

Abstract
Haploinsufficiency of Eya1 causes the branchio-oto-renal (BOR) syndrome, and abnormally high levels of Eya1 are linked to breast cancer progression and poor prognosis. Therefore, regulation of Eya1 activity is key to its tissue-specific functions and oncogenic activities. Here, we show that Eya1 is posttranslationally modified by ubiquitin and that its ubiquitination level is self-limited to prevent premature degradation. Eya1 has an evolutionarily conserved CDC4 phosphodegron (CPD) signal, a target site of glycogen synthase kinase 3 (GSK3) kinase and Fbw7 ubiquitin ligase, which is required for Eya1 ubiquitination. Genetic deletion of Fbw7 and pharmacological inhibition of GSK3 significantly decrease Eya1 ubiquitination. Conversely, activation of the phosphatidylinositol 3-kinase (PI3K)/Akt and the canonical Wnt signal suppresses Eya1 ubiquitination. Compound Eya1+/−; Wnt9b+/− mutants exhibit an increased penetrance of renal defect, indicating that they function in the same genetic pathway in vivo. Together, these findings reveal that the canonical Wnt and PI3K/Akt signal pathways restrain the GSK3/Fbw7-dependent Eya1 ubiquitination, and they further suggest that dysregulation of this novel axis contributes to tumorigenesis.
INTRODUCTION
The name of the eyes absent (eya) gene tells only a partial tale. Indeed, it is required for development of and is capable of inducing the ectopic Drosophila eye (1–3). The function of mammalian Eya family proteins, however, expands beyond organogenesis (4–6), from tumorigenesis (7–10) to tissue remodeling (11, 12) to innate immunity (13). The underlying mechanism of Eya1 pleotropic functions remains to be fully defined.
Haploinsufficiency of human EYA1 causes the branchio-oto-renal (BOR) syndrome, suggesting that normal organ development depends on a minimal critical level of Eya1 protein (4). The murine Eya1 heterozygous mutant (Eya1+/−), which produces approximately 43% of the normal level of Eya1 protein, exhibits the hearing defects but not the renal defects seen in human BOR patients (14). A further reduction to 38% results in renal hypoplasia, and 14% of the normal Eya1 level is incompatible with kidney formation in mouse mutants (14). On the other hand, abnormally high levels of human EYA1 are linked to breast cancer progression and poor prognosis (7). These observations suggest that cell type-specific Eya1 function may in part depend on its protein levels.
The ubiquitin-proteasome system is a sophisticated cellular machinery to selectively break down protein targets. A polyubiquitin chain with 4 or more ubiquitin moieties is often needed to efficiently usher covalently tagged targets to the proteasome degradation complex (15). Substrate selectivity depends mainly on each of hundreds of E3 ubiquitin ligases. For example, Fbw7 (F-box and WD repeat domain-containing 7, Cdc4) is a substrate recognition component of the SCF (SKP1, CUL1, and F-box protein)-type ubiquitin ligases. Fbw7 selectively targets a subset of ubiquitination substrates that have one or more consensus Cdc4 phosphodegron (CPD) sequences (16, 17). Phosphorylation of the CPD is a prerequisite for high-affinity Fbw7 binding and subsequent ubiquitination. Glycogen synthase kinase 3 (GSK3) is a major kinase that phosphorylates the CPD of most if not all Fbw7 substrates (18, 19). Together, GSK3 and Fbw7 ubiquitin ligase are two gatekeepers working together as a unit to instigate degradation of a selective group of protein targets.
Eya1 is posttranslational modified by small ubiquitin-related modifier (SUMO) (20). We find that the Eya1 N-terminal lysine 43 and 146 residues are SUMOylated. In addition, SUMOylation inhibits Eya1 activities (Y. Sun et al., submitted for publication). These studies suggest that Eya1 may also be ubiquitinated and that Eya1 ubiquitination by specific ubiquitin ligases may contribute to its diverse functions. Indeed, Sun et al. reported that the Eya1 protein level fluctuates during the cell cycle and that the late-M-phase Eya1 is actively targeted to the ubiquitin-dependent degradation pathway (21). Intriguingly, the rate of Eya1 proteolysis depends on the cell type, suggesting addition regulatory mechanism that might be independent of the cell cycle.
Here, we provide evidence that Eya1 protein is modified by ubiquitin at the conserved C-terminal phosphatase domain. The Eya1 ubiquitination level is limited by its intrinsic tyrosine phosphatase activity, which prevents premature degradation. Eya1 ubiquitination depends on its conserved CPD signal sequence and the GSK3/Fbw7 destruction complex. Both canonical Wnt and phosphatidylinositol 3-kinase (PI3K)/Akt signals restrain Eya1 ubiquitination. Thus, Eya1 may function as a node of multiple signal networks to promote organ development and tumor formation.
MATERIALS AND METHODS
Mouse lines and cell lines.
The Wnt9b+/− and Eya1+/− heterozygous mouse lines were described previously and were maintained in a mixed genetic background (4, 22). All animal studies were performed according to protocols reviewed and approved by the Institutional Animal Care and Use Committee at Boston Children's Hospital. Wild-type and FBW7−/− HCT116 cells were obtained from Bert Vogelstein (Johns Hopkins University), and cultured in McCoy's 5a medium (Invitrogen) with 10% fetal bovine serum. L cells and L-Wnt3a cells were gifts from Xi He (Boston Children's Hospital). L cells and L-Wnt3a cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 0.4 mg/ml G418 for L-Wnt3a cells to generate conditioned medium.
IP/IB.
A standard immunoprecipitation and immunoblotting (IP/IB) protocol was used with minor modifications. Specifically, high-stringency lysis buffer (300 mM NaCl, 0.5% NP-40, Tris HCl [pH 7.4], 0.5 mM EDTA) was used for ubiquitination assays, while low-salt lysis buffer (150 mM NaCl, 1% NP-40, Tris HCl [pH 7.4], 0.5 mM EDTA) was used for protein interaction assays. Proteins were separated by electrophoresis using 4 to 12% NuPAGE Novex bis-Tris gels (Invitrogen). Standard molecular cloning techniques were used to generate luciferase reporter and epitope-tagged Six1, Eya1, and Fbw7 expression constructs. The QuikChange II XL site-directed mutagenesis kit (Stratagene) was used to generate mutant variants. Hemagglutinin epitope-tagged ubiquitin (HA/UB) was a kind gift from Martin E. Dorf and Daniel Finley (Harvard Medical School). GSK3β and Dkk1 expression plasmids were from Xi He (Boston Children's Hospital). Antibodies were anti-HA (Covance), anti-Flag (Sigma), anti-Myc (Clontech), peroxidase-conjugated anti-Flag (Sigma), peroxidase-conjugated anti-HA (Roche), anti-GAPDH (anti-glyceraldehyde-3-phosphate dehydrogenase) (Santa Cruz) and anti-UB (Santa Cruz). Cycloheximide (CHX), MG132, GSK3β rabbit recombinant protein, and lithium chloride were from Sigma.
In vitro kinase assay.
HEK293 cells pretransfected with Flag/Eya1 were lysed with hypotonic buffer containing 50 mM Tris (pH 7.4), 0.5 mM dithiothreitol (DTT), 10 mM NaF, 1 mM Na3VO4, 5 nM okadaic acid, 5 nM calyculin A, 1 mM β-glycerophosphate, 2.5 mM Na4P2O7, protease inhibitor cocktail, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Flag/Eya1 was immunoprecipitated according to the protocol described above. Immunoprecipitated Flag/Eya1 was incubated with the active form of GSK3β rabbit recombinant protein (Sigma) in a kinase reaction buffer (20 mM HEPES [pH 8.0], 20 mM MgCl2, 2 mM DTT, 10 mM NaF, 1 mM Na3VO4, 5 nM okadaic acid, 5 nM calyculin A, 1 mM beta-glycerophosphate, 2.5 mM Na4P2O7, 1 mM EDTA, 25 μM ATP, and 5 μCi [γ-32P]ATP) at room temperature for 6 h. After incubation, the reactions were stopped by adding SDS sample buffer, and the products were resolved in a 4 to 12% NuPAGE Novex bis-Tris gradient gel.
Statistics.
The paired 2-tailed Student t test was used for analysis of differences between groups. Data are presented as means ± standard errors of the means (SEM) from at least three independent experiments. A P value of less than 0.05 was considered statistically significant.
RESULTS
The intrinsic phosphatase activity restricts ubiquitination and stabilizes Eya1.
To examine potential mechanisms of Eya1 protein stability, a Flag epitope-tagged Eya1 (Flag-Eya1) was transfected in HEK293 cells, and its expression level was monitored using a Flag-specific antibody. These cells were treated with MG132, a potent cell-permeative inhibitor of the ubiquitin-mediated proteasome complex. Compared to treatment of cells with dimethyl sulfoxide (DMSO) vehicle, MG132 treatment (12.5 μM for 16 h) resulted in a marked increase of the Flag/Eya1 protein level (Fig. 1A), suggesting that Eya1 protein is targeted, either directly or indirectly, by the ubiquitin-mediated protein degradation pathway. To measure the Eya1 protein half-life, these HEK293 cells was treated with cycloheximide (CHX) to inhibit the protein synthesis machinery (Fig. 1B). Resident proteins were chased over time to measure the persistence of endogenous proteins. Wild-type Eya1 (Flag/Eya1) did not change over an 8-hour pulse-chase period, indicating that its half-life was more than 8 h in these cells. In contrast, a phosphatase-dead mutant variant (Flag/D327A) with a point mutation in the catalytic motif (5, 23, 24) had nearly an 80% reduction within 4 h. Thus, Eya1 intrinsic phosphatase activity is required to its own stability via a possible ubiquitination mechanism.
FIG 1.

Eya1 is posttranslationally modified by ubiquitin. (A and B) A proteasome inhibitor, MG132, increases the Eya1 protein level. HEK293 cells that were stably transfected with a Flag-Eya1 expression vector were treated with MG132 (12.5 μM). The Flag/Eya1 protein level in total cell lysates was determined using a Flag-specific antibody after 16 h of MG132 treatment (A) or during a time course (B). GAPDH was used as a protein loading control. DMSO mock treatment was considered the starting point (0 h). (C and D) Eya1 is ubiquitinated in HEK293 cells. Flag/Eya1, Flag/D327A, and HA-tagged ubiquitin (HA/UB) were transiently coexpressed in HEK293 cells. An immunoprecipitation/immunoblotting (IP/IB) assay was done using the indicated antibodies or control IgG at 48 h after transfection. Numbers on the side of each panel indicate the ubiquitin chain length.
To examine whether Eya1 could be posttranslationally modified by ubiquitin, Flag/Eya1 and HA epitope-tagged ubiquitin (HA/UB) were coexpressed in HEK293 cells. Flag/Eya1 protein, regardless of its modification status, was immunoprecipitated under a high-stringency condition to discourage protein-protein interaction. The immunocomplex was resolved using a gradient gel and subsequently investigated using HA- as well as Flag-specific antibodies (Fig. 1C). A HA/UB antibody detected a series of high-molecular-weight species from the Flag/Eya1-specific immunocomplex but not from a normal IgG control, and some of these slow migrating bands displayed a characteristic incremental size increase of individual ubiquitin moieties (∼8.5 kDa). A large portion of ubiquitinated Eya1 had a single or two ubiquitin moieties (Fig. 1C). Indeed, mono- and diubiquitinated Flag/Eya1 were enriched from the HA/UB immunocomplex (Fig. 1D). The ubiquitination pattern of the D327A variant was therefore compared directly with that of the wild-type control (Fig. 1C and D). In comparison to the wild-type control, a weaker signal corresponding to monoubiquitination of D327A was detected, but a polyubiquitination signal with chains longer than 4 ubiquitin moieties was relatively stronger (Fig. 1C, lane 4). Since a long polyubiquitin chain with at least four ubiquitin moieties is needed to efficiently target substrates to the degradation complex (15), meagerly ubiquitinated Eya1 was unlikely to be degraded readily, consistent with the observation that it is relatively stable.
Eya1 has a conserved C-terminal tyrosine phosphatase domain from amino acid (aa) 320 to 591 (5, 23, 24). To dissect regions of Eya1 that were modified by ubiquitin, we characterized the ubiquitination potential of several Eya1 fragments. Fragment A (aa 1 to 326) had no detectable ubiquitination (Fig. 2A and B, Flag/Eya1-A), even though this fragment was highly expressed. Fragments B (Flag/Eya1-B, aa 1 to 497), C (Flag/Eya1-C, aa 316 to 497), and D (Flag/Eya1-D, aa 316 to 591), however, were heavily ubiquitinated. The conserved phosphatase domain is disrupted in fragments B and C. The intense signal of high-molecular-weight bands suggested that fragments B and C have longer ubiquitin chains than fragment D.
FIG 2.

The conserved Eya tyrosine phosphatase domain is ubiquitinated. (A and B) The C terminus of Eya1 is ubiquitinated. Flag-tagged Eya1 truncated fragments (as indicated) and HA/UB were coexpressed in HEK293 cells, and total cell lysates were subjected to an IP/IB assay at 48 h after transfection. Results are summarized in panel B. Lane 1, total cell lysate; lane 2, IP with control IgG; lane 3, IP with a Flag-specific antibody. (C) Ubiquitination patterns of several Flag-tagged Eya1 variants that were found in human branchio-oto-renal (BOR) syndrome. WT, wild type.
Taken together, these results indicate that Eya1 protein is likely targeted by the ubiquitin-mediated proteasome degradation pathway and that the intrinsic tyrosine phosphatase activity restricts ubiquitination and stabilizes Eya1.
Aberrant ubiquitination of human BOR disease forms of Eya1 variants.
To examine the potential pathophysiological significance of Eya1 ubiquitin modification, we analyzed a number of missense mutations within the conserved C-terminal domain identified in patients with BOR syndrome (9, 25, 26). Among them, the S487P and L505R point mutants have a significantly lower level of tyrosine phosphatase catalytic activity (23, 27). Both variants exhibited an increase of ubiquitination (Fig. 2C). Enzymatic activities of the L583P and E362K variants are unknown. While the ubiquitination status of L583P was similar to that of wild-type Eya1, the E362K variant exhibited the highest level of ubiquitination among all the mutant variants. Moreover, a truncated form of E362K was detected (Fig. 2C, last lane). We also noted that the G426S, D429G, and R440Q variants displayed a slight increase of ubiquitination, even though they may have comparable levels of tyrosine phosphatase activity in vitro (23, 27). Structural and site-directed mutator (SDM) analysis suggests that the S487P, G426S, D429G, and R440Q mutants have minimal impact on the overall structural integrity of the Eya1 protein (9). The aberrant modification patterns of diseased forms of Eya1, including S487P, L505R, and E362K, suggest that dysregulation of human EYA1 protein stability could contribute to BOR syndrome.
Eya1 is a target of GSK3 via the CPD signal.
The Eya1 protein sequence has an evolutionarily conserved motif that resembles both the GSK3 kinase phosphorylation consensus site and the CPD signal sequence (Fig. 3A). This prompted us to test whether Eya1 was targeted by the GSK3/Fbw7 axis. HA-tagged GSK3β (HA/GSK3β) was detected in the Flag/Eya1 immunocomplex, suggesting a potential association between Eya1 and GSK3β (Fig. 3B). The phosphatase mutant D327A variant had a lower expression level than wild-type Flag/Eya1. However, more GSK3β proteins were recruited into the Flag/Eya1(D327A) immunocomplex (Fig. 3B and C). Thus, both wild-type and phosphatase mutant Eya1 proteins interact with GSK3β, but the mutant appears to have a much higher affinity.
FIG 3.

GSK3β phosphorylates and enhances the ubiquitination level of Eya1. (A) Sequence alignment of Eya1 proteins reveals a conserved GSK3β consensus motif and the Cdc4 (also known as Fbw7) phosphodegron (CPD) signal sequence. (B and C) GSK3β, wild-type Eya1, and a phosphatase mutant D327A variant were transiently expressed in HEK293 cells, and total cell lysates were used for a co-IP/IB assay. Quantitative results are shown in panel C. *, P < 0.05 (n = 3). (D and E) An autoradiograph of radiolabeled Flag/Eya1 (top panel) and immunoblot (bottom panel) following an in vitro kinase reaction using recombinant GSK3β protein, immunopurified Flag/Eya1, and radiolabeled [γ-32P]ATP is shown in panel D. The relative density of 32P labeling was normalized against the input Flag/Eya1 level and presented in panel E. (F) Mutation of the GSK3β consensus motif/CPD signal sequence reduces the Eya1 ubiquitination level. Flag/Eya1 variants and HA/UB were transiently expressed in HEK293 cells, and the Flag/Eya1 ubiquitination level was determined using the indicated antibodies in an IP/IB assay. (G) GSK3β was coexpressed with Flag/Eya1 and HA/UB to assess its impact on Eya1 ubiquitination using an IP/IB assay. WT, wild type; con, control (SSSEIAS [aa 48 to 54] to AAAEIAS); M1, S152A/S156A; M2, S156A/T160A; M3, T160A/S164A.
To examine whether Eya1 served as a substrate of GSK3β kinase, immunopurified Flag/Eya1 protein was incubated with recombinant GSK3β protein in a kinase reaction buffer containing radioactive [γ-32P]ATP. Radioactively labeled Eya1 protein was detected by the subsequent autoradiography (Fig. 3D and E), indicating that Eya1 is phosphorylated by GSK3ß in vitro. There are a number of predicted GSK3 sites. To pinpoint the potential phosphorylation site(s), we mutated candidate serine (S) and threonine (T) residues to alanine (A) within the CPD motif (Fig. 3A). The double mutation S156A and T160A (M2), which impaired all possible GSK3 consensus sites, decreased GSK3β labeling. Mutations of S152A/S156A (M1) and T160/S164 (M3) only partially removed GSK3 consensus sites and consistently had minimal impact on the in vitro kinase assay (Fig. 3D and E). The ubiquitination patterns of these mutants were also examined (Fig. 3F). Both the M1 and M2 mutations reduced Eya1 ubiquitination levels, suggesting that S156 may be critical for Eya1 ubiquitination in cells. The SSSEIAS (aa 48 to 54)-to-AAAEIAS mutation was used as a control since the sequence is located outside the CPD motif. This mutant exhibited a level of ubiquitination similar to that of the wild-type control (Fig. 3F). Furthermore, overexpression of GSK3β significantly enhanced Eya1 ubiquitination in a dose-dependent manner (Fig. 3G). Thus, Eya1 is likely targeted by GSK3 and the ubiquitination pathway via the conserved CPD signal.
Fbw7 ubiquitin ligase interacts with Eya1.
Fbw7 is a canonical ubiquitin ligase that recognizes the phosphorylated CPD signal. Similar to the case for GSK3β (Fig. 3B), the Myc-tagged Fbw7 (Myc/Fbw7) was detected in the Flag/Eya1 immunocomplex, and the phosphatase mutant Eya1 variant (D327A) exhibited a much stronger interaction with Fbw7 than wild-type Eya1 (Fig. 4A). Expression of GSK3β significantly enhanced the interaction between Fbw7 and Eya1 (Fig. 4B). Furthermore, mutations of the CPD signal, including S152A/S156A (M1), S156A/T160A (M2), and to a lesser extent T160/S164A (M3), severely hindered Eya1 interaction with Fbw7 (Fig. 4C). Intriguingly, the N-terminal A fragment did not interact with Fbw7 despite the fact that it has the CPD motif (Fig. 4D). On the other hand, C-terminal fragments C and D interacted strongly with Myc/Fbw7. Potential interaction of fragment B could not be determined with certainty since it had low level of expression. To further examine whether recruitment of Fbw7 by Eya1 may contribute to the pathogenesis of BOR, we examined interaction between Fbw7 and a number of BOR point mutants (Fig. 4E). While most BOR mutants preserved the feature of interacting with Fbw7, the S487P and L583P variants had weaker interaction than the wild-type control. Potential interaction with E362K could not be assessed reliably due to its low expression level. Together, these findings suggest that both the N-terminal CPD motif and the C-terminal phosphatase domain of Eya1 are important for the recruitment of Fbw7 ubiquitin ligase.
FIG 4.

Eya1 interacts with Fbw7 ubiquitin ligase. (A and B) Wild-type (Flag/Eya1) and phosphatase mutant (Flag/D327A) Eya1 were coexpressed with Myc/Fbw7 in HEK293 cells with (B) or without (A) HA/GSK3β. Interaction between Eya1 and Fbw7 was examined by the IP/IB assay using the indicated antibodies. The numbers in panel A indicate relative levels. (C) Combinations of Flag/Eya1 variants, Myc/Fbw7, and HA/GSK3β were coexpressed and examined using the indicated antibodies in a co-IP/IB assay. M1, S152A/S156A; M2, S156A/T160A; M3, T160A/S164A. (D and E) Interactions between Fbw7 and Eya1 fragments (D) as well as BOR mutant variants (E) as assessed by an IP/IB assay. Asterisk, nonspecific signal.
Fbw7 is required for ubiquitination and degradation of endogenous Eya1.
Concordant with the effect of GSK3β (Fig. 3F), Myc/Fbw7 significantly enhanced the Flag/Eya1 ubiquitination level in HEK293 cells (Fig. 5A). To further examine whether Fbw7 was required, we used human FBW7 knockout mutant (FBW7−/−) and wild-type (FBW7+/+) HCT116 cells to measure the Eya1 ubiquitination level. Flag/Eya1 was ubiquitinated in wild-type HCT116 cells (Fig. 5B). The ubiquitination pattern and level were similar to those found in HEK293 cells (Fig. 1C). Notably, knockout of FBW7 substantially diminished Flag/Eya1 ubiquitination. Expression of an exogenous Myc-Fbw7 murine gene fully restored and resulted in a robust Flag/Eya1 ubiquitination in FBW7−/− knockout cells (Fig. 5B, third lane). Thus, Fbw7 is required for an efficient ubiquitination of Eya1, and the GSK3/Fbw7 destruction complex is able to usher Eya1 to the ubiquitination pathway.
FIG 5.

Fbw7 regulates endogenous the Eya1 ubiquitination level and half-life. (A and B) Flag/Eya1, HA/UB, and Myc/Fbw7 were coexpressed in HEK293 cells (A) or FBW7 knockout HCT116 cells (B), and the Flag/Eya1 ubiquitination level was analyzed using the indicated antibodies in an IP/IB assay. (C) Endogenous EYA1 protein levels in wild-type and FBW7−/− HCT116 cells. (D to G) Flag/Eya1 and variants were expressed in wild-type (D and E) and FBW7−/− (F and G) HCT116 cells. Cells were treated with cycloheximide (CHX) (20 μg/ml), and the Flag/Eya1 protein level was chased in a period of 6 h and quantified using ImageJ based on signal density and a protein loading control (GAPDH).
We next examined whether FBW7 influenced endogenous EYA1 protein stability and steady-state level. Wild-type and FBW7 knockout HCT116 cells were treated with MG132, and endogenous EYA1 protein levels were measured (Fig. 5C). Wild-type HCT116 cells had a much smaller amount of endogenous EYA1 protein than FBW7 knockout cells. MG132 treatment significantly enhanced EYA1 protein in wild-type but not mutant cells (Fig. 5C). The half-life of wild-type Flag/Eya1 in Myc/Fbw7-expresing wild-type HCT116 cells was between 2 and 4 h (Fig. 5D and E). Its half-life, however, exceeded 6 h in FBW7 knockout cells (Fig. 5F and G). Mutations of the CPD signal rendered Eya1 insensitive to FBW7 status in HCT116 cells and significantly increased the half-life of Eya1 protein (Fig. 5D to G). Taken together, these findings indicate that Fbw7 ubiquitin ligase is required for an efficient degradation of endogenous Eya1 protein.
The PI3K/Akt and canonical Wnt signal pathways antagonize Eya1 ubiquitination.
GSK3β is a downstream negative regulator of several signal pathways, including the phosphatidylinositol 3-kinase (PI3K)/Akt and the canonical Wnt signal. As expected, pharmacological inhibition of the PI3K/Akt signal pathway using LY294002 increased Eya1 ubiquitination in a dose-dependent manner (Fig. 6A). LiCl treatment, which mimics activation of the canonical Wnt signal pathway, decreased Eya1 ubiquitination in a dose-dependent manner (Fig. 6B), providing initial evidence that the Wnt signal inhibits Eya1 ubiquitination. To further examine this possibility, we assayed the Eya1 ubiquitination level in cells that were treated with the conditioned medium containing Wnt3a, a canonical Wnt ligand (28). Compared to the mock control, Wnt3a treatment reduced the Eya1 ubiquitination level, especially the long-chain polyubiquitination (Fig. 6C, second lane). Conversely, Dkk1 conditioned medium, which inhibits the canonical Wnt signal pathway (29–31), enhanced the Eya1 polyubiquitination level (Fig. 6C, third lane). Together, these findings suggest that the canonical Wnt and the PI3K/Akt signal pathways restrict the Eya1 ubiquitination level.
FIG 6.

The canonical Wnt signal pathway regulates the Eya1 ubiquitination level. (A) Flag/Eya1 ubiquitination levels in transiently transfected cells with an increasing amount of LY294002, a pharmacological inhibitor of the PI3K/Akt signal pathway. (B and C) Activation of the canonical Wnt signal pathway by LiCl (B) and Wnt3a (C) reduces Eya1 ubiquitination. Dkk1 (C), an antagonist of canonical Wnt signal, enhanced Eya1 ubiquitination. Flag/Eya1 and HA/UB-expressing cells were treated with LY294002 (A), LiCl (B), or Wnt or Dkk1 conditioned medium (C), followed by IP/IB analysis using the indicated antibodies.
We next wished to examine whether these upstream signaling pathway regulate Eya1 function in vivo. Eya1 is required for normal development of a number of organs, including the kidney (4), inner ear (32, 33), pituitary gland (5), and cardiovascular system (6). Amount these organs, kidney is particularly sensitive to levels of Eya1 activity, making kidney size as an attractive indicator of Eya1 physiological activities (14). Among many canonical Wnt ligands, Wnt9b expression in the developing ureteric bud epithelial cells is required for kidney development in a dose-dependent manner (22). We therefore used Wnt9b as a proxy of the canonical Wnt signal and focused on its potential genetic interaction with Eya1. The size of Eya1+/− heterozygous kidneys was comparable to that of the wild-type control (Fig. 7A and B). Heterozygous Wnt9b+/− mutant kidney was slightly smaller than controls (Fig. 7C). However, kidneys from compound Eya1+/−; Wnt9b+/− mutants were much smaller than these from the single heterozygous mutants (Fig. 7D). To further examine the potential synergistic interaction between Wnt9b and Eya1, the kidney-to-body weight ratio was used in order to minimize growth variation among individuals. As expected, the overall kidney/body weight ratio of heterozygous Eya1+/− mutants (n = 25) was comparable to that of wild-type controls (n = 20) (Fig. 7E). The ratio for Wnt9b+/− mutants was reduced (n = 20). More importantly, the ratio for compound Eya1+/−; Wnt9b+/− mutants (n = 20) was significant reduced compared to that for either Eya1+/− or Wnt9b+/− mutants. Given that Eya1+/− alone did not cause any significant kidney growth defect, the enhanced kidney phenotype observed in compound mutants strongly suggests that Wnt9b and Eya1 function in the same genetic pathway in vivo.
FIG 7.
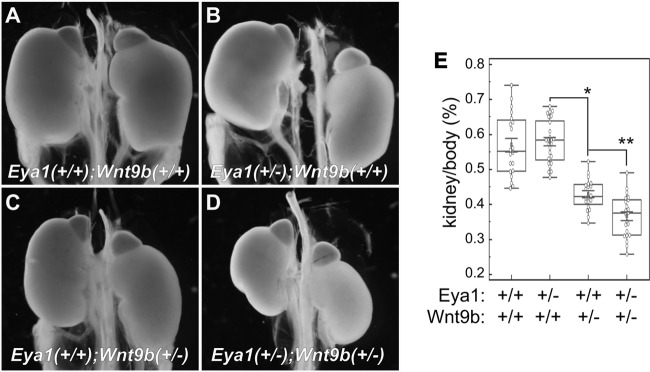
Wnt9b and Eya1 synergistically regulate kidney growth. (A to D) Wnt9b and Eya1 synergistically regulate kidney growth. Representative kidney images from newborn pups with different combinations of genotypes are shown. (E) Quantitative analysis of kidney/body weight ratio. *, P < 0.005; **, P < 0.001.
DISCUSSION
In this study, we showed that Eya1 protein is ubiquitinated at the C-terminal phosphatase domain and that its ubiquitination level is self-limited by the intrinsic tyrosine phosphatase activity. The conserved CPD signal is required for efficient ubiquitination and protein degradation of Eya1. We also present evidence that Eya1 is likely a substrate of both GSK3 kinase and Fbw7 ubiquitin ligase. In addition, activation of the upstream canonical Wnt signal and the PI3K/Akt signal restricts Eya1 ubiquitination. Taken together, these findings suggest that pleiotropic activities of Eya1 in organogenesis and tumorigenesis may depend on multiple upstream regulators, such as the canonical Wnt and PI3K/Akt signal pathways, via a phosphorylation-dependent ubiquitination mechanism (Fig. 8).
FIG 8.
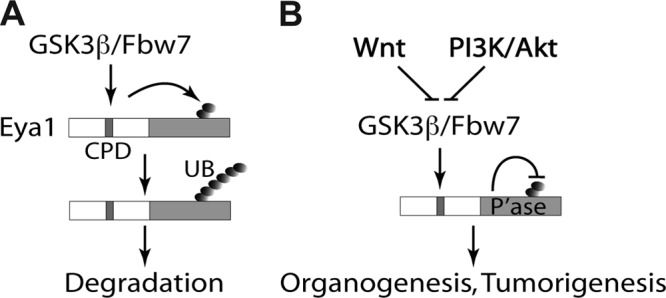
A proposed model of Eya1 ubiquitination and regulation. (A) A conserved N-terminal CPD signal facilitates Eya1 ubiquitination at the C-terminal tyrosine phosphatase domain. This is in part mediated by GSK3β kinase and Fbw7 ubiquitin ligase. (B) The canonical Wnt signal, PI3K/Akt signal, and Eya1 intrinsic tyrosine phosphatase activity restrict Eya1 ubiquitination.
The Eya1 protein sequence has an evolutionarily conserved CPD signal motif at the N terminus that serves as a candidate target site of GSK3 kinase (Fig. 3A). Molecular analyses suggest that the CPD signal is functionally required for efficient recruitment of the GSK/Fbw7 destruction complex and ubiquitination/degradation of Eya1. Paradoxically, the C-terminal Eya1 fragments interact with Fbw7 and are highly ubiquitinated (Fig. 2). These observations suggest that both the N-terminal CPD motif and the C-terminal phosphatase domain of Eya1 are involved in recruiting Fbw7 ubiquitin ligase. It is unclear how the Eya1 C-terminal domain recruits Fbw7. One possibility is that it has a yet-unidentified CPD motif(s). It is also possible that Fbw7 is recruited through an indirect mechanism. In addition to Fbw7, other ubiquitin ligases may also involved in Eya1 ubiquitination. For instance, sun et al. have shown that Eya1 is targeted by the anaphase-promoting complex (APC)/Cdh1 and that Eya1 is degraded in a cell cycle-dependent manner (21). Thus, ubiquitination and degradation of Eya1 may depend on sensitization of multiple signals through different ubiquitin ligases.
The results here indicate that Eya1 phosphatase activity restricts ubiquitination and promotes stability. This is supported by several observations. First, the phosphatase-dead point mutant has much shorter half-life than the wild-type control (Fig. 1B). Second, the Eya1 polyubiquitination level increases in the absence of phosphatase activity, and the ubiquitin chain length appears to be longer (Fig. 1C and D). Third, Eya1 fragments lacking the intact phosphatase activity also exhibit increased levels of polyubiquitination and instability (Fig. 2A). In addition, several Eya1 BOR point mutants with reduced phosphatase activity also exhibit increased ubiquitination (Fig. 2C). How Eya1 phosphatase activity may restrict its own ubiquitination level remains to be determined. We have observed that a phosphatase-dead Eya1 variant has an increased affinity toward GSK3 kinase and Fbw7 ubiquitin ligase, suggesting that recruitment of the GSK3/Fbw7 destruction complex may be involved in the feedback regulatory mechanism. Future identification and characterization of Eya1 substrates and cofactors are needed to better understand the underlying molecular mechanism.
Our findings suggest for the first time that the GSK3/Fbw7 destruction axis functions as a gatekeeper of Eya1. Eya1 is a canonical coactivator of Six family transcription factors (3, 5, 34). A high level of Six1 correlates to advanced stages of breast cancer (35–38), and coordinately expressed high levels of Eya (Eya1 or Eya2) and Six1 indicate a poor prognosis (7). In addition, we showed recently that Eya1 phosphatase activity promotes breast cancer cell migration and proliferation (10) (Sun et al., submitted). Fbw7 is a well-characterized tumor suppressor gene that selectively degrades a number of oncoproteins (18, 19). For example, Fbw7 binds and destabilizes c-Myc in a manner that depends on GSK3-mediated phosphorylation (39, 40). We have shown previously that the Six1/Eya1 transcription complex is required for c-Myc expression in vivo (5). Thus, Fbw7 mutant cancer cells would have increased stability as well as production of c-Myc oncoprotein. Because of the critical involvement of both Eya1 and Fbw7 in tumorigenesis, our findings underscore the need to profile expression patterns of both genes in tumor cells and the potential to increase efficacy to target both genes to slow down cancer cells.
In conclusion, posttranslational ubiquitin modification regulates the Eya1 transcription coactivator and phosphatase. Eya1 ubiquitination depends on GSK3 kinase and Fbw7 ubiquitin ligase but is limited by its own tyrosine phosphatase activity and upstream signals, including canonical Wnt and PI3K/Akt. Together, these findings uncover the novel Wnt/Eya1 and PI3K/Akt/Eya1 axes and further suggest that Eya1 is a critical node of signal networks important for organogenesis and tumorigenesis.
ACKNOWLEDGMENTS
We are grateful to Bert Vogelstein for the HCT116 and HCT116 FBW7−/− cell lines. We thank Xi He for providing Gsk3β and DKK1 expression plasmids and L-Wnt3a cells, Martin E. Dorf and Daniel Finley for the HA/UB expression plasmid, and Satoshi Kaneko for the IP kinase assay. We thank Ian Teng for technical support.
This work was supported by grants to X.L. from NIH/NIDCR (1R01DE019823), NIH/NIDDK (R01DK916451 and P50DK65298), and AHA (13GRNT16950006).
Footnotes
Published ahead of print 21 April 2014
REFERENCES
- 1.Bonini NM, Leiserson WM, Benzer S. 1993. The eyes absent gene: genetic control of cell survival and differentiation in the developing Drosophila eye. Cell 72:379–395. 10.1016/0092-8674(93)90115-7 [DOI] [PubMed] [Google Scholar]
- 2.Bonini NM, Bui QT, Gray-Board GL, Warrick JM. 1997. The Drosophila eyes absent gene directs ectopic eye formation in a pathway conserved between flies and vertebrates. Development 124:4819–4826 [DOI] [PubMed] [Google Scholar]
- 3.Pignoni F, Hu B, Zavitz KH, Xiao J, Garrity PA, Zipursky SL. 1997. The eye-specification proteins So and Eya form a complex and regulate multiple steps in Drosophila eye development. Cell 91:881–891. 10.1016/S0092-8674(00)80480-8 [DOI] [PubMed] [Google Scholar]
- 4.Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. 1999. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat. Genet. 23:113–117. 10.1038/12722 [DOI] [PubMed] [Google Scholar]
- 5.Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, Rosenfeld MG. 2003. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 426:247–254. 10.1038/nature02083 [DOI] [PubMed] [Google Scholar]
- 6.Guo C, Sun Y, Zhou B, Adam RM, Li X, Pu WT, Morrow BE, Moon A, Li X. 2011. A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J. Clin. Invest. 121:1585–1595. 10.1172/JCI44630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farabaugh SM, Micalizzi DS, Jedlicka P, Zhao R, Ford HL. 2012. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene 31:552–562. 10.1038/onc.2011.259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pandey RN, Rani R, Yeo EJ, Spencer M, Hu S, Lang RA, Hegde RS. 2010. The Eyes Absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene 29:3715–3722. 10.1038/onc.2010.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patrick AN, Cabrera JH, Smith AL, Chen XS, Ford HL, Zhao R. 2013. Structure-function analyses of the human SIX1-EYA2 complex reveal insights into metastasis and BOR syndrome. Nat. Struct. Mol. Biol. 20:447–453. 10.1038/nsmb.2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu K, Li Z, Cai S, Tian L, Chen K, Wang J, Hu J, Sun Y, Li X, Ertel A, Pestell RG. 2013. EYA1 phosphatase function is essential to drive breast cancer cell proliferation through cyclin D1. Cancer Res. 73:4488–4499. 10.1158/0008-5472.CAN-12-4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delgado-Olguin P, Huang Y, Li X, Christodoulou D, Seidman CE, Seidman JG, Tarakhovsky A, Bruneau BG. 2012. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat. Genet. 44:343–347. 10.1038/ng.1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SH, Yang DK, Choi BY, Lee YH, Kim SY, Jeong D, Hajjar RJ, Park WJ. 2009. The transcription factor Eya2 prevents pressure overload-induced adverse cardiac remodeling. J. Mol. Cell Cardiol. 46:596–605. 10.1016/j.yjmcc.2008.12.021 [DOI] [PubMed] [Google Scholar]
- 13.Okabe Y, Sano T, Nagata S. 2009. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature 460:520–524. 10.1038/nature08138 [DOI] [PubMed] [Google Scholar]
- 14.Sajithlal G, Zou D, Silvius D, Xu PX. 2005. Eya 1 acts as a critical regulator for specifying the metanephric mesenchyme. Dev. Biol. 284:323–336. 10.1016/j.ydbio.2005.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. 2000. Recognition of the polyubiquitin proteolytic signal. EMBO J. 19:94–102. 10.1093/emboj/19.1.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. 2001. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414:514–521. 10.1038/35107009 [DOI] [PubMed] [Google Scholar]
- 17.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, Xiao Y, Christie AL, Aster J, Settleman J, Gygi SP, Kung AL, Look T, Nakayama KI, DePinho RA, Wei W. 2011. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471:104–109. 10.1038/nature09732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welcker M, Clurman BE. 2008. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8:83–93. 10.1038/nrc2290 [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Inuzuka H, Zhong J, Wan L, Fukushima H, Sarkar FH, Wei W. 2012. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett. 586:1409–1418. 10.1016/j.febslet.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alkuraya FS, Saadi I, Lund JJ, Turbe-Doan A, Morton CC, Maas RL. 2006. SUMO1 haploinsufficiency leads to cleft lip and palate. Science 313:1751. 10.1126/science.1128406 [DOI] [PubMed] [Google Scholar]
- 21.Sun J, Karoulia Z, Wong EY, Ahmed M, Itoh K, Xu PX. 2013. The phosphatase-transcription activator EYA1 is targeted by anaphase-promoting complex/Cdh1 for degradation at M-to-G1 transition. Mol. Cell. Biol. 33:927–936. 10.1128/MCB.01516-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP. 2005. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 9:283–292. 10.1016/j.devcel.2005.05.016 [DOI] [PubMed] [Google Scholar]
- 23.Rayapureddi JP, Hegde RS. 2006. Branchio-oto-renal syndrome associated mutations in Eyes Absent 1 result in loss of phosphatase activity. FEBS Lett. 580:3853–3859. 10.1016/j.febslet.2006.06.009 [DOI] [PubMed] [Google Scholar]
- 24.Tootle TL, Silver SJ, Davies EL, Newman V, Latek RR, Mills IA, Selengut JD, Parlikar BE, Rebay I. 2003. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature 426:299–302. 10.1038/nature02097 [DOI] [PubMed] [Google Scholar]
- 25.Orten DJ, Fischer SM, Sorensen JL, Radhakrishna U, Cremers CW, Marres HA, Van Camp G, Welch KO, Smith RJ, Kimberling WJ. 2008. Branchio-oto-renal syndrome (BOR): novel mutations in the EYA1 gene, and a review of the mutational genetics of BOR. Hum. Mutat. 29:537–544. 10.1002/humu.20691 [DOI] [PubMed] [Google Scholar]
- 26.Krug P, Moriniere V, Marlin S, Koubi V, Gabriel HD, Colin E, Bonneau D, Salomon R, Antignac C, Heidet L. 2011. Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio-oto-renal syndrome calls into question the pathogenic role of SIX5 mutations. Hum. Mutat. 32:183–190. 10.1002/humu.21402 [DOI] [PubMed] [Google Scholar]
- 27.Musharraf A, Markschies N, Teichmann K, Pankratz S, Landgraf K, Englert C, Imhof D. 2008. Eyes absent proteins: characterization of substrate specificity and phosphatase activity of mutants associated with branchial, otic and renal anomalies. Chembiochem 9:2285–2294. 10.1002/cbic.200800224 [DOI] [PubMed] [Google Scholar]
- 28.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR, III, Nusse R. 2003. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423:448–452. 10.1038/nature01611 [DOI] [PubMed] [Google Scholar]
- 29.Semenov MV, Tamai K, Brott BK, Kuhl M, Sokol S, He X. 2001. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr. Biol. 11:951–961. 10.1016/S0960-9822(01)00290-1 [DOI] [PubMed] [Google Scholar]
- 30.Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C. 2002. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature 417:664–667. 10.1038/nature756 [DOI] [PubMed] [Google Scholar]
- 31.Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. 2001. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 411:321–325. 10.1038/35077108 [DOI] [PubMed] [Google Scholar]
- 32.Ahmed M, Wong EY, Sun J, Xu J, Wang F, Xu PX. 2012. Eya1-Six1 interaction is sufficient to induce hair cell fate in the cochlea by activating Atoh1 expression in cooperation with Sox2. Dev. Cell 22:377–390. 10.1016/j.devcel.2011.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmed M, Xu J, Xu PX. 2012. EYA1 and SIX1 drive the neuronal developmental program in cooperation with the SWI/SNF chromatin-remodeling complex and SOX2 in the mammalian inner ear. Development 139:1965–1977. 10.1242/dev.071670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Perissi V, Liu F, Rose DW, Rosenfeld MG. 2002. Tissue-specific regulation of retinal and pituitary precursor cell proliferation. Science 297:1180–1183. 10.1126/science.107326 [DOI] [PubMed] [Google Scholar]
- 35.Ford HL, Kabingu EN, Bump EA, Mutter GL, Pardee AB. 1998. Abrogation of the G2 cell cycle checkpoint associated with overexpression of HSIX1: a possible mechanism of breast carcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 95:12608–12613. 10.1073/pnas.95.21.12608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reichenberger KJ, Coletta RD, Schulte AP, Varella-Garcia M, Ford HL. 2005. Gene amplification is a mechanism of Six1 overexpression in breast cancer. Cancer Res. 65:2668–2675. 10.1158/0008-5472.CAN-04-4286 [DOI] [PubMed] [Google Scholar]
- 37.McCoy EL, Iwanaga R, Jedlicka P, Abbey NS, Chodosh LA, Heichman KA, Welm AL, Ford HL. 2009. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J. Clin. Invest. 119:2663–2677. 10.1172/JCI37691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Micalizzi DS, Christensen KL, Jedlicka P, Coletta RD, Baron AE, Harrell JC, Horwitz KB, Billheimer D, Heichman KA, Welm AL, Schiemann WP, Ford HL. 2009. The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-beta signaling. J. Clin. Invest. 119:2678–2690. 10.1172/JCI37815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE. 2004. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. U. S. A. 101:9085–9090. 10.1073/pnas.0402770101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. 2004. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23:2116–2125. 10.1038/sj.emboj.7600217 [DOI] [PMC free article] [PubMed] [Google Scholar]