

Abstract
Eukaryotic elongation factor 2 kinase (eEF2K) is the best-characterized member of the α-kinase family. Within this group, only eEF2K and myosin heavy chain kinases (MHCKs) have known substrates. Here we have studied the roles of specific residues, selected on the basis of structural data for MHCK A and TRPM7, in the function of eEF2K. Our data provide the first information regarding the basis of the substrate specificity of α-kinases, in particular the roles of residues in the so-called N/D loop, which appears to occupy a position in the structure of α-kinases similar to that of the activation loop in other kinases. Several mutations in the EEF2K gene occur in tumors, one of which (Arg303Cys) is at a highly conserved residue in the N/D loop. This mutation greatly enhances eEF2K activity and may be cytoprotective. Our data support the concept that the major autophosphorylation site (Thr348 in eEF2K) docks into a binding pocket to help create the kinase-competent conformation. This is similar to the situation for MHCK A and is consistent with this being a common feature of α-kinases.
INTRODUCTION
Eukaryotic elongation factor 2 kinase (eEF2K) is the best-characterized member of a small and still poorly understood family of protein kinases (generally termed α-kinases [1, 2]) that show little or no sequence homology with the main eukaryotic protein kinase superfamily. In stark contrast to the wealth of information about the structure and function of members of that family, there is a paucity of data on the α-kinases and, for example, although the structures of the catalytic domains of two of them have been determined (3, 4), very little work has been done to test the roles of specific residues that have been predicted, based on structural studies, to play important roles in their functions. No structural data are yet available for eEF2K itself.
eEF2K is the only calcium/calmodulin (CaM)-dependent α-kinase. It phosphorylates and inactivates eEF2, thereby slowing down translation elongation. In addition to activation by Ca2+/CaM, eEF2K is also subject to several other regulatory inputs, e.g., from the mammalian target of rapamycin complex 1 (mTORC1) or from signals such as cyclic AMP (reviewed in reference 5).
The three-dimensional structures of the catalytic domains of two α-kinases are available: those of mouse ChaK (also termed TRPM7 [3]) and Dictyostelium discoideum myosin heavy chain kinase A (MHCK A [4]). They reveal a number of structural similarities to the main protein kinase family, as well as substantial differences. In particular, while there is no direct equivalent of the activation loop (subdomain VII) of the canonical kinase group, which is involved in interactions with protein or peptide substrates, the structural studies suggest that a region termed the N/D loop may play an analogous role in α-kinases (1, 4, 6). However, the importance of this region and specific residues within it for substrate phosphorylation remains to be tested. In eEF2K, a feature at the C terminus, remote from the catalytic domain, is required for phosphorylation of eEF2 (7–9) but not for an artificial substrate, the MH-1 peptide (8, 9), whose sequence corresponds to that around the phosphorylation site for MHCKs in the myosin heavy chains in Dictyostelium.
eEF2K and other α-kinases undergo autophosphorylation at multiple sites (10–15). For eEF2K, this is an intramolecular reaction (14), and at extended times, several moles of phosphate are incorporated per mole of eEF2K protein (13, 15). However, in our studies, only two major sites of autophosphorylation were evident (13), suggesting that further autophosphorylation involves low-level phosphorylation at multiple sites. One of the major sites, a threonine C-terminal to the kinase domain (Thr348 in human eEF2K), is conserved in MHCK A and appears to play a critical role in kinase activation (11, 13). Structural and other studies from Côté's group suggest that, for MHCK A, the phosphothreonine docks into a phosphate-binding pocket to induce an active conformation (11).
A positively charged residue(s), C-terminal to the target phosphoacceptor, appears to play a crucial role in substrate recognition by MHCK A, MHCK B, and eEF2K, making these enzymes basophilic kinases (16). These data also show that their substrate specificities are distinct. The structural basis of this has not been explored.
Recent work has shown that eEF2K helps cancer cells to resist nutrient starvation and that ablating eEF2K impairs the growth of solid tumors (17). Furthermore, certain tumors express eEF2K at high levels.
It is important to gain a better understanding of these atypical protein kinases. Here, we have tested the functional effects of altering residues that, based on the structures of the catalytic domains of MHCK A and TRPM7 (Chak1), are predicted to play important roles in the function or regulation of eEF2K.
Our data provide the first detailed analysis for this α-kinase. In particular, they show that (i) residues in the N/D loop that are highly conserved are critical for activity and (ii) other residues in this loop that differ between eEF2K and MHCK A play a key role in substrate specificity. Our data also reveal that a mutation in this region that occurs in cancer cells enhances eEF2K activity. Our findings also identify residues involved in the basophilic specificity of eEF2K and indicate that a binding pocket for the autophosphorylated threonine does indeed play a key role in the activity/activation of eEF2K, as reported for MHCK A (11).
MATERIALS AND METHODS
Homology modeling.
The modeling server ModWeb (http://salilab.org/modweb), an integral module of ModBase (18, 19), was used to create the three-dimensional structure of the human eEF2K domain. ModBase relies on the homology modeling package MODELLER (20). The coordinates of the MHCK domain (PDB code 3LKH), based on the X-ray crystal structure of a related α-kinase domain, were identified and used for the template structure for the homology modeling. The modeling was restricted to the eEF2K residues 104 to 323, because the sequence identity of the template sequence was only sufficiently high (40%) in this region (which corresponds to residues 552 to 805 of MHCK A). The ZDope score was −0.49, and a small number of clashes indicated a reliable protein structure. Figure 1 was generated using UCSF Chimera 1.7.1 (21).
FIG 1.

Comparison of the eEF2K homology model and the MHCK A structure. (A) Front view of the homology model for eEF2K (purple) with the β-strands and α-helices numbered according to the sequence alignment with TRPM7-CAT. The kinase domain consists of small and large lobes, named the N and C lobes. N and C termini are indicated. (B) Front view of the superimposed homology model of eEF2K (purple) and the structure of MHCK A (blue) (PDB code 3lkm). Amino acid residues, AMP, and two phosphate (Pi) molecules (Pi1 and Pi2) are shown as sticks, and Mg2+ ions are shown as green spheres. The location of the Pi pocket, active site, and N/D loop in the context of the entire structure of MHCK A and the eEF2K homology model are highlighted by boxes labeled C to E, which are shown in greater detail in panels C to E. (C) A detailed view of the Pi pocket. The phosphate molecule (Pi1) interacts with the side chains of Arg252 (Arg734 in MHCK A), Thr254 (Thr736), and Lys205 (Lys684). This is indicated by the blue dashed lines. The salt bridge between Arg252 (Arg734) and Asp280 (Asp762) is shown as a black dotted line. (D) Detailed view of the active site. The residue Asp284 in the active site of eEF2K is located in a position similar to that of Asp766 in MHCK A. This residue in MHCK A forms hydrogen bonds with the phosphate molecule (Pi2), the α-phosphate of AMP, and Mg2+ in the active site pocket. These hydrogen bonds are shown as blue dashed lines. (E) Detail of the N/D loop. The position of Asn299 in eEF2K is essentially identical to that of Asn781 in MHCK A. Hydrogen bonds between Asn781 (Asn299) and Thr771 (Thr289), Gly776 (Asp294), Gly783 (Gly301), and Mg2+ are shown as blue dashed lines.
Chemicals and other reagents.
All chemicals and biochemicals were purchased from Sigma-Aldrich unless otherwise stated. [γ-32P]ATP was from PerkinElmer. Lambda protein phosphatase was purchased from New England BioLabs. Antiserum against anti-phospho-Thr348 eEF2K was generated by Eurogentec (13). The MH-1 peptide RKKFGESEKTKTKEFL (8) was synthesized by China Peptides. Glutathione-Sepharose high-performance affinity medium for purification of glutathione S-transferase (GST)-tagged eEF2K was purchased from GE Healthcare.
SDS-PAGE and immunoblotting.
SDS-PAGE was performed with the Bio-Rad Laboratories Protean 3 minslab gel system, using the standard procedure (22). Immunoblotting was performed by electrotransferring proteins resolved by SDS-PAGE onto nitrocellulose membranes. Membranes were then blocked in phosphate-buffered saline containing 0.05% (vol/vol) Tween 20 and 5% (wt/vol) skimmed milk powder for 1 h at room temperature (20 to 22°C). Membranes were probed with the indicated primary antibody overnight at 4°C. After incubation with fluorescently tagged secondary antibody, signals were detected by using a Li-Cor Odyssey imaging system.
Protein expression and mutagenesis.
The cDNA encoding human eEF2K was cloned into the vector pGEX-6P to allow efficient expression as a GST fusion protein in Escherichia coli. Point mutations were created using Pfu polymerase (Promega). The expression of GST-tagged eEF2K in E. coli Rosetta cells (Novagen) was performed as described previously (9).
Cell culture and treatment.
HEK 293 (human embryonic kidney) cells were cultured and transfected as described previously (23). Cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum, 2 mM glutamine, and 1% penicillin-streptomycin. For Western blot analyses, cells were extracted into buffer A, consisting of 50 mM β-glycerophosphate (pH 7.5), 1 mM EGTA, 1 mM EDTA, 1% (vol/vol) Triton X-100, 1 mM Na3VO4, 0.1% (vol/vol) 2-mercaptoethanol, and protease inhibitors (leupeptin, pepstatin, and phenylmethylsulfonyl fluoride, each at 1 μg/ml). Lysates were centrifuged at 13,000 rpm to remove debris. Protein concentrations in the resulting supernatants were determined as described previously (24).
Protein synthesis measurements.
Protein synthesis was measured by the SUnSET method as described previously (25). HEK 293 cells were transfected for 24 h. Cells were incubated with puromycin (1 μM final concentration) for 15 min. For Western blot analyses, cells were extracted into buffer A as described above in “Cell culture and treatment.” Where used, cycloheximide was added to a final concentration of 10 μg/ml. Lysates were analyzed by SDS-PAGE and Western blotting with an antipuromycin antibody. (25).
Assays for eEF2 kinase activity.
Assays of eEF2K activity were performed against purified eEF2 or MH-1 as described previously (9). Unless otherwise stated, MH-1 was used at a final concentration of 300 μM.
Dephosphorylation of recombinant eEF2K.
Recombinant eEF2K (1 μg of protein) was incubated at 30°C with λ phosphatase (approximately 200 units/20 μl of reaction mixture, where 1 unit corresponded to the amount of enzyme that hydrolyzes 1 μmol of p-nitrophenyl phosphate per min at pH 9.8 and 37°C). Dephosphorylated eEF2K was then diluted in autophosphorylation buffer.
Autophosphorylation of recombinant eEF2K.
Recombinant eEF2K (0.5 μg) was incubated with phosphorylation buffer containing Ca2+/CaM (9), and the reaction was initiated by adding [γ-32P]ATP (final concentration, 0.1 mM; 1 μCi per reaction mixture). Reaction mixtures were incubated at 30°C for the appropriate time (up to 10 min), and then SDS-PAGE sample buffer was added. Samples were immediately heated at 95°C for 5 min to denature the proteins and stop the reaction. Products were analyzed by SDS-PAGE (10% gel) and, after staining with Coomassie brilliant blue, the gels were placed into destain/fixing solution (50% [vol/vol] methanol and 10% [vol/vol] acetic acid). Gels were then placed on Whatman 3MM paper, covered with Saran wrap, and then dried on a vacuum gel dryer. Radioactivity was detected by using a phosphorimager (Typhoon; GE Healthcare). Assays were all also performed as described above but with unlabeled ATP (200 μM) to study a specific autophosphorylation site (Thr348). The samples were immediately heated at 95°C for 5 min to denature the proteins and stop the reaction. Products were analyzed by SDS-PAGE and Western blotting with phospho-specific antibody toward Thr348 on eEF2K.
Reproducibility and statistical analysis.
Numerical data are expressed as means ± standard errors of the means (SEM) for the indicated number of individual experiments. Wherever appropriate, the statistical significance of the data was assessed by two-way analysis of variance (ANOVA) followed by a Bonferroni post hoc test. For immunoblots and phosphorimages, data shown are typical of at least three entirely independent experiments.
RESULTS AND DISCUSSION
Comparative molecular modeling of human eEF2K.
The absence of a three-dimensional structure of eEF2K and the increasing interest in studying eEF2K prompted us to construct a homology model for it. Here we were interested primarily in the Pi pocket, the active site, and the N/D loop. To obtain a homology model of eEF2K (Fig. 1A), the crystallographic structure of MHCK A, available in the Protein Data Bank (PDB 3lkm) (4), was used as a template. The proteins are about 44% identical in their catalytic domain sequences. Several homology modeling tools were used (see Materials and Methods). The Ramachandran plot showed 82.9% of residues were in the most favorable regions, 14% in additional allowed regions, 2.1% in generously allowed regions, and 1% (only 2 amino acids) in the disallowed region. It is important to keep in mind that the template does have one region where there is a large insertion (corresponding to MHCK A residues 603 to 630) and another where there is a small insertion (residues 701 to 707). Consequently, the homology model may not be as accurate in these regions, although the high identity of the target template sequences in a number of other regions makes the whole model plausible.
Superposition of the eEF2K homology model with the known structure of MHCK A revealed a Cα root mean square deviation of 0.6 Å, indicating a close resemblance of the modeled domain with the catalytic domain of the MHCK A structure (4) (Fig. 1B). Most of the differences between the homology model and the X-ray crystal structure occur in the flexible loop regions corresponding to MHCK A residues 600 to 630 and 649 to 656 (Fig. 1B). Importantly, certain residues in the active site (Fig. 1C), the Pi pocket (Fig. 1D), and the N/D loop (Fig. 1E), are highly conserved and distant from the flexible regions. The Pi pocket is located in the loop after the α-helix C near the bottom of the C-terminal lobe and is formed by the conserved side chains of Lys205, Arg252, and the main chain carbonyl and side chain hydroxyl group of Thr254 (Fig. 1C). In addition, the salt bridge between Arg252 in the Pi pocket and Asp280 in the active site loop is conserved and anchors the catalytic loop in place. The invariant residue Asp184 lies at the back of the active site pocket (Fig. 1C). Asp284 is positioned in the active site in a location similar to that of Asp766 in the active site of MHCK A (Fig. 1D). Comparison of the structure of MHCK A and the eEF2K homology model showed that the N/D loop contains the highly conserved Asn299 in exactly the same position, where hydrogen bonds likely form with the side chain hydroxyl group of Thr289, the main chain carbonyl of Asp294, and the main chain amide of Gly301 (Fig. 1E). The side chain of Asp297 points outward, away from the N/D loop, in a fashion to similar to that of Leu779 in MHCK A. Interestingly, the region directly adjacent to the N/D loop, which forms an α-helix, varies between eEF2K and MHCK A. The locations of the positively charged residue Lys784 (Val302 in eEF2K) and Arg303 (Ala785 in MHCK A) differ; however, both point into solvent.
Based on these observations from the structure modeling, we created a suite of mutant proteins to allow us to explore the roles of specific residues in the function of eEF2K.
Residues involved in the proposed docking site for p-Thr348.
We began by investigating the autophosphorylation of eEF2K and its role in promoting its function. When expressed in E. coli, eEF2K is already substantially autophosphorylated at Thr348 (Fig. 2A, right hand lanes, with or without phosphatase treatment). This event is required for eEF2K function, as shown by the observation that mutating Thr348 to alanine abolishes eEF2K activity (13). Since E. coli does not contain CaM, it was possible that autophosphorylation of eEF2K is independent of Ca2+/CaM. To test this, GST-eEF2K from bacteria was again treated with λ phosphatase to dephosphorylate Thr348 (Fig. 2A) and then incubated with ATP with or without Ca2+/CaM. Analysis by Western blotting using an antibody specific for eEF2K phosphorylated on Thr348 (13) revealed that autophosphorylation is heavily dependent upon Ca2+/CaM, as observed for other substrates (eEF2 and MH-1 [9]). The autophosphorylation of eEF2K in E. coli presumably reflects the cumulative effect of a low basal rate of this process, resulting in the appreciable level of autophosphorylation seen in Fig. 2A.
FIG 2.

Effects of selected point mutations on the activity of eEF2K. (A) Recombinant GST-eEF2K was incubated with alkaline phosphatase (PPase) for 20 min at 30°C before incubating the mixture with nonradioactive ATP in the presence or absence of Ca2+/CaM for the times indicated in the figure. Samples were analyzed by SDS-PAGE and Western blotting using the indicated antibody. (B) Alignment of relevant regions of human eEF2K, eEF2K from selected other species, and MHCK isoforms; conserved residues of interest are indicated by black boxes. −/− in a sequence indicates a gap in the sequence. Hs, Homo sapiens; Mm, Mus musculus; Ac, Anolis carolinensis; Dr, Danio rerio; Ce, Caenorhabditis elegans; Dd, Dictyostelium discoideum. (C) WT and mutant eEF2K proteins were expressed as GST fusions in E. coli and purified, and their concentrations were carefully measured. Samples were analyzed by SDS-PAGE followed by Western blotting. In all cases, the same amounts of WT and mutant GST-eEF2K were used in all experiments. The level of autophosphorylation was measured using a phospho-specific antibody toward Thr348. (D) Recombinant GST-eEF2K or selected point mutants were incubated with alkaline phosphatase (PPase) for 20 min at 30°C, with samples taken for analysis by Western blotting (upper two sections) before incubation with Ca2+/CaM, as indicated, with ATP or [γ-32P]ATP for 10 min at 30°C (lower two sections). The reaction products were analyzed by SDS-PAGE and phosphorimaging or Western blotting using the indicated antibodies. (E) Recombinant GST-eEF2K was pretreated with alkaline phosphatase as described for panel D and then incubated with nonradioactive ATP in the presence of Ca2+/CaM for the indicated times; samples were analyzed by Western blotting using the indicated antibodies (eEF2K was detected using anti-GST). (F) Activities of selected point mutants of eEF2K were determined against eEF2 (without pretreating the GST-eEF2K with phosphatase). All assays were performed within the linear range of the assay. Numbers below each lane indicate the level of radiolabeling relative to that of WT eEF2K at 2 min. (G) Activities of selected point mutants against MH-1. All assays were performed within the linear range of the assay. Data are means ± SEM (n = 3). ****, P < 0.0001. (H) Activities of WT eEF2K, T348A, and T254A/T348A were assayed for MH-1 in the presence or absence of 5 mM NaH2PO4 for 10 min. Data are expressed for each mutant in the presence of phosphate relative to the same mutant tested without additional phosphate. Data are means ± SEM (n = 3). **, P < 0.01; ****, P < 0.0001. P values were derived using a two-way ANOVA for comparison with the WT followed by Tukey's multiple comparison post hoc tests.
Structural and biochemical studies on MHCK A indicate that Lys684, Arg734, and Thr736 help form the pocket into which the autophosphorylated threonine (Thr825) is found (the Pi pocket (Fig. 1C) (11). Mutating any of these residues (to alanine) almost abolished MHCK A activity (11). Thr825 corresponds to Thr348 in eEF2K. The residues forming the pocket in MHCK A are precisely conserved in eEF2K, as Lys205, Arg252, and Thr254 (Fig. 1C and 2B). We therefore tested the effects of mutating them on the autophosphorylation and activity of eEF2K; the mutations tested were K205A and R252M (both cause loss of positive charge) and T254A (causing removal of possible hydrogen bonding). Wild-type (WT) and mutant eEF2K proteins were expressed as GST fusions in E. coli and purified, and their concentrations were carefully measured; the same amounts of WT and mutant GST-eEF2K were used (verified by staining an SDS gel [e.g., Fig. 2C]). All three mutants were phosphorylated at Thr348 to similar final levels as with WT eEF2K (Fig. 2C), and so their differing activities do not arise from differences in autophosphorylation.
It was nevertheless possible that these mutations decreased the rate of autophosphorylation but not its final level. To assess this, we first treated eEF2K with λ phosphatase, which removed the phosphate from Thr348 (Fig. 2D, upper section). We then incubated WT or mutant eEF2K with radioactive ATP. As shown in Fig. 2D (lower section), all three mutants underwent autophosphorylation at much lower rates than WT eEF2K (Fig. 2D). Autophosphorylation of Thr348 in wild-type eEF2K preceded that of Ser445 (Fig. 2E), consistent with the idea that Thr348 is required for activation of eEF2K (in a similar way to Thr825 in MHCK A [11]), which can subsequently phosphorylate itself on other sites.
Although their final levels of autophosphorylation were similar to WT eEF2K (Fig. 2C), each of these mutants, when expressed in E. coli, showed markedly decreased activity against eEF2 (Fig. 2F) or MH-1 (Fig. 2G). This is consistent with the earlier conclusion (11) that the corresponding arginyl residue in MHCK A interacts with the autophosphorylated threonine to allow it to attain the active conformation.
It has previously been shown that adding orthophosphate can promote the activity of a mutant of MHCK A where Thr825 has been mutated, presumably by acting as a ligand for the phosphate binding pocket (11). Mutating the corresponding residue in eEF2K, Thr348, to alanine significantly reduces catalytic activity. The dramatically lower activity of eEF2K[T348A] toward MH-1 (13) was significantly increased by the addition of NaH2PO4 to the assay mixture (Fig. 2H). Mutating Thr254 to Ala blocked the ability of NaH2PO4 to activate the T348A mutant (Fig. 2H), supporting the interpretation that this residue plays an important role in binding phospho-Thr348.
Mutation of Asp284, a potential phosphoacceptor residue, abolishes eEF2K activity.
The crystal structure of MHCK A revealed that a highly conserved aspartate (Asp766) in the active site is phosphorylated (4), perhaps as part of its catalytic mechanism. An aspartate also occurs at this position in eEF2K (Asp284 [Fig. 1B and 2B]), changing it to a similar but uncharged asparagine, which cannot accept a phosphate group, greatly decreased the level of autophosphorylation (Fig. 3A, inset) and, probably as a result, activities against MH-1 (Fig. 3A) and eEF2 (Fig. 3B). Adjacent to this, Asp280 in eEF2K corresponds to an acidic residue (usually Asp) in other α-kinases (Fig. 1 and 2B). eEF2K[D280A] showed a decreased level and rate of autophosphorylation (Fig. 2C and D) and lower activities against eEF2 and MH-1 (Fig. 3C and D). Thus, both aspartates in this part of eEF2K are critical for activity.
FIG 3.

Activities of point mutants of eEF2K. (A) Activities of WT and D284N GST-eEF2K against the MH-1 peptide. Data are means ± SEM (n = 3). ***, P < 0.001; ****, P < 0.0001. (Inset) Levels of autophosphorylation of WT and D284N. (B) Activities of WT and D284N eEF2K against eEF2. Assays were performed within the linear range. Samples were analyzed by SDS-PAGE and phosphorimaging. The gel was also stained with Coomassie brilliant blue to confirm equal amounts of eEF2. (C) Activities of selected point mutants against MH-1. All assays were performed within the linear range of the assay. Data are means ± SEM (n = 3). **, P < 0.01; ****, P < 0.0001. P values were obtained using a two-way ANOVA compared with the WT followed by Bonferroni post hoc tests. (D) Activities of WT (lane 1) and D280A (lane 4) forms of eEF2K were determined against eEF2. All assays were performed within the linear range of the assay. Samples were analyzed by SDS-PAGE and autoradiography. The gel was also stained with Coomassie brilliant blue to confirm equal amounts of eEF2.
Residues adjacent to the autophosphorylation site at Thr348.
The only known substrates for eEF2K, eEF2 and MH-1, each contain a basic residue at the +3 position relative to the phospho-acceptor (Fig. 4A); interestingly, the residue at +3 relative to the main autophosphorylation site in eEF2K, Thr348, is also a basic one (Arg351), and this is conserved in vertebrate eEF2K sequences (Fig. 4B). To test its importance for autophosphorylation of eEF2K, this arginyl residue was mutated to methionine. Since Arg351 is part of the epitope for the phospho-specific antibody, this reagent cannot be used to assess the level of p-Thr348 in the mutant proteins. We therefore treated the purified proteins with λ phosphatase and allowed them to undergo autophosphorylation in vitro. As shown in Fig. 4C, eEF2K[R351M] showed much lower efficiency of (re)autophosphorylation (32P labeling) than WT eEF2K. These findings support the idea that a C-terminal basic residue is important for substrate recognition by eEF2K, consistent with data obtained using synthetic peptide substrates (16). The eEF2K[R351M] protein expressed in E. coli also showed similar activity to WT eEF2K against eEF2 (Fig. 4D) but decreased activity against MH-1 (Fig. 4E), suggesting this residue is less important for trans phosphorylation than for autophosphorylation.
FIG 4.

Effects of mutations at residues potentially involved in the autophosphorylation of eEF2K. (A) The residue phosphorylated is shown in bold, positively charged residues are underlined, and the residues at +1 and +3 are italicized and denoted by arrows. The phosphorylated residue (Thr56 in eEF2K) is also indicated by an arrow. The phosphorylation sites in eEF2 and MH-1 were identified previously (reference 16 and 29, respectively). (B) Alignment of relevant regions of eEF2K sequences from selected species and Dictyostelium MHCK isoforms; conserved residues of interest are indicated by black boxes. −/− indicates a gap in the sequence. Abbreviations for species are given in the legend to Fig. 2. The arrow denotes the basic residue present at +3 relative to the autophosphorylation site. (C) Recombinant GST-eEF2K or selected point mutants were incubated with alkaline phosphatase (PPase) for 20 min at 30°C, and samples were taken for Western blot analysis prior to incubating the phosphatase-treated eEF2K with Ca2+/CaM, as indicated, with ATP or [γ-32P]ATP for 10 min at 30°C. Samples were analyzed by SDS-PAGE and phosphorimaging or Western blotting using the indicated antibodies. The upper part shows data for phosphatase-treated sample; the lower part shows data for incubation of the material with radioactive ATP. (D) Activities of selected point mutants of eEF2K were determined against eEF2 (without pretreating the GST-eEF2K with phosphatase). All assays were performed within the linear range of the assay. Numbers below each lane indicate the level of radiolabeling relative to that of WT eEF2K at 2 min. (E) Activities of selected point mutants against the MH-1 peptide. All assays were performed within the linear range of the assay. Data are means ± SEM (n = 3). **, P < 0.01; ****, P < 0.0001. (F) Level of autophosphorylation for WT eEF2K and the E183A/D184A mutant determined by SDS-PAGE and Western blotting using a phospho-Thr348 antibody. (G) The activities of WT and the E183A/D184A mutant eEF2K against various concentrations of the MH-1 peptide. Data are means ± SEM (n = 3). **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Given that they are conserved in eEF2K sequences from diverse species (Fig. 4B), the hydrophobic residues immediately C-terminal to the autophosphorylation site may also be important for this reaction; in eEF2K, these are Ile349 and Leu350 (Fig. 4B). The first of these is also a hydrophobic residue (Ile, Leu, Met, Val, or, in one case, Phe) in other α-kinases; the second is nearly always a nonpolar residue (Fig. 4B). The eEF2K[I349A/L350A] mutant showed a reduced ability to undergo (re)autophosphorylation following phosphatase treatment (Fig. 4C) and, to a lesser extent, lower activity against eEF2 or MH-1 (Fig. 4D and E). Although the data suggest that these mutations decrease the activity of eEF2K directly, we cannot exclude that this reflects, at least in part, alterations in its level of autophosphorylation, which we cannot assess using the phospho-specific antibody for Thr348.
Residues that may interact with the C-terminal basic residue.
The finding that a C-terminal positively charged residue is important for the activity of eEF2K in autophosphorylation (this study) and for phosphorylation of peptide substrates (16) raises the question of how eEF2K recognizes such residues in its substrates. Structural data and modeling studies on MHCK A suggest that the basic residue at +3 in the substrate for MHCK A may interact electrostatically with a highly conserved aspartate (4, 16) (Asp184 in human eEF2K). There is another negatively charged residue in this part of eEF2K, Glu183 (Fig. 4B). We mutated both Glu183 and Asp184 to alanines. This had little effect on the final level of autophosphorylation at Thr348 (Fig. 4F) but did impair the rate of autophosphorylation following phosphatase pretreatment (Fig. 4C). These mutations also decreased the activity of eEF2K against MH-1 (Fig. 4G). The lower activity of the double mutant against MH-1 was apparent across a range of substrate concentrations (Fig. 4G). These data clearly support the conclusion that these acidic residues play a role in peptide substrate recognition. These mutations had less effect on activity against eEF2 (Fig. 4D), perhaps reflecting an additional role for the extreme C terminus of eEF2K in phosphorylation of its physiological substrate (8, 9), which might partly compensate for mutations in the N/D loop.
The N/D loop plays a crucial role in the activity and substrate specificity of eEF2K.
There are no experimental data regarding what determines the substrate specificities of the catalytic domains of the different α-kinases. Structural studies (3, 4) have indicated that the N/D loop may play a role similar to that of the activation loop in members of the main protein kinase superfamily, which undergoes a conformational change that leads to kinase activation and plays a key role in binding to protein/peptide substrates. There is no evidence that any residue in this loop in eEF2K is phosphorylated. To explore the role of the N/D loop in the activity of eEF2K, we made and tested selected mutants. We were particularly interested in identifying mutations that altered the relative levels of activity of eEF2K against eEF2 or MH-1, as these may give insights into the basis of its specificity; mutations that affect activity against both substrates to similar extents may reflect a role for the residue under study in allowing eEF2K to adopt its active conformation.
Asn299 is eEF2K corresponds to a residue that is part of a motif (Asn-Leu-Gly) that is conserved in other α-kinases, e.g., MHCK A and TRPM7. The conformation of this region differs in the crystal structures of MHCK A and TRPM7 (3, 4). In the former, the side chain of this Asn (781) is directed inwards (toward the N/D loop), whereas in TRPM7 it points outward and is solvent accessible. It has been suggested (3) that in TRPM7 the side chain of this residue (Asn1795) may form hydrogen bonds with the protein substrate and that another Asn in the same region (Asn1731) likewise contributes to substrate binding. In eEF2K, these residues correspond to Asn242 and Asn299 (Fig. 1E and 5A). Strikingly, mutation of either residue in eEF2K to alanine completely abolished activity against MH-1 (Fig. 5B). Mutation of the corresponding asparagine residues in the kinase domain of TRPM7 also markedly decreased the activity, although less so than for eEF2K (26).
FIG 5.

Mutations at asparagine residues in the N/D loop of eEF2K. (A) Alignment of relevant regions of selected eEF2K sequences and certain other α-kinases, with conserved residues of interest indicated by black boxes. −/− indicates a gap in the sequence. Abbreviations for species are given in the legend to Fig. 2. (B) Activities of selected point mutants against MH-1. All assays were performed within the linear range of the assay. Data are means ± SEM (n = 3). ****, P < 0.0001. (C) Activities of selected point mutants against eEF2. (D) The activities of WT and N299A against increasing concentrations of eEF2. (E and F) Level of autophosphorylation determined by SDS-PAGE and Western blot using the phospho-Thr348 antibody. (F) HEK293 cells were transfected with vectors encoding FLAG-tagged wild-type eEF2K or the N242A, N299A, or K170M mutants. Twenty-four hours later, cells were lysed, and equal amounts of protein were analyzed for p-eEF2, phospho-Thr348, FLAG (for eEF2K expression), or actin (loading control). The figures below each lane indicate the level of eEF2 phosphorylation, normalized to the signal for FLAG, as a ratio.
In the case of eEF2 as a substrate, eEF2K[N242A] showed no detectable activity, while eEF2K[N299A] did exhibit a very low level of function when tested at high eEF2 concentrations (Fig. 5C and D). This contrasts with its complete lack of activity against MH-1. The fact that appreciable activity is still seen at high eEF2 concentrations indicates that the Asn299Ala mutation may increase the Km of eEF2K for eEF2, whereas it eliminates activity against MH-1. The level of autophosphorylation of Thr348 was very low in each of these mutants (Fig. 5E), which may explain their decreased activities. Some autophosphorylation was detectable upon long exposure of the immunoblots and was stronger for eEF2K[N299A] than for eEF2K[N242A] (Fig. 5F). Nonetheless, the complete loss of activity of eEF2K[N299A] against MH-1, which does retain some activity against eEF2, suggests a role for this region of eEF2K in substrate binding and substrate specificity, as suggested previously (3). It should be noted that the Km of eEF2K for MH-1 is high (in our hands, about 350 μM; previously it was estimated at 660 μM [8]).
It was important to assess whether these mutants also showed reduced activities against eEF2 when expressed in mammalian cells. Figure 5G shows that levels of phosphorylated eEF2 were much lower in cells ectopically expressing either eEF2K[N242A] or eEF2K[N299A] relative to cells overexpressing the wild-type kinase, even though the expression levels of the mutants were at least as high as that of WT eEF2K. Indeed, eEF2 phosphorylation in cells expressing these mutants was similar to that observed in cells expressing eEF2K[K170M], which is essentially inactive (9). The level of autophosphorylation of Thr348 was also greatly decreased in these mutants, consistent with their strongly impaired activity (Fig. 5G).
While several residues within the N/D loop are strongly, or even universally, conserved between α-kinases, others differ substantially (Fig. 6A). For example, Gly1777, Lys1785, and Ala1786 in MHCK A are replaced in eEF2K by residues with very different side chains, i.e., Asp297, Val302, and Arg303, respectively. Since this feature seems to correspond to the activation loop in mainstream kinases, these residues might be involved in substrate recognition/specificity in eEF2K and perhaps other α-kinases. The levels of phosphorylation at Thr348 for the D297A and V302K/R303A variants were at least as high as for WT eEF2K (Fig. 6B). eEF2K[D297A] also showed much higher rates of (re)autophosphorylation at Thr348, consistent with elevated catalytic activity (Fig. 6C). eEF2K[D297A] showed markedly enhanced activity against MH-1 (Fig. 6D) and a sizeable increase in activity against eEF2 (Fig. 6E).
FIG 6.

Mutations at residues in the N/D loop region of eEF2K alter its substrate specificity. (A) Alignment of N/D loop regions from eEF2K from different species against MHCK isoforms, with conserved residues of interest indicated by black boxes. Abbreviations for species are given in the legend to Fig. 2. (B) Recombinant GST-eEF2K and selected mutants were analyzed by SDS-PAGE and Western blotting to determine the level of autophosphorylation using phospho-Thr348 antibody. (C) Recombinant GST-eEF2K or selected point mutants were incubated with alkaline phosphatase (PPase) for 20 min at 30°C before incubation with Ca2+/CaM, as indicated with ATP or [γ-32P]ATP for 10 min at 30°C. Samples were analyzed by SDS-PAGE and phosphorimaging or Western blotting using the indicated antibodies. (D) Activities of selected point mutants against the MH-1 peptide. All assays were performed within the linear range of the assay. Data are means ± SEM (n = 3). ***, P < 0.001; ****, P < 0.0001. (E) Activities of selected point mutants of eEF2K against EF2 were determined. All assays were performed within the linear range of the assay. Samples were analyzed by SDS-PAGE and phosphorimaging. The labeling of eEF2 at the 5-min time point was quantified (values below each set of assays; normalized to labeling of eEF2 by WT eEF2K). (F) Where indicated, HEK293 cells were transfected with vectors encoding WT eEF2K or the D297L mutant. In some cases, cells were treated with MG132 (10 μM, 16 h). Forty hours after transfection, cell lysates were prepared and analyzed by Western blotting using the indicated antibodies. (G) Results of an experiment similar to that shown in panel F, except that protein synthesis was assessed using the SUnSET method. In one case, cells were treated with cycloheximide (CHX) to inhibit protein synthesis (in order to provide a baseline for assessing the true rate of protein synthesis). Mock indicates cells were treated in a similar way as when transfected but no DNA was added. Detection of new protein was achieved using antipuromycin antibodies; the membrane was reprobed with an anti-FLAG antibody. Numbers below each lane indicate relative levels of puromycin incorporation (protein synthesis [P.S.]), determined after subtracting the signal for CHX-treated cells. (H) Recombinant WT GST-eEF2K and the D297L mutant were analyzed by SDS-PAGE and Western blotting using phospho-Thr348 or phospho-Thr445 antibodies. Total amounts were assessed using anti-GST antibody.
To further test their roles, we created mutants in which one or more residues in this region of eEF2K were replaced by the residue found at that position in MHCK A. For example, we initially created eEF2K[D297L] and, as a control, also eEF2K[D297A]. We also made eEF2K[V302K/R303A]. These mutants all showed levels of phosphorylation at Thr348 similar to that of WT eEF2K (Fig. 6B). eEF2K[D297L] showed much higher activity than WT eEF2K against MH-1 (Fig. 6D) and increased activity against eEF2 (Fig. 6E). To study further its activity against its natural substrate, eEF2, we compared the levels of eEF2 phosphorylation in cells ectopically expressing wild-type eEF2K or the D297L mutant. The latter was expressed at much lower levels than wild-type eEF2K (Fig. 6F). Since high activity can lead to degradation of eEF2K by a proteasome-dependent mechanism (27), the low expression level could result from its degradation. We therefore sought to stabilize the enzyme by treating cells with the proteasome inhibitor MG132. This increased the levels of eEF2K[D297L] without affecting the levels of the wild-type protein, but eEF2K[D297L] was still expressed at lower levels. This likely reflects the previously reported ability of active eEF2K (which negatively regulates translation elongation) to inhibit its own expression (or expression of a cotransfected reporter [28]). Despite these differences in expression levels, it is clear that eEF2K[D297L] causes a level of phosphorylated eEF2 that is at least as high as seen in cells ectopically expressing wild-type eEF2K. This indicates that the D297L mutant is considerably more active than the wild-type kinase when expressed in human cells. Consistent with this, eEF2K[D297L] showed a greater level of autophosphorylation than wild-type eEF2K when its levels of expression were taken into account (Fig. 6F). We employed a recently described nonradioactive labeling method to assess rates of protein synthesis (the SUnSET method [25]). This revealed that, although the eEF2K[D297L] mutant was expressed at lower levels than wild-type eEF2K, it caused a greater inhibition of protein synthesis, consistent with its higher intrinsic activity (Fig. 6G).
We also noted that eEF2K[D297L] showed partial retardation on an SDS-PAGE gel (Fig. 6B), which could indicate higher autophosphorylation; consistent with this, and with its increased catalytic activity, the level of phosphorylation of the other major autophosphorylation site (Ser445 [13]) was much higher for this mutant than for WT eEF2K (Fig. 6H).
The above data show that changing D297 in eEF2K to the residue found at the corresponding position in MHCK A greatly enhances the activity of eEF2K against MH-1, which is based upon the natural substrate of MHCK A. This observation lends strong support to the concept that certain residues in the N/D loop play an important role in determining substrate specificity. eEF2K[D297A] also showed increased activity against MH-1, but the effect was smaller than for eEF2K[D297L] (Fig. 6D). To examine this further, we tested the activities of eEF2K, eEF2K[D297A], and eEF2K[D297L] by using a range of concentrations of MH-1.
The data clearly showed that the activity of the mutants, especially eEF2K[D297L], was strikingly increased at low MH-1 concentrations (Fig. 7A), i.e., that they likely had a lower Km for MH-1. We therefore directly compared the activity of eEF2K[D297L] with that of WT eEF2K at a single concentration of MH-1 (50 μM), which is lower than that used in our standard assay (300 μM). This revealed that, at this lower concentration, eEF2K[D297L] showed much higher activity against MH-1 than did WT eEF2K (Fig. 7B). Detailed analysis of the activities of these eEF2K proteins across a range of substrate concentrations revealed that eEF2K[D297A] and eEF2K[D297L] indeed showed lower Km values for MH-1 (110 μM and 240 μM, respectively) than did WT eEF2K (350 μM). The D297A and D297L mutants also showed much higher Vmax values (330% and 440% of wild-type eEF2K, respectively).
FIG 7.

Asp297 plays a key role in the substrate specificity of eEF2K. (A) Activities of selected eEF2K mutants against different concentrations of MH-1, as indicated. All assays were performed within the linear range of the assay. Data are means ± SEM. (B) Time course of WT and D297L against MH-1 at a low MH-1 concentration (50 μM). Data are means ± SEM (n = 3). (C) Activities of WT and D297L/V302K/R303A eEF2K against different concentrations of MH-1. All assays were performed within the linear range of the assay. Data are means ± SEM (n = 3).*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
In contrast, eEF2K[V302K/R303A] showed activity similar to WT eEF2K against MH-1 and eEF2 (Fig. 6D and E). To determine whether mutating Val302 and Arg303 might only affect activity in the context of having the MHCK A residue at position 297, we created mutants in which the D297A or D297L mutation was introduced together with V302K and R303A. Strikingly, these triple mutants showed enhanced activity against MH-1, as observed for the D297 mutations alone (Fig. 6D), but no increase in activity against eEF2 itself (Fig. 6E). The D297L/V302K/R303A triple mutant showed a pronounced increase in activity against MH-1 across a range of substrate concentrations, suggesting a lower Km for this substrate (Fig. 7C). Further analysis revealed it does show a much lower Km for MH-1 (90 μM) and a moderate increase in its Vmax (150% of that of wild-type eEF2K). Thus, by introducing these three mutations replacing residues in eEF2K by the corresponding ones in MHCK A, we were able to engineer the specificity of eEF2K to favor phosphorylation of MH-1, which is based on the physiological substrate for MHCK A.
To explore this further, we created mutations at other residues in the same region (Fig. 5A and 7). Thus, we made eEF2K[E290P], [T293K], and [T293K/D294G], thereby altering the residue in eEF2K to the one at that position in MHCK A (and also [T293A], a less radical change). All four mutants showed broadly similar final levels of autophosphorylation at Thr348 (Fig. 8A). After dephosphorylation by λ phosphatase, eEF2K[E290P] and [T293A], but not eEF2K[T293K/D294G], showed faster (re)autophosphorylation than WT eEF2K (Fig. 8B). eEF2K[E290P] and [T293A] showed increased activity against MH-1 relative to WT eEF2K (Fig. 8C) but no change in activity against eEF2 (Fig. 8D). eEF2K[T293K/D294G] actually showed decreased activity against MH-1 and eEF2. Overall, these data support the conclusion that residues in this region of eEF2K play an important role in determining substrate preference, although it is not always the case that altering multiple residues to those found in MHCK A simply increases activity against MH-1 (e.g., data for eEF2K[T293K/D294G]). This was not surprising, given that there are other differences between the two proteins in this region (Fig. 6A). Indeed, mutating only Thr293 to the residue in MHCK A (creating eEF2K[T293K]) yields a protein with substantially higher activity against MH-1 not only but also eEF2 (Fig. 8E and F). This further supported the notion that this region is critical for activity against protein and peptide substrates. With the MH-1 peptide, eEF2K[T293K] showed a slightly lower Km and considerably higher Vmax than wild-type eEF2K (Km, 274 μM; Vmax, 385% of wild-type eEF2K).
FIG 8.

Mutations at certain residues in the N/D loop of eEF2K differentially affect the activities against MH-1 and eEF2. (A) Recombinant GST-eEF2K and selected mutants were analyzed by SDS-PAGE and Western blotting to determine the level of autophosphorylation. (B) Recombinant GST-eEF2K or selected point mutants were incubated with alkaline phosphatase (PPase) for 20 min at 30°C before incubation with Ca2+/CaM, with [γ-32P]ATP or nonradioactive ATP (bottom section), for 10 min at 30°C. Samples were analyzed by SDS-PAGE and phosphorimaging or immunoblotting, as appropriate. (C) Activities of selected point mutants against the MH-1 peptide. Assays were performed within the linear range. Data are means ± SEM (n = 3). (D) Activities of selected point mutants against eEF2. Assays were performed within the linear range. (E) Activities of WT and eEF2K[T293K] against different concentrations of MH-1, as indicated. Assays were performed within the linear range. Data are means ± SEM (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (F) Activities of WT and T293K eEF2K against eEF2. Assays were performed within the linear range. The figure shows a phosphorimage.
A mutation in the N/D loop found in cancer cells enhances eEF2K activity.
Recent data showed that eEF2K exerts a cytoprotective role in cancer cells, decreasing cell death under situations of nutrient deprivation (17). Mutations in the EEF2K gene have been reported for cancer cells from solid tumors (COSMIC database). These include R303C, which lies close to the N/D loop. The R303C mutation did not alter the final level of autophosphorylation at Thr348 (Fig. 9A). However, strikingly, the eEF2K[R303C] protein expressed in E. coli showed markedly increased activity against either MH-1 or eEF2 (Fig. 9B and C). This single mutation, to a residue not corresponding to that found in another α-kinase, enhances overall activity, rather than altering substrate selection, and may serve to protect tumor cells against nutrient deprivation.
FIG 9.
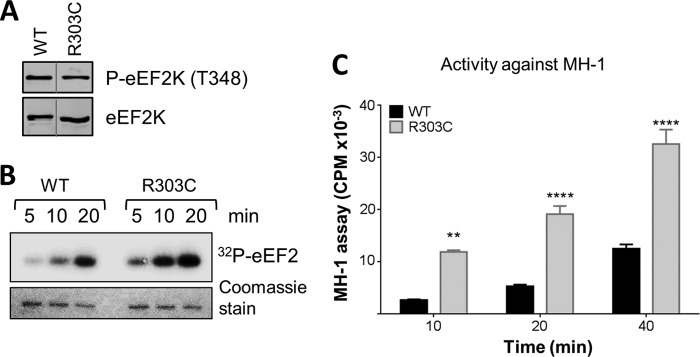
The R303C mutation, which occurs in cancer cells, enhances eEF2K activity. (A) Recombinant GST-eEF2K (WT) and R303C were analyzed by SDS-PAGE and Western blotting to determine the level of autophosphorylation, which was determined with a phospho-Thr348 antibody. (B) Activities of WT and R303C were determined against eEF2. All assays were performed within the linear range of the assay. (C) Activities of selected point mutants against the MH-1 peptide. All assays were performed within the linear range of the assay for WT eEF2K. Data are means ± SEM (n = 3). **, P < 0.001; ****, P < 0.0001.
Concluding remarks.
Our data provide substantial new insights into the structural organization and function of eEF2K, one of the so-far poorly characterized α-kinases. First, our data are consistent with the idea, previously suggested by Crawley et al. (11), that the major autophosphorylation site (Thr348 in eEF2K) docks into a conserved binding pocket to stabilize an active conformation. Given that the autophosphorylation site is conserved in several other α-kinases (11), this may be a widespread feature of this group of enzymes.
Second, our data provide the first insights into the molecular basis of the specificity of α-kinases. Indeed, in most cases no physiological substrates have been identified (other than the kinase itself via autophosphorylation). Because two substrates are known for eEF2K, we were able to explore the basis of its specificity. Our data suggest that the basic residue C-terminal to the acceptor residue (the main site in eEF2 being Thr56 [29[) may fit into a binding pocket in eEF2K, consistent with the conservation of the relevant residues across eEF2K proteins in diverse species, from budding yeast to mammals, and the fact that the main autophosphorylation sites in eEF2K, Thr348 and Ser445, also have a C-terminal basic residue (13).
In members of the main protein kinase superfamily, the activation loop plays an important role in their substrate specificities. It corresponds in the α-kinases to the N/D loop or glycine-rich motif (3, 4). Our data showed that residues in the N/D loop are either critical for activity (e.g., Asn299) or modify substrate specificity, providing the first evidence for the structural basis of this feature of α-kinases and for the idea that it serves an analogous role to the activation loop in mainstream kinases. Notably, mutating certain residues in eEF2K to the corresponding residue(s) in MHCK A enhanced activity against MH-1 (a peptide based on the physiological substrate for this α-kinase) relative to eEF2. Thus, it is clear that residues in the N/D loop play an important role in the substrate specificity of eEF2K and, likely, other α-kinases.
Finally, a mutation in this region that occurs in solid tumors enhances the activity of eEF2K against eEF2. Given that eEF2K helps protect cancer cells against nutrient starvation (17), this mutation may help to confer a survival advantage for such cells within tumors.
ACKNOWLEDGMENT
These studies were funded by a Programme Grant from the Wellcome Trust (to C.G.P.; 086688/Z/08/Z).
Footnotes
Published ahead of print 14 April 2014
REFERENCES
- 1.Middelbeek J, Clark K, Venselaar H, Huynen MA, van Leeuwen FN. 2010. The alpha-kinase family: an exceptional branch on the protein kinase tree. Cell Mol. Life Sci. 67:875–890. 10.1007/s00018-009-0215-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryazanov AG, Ward MD, Mendola CE, Pavur KS, Dorovkov MV, Wiedmann M, Erdjument-Bromage H, Tempst P, Parmer TG, Prostko CR, Germino FJ, Hait WN. 1997. Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc. Natl. Acad. Sci. U. S. A. 94:4884–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamaguchi H, Matsushita M, Nairn AC, Kuriyan J. 2001. Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol. Cell 7:1047–1057 http://dx.doi.org/10.1016/S1097-2765(01)00256-8 [DOI] [PubMed] [Google Scholar]
- 4.Ye Q, Crawley SW, Yang Y, Cote GP, Jia Z. 2010. Crystal structure of the alpha-kinase domain of Dictyostelium myosin heavy chain kinase A. Sci. Signal. 3:ra17. 10.1126/scisignal.2000525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Proud CG. 2007. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem. J. 403:217–234. 10.1042/BJ20070024 [DOI] [PubMed] [Google Scholar]
- 6.Drennan D, Ryazanov AG. 2004. Alpha-kinases: analysis of the family and comparison with conventional protein kinases. Prog. Biophys. Mol. Biol. 85:1–32 http://dx.doi.org/10.1016/S0079-6107(03)00060-9 [DOI] [PubMed] [Google Scholar]
- 7.Diggle TA, Seehra CK, Hase S, Redpath NT. 1999. Analysis of the domain structure of elongation factor-2 kinase by mutagenesis. FEBS Lett. 457:189–192 http://dx.doi.org/10.1016/S0014-5793(99)01034-0 [DOI] [PubMed] [Google Scholar]
- 8.Pavur KS, Petrov AN, Ryazanov AG. 2000. Mapping the functional domains of elongation factor-2 kinase. Biochemistry 39:12216–12224. 10.1021/bi0007270 [DOI] [PubMed] [Google Scholar]
- 9.Pigott CR, Mikolajek H, Moore CE, Finn SJ, Phippen CW, Werner JM, Proud CG. 2012. Insights into the regulation of eukaryotic elongation factor 2 kinase and the interplay between its domains. Biochem. J. 442:105–118. 10.1042/BJ20111536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark K, Middelbeek J, Morrice NA, Figdor CG, Lasonder E, van Leeuwen FN. 2008. Massive autophosphorylation of the Ser/Thr-rich domain controls protein kinase activity of TRPM6 and TRPM7. PLoS One. 3(3):e1876. 10.1371/journal.pone.0001876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crawley SW, Gharaei MS, Ye Q, Yang Y, Raveh B, London N, Schueler-Furman O, Jia Z, Cote GP. 2011. Autophosphorylation activates Dictyostelium myosin II heavy chain kinase A by providing a ligand for an allosteric binding site in the alpha-kinase domain. J. Biol. Chem. 286:2607–2616. 10.1074/jbc.M110.177014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitsui K, Brady M, Palfrey HC, Nairn AC. 1993. Purification and characterization of calmodulin-dependent protein kinase III from rabbit reticulocytes and rat pancreas. J. Biol. Chem. 268:13422–13433 [PubMed] [Google Scholar]
- 13.Pyr Dit Ruys S, Wang X, Smith EM, Herinckx G, Hussain N, Rider MH, Vertommen D, Proud CG. 2012. Identification of autophosphorylation sites in eukaryotic elongation factor-2 kinase. Biochem. J. 442:681–692. 10.1042/BJ20111530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redpath NT, Proud CG. 1993. Purification and phosphorylation of elongation factor-2 kinase from rabbit reticulocytes. Eur. J. Biochem. 212:511–520. 10.1111/j.1432-1033.1993.tb17688.x [DOI] [PubMed] [Google Scholar]
- 15.Tavares CD, O'Brien JP, Abramczyk O, Devkota AK, Shores KS, Ferguson SB, Kaoud TS, Warthaka M, Marshall KD, Keller KM, Zhang Y, Brodbelt JS, Ozpolat B, Dalby KN. 2012. Calcium/calmodulin stimulates the autophosphorylation of elongation factor 2 kinase on Thr-348 and Ser-500 to regulate its activity and calcium dependence. Biochemistry 51:2232–2245. 10.1021/bi201788e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawley SW, Cote GP. 2008. Determinants for substrate phosphorylation by Dictyostelium myosin II heavy chain kinases A and B and eukaryotic elongation factor-2 kinase. Biochim. Biophys. Acta 1784:908–915. 10.1016/j.bbapap.2008.03.001 [DOI] [PubMed] [Google Scholar]
- 17.Leprivier G, Remke M, Rotblat B, Dubuc A, Mateo AR, Kool M, Agnihotri S, El-Naggar A, Yu B, Prakash SS, Faubert B, Bridon G, Tognon CE, Mathers J, Thomas R, Li A, Barokas A, Kwok B, Bowden M, Smith S, Wu X, Korshunov A, Hielscher T, Northcott PA, Galpin JD, Ahern CA, Wang Y, McCabe MG, Collins VP, Jones RG, Pollak M, Delattre O, Gleave ME, Jan E, Pfister SM, Proud CG, Derry WB, Taylor MD, Sorensen PH. 2013. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153:1064–1079. 10.1016/j.cell.2013.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eswar N, John B, Mirkovic N, Fiser A, Ilyin VA, Pieper U, Stuart AC, Marti-Renom MA, Madhusudhan MS, Yerkovich B, Sali A. 2003. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31:3375–3380. 10.1093/nar/gkg543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pieper U, Webb BM, Barkan DT, Schneidman-Duhovny D, Schlessinger A, Braberg H, Yang Z, Meng EC, Pettersen EF, Huang CC, Datta RS, Sampathkumar P, Madhusudhan MS, Sjolander K, Ferrin TE, Burley SK, Sali A. 2011. ModBase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 39:D465–D474. 10.1093/nar/gkt1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eswar N, Eramian D, Webb B, Shen MY, Sali A. 2008. Protein structure modeling with MODELLER. Methods Mol. Biol. 426:145–159. 10.1007/978-1-60327-058-8_8 [DOI] [PubMed] [Google Scholar]
- 21.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 22.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 23.Hall-Jackson CA, Cross DA, Morrice N, Smythe C. 1999. ATR is a caffeine-sensitive, DNA-activated protein kinase with a substrate specificity distinct from DNA-PK. Oncogene 18:6707–6713 [DOI] [PubMed] [Google Scholar]
- 24.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 77:248–254 [DOI] [PubMed] [Google Scholar]
- 25.Schmidt EK, Clavarino G, Ceppi M, Pierre P. 2009. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6:275–277. 10.1038/nmeth.1314 [DOI] [PubMed] [Google Scholar]
- 26.Matsushita M, Kozak JA, Shimizu Y, McLachlin DT, Yamaguchi H, Wei FY, Tomizawa K, Matsui H, Chait BT, Cahalan MD, Nairn AC. 2005. Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1. J. Biol. Chem. 280:20793–20803. 10.1074/jbc.M413671200 [DOI] [PubMed] [Google Scholar]
- 27.Kruiswijk F, Yuniati L, Magliozzi R, Low TY, Lim R, Bolder R, Mohammed S, Proud CG, Heck AJ, Pagano M, Guardavaccaro D. 2012. Coupled activation and degradation of eEF2K regulates protein synthesis in response to genotoxic stress. Sci. Signal. 5:ra40. 10.1126/scisignal.2002718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diggle TA, Subkhankulova T, Lilley KS, Shikotra N, Willis AE, Redpath NT. 2001. Phosphorylation of elongation factor-2 kinase on serine 499 by cAMP-dependent protein kinase induces Ca2+/calmodulin-independent activity. Biochem. J. 353:621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price NT, Redpath NT, Severinov KV, Campbell DG, Russell JM, Proud CG. 1991. Identification of the phosphorylation sites in elongation factor-2 from rabbit reticulocytes. FEBS Lett. 282:253–258. 10.1016/0014-5793(91)80489-P [DOI] [PubMed] [Google Scholar]