

Abstract
The RNA-binding protein HuR binds at 3′ untranslated regions (UTRs) of target transcripts, thereby protecting them against degradation. We show that HuR directly interacts with cellular retinoic acid-binding protein 2 (CRABP2), a protein known to transport RA from the cytosol to the nuclear retinoic acid receptor (RAR). Association with CRABP2 dramatically increases the affinity of HuR toward target mRNAs and enhances the stability of such transcripts, including that of Apaf-1, the major protein in the apoptosome. We show further that its cooperation with HuR contributes to the ability of CRABP2 to suppress carcinoma cell proliferation. The data show that CRABP2 displays antioncogenic activities both by cooperating with RAR and by stabilizing antiproliferative HuR target transcripts. The observation that CRABP2 controls mRNA stabilization by HuR reveals that in parallel to participating in transcriptional regulation, the protein is closely involved in posttranscriptional regulation of gene expression.
INTRODUCTION
The vitamin A metabolite retinoic acid (RA) regulates transcription by activating two classes of nuclear receptors: the retinoic acid receptors (RARs) (1) and the peroxisome proliferator-activated receptor β/δ (PPARβ/δ) (2, 3). RA also associates in cells with intracellular lipid-binding proteins (iLBPs) (4, 5). Two iLBPs, cellular RA-binding protein 2 (CRABP2) and fatty acid-binding protein 5 (FABP5), support the biological activities of RA by transporting it from the cytosol to cognate nuclear receptors in the nucleus. In the absence of ligands, iLBPs are cytosolic, and upon binding ligand, a nuclear localization signal (NLS) is activated and they translocate to the nucleus (2, 6, 7). Hence, CRABP2 delivers RA to RAR and FABP5 shuttles it to PPARβ/δ. These binding proteins thus facilitate the ligation and markedly enhance the transcriptional activities of the respective receptors (6, 8–10). The involvement of RA signaling in cancer is complex. While activation of RARs triggers cell cycle arrest, apoptosis, and differentiation and thus suppresses tumor growth (9, 11–14), activation of PPARβ/δ results in enhanced proliferation and survival and can promote tumor development (2, 15–17). Consequently, RA suppresses growth of carcinomas in which CRABP2 is highly expressed, leading to efficient activation of RAR, but promotes the development of tumors in which the CRABP2/FABP5 ratio is low, resulting in diversion of RA to PPARβ/δ (2, 18–20). Available information indeed indicates that by targeting RA to RARs, CRABP2 displays potent antioncogenic activities (2, 9, 12, 13, 18, 19). The reports that CRABP2 expression is markedly downregulated in various cancers further suggest that its loss contributes to tumor development (21–24).
Surprisingly, we previously found that in addition to promoting the transcriptional activity of RAR, expression of CRABP2 in mammary carcinoma cells increases the levels of mRNAs that are not encoded by RAR target genes and that the effect is exerted even in the absence of RA. For example, CRABP2 expression was found to markedly increase the level of mRNA for apoptotic peptidase-activating factor 1 (Apaf-1), the major protein of the apoptosome (12, 18). Consequently, CRABP2 displays proapoptotic activities in the absence of its ligand (12). These observations raise the possibility that in addition to cooperating with RAR in transcriptional regulation, CRABP2 regulates gene expression and exert tumor-suppressive activities by an additional, RA-independent function. One possibility is that CRABP2 is involved in posttranscriptional regulation of mRNAs.
One of the best-characterized proteins involved in posttranscriptional regulation of gene expression in animals is HuR, a ubiquitously expressed member of the ELAV/Hu family of RNA-binding proteins (25). In the nucleus, HuR is involved in various functions, including RNA splicing and nuclear export. In the cytosol, it binds to AU-rich elements (ARE) in 3′ untranslated regions (UTRs) of target mRNAs, thereby protecting them against degradation (26–29). By regulating the levels of its target mRNAs, HuR is involved in key biological processes, including cell cycle progression, apoptosis, immune function, inflammation, and carcinogenesis (25, 30, 31).
Here we show that CRABP2 directly interacts with HuR and markedly increases its affinity for some target transcripts, thereby enhancing their stability and increasing their expression levels. Binding of RA triggers dissociation of the CRABP2-HuR complex and induces CRABP2 to undergo a transient nuclear translocation, following which it returns to the extranuclear milieu and reassociates with HuR. We show further that the antioncogenic activity of CRABP2 partially stems from its cooperation with HuR and that HuR is critical for enabling CRABP2 to enhance apoptosis in mammary carcinoma cells.
MATERIALS AND METHODS
Cells.
The M2−/− cell line was generated from tumors that arose in MMTV-neu/CRABP2-null mice (18). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 4.5 g/liter of glucose, 4.5 g/liter of l-glutamine, 10% fetal bovine serum (FBS; Atlanta Biologicals), 100 IU/ml of penicillin, and 100 μg/ml of streptomycin.
Reagents.
RA was purchased from Calbiochem. Actinomycin D and etoposide were from Sigma-Aldrich. Antibodies against HuR (3A2; sc-5261), actin (I-19, sc-1616), and tubulin (H-235, sc-9104) were from Santa Cruz Biotechnology, Inc. Antibodies against caspase 3 (9665) and Apaf-1 (8723) were from Cell Signaling Technology, Inc. Antibodies against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were from Abcam (ab9485). Antibody against CRABP2 was a gift from Cecile Rochette-Egly (IGBMC, Strasbourg, France). LE540 was a gift from Hiroyuki Kagechika (Tokyo Medical and Dental University). Transfections were carried out using PolyFect (Qiagen). Small interfering RNAs (siRNAs) were purchased from Ambion.
Vectors.
A mammalian expression vector harboring cDNA encoding human CRABP2 (hCRABP2) with enhanced green fluorescent protein (EGFP) fused to the protein's N terminus (pEGFP-C2 vector) was previously described (9). hCRABP2ΔNLS with an N-terminal EGFP tag was generated by replacing residues K20, R29, and K30 of EGFP-hCRABP2 with alanines using the QuikChange 2 XL site-directed mutagenesis kit (Stratagene). pSG5 vectors harboring cDNA encoding hCRABP2 and hCRABP2ΔNLS were previously described (6, 32). A vector encoding Flag-tagged CRABP2 was generated by inserting cDNA for human CRABP2 into BamHI and EcoRI sites of pCMV-3Tag-1 vector with 3 Flag tag coding sequences in frame. Adenovirus encoding hCRABP2 in pAD5 was prepared by the Gene Transfer Vector Core (University of Iowa, Iowa City, IA).
Transactivation assays were carried out as previously described (32). For analysis of the Apaf1 and Elavl1 promoters, the upstream 2-kb promoter fragments of each gene were PCR amplified using Platinum Pfx DNA polymerase (Invitrogen) and subcloned into NheI and Hind 2I sites of pGL3-basic luciferase vector.
Lentiviral shRNA production.
pLKO.1 vectors harboring short hairpin RNAs (shRNAs) (Elavl1, TRCN0000112088; CRABP2, TRCN0000021373; ELAVL1, TRCN0000017277; and EGFP, RHS4459) were from Open Biosystems; pLKO.1 vector harboring luciferase shRNA (SHC007) was from Sigma-Aldrich. Lentiviruses were produced in HEK293T cells and target cells were infected using standard protocols.
Real-time quantitative PCR (qPCR) was performed using a StepOnePlus real-time PCR system with the following TaqMan probes: Apaf1, Mm01223702_m1; Casp7, Mm00432324_m1; Elavl1, Mm00516012_m1; Rarb, Mm01319677_m1; ACTB, Hs99999903_m1; APAF1, Hs00559441_m1; BTG2, Hs00198887_m1; CASP7, Hs00169152_m1; CRABP2, Hs00275636_m1; ELAVL1, Hs00171309_m1; and 18S RNA, 4352930E (Applied Biosystems). Levels of mRNAs were normalized to 18S rRNA using the threshold cycle (ΔΔCT) method (Applied Biosystems technical bulletin no. 2).
3′ UTR luciferase reporter assays.
The Apaf1 and Elavl1 3′ UTRs (4,419 to 6,557 bp and 1,214 to 6,030 bp downstream of transcription start sites, respectively) were cloned downstream of a luciferase reporter gene in pGL3 vector modified to include a minimal prolactin promoter. Putative ARE were deleted using the QuikChange 2 XL site-directed mutagenesis kit (Stratagene). Cells were cotransfected with the reporter, a vector encoding β-galactosidase, and an empty vector or vector encoding CRABP2. Forty-eight hours posttransfection, luciferase activity was measured using the luciferase assay system (Promega) and normalized to β-galactosidase activity.
Ribonucleoprotein immunoprecipitations (RIP) were performed as described previously (33). Semiquantitative PCR was performed using the following primer sequences: Elavl1, CGCCTGCTAGGCGGTTTGGA (forward) and CCCAGGCGGTAGCCGTTCAG (reverse); Apaf1, GATGGCAGGCTGCGGCAAGT (forward) and ACACGGAGGCGGTCTTTGGC (reverse); Actb, CCACCATGTACCCAGGCATT (forward) and AGGGTGTAAAACGCAGCTCA (reverse); and Gapdh, GGTTGTCTCCTGCGACTTCA (forward) and TAGGGCCTCTCTTGCTCAGT (reverse).
Confocal fluorescence microscopy.
Cells, cultured in DMEM containing 5% delipidated FBS, were transfected with pCMV-3Tag-1 vector encoding Flag-CRABP2. Cells were treated with 2 μM CGP-75415A (Sigma-Aldrich) for 2 h, fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS), and permeabilized with 0.2% Triton X-100. Endogenous CRABP2 in MCF-7 cells and Flag-tagged CRABP2 in M2−/− cells were visualized by immunostaining. Antibodies included anti-CRABP2 (Millipore; MAB5488), anti-Flag (Sigma-Aldrich; F1804), and anti-HuR (Millipore; 07-468). Cells were imaged using an LSM510 confocal microscope (Leica).
Proteins.
His-CRABP2 was expressed in Escherichia coli BL21 and purified as previously described (34). Protein viability was assessed by monitoring its ability to bind RA as described previously (35). Glutathione S-transferase (GST)–HuR was expressed in E. coli DH5α, partially purified as described previously (36).
Fluorescence anisotropy titrations.
RNA was synthesized and labeled with fluorescein at the 5′ end by Dharmacon. Fluorescence anisotropy titrations were carried out using a Photon Technology International Quantamaster spectrofluorometer equipped with Glan-Thompson polarizers. Labeled mRNA (0.1 μM) was placed in a cuvette and titrated with GST-HuR, His-CRABP2, or CRABP2 precomplexed with HuR at a 4:1 molar ratio in the absence or presence of RA. Fluorescence anisotropy (λexcitation = 494 nm; λemission = 518 nm) was measured.
Flow cytometry was performed as previously described (13) and analyzed on a BD Biosciences LSR 2 at the Case Comprehensive Cancer Center Cytometry and Imaging Microscopy Core Facility.
RESULTS
CRABP2 upregulates the expression of Apaf1 and Elavl1 independently of its cooperation with RAR.
The observations that CRABP2 induces the expression of Apaf1 in the absence of RA (12) suggest that the protein may have biological activities other than to deliver RA to RAR. To examine this notion, CRABP2-K20A/R29A/K30A, a mutant that lacks the nuclear localization signal (NLS) of the protein (CRABP2ΔNLS), was used. This mutant folds properly and, similarly to the wild-type (WT) protein, binds RA with nanomolar affinity, but it does not translocate to the nucleus in response to ligand binding (6). In the absence of RA, both the WT protein and its NLS mutant are cytosolic (6, 9, 37). A mammary carcinoma cell line derived from tumors that arose in transgenic MMTV-neu mice, a well-established mouse model of breast cancer (38), was used to examine whether CRABP2ΔNLS can enhance the transcriptional activity of RAR. The specific cell line used in these studies was generated from tumors that developed in MMTV-neu mice in which expression of CRABP2 was ablated, and accordingly, they completely lack the protein (M2−/− cells [18]). M2−/− cell lines that stably express EGFP, EGFP-CRABP2, or EGFP-CRABP2ΔNLS were generated (Fig. 1A), and the effects of the proteins on RA-induced activation of RAR were monitored by transactivation assays using a luciferase reporter driven by an RAR response element (RARE). When cells were cultured in medium containing delipidated serum, neither CRABP2 nor its ΔNLS mutant had any effect of the transcriptional activity of RAR (Fig. 1C), demonstrating efficient depletion of RA. In the presence of RA, CRABP2 enhanced reporter activation but the CRABP2ΔNLS had no effect on RA-induced activation of RAR (Fig. 1B). The data thus demonstrate that the mutant does not cooperate with the receptor.
FIG 1.

Apo-CRABP2 upregulates Apaf1 and Elavl1 mRNAs. (A) Immunoblots demonstrating stable overexpression of EGFP-CRABP2 (2) and EGFP-CRABP2ΔNLS (2ΔNLS) in M2−/− cells. (B) M2−/− cell lines stably expressing denoted proteins were cotransfected with a RARE-driven luciferase reporter gene and a vector encoding β-galactosidase. Cells were cultured for 48 h in delipidated media, and luciferase activity was assayed. Data are means ± SDs (n = 3). (C) M2−/− cell lines stably expressing denoted proteins were cotransfected with a RARE-driven luciferase reporter and a vector encoding β-galactosidase. Cells were treated with vehicle or 20 nM RA for 16 h, and luciferase activity was measured and normalized to β-galactosidase activity. Data are means ± standard errors of the means (n = 3). *, P ≤ 0.01 versus RA-treated EGFP-expressing control, determined using two-tailed student t test. (D and E) MCF-7 cells were infected with lentiviruses containing vector harboring shRNA targeting CRABP2 (shCRABP2) or luciferase (shLuc). Three days postinfection, cells were harvested and levels of the indicated mRNAs (D) and proteins (E) were assessed by qPCR and immunoblotting, respectively. *, P ≤ 0.01; #, P = 0.037; @, P = 0.077 (all versus shLuc, determined using two-tailed Student t test). (F) M2−/− cells were transfected with an empty vector (e.v.) or vectors harboring cDNA for CRABP2 or CRABP2ΔNLS. Levels of Apaf1 and Elavl1 mRNAs were measured by qPCR. Data are mean ± standard errors of the means (n = 3). *, P ≤ 0.01 versus corresponding e.v. control, calculated using a two-tailed Student t test. (Inset) Immunoblots demonstrating CRABP2 expression in M2−/− cells transfected with e.v. or vectors harboring cDNA for indicated proteins. (G and H) M2−/− cells were transfected with e.v. or vectors harboring cDNA for CRABP2 or CRABP2ΔNLS. (G) Levels of Apaf-1 and HuR protein were assessed by immunoblotting. (H) Immunoblots were quantitated; data are means ± standard errors of the means (n = 3). *, P ≤ 0.01, and #, P = 0.034, both versus corresponding e.v. control and calculated using a two-tailed Student t test. (I) M2−/− cells stably overexpressing EGFP-CRABP2 were treated with RA in the absence or presence of the RAR antagonist LE540 (1 μM each for 4 h). Rarb, Apaf1, and Elavl1 mRNAs were measured by qPCR. Data are means ± standard errors of the means (n = 3). *, P ≤ 0.01 versus vehicle control, calculated using a two-tailed student t test. (J) M2−/− cells stably overexpressing EGFP-CRABP2 were transfected with luciferase reporter constructs driven by 2 kb of the proximal promoters of Apaf1 or Elavl1 or by an RARE. Cells were treated with 100 nM RA for 16 h, and luciferase activity was measured. Data are means ± standard errors of the means (n = 3). *, P ≤ 0.01 versus corresponding vehicle control, calculated using a two-tailed Student t test. (K) M2−/− cells stably overexpressing EGFP-CRABP2 were treated with 10 μg/ml of cycloheximide for the indicated times. The level of HuR protein was assessed by immunoblotting. (Top) Immunoblot showing HuR protein expression levels in M2−/− cell lines stably overexpressing CRABP2; (bottom) immunoblot showing HuR protein levels after treatment with cycloheximide.
Reducing the expression of CRABP2 in MCF-7 breast cancer cells, which express a high level of CRABP2 (12), decreased the levels of APAF1 mRNA and protein (Fig. 1D and E). Correspondingly, ectopic expression of CRABP2 in M2−/− cells, which do not express the protein, increased the level of Apaf1 mRNA and protein (Fig. 1F to H). Notably, ectopic expression of either WT CRABP2 or CRABP2ΔNLS increased the levels of Apaf1 mRNA and protein to similar extents (Fig. 1F to H). These observations suggest that CRABP2 upregulates Apaf1 by a mechanism unrelated to its ability to promote the transcriptional activity of RAR. In support of this conclusion, silencing CRABP2 in MCF-7 cells had no effect on the established RAR target gene BTG2 (Fig. 1D), and treatment of M2−/− cells stably expressing CRABP2 with RA induced expression of the well-established RAR target gene Rarb but had no effect on Apaf1 mRNA (Fig. 1I). Moreover, while the pan-RAR antagonist LE540 attenuated the ability of RA to upregulate Rarb, the compound had no effect on expression of Apaf1 (Fig. 1I). Correspondingly, transactivation assays showed that RA effectively induced the expression of a luciferase reporter gene driven by an RARE but had no effect on a luciferase reporter driven by the proximal promoter of Apaf1 (Fig. 1J). The data thus clearly show that Apaf1 does not constitute RAR target gene.
These observations raise the possibility that CRABP2 elevates the level of Apaf1 mRNA by increasing the stability of this transcript. We therefore wondered whether CRABP2 functions in conjunction with HuR (encoded by the Elavl1 gene), an RNA-binding protein which is arguably the best-characterized regulator of mRNA stability in animals (25, 39). Remarkably, downregulation of CRABP2 expression in MCF-7 cells decreased the levels of HuR mRNA (Fig. 1D) and protein (Fig. 1E), and in M2−/− cells, ectopic expression of either WT CRABP2 or CRABP2ΔNLS cells increased HuR expression (Fig. 1F to H). Similar to the case with Apaf1, the expression of Elavl1 was not affected either by RA or by an RAR antagonist (Fig. 1I), and RA did not affect the expression of a luciferase reporter driven by the Elavl1 promoter (Fig. 1J). Hence, expression of both Apaf1 and Elavl1 is regulated by CRABP2 but not through transactivation of RAR.
The observations that CRABP2 upregulates the expression of both mRNAs and proteins suggest that the effect is mediated by stabilization of the mRNAs and not the proteins. Indeed, while overexpression of CRABP2 in M2−/− cells increased the level of HuR (Fig. 1K, top), monitoring the time course of protein degradation following treatment with protein synthesis inhibitor cycloheximide showed that CRABP2 did not have a discernible effect on the stability of HuR (Fig. 1K, bottom).
CRABP2 stabilizes Apaf1 and Elavl1 mRNA in an HuR-dependent manner.
The possibility that CRABP2 upregulates the expression of Apaf-1 and HuR by stabilizing their mRNAs was then evaluated. M2−/− cells were transfected with an empty vector or a vector encoding CRABP2 (Fig. 2A) and treated with transcription inhibitor actinomycin D, and the rates of degradation of Apaf1 and Elavl1 mRNAs were monitored. The mean half-lives of the Apaf1 and Elavl1 mRNAs were found to be 1.81 ± 0.08 and 2.44 ± 0.27 h, respectively, in the absence of CRABP2 and increased to 6.94 ± 0.9 and 6.23 ± 0.38 h, respectively, in cells expressing CRABP2 (Fig. 2B and C). In contrast, CRABP2 had no effect on the stability of Gapdh mRNA (Fig. 2D), indicating that the effect of CRABP2 on the half-life of the Apaf1 and Elavl1 mRNAs is specific to a subset of mRNAs. Considering that CRABP2 does not contain a recognizable RNA-binding motif, we wondered whether its ability to stabilize mRNAs involves cooperation with HuR. In agreement with this notion, decreasing the expression of HuR (Fig. 2E, inset) reduced the level of Apaf1 mRNA and completely abolished the ability of CRABP2 to upregulate the expression of this gene (Fig. 2E). Reducing the expression of HuR also reduced Apaf1 mRNA levels in MCF-7 mammary carcinoma cells (Fig. 2F).
FIG 2.

HuR mediates the ability of CRABP2 to stabilize Apaf1 and Elavl1 mRNAs. (A) Immunoblot demonstrating overexpression of CRABP2. (B to D) M2−/− cells were transfected with e.v. or vector encoding CRABP2 and treated with actinomycin D (2.5 μg/ml). Levels of Apaf1 (B), Elavl1 (C), and Gapdh (D) mRNAs at various time points following treatment were measured by qPCR. Data were normalized to corresponding values at time zero. Data are means ± standard errors of the means (n = 3). (Inset) mRNA levels of Apaf1, Elavl1, and Gapdh mRNAs in the absence and presence of CRABP2 overexpression at time zero. (E) M2−/− cells were transfected with scrambled siRNA (siScrm) or siRNA targeting Elavl1 (siElavl1). Twenty-four hours later, cells were infected with control adenovirus (Ad0) or adenovirus encoding CRABP2 (Ad2). Forty-eight hours postinfection, Apaf1 mRNA levels were assessed by qPCR. Data are means ± standard errors of the means (n = 3). *, P < 0.01 versus cells expressing siScrm and Ad0 by two-tailed Student t test. (Inset) Immunoblots demonstrating decreased expression of HuR in cells expressing siElavl1 and increased expression of CRABP2 upon infection with Ad2. (F) MCF-7 cells were infected with lentiviruses containing vector harboring shRNA targeting ELAVL1 (shELAVL1) or luciferase (shLuc). Three days postinfection, cells were harvested and levels of indicated mRNAs were assessed by qPCR. *, P ≤ 0.01 versus shLuc by two-tailed Student t test. (G and H) M2−/− cells were infected with lentiviruses containing vector harboring shRNAs targeting Elavl1 (shElavl1) or EGFP (shEGFP) and transfected with e.v. or a vector encoding CRABP2 and luciferase reporter harboring the Apaf1 (G) or Elavl1 (H) 3′ UTR. β-Galactosidase was used as a transfection control. Data were normalized to corresponding e.v./shEGFP-expressing cells. Data are means ± standard errors of the means (n = 3). *, P ≤ 0.01, and #, P = 0.045, both versus e.v./shEGFP control by two-tailed Student t test. (Inset) Immunoblots demonstrating reduced expression of HuR and overexpression of CRABP2.
HuR stabilizes target transcripts by interacting with specific sequences within their 3′ UTRs (26–29). To assess whether Apaf1 and Elavl1 comprise targets for HuR and to examine whether HuR cooperates with CRABP2 in stabilizing their mRNAs, the entire 3′ UTRs of Apaf1 and Elavl1 were cloned downstream of a luciferase reporter gene. The reporters were transfected into M2−/− cells, and the effect of modulating the expression of CRABP2 and HuR on luciferase activity was monitored. Similar to the response of the endogenous transcripts, ectopic expression of CRABP2 upregulated the expression of luciferase reporters containing either the Apaf1 or the Elavl1 3′ UTRs, and decreasing the expression of HuR downregulated the basal levels of both reporter genes and abolished the ability of CRABP2 to upregulate their expression (Fig. 2G and H). We noted that overexpression of CRABP2 was somewhat lower in cells expressing Elavl1 shRNA. This likely reflects a lower expression efficiency resulting from the multivector transfection of these cells. Nevertheless, the observation that, upon decreasing HuR levels, ectopic expression of CRABP2 had no effect on reporter expression indicates that HuR is critical for enabling modulation of the levels of the Apaf1 and the Elavl1 transcripts by CRABP2.
It was previously reported that HuR stabilizes its own mRNA (40). However, the Apaf1 transcript is a novel target, and the location of HuR-binding sequences within the gene's 3′ UTR is unknown. Inspection of the 3′ UTR of the Apaf1 mRNA revealed 3 potential ARE (Fig. 3A). To identify ARE responsible for mediating the ability of CRABP2 and HuR to stabilize this mRNA, the 3 elements were individually deleted from the luciferase reporter containing the 3′ UTR of Apaf1 and reporter assays were carried out. Deletion of putative ARE 1 and 2 had little effect on reporter activity, but deletion of the ARE at site 3 abolished the ability of CRABP2 to upregulate the expression of the reporter (Fig. 3B). Site 3 thus appears to contain the sequence through which HuR and CRABP2 stabilize the Apaf1 transcript.
FIG 3.

HuR mediates the association of CRABP2 with mRNA. (A) Diagram of the luciferase reporter harboring the Apaf1 3′ UTR. Putative ARE are underlined. (B) M2−/− cells were transfected with an e.v. or a vector encoding CRABP2 and the luciferase reporter harboring the Apaf1 3′ UTR or counterparts lacking the indicated putative HuR binding sites (Δ1, Δ2, and Δ3). β-Galactosidase was used as a transfection control. Data were normalized to luciferase activity in cells transfected with e.v. and the WT luciferase reporter. Data are means ± standard errors of the means (n = 3). *, P ≤ 0.01, and ‡, P = 0.025, both versus e.v. control by a two-tailed Student t test. (C) Coomassie blue-stained gels visualizing recombinant, bacterially expressed, and purified GST-HuR. (D) Fluorescein-labeled RNA containing 39 bases corresponding to site 3 in the Apaf-1 3′ UTR (WT RNA) or fluorescein-labeled RNA with the AUUUA of site 3 deleted (mutant RNA) was titrated with recombinant GST-tagged HuR. Progress of titrations was followed by monitoring the increase in the fluorescence anisotropy of the labeled RNA (λexcitation = 494 nm; λemission = 518 nm). Data representative of 3 independent experiments are shown. (E) HuR and CRABP2 were immunoprecipitated from lysates of M2−/− cells stably expressing EGFP-CRABP2 (Fig. 1A). Apaf1 and Elavl1 mRNAs that coprecipitated with the proteins were assessed by semiquantitative PCR. (F) M2−/− cells that stably overexpress CRABP2 were infected with lentiviruses harboring vectors encoding shRNA targeting Elavl1 (shElavl1) or luciferase (shLuc), and stable cell lines were generated. (Top) Immunoblot demonstrating HuR levels in cells expressing shLuc or shElavl1. Actin was used as loading control. (Bottom) CRABP2 was immunoprecipitated and RNAs that coprecipitated with the protein were assessed by semiquantitative PCR.
Fluorescence anisotropy titrations were then used to directly examine whether site 3 of the Apaf1 mRNA is the HuR binding site. GST-tagged HuR was expressed in E. coli and purified (Fig. 3C). A 39-nucleotide-long RNA containing the CRABP2-responsive sequence of the Apaf-1 3′ UTR (Fig. 3A and B) was covalently labeled with the fluorescent probe fluorescein, and its ability to bind HuR was examined. Fluorescence anisotropy reports on the rotational volume of a fluorophore, and thus on the size of complexes containing it, and they have been widely used to monitor molecular associations (41). Titration of the RNA with GST-HuR resulted in a saturable increase in fluorescence anisotropy, demonstrating protein-RNA association (Fig. 3D). Analysis of the data (35) showed that the Kd (dissociation constant) for the interaction between the RNA and HuR complexes is 413 ± 147 nM (mean ± standard deviation [SD]; n = 3). Deletion of site 3 of the mRNA from the 39-nucleotide-long RNA markedly decreased the affinity of the protein for the RNA (Fig. 3D), indicating that this site is the primary binding site for HuR.
Ribonucleoprotein immunoprecipitation (RIP) assays were carried out to further examine whether HuR and CRABP2 associate with the Apaf1 or Elav1 mRNA. Both mRNAs coprecipitated with either HuR or with CRABP2, indicating that both proteins are bound to these transcripts in cells (Fig. 3E). To examine the effect of HuR on the ability of CRABP2 to associate with target transcripts, a derivative of the M2−/− cell line that stably express CRABP2 (Fig. 1A) in which the expression HuR was stably reduced was generated (Fig. 3F, top). Reducing the expression of HuR markedly decreased the association of CRABP2 with the Apaf1 mRNA (Fig. 3F, bottom), indicating that HuR mediates the association of CRABP2 with this transcript.
Apo-CRABP2 interacts with HuR and enhances its affinity for target mRNAs.
The observations that HuR is required for binding of CRABP2 to target transcripts and upregulate their expression suggest that the two proteins associate with each other. Indeed, immunoprecipitation assays showed that HuR and CRABP2 coprecipitate from MCF-7 cell lysates (Fig. 4A and B). Treatment of cell lysates with RNase prior to precipitation did not inhibit the association, indicating that RNA binding is not required for the interactions (Fig. 4A and B).
FIG 4.

Apo-CRABP2 associates with HuR and enhances its affinity for target mRNAs. For all experiments, cells were cultured in delipidated medium for 48 h to deplete retinoid stores. (A) Lysates from MCF-7 cells were treated with vehicle or RNase (100 μg/ml), CRABP2 was immunoprecipitated, and precipitates were immunoblotted for the presence of CRABP2 and HuR. (B) Lysates from MCF-7 cells were treated with vehicle or RNase, HuR was immunoprecipitated, and precipitates were immunoblotted for the presence of HuR and CRABP2. (C) M2−/− cells were transfected with a vector encoding Flag-CRABP2 and treated or not with CGP74514A (2 μM for 2 h). Flag-CRABP2 and HuR were detected by immunostaining, and cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to visualize nuclei. Bars, 5 μm. (D) MCF-7 cells and M2−/− cells expressing Flag-tagged CRABP2 were treated with CGP-74514A (2 μM for 2 h). Endogenous CRABP2 in MCF-7 cells and Flag-tagged CRABP2 in M2−/− cells were visualized by immunostaining using CRABP2 and Flag antibodies, respectively. Cells were also immunostained for HuR and counterstained with DAPI. Confocal fluorescence microscopy was used to visualize cells. Bars, 5 μm. (E) HuR was immunoprecipitated from M2−/− cells stably overexpressing EGFP or EGFP-CRABP2 (Fig. 1A). Apaf1, Elavl1, and Actb mRNAs coprecipitating with HuR were detected by semiquantitative PCR. (F) Coomassie blue-stained gels visualizing recombinant, bacterially expressed, and purified His-CRABP2 and His-CRABP2ΔNLS. (G) Fluorescein-labeled RNA containing 39 bases corresponding to site 3 in the Apaf-1 3′ UTR was titrated with recombinant GST-tagged HuR alone or in complex with CRABP2 or CRABP2ΔNLS. To ensure saturation, CRABP2s were precomplexed with HuR at a 4:1 molar ratio. Progress of titrations was followed by monitoring the increase in the fluorescence anisotropy of the labeled RNA (λexcitation = 494 nm; λemission = 518 nm). Data representative of 3 independent experiments are shown. (Inset) Fluorescein-labeled RNA was titrated with CRABP2 or CRABP2ΔNLS.
Confocal fluorescence microscopy was used to examine if CRABP2 and HuR colocalize in cells. It was previously reported that in the absence of RA, CRABP2 is located in the extranuclear milieu, where it appears to be associated with endoplasmic reticulum (ER). However, the means by which this highly soluble protein is localized at the ER is unknown (6, 9, 37). In agreement with the previous reports, in the absence of retinoids, both endogenous CRABP2 in MCF-7 cells and ectopically expressed CRABP2 in M2−/− cells were present in the extranuclear milieu (Fig. 4C and D). HuR shuttles between the nucleus and the cytosol, and it is present predominantly in the nucleus in cell cycle phases other than late mitosis (42, 43) (Fig. 4C). The cytonuclear shuttling of HuR is regulated by cyclin-dependent kinase 1 (Cdk1)-catalyzed phosphorylation, and inhibition of the kinase results in retention of the protein in the cytosol (44) (Fig. 4C). To increase the level of HuR in the cytosol in resting cells and enhance microscopic examination of possible colocalization between CRABP2 and HuR, cells were treated with the Cdk1 inhibitor CGP-74514A prior to imaging. Immunostaining showed that both endogenously expressed and ectopically overexpressed CRABP2 extensively colocalized with HuR (Fig. 4D).
Two approaches were taken to examine whether association with CRABP2 affects the interactions of HuR with target transcripts. RIP assays showed that expression of CRABP2 increased the efficiency by which HuR binds both the Apaf1 and the Elavl1 mRNAs (Fig. 4E). The apparent enhancement of the association of HuR with these mRNAs may result either from increased affinity toward the transcripts or from increased cellular levels brought about by CRABP2 expression (Fig. 1C to E). Fluorescence anisotropy titrations were then used to directly examine whether CRABP2 modulates the mRNA-binding affinity of HuR. Histidine-tagged CRABP2 or CRABP2ΔNLS were expressed in E. coli and purified (Fig. 4F). A 39-nucleotide-long RNA containing the CRABP2-responsive sequence of the Apaf-1 3′ UTR (Fig. 3A and B) was covalently labeled with the fluorescent probe fluorescein and titrated with recombinant HuR, CRABP2-bound HuR, or CRABP2 alone, and fluorescence anisotropy was monitored (Fig. 4G). Titration of the RNA with either CRABP2 or CRABP2ΔNLS did not affect the fluorescence anisotropy of the RNA (Fig. 4G, inset), indicating that these proteins do not directly associate with the RNA. However, titrations with HuR complexed with either CRABP2 or its ΔNLS mutant displayed a markedly steeper curve and earlier saturation than for binding of HuR alone, demonstrating a dramatic increase in binding affinity (Fig. 4G). Analyses of the data (35) revealed that the Kd for the HuR-RNA association decreased from 413 ± 147 nM, which characterizes the interactions of the RNA with HuR alone, to <0.1 nM, observed in the presence of CRABP2. Note that the observed binding affinity was too high for accurate measurements using this assay, and thus, this value reflects an upper limit. The data thus strikingly show that HuR directly binds both CRABP2 and CRABP2ΔNLS and that both CRABP2 and its ΔNLS mutant increase the affinity of HuR toward the transcript by more than 3 orders of magnitude.
RA triggers transient dissociation of CRABP2 from HuR and target transcripts.
In vitro assays were carried out to examine whether the formation of the HuR-CRABP2 complex is sensitive to RA. Bacterially expressed recombinant GST-HuR was immobilized on glutathione-Sepharose beads and incubated with recombinant His-CRABP2 in the absence or presence of increasing concentrations of RA. Beads were precipitated, and CRABP2 that coprecipitated with HuR was visualized by Coomassie blue staining (Fig. 5A). The data showed that CRABP2 efficiently coprecipitated with HuR in the absence but not in the presence of RA, indicating that the protein dissociates from HuR when ligated. In support of this conclusion, fluorescence anisotropy titrations of the HuR-binding region of the Apaf1 3′ UTR showed that RA markedly decreased the affinity of HuR toward the RNA (Fig. 5B). In the presence of RA, the Kd that characterizes the HuR-RNA interactions was found to be 178 ± 46 nM, similar to that of the association of the RNA with HuR alone. These data thus demonstrate that HuR directly binds CRABP2 both in solution and on target RNAs and that CRABP2 dissociates from HuR in the presence of RA.
FIG 5.

RA triggers transient dissociation of CRABP2 from HuR and target transcripts. In all experiments, cells were cultured in delipidated medium for 48 h to deplete retinoid stores. (A) Recombinant, bacterially expressed GST-HuR was immobilized on glutathione-Sepharose beads. Beads were incubated in a buffer containing 1.25 μM recombinant His-CRABP2 in the presence of denoted concentrations of RA. Beads were precipitated and proteins in precipitates visualized by Coomassie blue staining. (B) Fluorescein-labeled RNA containing 39 bases corresponding to site 3 in the Apaf-1 3′ UTR was titrated with His-CRABP2 precomplexed with GST-HuR in the presence or absence of a 2-fold molar excess of RA. Progress of titrations was followed by monitoring the increase in the fluorescence anisotropy of the labeled RNA. Data representative of 3 independent experiments are shown. (C) M2−/− cells were transfected with a vector encoding Flag-CRABP2 and pretreated with CGP-74514A (2 μM for 2 h) prior to treatment with RA (1 μM) for the indicated times. Flag-CRABP2 and HuR were detected by immunostaining, and cells were counterstained with DAPI. Cells were visualized by confocal fluorescence microscopy. Bars, 10 μm. (D) MCF-7 cells were treated with 1 μM RA for the indicated times, and CRABP2 was precipitated. Precipitates were immunoblotted for CRABP2 and HuR. (E) MCF-7 cells were treated with 1 μM RA for the indicated times, and HuR was precipitated. Precipitates were immunoblotted for HuR and CRABP2. (F) M2−/− cells that stably express CRABP2 (Fig. 1A) were treated with 1 μM RA for the indicated times. HuR and CRABP2 were immunoprecipitated, and the presence of Apaf1, Elavl1, and Gapdh mRNAs in precipitates was assessed by semiquantitative PCR.
In agreement with previous reports (6, 9, 37), a 30-min treatment of M2−/− cells ectopically expressing CRABP2 with RA resulted in a massive nuclear localization of the protein (Fig. 5C). However, the residence of CRABP2 in the nucleus was found to be short lived, and the protein returned to the extranuclear milieu, where it again colocalized with HuR 90 min after RA treatment (Fig. 5C). The cytonuclear shuttling behavior of CRABP2 was similar in the presence of the protein synthesis inhibitor cycloheximide, indicating that the protein indeed undergoes RA-induced reversible cytonuclear shuttling and that the observations do not reflect de novo protein synthesis (data not shown). Moreover, coimmunoprecipitation assays showed that in cells, a 30-min treatment with RA results in complete dissociation of CRABP2 from HuR and that the complex reforms 90 min posttreatment (Fig. 5D and E). RIP experiments similarly showed that RA induced dissociation of CRABP2 from both the Apaf1 and Elavl1 mRNAs but that the effect was transient and CRABP2 regained its RNA-binding capacity 90 min following treatment (Fig. 5F). Taken together, the data indicate that the residence time of CRABP2 in the nucleus is short and hence that the ability of the protein to deliver RA to the nucleus does not significantly interfere with its ability to stabilize mRNA in conjunction with HuR. The observations that RA treatment did not significantly affect RNA binding by HuR likely reflect that the rate of dissociation of HuR from target transcript is slow and does not proceed to a significant extent during the short residence time of CRABP2 in the nucleus (Fig. 5F).
CRABP2 enhances apoptosis and suppresses carcinoma cell growth through its cooperation with HuR.
It has been reported that HuR displays antiproliferative activities (45–47). In agreement, reducing the expression of HuR facilitated the growth of M2−/− cells (Fig. 6A). It has also been reported that CRABP2 suppresses the growth of various carcinomas (2, 9, 12, 13, 18, 19). In accordance, overexpression of CRABP2 inhibited cell growth both in parental cells and in cells in which the expression of HuR was reduced (Fig. 5A). In these experiments, which were carried out in retinoid-depleted cells, downregulation of HuR enhanced cell growth to similar extents in cells that do not express and that ectopically express CRABP2 (Fig. 6A). In the presence of RA, both CRABP2 and CRABP2ΔNLS suppressed cell growth, but the latter was less efficacious than the WT protein in exerting the effect (Fig. 6B). These observations suggest that CRABP2 inhibits proliferation by two distinct mechanisms and that one of these but not the other depends on its cooperation with RAR. In agreement with this notion, in the absence of retinoids, CRABP2 and CRABP2ΔNLS inhibited proliferation with similar efficacies (Fig. 6C).
FIG 6.

CRABP2 enhances apoptosis by cooperating with HuR. In all experiments, cells were cultured in delipidated medium for 48 h to deplete retinoid stores. (A) M2−/− cells stably expressing EGFP or EGFP-CRABP2 and harboring shRNAs targeting Elavl1 (shElavl1) or luciferase (shLuc) were plated at 35,000 cell/well and counted after 4 days of growth. Data are means ± standard errors of the means (n = 3). Statistical analysis was carried out by one-way analysis of variance and Bonferroni's post hoc test. *, P < 0.01 versus cells expressing EGFP and shLuc; #, P < 0.01 versus cells expressing EGFP and shElavl1. (Inset) The number of shElavl1-expressing cells was normalized to the corresponding shLuc control. (B) Growth of M2−/− cells stably overexpressing the indicated proteins in delipidated medium supplemented with 200 nM RA. Data are means ± standard errors of the means (n = 3). *, P < 0.01 versus EGFP control cells and, on day 3, P < 0.01 comparing cells expressing EGFP-CRABP2 versus EGFP-2ΔNLS. Analysis was carried out by one-way analysis of variance and Bonferroni's post hoc test. (C) Growth of M2−/− cells stably overexpressing the indicated proteins in delipidated medium. Data are means ± standard errors of the means (n = 3). *, P ≤ 0.01 versus control EGFP-expressing cells using one-way analysis of variance with Bonferroni's post hoc test. P > 0.1 for EGFP-CRABP2 versus EGFP-2ΔNLS on days 2 and 3, as calculated by two-tailed Student t test. (D and E) M2−/− cells were transfected with vectors encoding EGFP-CRABP2 or EGFP-CRABP2ΔNLS. Cells were treated with etoposide (10 μM for 48 h). (D) Apoptosis was evaluated by FACS to quantitate percentage of cells in G1. The experiment was repeated, with similar results. (E) Apoptosis was evaluated by using immunoblotting to monitor cleavage of caspase 3. The experiment was repeated, with similar results. (F) M2−/− cells were stably transfected with EGFP or EGFP-CRABP2 and treated with etoposide (10 μM for 48 h), and apoptosis was assessed by monitoring PARP cleavage by immunoblotting. (G) (Top) M2−/− cells stably expressing EGFP or EGFP-CRABP2 were infected with lentivirus harboring shRNA targeting either Elavl1 (shElavl1) or luciferase (shLuc). Cells were then treated with etoposide (10 μM for 48 h), and apoptosis was evaluated by immunoblotting monitoring caspase 3 cleavage. (Bottom) Quantitation of immunoblots. The experiment was repeated, with similar results. (H) MCF-7 cells were infected with lentiviruses containing vector harboring shRNA targeting CRABP2 (shCRABP2) or luciferase (shLuc). Three days postinfection, cells were harvested and the level of CASP7 mRNA was assessed by qPCR. *, P ≤ 0.01 versus shLuc, determined using two-tailed Student t test. See Fig. 1D and E for knockdown of CRABP2. (I) M2−/− cells were infected with lentiviruses containing vector harboring shRNA targeting Elavl1 (shElavl1) or luciferase (shLuc). Forty-eight hours later, cells were infected with control adenovirus (Ad0) or adenovirus encoding CRABP2 (Ad2). Forty-eight hours after adenoviral infection, RNA was extracted and Casp7 mRNA levels were assessed by qPCR. Data are means ± standard errors of the means (n = 3). *, P < 0.01 versus cells expressing shLuc and Ad0 by two-tailed Student t test. (Inset) Immunoblots demonstrating decreased expression of HuR in cells expressing shElavl1 and increased expression of CRABP2 upon infection with Ad2.
To examine the basis for the growth-inhibitory activity of CRABP2, M2−/− cells expressing either CRABP2 or CRABP2ΔNLS were treated with the proapoptotic agent etoposide (48). Apoptotic responses were monitored by fluorescence-activated cell sorting (FACS) to measure the fraction of cells undergoing DNA fragmentation (Fig. 6D) and by assessing etoposide-induced cleavage of caspase 3 (Fig. 6E) and poly(ADP-ribose) polymerase (PARP) (Fig. 6F). Both CRABP2 and CRABP2ΔNLS enhanced etoposide-induced apoptosis, and they exerted similar effects in this capacity. The data thus indicate that the ability of CRABP2 to sensitize cells to apoptotic stimuli does not require cooperation with RAR. In agreement with this notion, decreasing the expression of HuR abolished the ability of CRABP2 to sensitize cells to etoposide-induced apoptosis (Fig. 6G).
It is unlikely that the only genes regulated by HuR in cooperation with CRABP2 are Apaf1 and Elavl1. Considering the proapoptotic activities of the HuR-CRABP2 complex, we examined its effect on the mRNA for caspase 7. Decreasing the expression of CRABP2 in MCF-7 cells reduced the level of CASP7 mRNA (Fig. 6H), and overexpression of CRABP2 in M2−/− cells upregulated Casp7 mRNA (Fig. 6I). Notably, downregulation of HuR in M2−/− cells completely abolished the ability of CRABP2 to increase the level of this transcript (Fig. 6I). The data thus indicate that expression of caspase 7 is cooperatively regulated by HuR and CRABP2. The full spectrum of genes regulated by the CRABP2-HuR complex remains to be examined.
DISCUSSION
The observations suggest the following model (Fig. 7): in the context of some transcripts, apo-CRABP2 associates with mRNA-bound HuR. The association considerably enhances the RNA-binding affinity of HuR, promoting the stability and increasing the levels of such target transcripts, including Apaf1, Elavl1, and Casp7 mRNAs. RA binding by CRABP2 triggers dissociation from HuR and induces its translocation to the nucleus, where it delivers RA to RAR. The residence of CRABP2 in the nucleus is short lived, and following ligand delivery, the protein rapidly exits the nucleus and reassociates with HuR and target transcripts.
FIG 7.
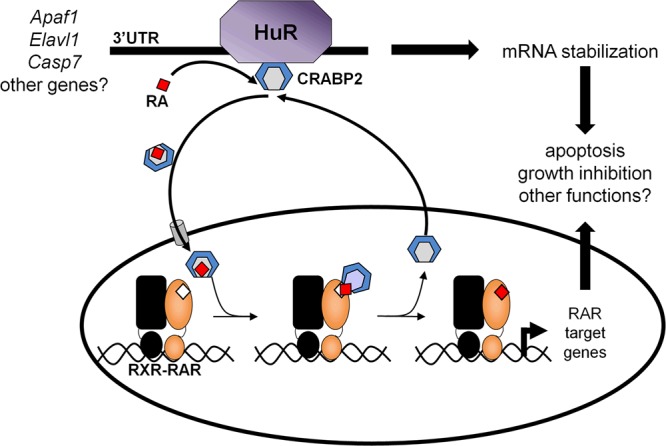
Model for the parallel involvement of CRABP2 in HuR-mediated mRNA stabilization and in RAR-mediated transcriptional regulation. Apo-CRABP2 directly interacts with HuR, dramatically increases its RNA-binding affinity, and thereby enhances the stability of HuR-targeted transcripts. Ligation of CRABP2 by RA triggers its dissociation from HuR and induces the protein to mobilize to the nucleus, where it delivers RA to RAR, enhancing the transcriptional activity of the receptor. The residence time of CRABP2 in the nucleus is short, and following ligand delivery, the protein rapidly returns to the extranuclear milieu and reassociates with HuR. The data suggest that the anticarcinogenic activity of CRABP2 is exerted both by promoting HuR-mediated stabilization of transcripts for proteins that suppress proliferation and by enhancing RA-induced RAR-mediated transcriptional upregulation of growth-inhibitory genes.
The structural features of CRABP2 and HuR that mediate their interactions, the mechanism by which CRABP2 enhances the affinity of HuR toward target mRNAs, and the structural basis for the RA responsiveness of the complex remain to be elucidated. The observation that apo- but not holo-CRABP2 binds to HuR suggests that the CRABP2 residues that mediate the interactions are located in a region that can sense ligand binding. One such region is the protein's helix-loop-helix domain, which contains its ligand-controlled NLS (6). Another RA-responsive region is the RAR interaction domain of CRABP2, comprised of residues Q75, P81, and K102 (32). The report that RA-induced SUMOylation of K102 allows CRABP2 to dissociate from the ER and mobilize to the nucleus in response to RA (37) raises the possibility that K102 is involved in the association of CRABP2 with HuR.
It is well established that CRABP2 enables transcriptional activation by RAR by delivering RA directly to the receptor in the nucleus. The data presented here surprisingly reveal that CRABP2 also functions by cooperating with HuR to enhance mRNA stability. The observations that the RA-induced nuclear translocation of CRABP2 is short lived and that the protein rapidly returns to the cytosol and reassociates with HuR suggest that CRABP2 can exert its two functions in parallel. In support of this notion, RA treatment does not affect the levels of either the Apaf-1 or the HuR transcripts (Fig. 1I) or the interactions between HuR and target transcripts (Fig. 5F). The mechanism by which CRABP2 is exported from the nucleus remains to be clarified. Notably, while a nuclear export signal has been identified in other intracellular lipid-binding proteins, CRABP2 does not appear to contain such a signal (7, 49). An intriguing possibility is that following delivery of RA to RAR, CRABP2 associates with nuclear HuR and leaves the nucleus in complex with this protein.
The role that HuR plays in cancer cell biology is incompletely understood. It has been reported that it stabilizes some mRNAs involved in cell proliferation (31). However, it was also shown that HuR is required for apoptosis (47), that it sensitizes cells to DNA-damaging agents (45), and that it inhibits tumor growth in a mouse model (46). In addition, low expression levels of HuR were found to be predictive of a higher risk of breast cancer recurrence (50). In agreement with these observations, we show here that HuR stabilizes the transcripts of Apaf-1 and caspase 7, proteins closely involved in apoptotic responses, and that reducing the expression of HuR facilitates cell growth. We show further that the stability of the Apaf1 transcript is enhanced by the cooperation of HuR with CRABP2 and that the two proteins, working in concert, suppress the growth of mammary carcinoma cells and potently enhance the cellular response to an apoptotic agent. While the spectrum of antiproliferative genes whose expression is regulated by HuR in cooperation with CRABP2 remains to be identified, the data establish that the tumor suppressive activity of CRABP2 are exerted both by its ability to deliver RA to RAR, resulting in induction of RAR-targeted growth inhibitory genes, and by its involvement in HuR-mediated stabilization of proapoptotic transcripts. Modulation of the interactions of HuR with mRNAs may comprise a novel strategy for suppressing carcinoma cell growth.
ACKNOWLEDGMENTS
We thank Ann Koehler for assistance with purification of recombinant proteins, Hua Lou (Case Western Reserve University) and Imed Gallouzi (McGill University) for HuR vectors, Cecile Rochette-Egly (IGBMC, Strasbourg, France) for CRABP2 antibodies, and Hiroyuki Kagechika (Tokyo Medical and Dental University) for LE540.
This work was supported by NIH grants DK060684 and CA166955 to N.N. A.C.V. was partially supported by NIH grant 5T32GM008803. The Cytometry and Imaging Microscopy Core Facility of the Case Comprehensive Cancer Center is supported by P30 CA43703.
Footnotes
Published ahead of print 31 March 2014
REFERENCES
- 1.Chambon P. 1996. A decade of molecular biology of retinoic acid receptors. FASEB J. 10:940–954 [PubMed] [Google Scholar]
- 2.Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. 2007. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 129:723–733. 10.1016/j.cell.2007.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaw N, Elholm M, Noy N. 2003. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor beta/delta. J. Biol. Chem. 278:41589–41592. 10.1074/jbc.C300368200 [DOI] [PubMed] [Google Scholar]
- 4.Storch J, Corsico B. 2008. The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu. Rev. Nutr. 28:73–95. 10.1146/annurev.nutr.27.061406.093710 [DOI] [PubMed] [Google Scholar]
- 5.Noy N. 2000. Retinoid-binding proteins: mediators of retinoid action. Biochem. J. 348(Part 3):481–495 [PMC free article] [PubMed] [Google Scholar]
- 6.Sessler RJ, Noy N. 2005. A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Mol. Cell 18:343–353. 10.1016/j.molcel.2005.03.026 [DOI] [PubMed] [Google Scholar]
- 7.Ayers SD, Nedrow KL, Gillilan RE, Noy N. 2007. Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPARgamma by FABP4. Biochemistry 46:6744–6752. 10.1021/bi700047a [DOI] [PubMed] [Google Scholar]
- 8.Dong D, Ruuska SE, Levinthal DJ, Noy N. 1999. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J. Biol. Chem. 274:23695–23698. 10.1074/jbc.274.34.23695 [DOI] [PubMed] [Google Scholar]
- 9.Budhu AS, Noy N. 2002. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol. Cell. Biol. 22:2632–2641. 10.1128/MCB.22.8.2632-2641.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan NS, Shaw NS, Vinckenbosch N, Liu P, Yasmin R, Desvergne B, Wahli W, Noy N. 2002. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 22:5114–5127. 10.1128/MCB.22.14.5114-5127.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soprano DR, Qin P, Soprano KJ. 2004. Retinoic acid receptors and cancers. Annu. Rev. Nutr. 24:201–221. 10.1146/annurev.nutr.24.012003.132407 [DOI] [PubMed] [Google Scholar]
- 12.Donato LJ, Noy N. 2005. Suppression of mammary carcinoma growth by retinoic acid: proapoptotic genes are targets for retinoic acid receptor and cellular retinoic acid-binding protein II signaling. Cancer Res. 65:8193–8199. 10.1158/0008-5472.CAN-05-1177 [DOI] [PubMed] [Google Scholar]
- 13.Donato LJ, Suh JH, Noy N. 2007. Suppression of mammary carcinoma cell growth by retinoic acid: the cell cycle control gene Btg2 is a direct target for retinoic acid receptor signaling. Cancer Res. 67:609–615. 10.1158/0008-5472.CAN-06-0989 [DOI] [PubMed] [Google Scholar]
- 14.Noy N. 2010. Between death and survival: retinoic acid in regulation of apoptosis. Annu. Rev. Nutr. 30:201–217. 10.1146/annurev.nutr.28.061807.155509 [DOI] [PubMed] [Google Scholar]
- 15.Di-Poï N, Michalik L, Tan NS, Desvergne B, Wahli W. 2003. The anti-apoptotic role of PPARbeta contributes to efficient skin wound healing. J. Steroid Biochem. Mol. Biol. 85:257–265. 10.1016/S0960-0760(03)00215-2 [DOI] [PubMed] [Google Scholar]
- 16.Di-Poï N, Tan NS, Michalik L, Wahli W, Desvergne B. 2002. Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol. Cell 10:721–733. 10.1016/S1097-2765(02)00646-9 [DOI] [PubMed] [Google Scholar]
- 17.Wang D, Wang H, Guo Y, Ning W, Katkuri S, Wahli W, Desvergne B, Dey SK, DuBois RN. 2006. Crosstalk between peroxisome proliferator-activated receptor delta and VEGF stimulates cancer progression. Proc. Natl. Acad. Sci. U. S. A. 103:19069–19074. 10.1073/pnas.0607948103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schug TT, Berry DC, Toshkov IA, Cheng L, Nikitin AY, Noy N. 2008. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARbeta/delta to RAR. Proc. Natl. Acad. Sci. U. S. A. 105:7546–7551. 10.1073/pnas.0709981105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manor D, Shmidt EN, Budhu A, Flesken-Nikitin A, Zgola M, Page R, Nikitin AY, Noy N. 2003. Mammary carcinoma suppression by cellular retinoic acid binding protein-II. Cancer Res. 63:4426–4433 [PubMed] [Google Scholar]
- 20.Levi L, Lobo G, Doud MK, von Lintig J, Seachrist D, Tochtrop GP, Noy N. 2013. Genetic ablation of the fatty acid binding protein FABP5 suppresses HER2-induced mammary tumorigenesis. Cancer Res. 73:4770. 10.1158/0008-5472.CAN-13-0384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campos B, Warta R, Chaisaingmongkol J, Geiselhart L, Popanda O, Hartmann C, von Deimling A, Unterberg A, Plass C, Schmezer P, Herold-Mende C. 2012. Epigenetically mediated downregulation of the differentiation-promoting chaperon protein CRABP2 in astrocytic gliomas. Int. J. Cancer 131:1963–1968. 10.1002/ijc.27446 [DOI] [PubMed] [Google Scholar]
- 22.Calmon MF, Rodrigues RV, Kaneto CM, Moura RP, Silva SD, Mota LD, Pinheiro DG, Torres C, de Carvalho AF, Cury PM, Nunes FD, Nishimoto IN, Soares FA, da Silva AM, Kowalski LP, Brentani H, Zanelli CF, Silva WA, Jr, Rahal P, Tajara EH, Carraro DM, Camargo AA, Valentini SR. 2009. Epigenetic silencing of CRABP2 and MX1 in head and neck tumors. Neoplasia 11:1329–1339 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta S, Pramanik D, Mukherjee R, Campbell NR, Elumalai S, de Wilde RF, Hong SM, Goggins MG, De Jesus-Acosta A, Laheru D, Maitra A. 2012. Molecular determinants of retinoic acid sensitivity in pancreatic cancer. Clin. Cancer Res. 18:280–289. 10.1158/1078-0432.CCR-11-2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu YS, Wang Q, Ma JX, Yang XH, Wu ML, Zhang KL, Kong QY, Chen XY, Sun Y, Chen NN, Shu XH, Li H, Liu J. 2012. CRABP-II methylation: a critical determinant of retinoic acid resistance of medulloblastoma cells. Mol. Oncol. 6:48–61. 10.1016/j.molonc.2011.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinman MN, Lou H. 2008. Diverse molecular functions of Hu proteins. Cell. Mol. Life Sci. 65:3168–3181. 10.1007/s00018-008-8252-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myer VE, Fan XC, Steitz JA. 1997. Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J. 16:2130–2139. 10.1093/emboj/16.8.2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brennan CM, Steitz JA. 2001. HuR and mRNA stability. Cell. Mol. Life Sci. 58:266–277. 10.1007/PL00000854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen CY, Xu N, Shyu AB. 2002. Highly selective actions of HuR in antagonizing AU-rich element-mediated mRNA destabilization. Mol. Cell. Biol. 22:7268–7278. 10.1128/MCB.22.20.7268-7278.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.López de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. 2004. Identification of a target RNA motif for RNA-binding protein HuR. Proc. Natl. Acad. Sci. U. S. A. 101:2987–2992. 10.1073/pnas.0306453101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh M, Aguila HL, Michaud J, Ai Y, Wu MT, Hemmes A, Ristimaki A, Guo C, Furneaux H, Hla T. 2009. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J. Clin. Invest. 119:3530–3543. 10.1172/JCI38263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdelmohsen K, Gorospe M. 2010. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip. Rev. RNA 1:214–229. 10.1002/wrna.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Budhu A, Gillilan R, Noy N. 2001. Localization of the RAR interaction domain of cellular retinoic acid binding protein-II. J. Mol. Biol. 305:939–949. 10.1006/jmbi.2000.4340 [DOI] [PubMed] [Google Scholar]
- 33.Keene JD, Tenenbaum SA. 2002. Eukaryotic mRNPs may represent posttranscriptional operons. Mol. Cell 9:1161–1167. 10.1016/S1097-2765(02)00559-2 [DOI] [PubMed] [Google Scholar]
- 34.Donato LJ, Noy N. 2006. A fluorescence-based method for analyzing retinoic acid in biological samples. Anal. Biochem. 357:249–256. 10.1016/j.ab.2006.07.020 [DOI] [PubMed] [Google Scholar]
- 35.Norris AW, Cheng L, Giguere V, Rosenberger M, Li E. 1994. Measurement of subnanomolar retinoic acid binding affinities for cellular retinoic acid binding proteins by fluorometric titration. Biochim. Biophys. Acta 1209:10–18. 10.1016/0167-4838(94)90130-9 [DOI] [PubMed] [Google Scholar]
- 36.Giles KM, Daly JM, Beveridge DJ, Thomson AM, Voon DC, Furneaux HM, Jazayeri JA, Leedman PJ. 2003. The 3′-untranslated region of p21WAF1 mRNA is a composite cis-acting sequence bound by RNA-binding proteins from breast cancer cells, including HuR and poly(C)-binding protein. J. Biol. Chem. 278:2937–2946. 10.1074/jbc.M208439200 [DOI] [PubMed] [Google Scholar]
- 37.Majumdar A, Petrescu AD, Xiong Y, Noy N. 2011. Nuclear translocation of cellular retinoic acid-binding protein II is regulated by retinoic acid-controlled SUMOylation. J. Biol. Chem. 286:42749–42757. 10.1074/jbc.M111.293464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. 1992. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc. Natl. Acad. Sci. U. S. A. 89:10578–10582. 10.1073/pnas.89.22.10578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barreau C, Paillard L, Osborne HB. 2005. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33:7138–7150. 10.1093/nar/gki1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Ahmadi W, Al-Ghamdi M, Al-Haj L, Al-Saif M, Khabar KS. 2009. Alternative polyadenylation variants of the RNA binding protein, HuR: abundance, role of AU-rich elements and auto-regulation. Nucleic Acids Res. 37:3612–3624. 10.1093/nar/gkp223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lakowicz JR. 1983. Principles of fluorescence spectroscopy. Plenum Press, New York, NY [Google Scholar]
- 42.Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M. 2000. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 19:2340–2350. 10.1093/emboj/19.10.2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pullmann R, Jr, Juhaszova M, Lopez de Silanes I, Kawai T, Mazan-Mamczarz K, Halushka MK, Gorospe M. 2005. Enhanced proliferation of cultured human vascular smooth muscle cells linked to increased function of RNA-binding protein HuR. J. Biol. Chem. 280:22819–22826. 10.1074/jbc.M501106200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HH, Abdelmohsen K, Lal A, Pullmann R, Jr, Yang X, Galban S, Srikantan S, Martindale JL, Blethrow J, Shokat KM, Gorospe M. 2008. Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev. 22:1804–1815. 10.1101/gad.1645808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Latorre E, Tebaldi T, Viero G, Sparta AM, Quattrone A, Provenzani A. 2012. Downregulation of HuR as a new mechanism of doxorubicin resistance in breast cancer cells. Mol. Cancer 11:13. 10.1186/1476-4598-11-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gubin MM, Calaluce R, Davis JW, Magee JD, Strouse CS, Shaw DP, Ma L, Brown A, Hoffman T, Rold TL, Atasoy U. 2010. Overexpression of the RNA binding protein HuR impairs tumor growth in triple negative breast cancer associated with deficient angiogenesis. Cell Cycle 9:3337–3346. 10.4161/cc.9.16.12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mazroui R, Di Marco S, Clair E, von Roretz C, Tenenbaum SA, Keene JD, Saleh M, Gallouzi IE. 2008. Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J. Cell Biol. 180:113–127. 10.1083/jcb.200709030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Minocha A, Long BH. 1984. Inhibition of the DNA catenation activity of type II topoisomerase by VP16-213 and VM26. Biochem. Biophys. Res. Commun. 122:165–170. 10.1016/0006-291X(84)90454-6 [DOI] [PubMed] [Google Scholar]
- 49.Gillilan RE, Ayers SD, Noy N. 2007. Structural basis for activation of fatty acid-binding protein 4. J. Mol. Biol. 372:1246–1260. 10.1016/j.jmb.2007.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ortega AD, Sala S, Espinosa E, Gonzalez-Baron M, Cuezva JM. 2008. HuR and the bioenergetic signature of breast cancer: a low tumor expression of the RNA-binding protein predicts a higher risk of disease recurrence. Carcinogenesis 29:2053–2061. 10.1093/carcin/bgn185 [DOI] [PubMed] [Google Scholar]