higgs A, Dimattia A, Patel S, Woodgett JR, Beier F. 2011. Deletion of glycogen synthase kinase-3β in cartilage results in up-regulation of glycogen synthase kinase-3α protein expression. Endocrinology 152:1755–1766. 10.1210/en.2010-1412 [DOI] [PubMed] [Google Scholar]
- 38.Umoh JU, Sampaio AV, Welch I, Pitelka V, Goldberg HA, Underhill TM, Holdsworth DW. 2009. In vivo micro-CT analysis of bone remodeling in a rat calvarial defect model. Phys. Med. Biol. 54:2147–2161. 10.1088/0031-9155/54/7/020 [DOI] [PubMed] [Google Scholar]
- 39.Agoston H, Khan S, James CG, Gillespie JR, Serra R, Stanton LA, Beier F. 2007. C-type natriuretic peptide regulates endochondral bone growth through p38 MAP kinase-dependent and -independent pathways. BMC Dev. Biol. 7:18. 10.1186/1471-213X-7-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ulici V, Hoenselaar KD, Gillespie JR, Beier F. 2008. The PI3K pathway regulates endochondral bone growth through control of hypertrophic chondrocyte differentiation. BMC Dev. Biol. 8:40. 10.1186/1471-213X-8-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dick FA. 2007. Structure-function analysis of the retinoblastoma tumor suppressor protein: is the whole a sum of its parts? Cell Div. 2:26. 10.1186/1747-1028-2-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dick FA, Rubin SM. 2013. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 14:297–306. 10.1038/nrm3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bourgo RJ, Thangavel C, Ertel A, Bergseid J, McClendon AK, Wilkens L, Witkiewicz AK, Wang JY, Knudsen ES. 2011. RB restricts DNA damage-initiated tumorigenesis through an LXCXE-dependent mechanism of transcriptional control. Mol. Cell 43:663–672. 10.1016/j.molcel.2011.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D. 1998. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc. Natl. Acad. Sci. U. S. A. 95:10493–10498. 10.1073/pnas.95.18.10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo RX, Postigo AA, Dean DC. 1998. Rb interacts with histone deacetylase to repress transcription. Cell 92:463–473. 10.1016/S0092-8674(00)80940-X [DOI] [PubMed] [Google Scholar]
- 46.Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A. 1998. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391:601–605. 10.1038/35410 [DOI] [PubMed] [Google Scholar]
- 47.Fine DA, Rozenblatt-Rosen O, Padi M, Korkhin A, James RL, Adelmant G, Yoon R, Guo L, Berrios C, Zhang Y, Calderwood MA, Velmurgan S, Cheng J, Marto JA, Hill DE, Cusick ME, Vidal M, Florens L, Washburn MP, Litovchick L, DeCaprio JA. 2012. Identification of FAM111A as an SV40 host range restriction and adenovirus helper factor. PLoS Pathog. 8:e1002949. 10.1371/journal.ppat.1002949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nor Rashid N, Yusof R, Watson RJ. 2011. Disruption of repressive p130-DREAM complexes by human papillomavirus 16 E6/E7 oncoproteins is required for cell-cycle progression in cervical cancer cells. J. Gen. Virol. 92:2620–2627. 10.1099/vir.0.035352-0 [DOI] [PubMed] [Google Scholar]
- 49.Dick FA, Sailhamer E, Dyson NJ. 2000. Mutagenesis of the pRB pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins. Mol. Cell. Biol. 20:3715–3727. 10.1128/MCB.20.10.3715-3727.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moberg K, Starz MA, Lees JA. 1996. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol. Cell. Biol. 16:1436–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takahashi Y, Rayman JB, Dynlacht BD. 2000. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 14:804–816 [PMC free article] [PubMed] [Google Scholar]
- 52.Wells J, Boyd K, Fry C, Bartley S, Farnham P. 2000. Target gene specificity of E2F and pocket protein family members in living cells. Mol. Cell. Biol. 20:5797–5807. 10.1128/MCB.20.16.5797-5807.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dannenberg J-H, van Rossum A, Schuijff L, te Riele H. 2000. Ablation of the retinoblastoma gene family deregulates G1 control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 14:3051–3064. 10.1101/gad.847700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sage J, Mulligan G, Attardi L, Miller A, Chen S, Williams B, Theodorou E, Jacks T. 2000. Targeted disruption of the three Rb-related genes leads to loss of G1 control and immortalization. Genes Dev. 14:3037–3050. 10.1101/gad.843200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rossi F, MacLean HE, Yuan W, Francis RO, Semenova E, Lin CS, Kronenberg HM, Cobrinik D. 2002. p107 and p130 Coordinately regulate proliferation, Cbfa1 expression, and hypertrophic differentiation during endochondral bone development. Dev. Biol. 247:271–285. 10.1006/dbio.2002.0691 [DOI] [PubMed] [Google Scholar]
- 56.Henley SA, Dick FA. 2012. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 7:10. 10.1186/1747-1028-7-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sim CK, Perry S, Tharadra SK, Lipsick JS, Ray A. 2012. Epigenetic regulation of olfactory receptor gene expression by the Myb-MuvB/dREAM complex. Genes Dev. 26:2483–2498. 10.1101/gad.201665.112 [DOI] [PMC free article] [PubMed] [Google Scholar]