

Abstract
The C-terminal domain of the RNA polymerase II largest subunit (the Rpb1 CTD) is composed of tandem heptad repeats of the consensus sequence Y1S2P3T4S5P6S7. We reported previously that Thr 4 is phosphorylated and functions in histone mRNA 3′-end formation in chicken DT40 cells. Here, we have extended our studies on Thr 4 and to other CTD mutations by using these cells. We found that an Rpb1 derivative containing only the N-terminal half of the CTD, as well as a similar derivative containing all-consensus repeats (26r), conferred full viability, while the C-terminal half, with more-divergent repeats, did not, reflecting a strong and specific defect in snRNA 3′-end formation. Mutation in 26r of all Ser 2 (S2A) or Ser 5 (S5A) residues resulted in lethality, while Ser 7 (S7A) mutants were fully viable. While S2A and S5A cells displayed defects in transcription and RNA processing, S7A cells behaved identically to 26r cells in all respects. Finally, we found that Thr 4 was phosphorylated by cyclin-dependent kinase 9 in cells and dephosphorylated both in vitro and in vivo by the phosphatase Fcp1.
INTRODUCTION
RNA polymerase II (RNAP II) consists of 12 subunits and transcribes all mRNA and many noncoding RNA genes. Rpb1, the largest subunit, possesses a unique C-terminal domain (CTD) that consists of tandem heptad repeats, with a consensus sequence of Tyr-Ser-Pro-Thr-Ser-Pro-Ser (Y1S2P3T4S5P6S7). The number of repeats, generally reflecting the complexity of the organism, ranges from 26 in yeast to 52 in vertebrates. The CTD plays important roles in connecting transcription with all the steps of RNA production, and these activities are modulated by posttranslational modification, mainly phosphorylation. Thus, the CTD can be envisioned as functioning as a platform to recruit factors needed for proper RNA synthesis and maturation (for a review, see references 1 to 4).
The CTD is subject to extensive phosphorylation. All five hydroxylated amino acids are potential phosphorylation sites, and several CTD kinases and phosphatases have been described (3, 5). Briefly, Ser 5 and Ser 7 are phosphorylated by cyclin-dependent kinase 7 (CDK7), a component of TFIIH (6, 7), whereas Ser 2 is phosphorylated by CDK9 (P-TEFb) (8, 9) and by CDK12/13 for a subset of genes (10, 11). Tyr 1 can be phosphorylated in mammals by the c-Abl kinase (12) and by an unknown kinase in yeast (13). While Thr 4 phosphorylation is blocked by specific CDK9 inhibitors (14, 15), in vitro and in vivo assays have provided evidence that Thr 4 can be phosphorylated by Polo-like kinase 3 (15). The CTD is dephosphorylated by several phosphatases. Fcp1 has been reported to dephosphorylate Ser 2 and Ser 5, with a preference toward Ser 2 (8, 16), and Ssu72 can dephosphorylate both Ser 5 and Ser 7 (17–20). While Rtr1/RPAP2 has been reported to dephosphorylate Ser 5 (21, 22), structural and enzymatic studies have questioned whether Rtr1/RPAP2 indeed possesses phosphatase activity (23).
The CTD phosphorylation pattern corresponds in general to the position of RNAP II along a transcribed gene. Several genome-wide chromatin immunoprecipitation (ChIP) analyses have provided evidence that early during transcription, the CTD is phosphorylated on Ser 5 and Ser 7, with Ser 5 gradually removed during elongation, while Ser 2 and Thr 4 phosphorylation increases as RNAP II progresses along the gene (15, 17, 24–26). All phosphorylation is then “cleared” at or around transcription termination, which prepares RNAP II for reinitiation. While the majority of these studies have been performed in yeast, this general pattern of CTD phosphorylation is thought to be universal throughout eukaryotes (reviewed in references 2, 4, and 5).
This temporal pattern of CTD phosphorylation helps to link transcription with RNA processing events. For example, Ser 5 phosphorylation facilitates capping enzyme recruitment and indeed enhances the capping reaction (27–29). Splicing factors, such as Prp40 and U2AF65, bind to the phosphorylated CTD, which facilitates recruitment and/or activation of the splicing machinery (30, 31). Recruitment of several mRNA 3′-end processing factors to the vicinity of the nascent RNA (32) and 3′-processing sites (33) is enhanced by Ser 2 phosphorylation. Ser 7 phosphorylation has been implicated in recruitment of the Integrator complex, which functions in snRNA 3′-end formation (34), while Thr 4 is important for efficient recruitment of 3′-processing factors to histone genes (14) and for transcription elongation (15).
The requirement of the phosphorylatable CTD residues for viability varies among species. For example, in Schizosaccharomyces pombe, only Ser 5 is essential (29), whereas in Saccharomyces cerevisiae, Tyr 1 and Ser 2, as well as Ser 5, appear to be necessary for viability (35, 36). Although Thr 4 and Ser 7 residues are dispensable for yeast cell viability (36), this may not to be the case in higher eukaryotes. For example, mutation of Ser 7 residues to Ala was shown to compromise cell viability (37), while mutation of Thr 4 to Val in chicken cells (14) or to Ala in human cells (15) was lethal.
In this report, we examine functions of various CTD residues by using the chicken DT40 cell system we described previously to analyze the function of Thr 4 (14), and we also extend our analysis of Thr 4 phosphorylation. We show that while an Rpb1 derivative containing only the consensus-rich N-terminal half of the CTD, as well as a similar all-consensus derivative (26r), conferred full viability, the more divergent C-terminal half did not, and we show that this reflects a strong and specific defect in snRNA 3′-end formation. We found that substitution of all Ser 2 or Ser 5 residues with Ala in the 26r derivative was lethal, reflecting defects in transcription and mRNA processing. Unexpectedly, a strain expressing a similar derivative with all Ser 7 residues mutated to Ala displayed no detectable defects, including in snRNA expression, and was fully viable. Finally, extending our analysis of Thr 4 phosphorylation, we provide additional evidence that CDK9 phosphorylates Thr 4 and that the Ser 2 phosphatase Fcp1 is responsible for Thr 4 dephosphorylation.
MATERIALS AND METHODS
Cell culture and cloning.
DT40 cells and HEK293 cells were cultured at 37°C with 5% CO2 in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and 1% chicken serum and in Dulbecco's modified Eagle's medium containing 10% FBS, respectively.
Rpb1 CTD derivatives were cloned as previously described (14). Briefly, a fragment of β-actin promoter and FLAG tag was inserted into pBlueScript containing a neomycin resistance gene. The human Rpb1 body without the CTD was inserted behind the FLAG tag, and various CTD fragments were inserted directly 3′ to the Rpb1 body.
Small interfering RNAs (siRNAs) that have been previously described (38) to knock down CDK9 mRNA targeted the following sequences: TAGGGACATGAAGGCTGCTAA, CAACTTGATTGAGATTTGTCG, and AAGGGTAGTATATACCTGGTG.
Fcp1 knockdown shRNA constructs were generated with DNA oligonucleotides targeting the following sequences: AAGAGGAAGCTGAATGAAGAGGA, AAGTATGACCGCTACCTCAACAA, and AATCATTCTCGAGGCACTGAGGT in Fcp1. Synthesized oligonucleotides were cloned into the HuSH pRS vector (Origene) and verified by sequencing.
Transfection was performed with Lipofectamine 2000 (Invitrogen) as described by the manusfacturer's manual. Briefly, HEK293 cells (10 to 20% confluence) were transfected with siRNA or shRNA for 30 to 72 h. Cell lysates were analyzed using Western blotting.
Complementation and construction of stable cell lines.
Procedures for complementation assays and for constructing stable cell lines were as previously described (14). Briefly, 107 cells were transfected with ∼15 μg of linearized DNA and selected in the presence of the appropriate antibiotics. Surviving cell clones were isolated and further analyzed using Western blotting.
Western blotting.
Cells lysates were resolved by SDS-PAGE with the indicated percentages of acrylamide. Western blotting was performed using standard protocols. Briefly, protein samples were transferred to a nitrocellulose membrane, blocked in 5% milk, and incubated with primary antibodies diluted in PBST (phosphate-buffered saline with 0.1% Tween 20). Membranes were then washed in PBST and incubated with appropriate secondary antibodies. For quantification, Western blots were analyzed by using ImageJ. Antibodies used in this study were as follows: FLAG tag (M2; Sigma), actin (Sigma), phospho-Ser 7 CTD heptad (4E12; Millipore), phospho-Thr 4 CTD heptad (Novus), phosphor-Ser 5 (3E8; Millipore), phospho-Ser 5 (H14; Covance), phospho-Ser 2 (H5; Covance), phospho-Ser 2 (3E10; Millipore), glutathione S-transferase (GST) tag (Invitrogen), Rpb1 (N20; Santa Cruz Biotechnology), Fcp1 (Bethyl Laboratories), Rpb2 (biorbyt), U2AF65 (Sigma), histone H3 (Abcam), and CDK9 (Cell Signaling).
RT-qPCR.
RNA was extracted using TRIzol (Invitrogen) and further treated with DNase I. Reverse transcription (RT) and quantitative PCR (qPCR) analysis were performed as previously described (14). Primer sequences are available upon request.
In vivo labeling of nascent RNA and nuclear run-on and slot blotting assays.
In vivo [3H]uridine labeling and nuclear run-on (NRO) assays were performed as previously described (14).
RNase protection assay.
The RNase protection assay was performed as described previously (39).
ChIP.
Cells were grown to 70% confluence (∼2 × 106/ml), cross-linked with 1% formaldehyde for 10 min, and processed for ChIP as previously described (14). ChIP was performed using antibody against FLAG tag (M2; Sigma). Primer sequences we used are available upon request.
Immunoprecipitation.
A total of ∼2 × 107 cells were lysed in 1 ml RIPA buffer containing protease inhibitors. After removal of debris from sonicated lysates by centrifugation, Rpb1 was immunoprecipitated using FLAG antibody at 4°C for 1 h. Beads were washed with RIPA buffer and resuspended in 1× SDS sample buffer.
Subcellular fractionation.
A total of ∼2 × 107 cells were lysed in 0.5 ml of RSB100 (50 mM Tris-HCl [pH 7.4], 100 mM NaCl) containing 40 μg/ml digitonin and incubated on ice for 5 min. The cytoplasmic fraction was separated from the nuclear fraction by centrifugation, and the nuclear fraction was resuspended in 0.5 ml of RSB100 containing 0.5% Triton X-100. Soluble nuclear proteins were further separated from insoluble chromatin-bound proteins by centrifugation. Insoluble chromatin-bound pellets were resuspended in RSB100 (0.5% Triton X-100) and sonicated.
In vitro kinase and phosphatase assays.
CDK9 protein complexes were expressed in insect cells as described previously (40) and purified using Ni-nitrilotriacetic acid–agarose (Qiagen). A GST-CTD fusion was expressed in Escherichia coli as previously described (41) and purified using glutathione-Sepharose 4B (GE Healthcare). A 0.75-μg aliquot of GST-CTD was phosphorylated by CDK9 complexes at 37°C for 1 h in a kinase buffer (25 mM HEPES [pH 7.5], 10 mM MgCl2, 150 mM NaCl, 1 mM ATP). In vitro phosphorylation of GST-CTD by HeLa nuclear extract was performed as previously described (41). Baculoviruses expressing CDK9/cyclin T were gifts from Robert Fisher (Mt. Sinai).
For phosphatase assays, S. cerevisiae Fcp1 and a catalytic mutant Fcp1 (M271E) were expressed in E. coli and purified as described previously (42). Two hundred nanograms of GST-CTD, phosphorylated by the HeLa nuclear extract, was incubated with Fcp1 proteins in a buffer containing 50 mM Tris-acetate (pH 5.5) and 10 mM MgCl2 at 30°C for 90 min. A vector expressing functional Fcp1 (residues 168 to 606) [Fcp1(168–606)] was a gift from Patrick Cramer (Munich, Germany). Based on previous mutational studies (43), a point mutation was introduced into the Fcp1(168–606) expression vector to create the catalytic mutant Fcp1(M271E).
RESULTS
Genetic complementation analysis revealed distinct requirements of CTD residues.
To investigate genetically the functions of the CTD in vertebrate cells, we constructed a number of FLAG-tagged Rpb1 derivatives with various CTD mutations (Table 1) and used an Rpb1 conditional knockout chicken cell line (tetracycline [Tet] sensitive) described previously, DT40-Rpb1 (14), to study the phenotypes of the Rpb1 mutants (see Fig. S1 in the supplemental material for a schematic diagram of cell line construction). The vertebrate CTD, which is almost invariant among species (the amino acid sequence of the zebrafish CTD is 97% identical to that in humans), contains 52 tandem heptads (44). The N-terminal heptads deviate little from the consensus sequence, while the C-terminal repeats display more variation (Fig. 1A). The complete chicken RPB1 gene has proven impossible to isolate, and the sequence in the chicken genome does not include the CTD. We obtained the majority of the gene, however, which is 97% identical to the human sequence, and the CTD is 100% identical through the first 22 heptads repeats (see Fig. S2 in the supplemental material).
TABLE 1.
Viability of Rpb1-CTD mutantsa
| Rpb1-CTD mutant | Viable? |
|---|---|
| Rpb1(1–52) | Yes |
| Rpb1(1–26) | Yes |
| Rpb1(27–52) | No |
| 26r (YSPTSPS)26 | Yes |
| 19r (YSPTSPS)19 | No |
| S2A (YAPTSPS)26 | No |
| T4V (YSPVSPS)30 | No |
| S5A (YSPTAPS)28 | No |
| S7A (YSPTSPA)30 | Yes |
Cells were selected in the presence of tetracycline (1 μg/ml). Surviving cell clones were isolated, and the identity of these cells was confirmed using Western blotting with an anti-FLAG antibody. Multiple verified clones were isolated for the viable derivatives, and no clones were detected for any of the nonviable constructs. The numbers following the sequences in parentheses indicate the numbers of heptads. Each construct contained an N-terminal FLAG tag and the natural 10-residue C-terminal sequence (ISPDDSDEEN) at its C terminus.
FIG 1.

CTD mutants and growth properties. (A) The CTD composition of human Rpb1. The CTD of human Rpb1 consists of 52 tandem heptapeptide repeats. Twenty-one out of 52 repeats are composed of conserved consensus Y1S2P3T4S5P6S7 repeats, and the remaining 31 repeats are less conserved. The Rpb1(1–26) expression vector contains the first half of the CTD, whereas Rpb1(27–52) contains the second half. The consensus repeats, less-conserved repeats, Rpb1 bodies without CTDs (gray boxes), and C-terminal 10-amino-acid motif are shown. (B) Growth curves of various mutant cell lines cultured in medium containing Tet. Averages from two independent experiments were plotted. Cells able to reach full confluence were split every 2 days. (C) Western blot showing the protein expression profiles of the various CTD mutants. Samples were treated with Tet (1 μg/ml) for 24 h.
We showed previously that an Rpb1 derivative (26r) containing 26 all-consensus repeats plus the very-C-terminal 10 residues, which are important for Rpb1 stability (45, 46), was able to completely rescue viability of DT40-Rpb1 cells in the presence of Tet (14). To determine whether the natural N-terminal or C-terminal 26 repeats are also sufficient for viability, two CTD derivatives, Rpb1(1–26) and Rpb1(27–52), were constructed (Fig. 1A). Rpb1(1–26) contains the first 26 heptads plus the C-terminal 10 residues, while Rpb1(27–52) consists of the second half of the CTD, including the C-terminal 10 residues. Analogous to our studies with Thr 4 (14), we also mutated all Ser residues to Ala, constructing Rpb1 derivatives with 26 repeats of YAPTSPS (S2A), 28 repeats of YSPTAPS (S5A), or 30 repeats of YSPTSPA (S7A). While as mentioned above 26 all-consensus repeats are sufficient to confer full viability, we also constructed an Rpb1 derivative with 19 YSPTSPS repeats (19r) to determine whether this lower number of repeats also confers viability.
We first asked if cells expressing these mutant Rpb1 derivatives are viable in the absence of wild-type Rpb1. Plasmids carrying the Rpb1 mutant genes were introduced into DT40-Rpb1 cells, and the transfected cells were grown in the presence of Tet. The identity of surviving cell colonies was confirmed by Western blotting with anti-FLAG antibodies (results not shown). As shown in Table 1, Rpb1(1–26) allowed growth in Tet, but Rpb1(27–52) failed to give rise to Tet-resistant colonies. As shown previously (14), 26r cells were fully viable, but the 19r cells were not, in line with previous observations that nearly half of the CTD is required for viability in yeast (35) and mouse (47) cells. Significantly, cells expressing the Rpb1 derivatives S2A and S5A were inviable, as we observed previously with T4V cells (14). However, somewhat unexpectedly, the S7A cells were fully viable. Together, these results revealed that the length and conservation of the CTD repeats are crucial determinants for cell viability and that Ser 2 and Ser 5 and Thr 4 are essential for viability, while Ser 7 is not. Notably, however, it was recently shown that budding yeast expressing a full-length S2A mutant Rpb1 are viable (48), contrary to an earlier study showing lethality of S2A in yeast harboring an Rpb1 with a truncated CTD (35). Thus, it is possible that a synthetic phenotype from the combination of a mutation and CTD length contributed to the inviability of cells expressing the Ser 2, Ser 5, and/or Thr 4 Rpb1 mutant derivatives.
To investigate further the phenotypes of the above mutant CTDs, we established stable DT40-Rpb1 cell lines expressing each of the Rpb1 derivatives described above and analyzed how these mutants affect cell growth, transcription, and RNA processing. In agreement with the results of the complementation analysis above, stable cell lines expressing wild-type Rpb1, Rpb1(1–52), Rpb1(1–26), 26r, and S7A were viable in medium containing Tet, although Rpb1(1–26), 26r, and S7A displayed slightly slower growth rates than Rpb1(1–52) cells (Fig. 1B, left). Cells expressing Rpb1(27–52) ceased growth ∼48 h after Tet addition, and S2A, T4V, and S5A cells all stopped growing after ∼24 h of Tet treatment (Fig. 1B, right). All Rpb1 derivatives were expressed at similar levels in these cell lines (Fig. 1C).
CTD mutations differentially impact transcription and RNA processing.
We next examined the effects of the CTD mutations on transcription, splicing, and 3′ processing, initially by using an inducible endogenous gene, Egr1. Cells were cultured in the presence of Tet for 24 h, and Egr1 was induced by addition of ionomycin and phorbol 12-myristate 13-acetate (PMA). Following a short 20-min induction, Egr1 mRNA levels in 26r cells were strikingly increased, by about 300-fold as measured by RT-qPCR (Fig. 2A) (14). In contrast, Egr1 induction levels were 4- to 5-fold lower in S2A and S5A cells. Small decreases in Egr1 mRNA levels were observed in Rpb1(1–26), Rpb1(27–52), and S7A cells, but compared to Rpb1(1–52) cells the decreases were not significant (Fig. 2B). In the parental DT40-Rpb1 cells, Egr1 induction was barely detected after 24 h of growth in Tet, indicating that the observed Egr1 induction was not due to residual levels of wild-type Rpb1. We next used Egr1 to examine whether the Rpb1 CTD derivatives affected splicing and 3′-end processing. RT-qPCR assays designed to detect spliced and unspliced RNAs revealed that ∼2-fold more unspliced Egr1 mRNA was detected in S2A and S5A cells than in 26r cells (Fig. 2C). Similarly, probes to distinguish total and 3′-uncleaved RNAs detected 4- to 6-fold more uncleaved Egr1 RNA in these cells (Fig. 2D). More uncleaved Egr1 RNA was detected in S2A cells than S5A cells, consistent with the known importance of Ser 2 phosphorylation in 3′ cleavage (see above). Splicing and 3′ processing were not affected detectably in any of the other mutants, although it is notable that 3′ processing but not splicing appeared somewhat more efficient in Rpb1(1–52) than in 26r or any of the other derivatives (Fig. 2D).
FIG 2.

Impact of CTD mutations on Egr1 transcription, splicing and 3′-end processing. (A) 26r cells were cultured in medium containing 1 μg/ml tet for 24 h, and then treated with dimethyl sulfoxide (DMSO) or ionomycin and PMA for 20 min to induce Egr1 expression. Egr1 induction levels were measured by RT-qPCR and are plotted relative to the noninduced control (DMSO treatment) (n = 3). (B) Analysis of Egr1 induction levels in various cell lines. Cells were treated with Tet for 24 h and induced with ionomycin and PMA for 20 min. Egr1 mRNA levels were measured using RT-qPCR and are plotted relative to levels in 26r cells (n = 3). (C) Cells were treated as described for panel B. The ratios of unspliced to spliced Egr1 mRNA were determined using RT-qPCR and are plotted relative to the ratios for 26r cells. The diagram depicts the two-exon Egr1 gene. The two arrows depict the primers used to detect spliced products, and the primer set (top) detected unspliced Egr1 (n = 3). (D) Cells were treated as described for panel B. Ratios of uncleaved to total Egr1 mRNA were measured using RT-qPCR and are plotted relative to the ratio detected in 26r cells. The left primer set was used to measure total Egr1 mRNA, and the right set detected uncleaved RNA (n = 3). Error bars indicate standard deviations.
Given that Rpb1(27–52) cells are inviable, it was somewhat unexpected that we did not observe significant changes in Egr1 transcription and/or RNA processing. To investigate if overall transcription and/or polyadenylation were affected in Rpb1(27–52) cells, we performed in vivo labeling with [3H]uridine and found that newly synthesized nonpolyadenylated and polyadenylated RNA levels in Rpb1(27–52) cells were similar to 26r and Rpb1(1–52) cells (Fig. 3A), indicating that these processes were not impaired by deletion of the N-terminal half of the CTD. In agreement with the full viability of S7A cells (Table 1 and Fig. 1B), overall transcription and polyadenylation in these cells were also comparable to Rpb1(1–52) and 26r cells (Fig. 3A).
FIG 3.

Analysis of transcription/polyadenylation in CTD mutant cells. (A) Cells were treated with Tet for 40 h, and nascent RNA was labeled with [3H]uridine for 30 min. Extracted RNA was separated into nonpolyadenylated (non-polyA) and polyadenylated (polyA) fractions. Non-poly(A) and poly(A) RNAs were quantified by scintillation counting, and the counts per minute relative to 26r cells were plotted [n = 3 for Rpb1(1–52) and S7A samples; n = 2 for the Rpb1(27–52) sample]. (B) Cells were treated with Tet for 24 h. ChIP was performed as described in the text. The distribution of FLAG-tagged Rpb1 on the Rplp1 gene was determined using qPCR. The diagrams depict the genes analyzed. Thick lines represent genes, and dashed lines display transcripts. The triangle denotes the 3′-cleavage site. For Rplp1, amplicon B is at the TSS and amplicon D covers the 3′-cleavage site (n = 3). (C) ChIP was performed for the β-actin (Actb) gene as described for the genes in panel B. Amplicon A is at the TSS, and amplicon C covers the 3′-cleavage site (n = 3). (D) ChIP analysis results of Rpb1 recruitment for histone H2A, U1, and U2 genes. For H2A, Rpb1 levels at the TSS and 3′-cleavage site were measured, whereas for the U1 and U2 genes, Rpb1 levels on the gene body were determined. ChIP was performed as described for panel B (n = 3).
The above data provided evidence that S2A and S5A cells are defective in Egr1 transcription and/or RNA processing. To investigate this further, the effects of S2A and S5A mutations on RNAP II levels for several highly expressed genes were examined via Rpb1 ChIP assays. Consistent with the Egr1 results, both S2A and S5A cells displayed impaired RNAP II occupancy on several genes, a ribosomal protein gene, Rplp1 (Fig. 3B), β-actin (Fig. 3C), and a histone H2A gene (Fig. 3D). Compared to 26r cells, decreased levels of Rpb1 were observed at the transcription start sites, coding regions, and 3′-cleavage sites of all three genes in both S2A and S5A cells, more so in the S2A cells. In line with the decreased occupancy of Rpb1-S5A, NRO assays revealed significantly reduced transcription of histone genes (H2A and H2B) and a marginal reduction of Rplp1 (Fig. 4A). NRO with S2A cells also revealed reduced transcription, but this was difficult to interpret conclusively, as controls (e.g., 18S and 5S RNAs) were also reduced (data not shown), likely reflecting an indirect effect, for example, an effect stemming from the inviability of the cells. The decrease in RNAP II occupancy or NRO signals was not due to defective RNAP II assembly, as Rpb2 equally coimmunoprecipitated with Rpb1 in S2A and S5A cells (Fig. 4B). Consistent with the ChIP results, subcellular fractionation showed that less S2A and S5A Rpb1 was bound to chromatin, whereas soluble nuclear Rpb1-S2A and Rpb1-S5A protein levels were similar to Rpb1-26r (Fig. 4C), indicating that nuclear import was not affected by CTD mutations but that chromatin association, i.e., transcription, was.
FIG 4.
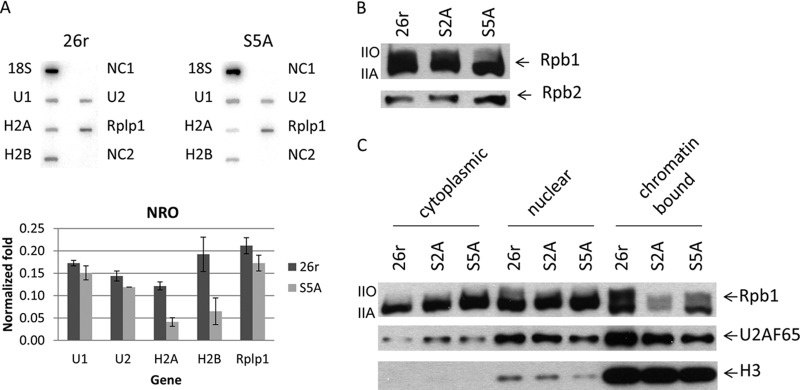
Effects of CTD mutations on transcription, RNAP II assembly, and subcellular distribution. (A) Cells were treated with Tet for 40 h, and nuclei were harvested. NRO assays were performed as described in the text. Slot blots contained the indicated DNA probes (top panel). NC1 and NC1 are two negative controls. Signals from each gene were normalized to 18S RNA and plotted (bottom panel) (n = 2). Error bars indicate standard deviations. (B) Rpb1 proteins were immunoprecipitated from cells cultured in the presence of Tet for 24 h. The content of Rpb2 in the RNAP II complex was determined by Western blotting. (C) Subcellular fractionation was performed in cells treated with Tet for 24 h. Rpb1 localization was determined by Western blotting. Nuclear protein U2AF65 and chromatin-bound histone H3 protein served as controls for fractionation.
We also examined RNAP II levels for the U1 and U2 snRNA genes. Strikingly, Rpb1 levels for these genes were reduced strongly in S2A cells but not significantly in S5A cells (Fig. 3D), and NRO assay results were consistent with this (Fig. 4A), suggesting that Ser 5 is unnecessary for U1 and U2 transcription. However, given that Ser 5 functions in facilitating capping enzyme recruitment (27), it is possible that Ser 5 is required for other aspects of U snRNA gene expression, and indeed that appears to be the case (see below). Ser 7 (and Ser 5) phosphorylation was unaffected in S2A cells, indicating that the strong decrease in snRNA transcription was not due to a defect in this modification (see Fig. S3 in the supplemental material). Note also that neither Ser 2 nor Ser 5 phosphorylation was affected in S7A cells (37) (see Fig. S3B), consistent with the lack of phenotype of the S7A mutation.
Distinct requirements for snRNA expression.
We next examined snRNA expression in the mutant cells in more detail. Ser 7 phosphorylation has been shown to play an important role in snRNA gene expression and processing (34, 49) and to be present on transcribing RNAP II on many genes (37). It was unexpected therefore that S7A cells were fully viable and displayed no defects in overall transcription (see above). To investigate the role of Ser 7 in snRNA expression in DT40-Rpb1 cells, we first performed NRO assays to determine the effect of the S7A mutation on snRNA transcription. Essentially identical levels of U1 and U2 transcription were detected in Rpb1(1–52), 26r, and S7A cells (Fig. 5A). We also measured steady-state levels of U1 and U2 snRNA in cells treated with Tet for 6 days. RT-qPCR with total cellular RNA demonstrated that the levels of U1 and U2 snRNA, in two independent S7A cell lines, were equivalent to those in Rpb1(1–52) and 26r cells (Fig. 5B). We also examined U2 snRNA levels by RT-qPCR in several of the mutant cell lines. Total U2 snRNA levels were sharply reduced in both S2A and, importantly, S5A cells and to a lesser degree in Rpb1(27–52 cells). U2 snRNA levels were not reduced, and indeed were very slightly increased, in S7A cells (Fig. 5C). We also measured U2 snRNA 3′-end formation, again by RT-qPCR, as depicted in Fig. 5D. S7A cells were essentially identical to 26r cells, and a modest increase in the ratio of uncleaved to total U2 snRNA (∼2-fold) was observed in S5A cells. However, a greater increase (∼6-fold) was detected in S2A cells, and a striking ∼14-fold increase was observed in Rpb1(27–52) cells (Fig. 5D). It is possible that this defect explains, at least in part, the inviability of Rpb1(27–52) cells. Very much the same conclusions were drawn from analysis of U1 snRNA expression (Fig. 5E and F). It is intriguing that Rpb1(27–52) cells, but not S7A cells, were defective in snRNA 3′-end formation. Below, we discuss these results and how they might be reconciled with previous findings.
FIG 5.

Effects of CTD mutations on snRNA expression. (A) Effects of the S7A mutation on snRNA transcription were analyzed in nuclear run-on experiments. Nuclei were harvested from cells treated with Tet for 40 h. Nascent RNA was labeled with [32P]UTP for 30 min and purified and analyzed in a slot blot assay with the DNA oligonucleotides indicated on the top panel. NC, negative control (antisense U1). Signals from each gene were normalized to 18S RNA and plotted (bottom panel) (n = 3). (B) Steady-state levels of U1 and U2 snRNAs in two independent cell lines expressing Rpb1-S7A compared with Rpb1(1–52) and 26r cells. Cells were treated with Tet for 6 days. Extracted RNA was analyzed using RT-qPCR. Levels of U1 and U2 RNA were normalized to 18S RNA, and values relative to 26r cells were plotted (n = 3). (C) Total U2 snRNA levels in Rpb1(27–52), S2A, and S5A cells. Cells were treated with Tet for 40 h. Levels of total U2 snRNA were measured using RT-qPCR and plotted relative to levels in 26r cells (n = 3). (D) U2 snRNA 3′ cleavage in Rpb1(27–52) and S2A cells. Cells were treated as described for panel C. Ratios of uncleaved versus total U2 snRNA were measured using RT-qPCR and plotted relative to levels in 26r cells (n = 3). (E) Total U1 snRNA levels were analyzed as described for panel C (n = 3). (F) U1 snRNA 3′-end cleavage efficiency was measured as described for panel D. Diagrams show snRNA transcripts. Triangles and arrows denote 3′-end cleavage sites and primers, respectively. The top primer set was used to detect uncleaved snRNA, and the bottom set was for total snRNA (n = 3). Error bars indicate standard deviations.
Thr 4 is phosphorylated by CDK9 and dephosphorylated by Fcp1.
The kinases and phosphatases responsible for the reversible phosphorylation of Ser 2, 5, and 7 have been well studied, but less is known about the enzymes responsible for Thr 4 modification. We showed previously that inhibition of CDK9 prevented Thr 4 phosphorylation in vivo (14), while a Polo-like kinase (Plk3) has also been implicated in this modification (15). Nothing is known about what enzyme dephosphorylates Thr 4. To investigate further the role of CDK9 in Thr 4 phosphorylation, we performed an in vitro kinase assay, using bacterially expressed and purified GST-CTD as a substrate and recombinant CDK9 isolated from baculovirus-infected insect cells (see Fig. S4 in the supplemental material for SDS gels of purified proteins). As shown in Fig. S5 in the supplemental material, CDK9 phosphorylated Thr 4, in a concentration-dependent manner, as judged by Western blotting with a Thr 4-P-specific antibody. Phosphorylation was inhibited by the CDK9 inhibitors DRB and flavopiridol (see Fig. S5), ruling out the possibility that phosphorylation was mediated by a contaminating activity in the CDK9 preparation. However, Ser 2, Ser 5, and Ser 7 were also phosphorylated by CDK9 (see Fig. S5). This promiscuous behavior of CDK9 in in in vitro assay prompted us to investigate the role of CDK9 in Thr 4 phosphorylation in vivo. We found that siRNA-mediated CDK9 knockdown in HEK293 cells, using three different siRNAs, indeed decreased Thr 4 phosphorylation relative to control siRNA-treated cells (Fig. 6A). Together, our results further support the involvement of CDK9 in Thr 4 phosphorylation.
FIG 6.

Phosphorylation and dephosphorylation of Thr 4. (A) CDK9 knockdown decreased phosphorylation on Thr 4. HEK293 cells were transfected with control siRNA and siRNAs targeting CDK9. Cell lysates were analyzed by Western blotting (left panel). CDK9 and P-Thr 4 levels were quantified after CDK9 knockdown (right panel). CDK9 protein levels were normalized to actin (right, bottom panel), and P-Thr 4 levels were normalized to Rpb1 (right, upper panel) (n = 2). (B) In vitro dephosphorylation of Thr 4. GST-CTD was phosphorylated by HeLa nuclear extract and incubated with Fcp1 or Fcp(M271E). Samples were analyzed by Western blotting with antibodies that detected P-Thr 4 (top panel) or P-Ser 2 or P-Ser 5 (bottom panels). (C) Knockdown of Fcp1 causes increased levels of P-Thr4. HEK293 cells were transfected with vectors expressing one of three shRNAs targeting Fcp1 or an shRNA targeting GFP (con). Cell lysates were analyzed by Western blotting with the indicated antibodies (left panel). Fcp1 and P-Thr4 levels were quantified after knockdown (right panel). Fcp1 protein levels were normalized to actin (right, bottom panel), and P-Thr4 was normalized to total Rpb1 (right, upper panel). Ratios relative to control were plotted (n = 3). Error bars indicate standard deviations. (D) HEK293 cells, transfected with plasmids carrying shRNAs, were subjected to subcellular fractionation. Cell lysates were analyzed using Western blotting. Four times more of the free chromatin unbound fraction was loaded compared to the chromatin-bound fraction. Histone H3 protein levels served as controls for the subcellular fractionation assay.
We next wished to identify the phosphatase responsible for Thr 4 dephosphorylation. As mentioned in the introduction, the two major CTD phosphatases are Fcp1 and Ssu72. Given that Fcp1 is known to dephosphorylate Ser 2 (8, 16), that globally, Thr 4 phosphorylation peaks toward the 3′ ends of genes (15), and that preliminary experiments provided no evidence that Ssu72 could dephosphorylate Thr 4 (results not shown), we reasoned that Fcp1 could be the Thr 4 phosphatase. To investigate this possibility, we first performed in vitro phosphatase assays, using recombinant Fcp1 (residues 168 to 606) (42) and a catalytically inactive derivative (M271E) (43) purified from bacterial cells and GST-CTD phosphorylated in HeLa nuclear extract and repurified (see Fig. S4 in the supplemental material for the SDS gels). The results, again from Western blotting, demonstrated that Thr 4, as well as Ser 2 and Ser 5, was dephosphorylated by wild-type Fcp1 but not by the M271E derivative (Fig. 6B). We next asked whether Fcp1 can dephosphorylate Thr 4 in vivo. To this end, we designed and utilized three distinct shRNAs to target Fcp1 mRNA in HEK293 cells and then measured their effect, relative to a control shRNA, on Thr 4 phosphorylation (Fig. 6C). Although with all three shRNAs, Fcp1 protein levels (and mRNA levels [see Fig. S6 in the supplemental material]) were only modestly reduced (protein levels were decreased ∼3-fold in all cases), Thr 4 phosphorylation levels were increased ∼2-fold by all three shRNAs. To extend these results, we examined P-Thr 4 levels in soluble nuclear and chromatin fractions following Fcp1 knockdown, again by Western blotting. As shown in Fig. 6D, ∼2-fold more P-Thr 4 was detected in both free and chromatin-bound Rpb1 after Fcp1 knockdown. Note that some P-Thr 4 was detected in the hypophosphorylated IIA form in the free Rpb1. This likely reflected some incompletely Thr 4-dephosphorylated Rpb1, but we cannot rule out cross-reactivity with the large amount of unphosphorylated IIA isoform present in this fraction. Most importantly, our data provide strong evidence that Fcp1 dephosphorylates Thr 4, extending its previously described role in Ser 2, and possibly Ser 5, dephosphorylation.
DISCUSSION
In this paper, we described experiments analyzing properties of the RNAP II CTD, primarily by utilizing genetically tractable chicken DT40 cells. For example, we found that the first half of the CTD, consisting of mostly all-consensus repeats, as well as Ser 2, Thr 4 (14), and Ser 5 residues, were essential for cell survival, while Ser 7 was not. Further analysis of these cells showed that, consistent with some but not all previous studies, Ser 2 and Ser 5 were required for optimal transcription and/or RNA splicing/polyadenylation. In contrast with expectations from previous studies, we did not observe any defects in snRNA gene expression in Rpb1-S7A cells. Instead, significant defects in snRNA expression, and specifically in 3′ processing, were detected in cells expressing Rpb1(27–52), which consists predominantly of nonconsensus repeats. Finally, we reported previously that CDK9 was required for Thr 4 phosphorylation in DT40 cells (14), and we showed here that CDK9 knockdown decreases Thr 4 phosphorylation in HEK293 cells, strengthening the case that Thr 4 is a direct CDK 9 target. We also provided evidence that Fcp1 dephosphorylates Thr 4 in vitro and in vivo. Below, we discuss the significance of these findings, especially regarding how they compare and contrast with those from previous related studies.
Our experiments have shown striking differences in the behavior of the N- and C-terminal halves of the CTD. Despite the fact the CTD contains consensus repeats that are highly conserved from yeast to humans, and it is nearly invariant in vertebrates, it is well known from studies in yeast (35), mouse (47), and chicken (14) cells that only ∼50% of the CTD is sufficient to confer cell viability. Furthermore, in human cells, when the α-amanitin system was used, both the N-terminal consensus-rich and the more divergent C-terminal half tended to behave similarly in functional assays (50), although in some cases modest differences were observed (51, 52). In agreement with this, we showed here that the N-terminal half of the CTD behaved essentially identically to the full-length CTD with respect to cell viability and in all functional assays, with the exception that 3′ processing of transcripts from the inducible Egr1 gene was slightly reduced. Cells expressing Rpb1(27–52) likewise behaved similarly to wild-type cells in assays measuring transcription, splicing, and polyadenylation. But surprisingly, these cells were completely inviable, revealing that these heptads cannot perform an essential function(s) of the CTD. As discussed below, this is likely to be the observed defect in snRNA 3′ processing.
Our studies have provided evidence that Ser 2 and Ser 5 are important for efficient mRNA splicing and 3′ processing. Previously, 3′-processing defects were observed in human cells upon CDK7 inhibition (6, 53) and in cells expressing an α-amanitin-resistant Rpb1-S5A (34). The 3′-processing defects were documented upon CDK9 deletion in yeast (54) or inhibition in Drosophila melanogaster (33), and both splicing and 3′-processing defects were observed in Xenpus laevis oocytes treated with CDK9 inhibitors (55) and in human cells expressing an α-amanitin-resistant Rpb1-S2A derivative (56). Significantly, we also found that Ser 5 phosphorylation, in addition to its involvement in 3′ processing, is also required for proper splicing. Although it is perhaps not surprising that Ser 5 phosphorylation functions to enhance splicing, for example, by facilitating capping to stimulate splicing of the first intron (57), to our knowledge ours is the first demonstration that this is the case.
Our results also extend studies examining the role of Ser 2 and Ser 5 phosphorylation on transcription. Transcription in cells expressing the Rpb1-S5A derivative, as measured by Pol II ChIP and NRO assays, was not as efficient as in the corresponding Rpb1-26r cells. This is consistent with previous reports that Cdk7 inhibition results in a decrease of Rpb1 levels on examined genes at the transcription start site (TSS), coding region, and 3′ end (6, 53), and our Rpb1-S5A ChIP assays extended these results by providing evidence that the effects of Cdk7 inhibition were indeed due to inhibition of Ser 5 phosphorylation. Our ChIP analyses also showed that Rpb1-S2A levels were decreased, in fact more severely than Rpb1-S5A, all along the length of the genes examined as well as in the chromatin fraction. This contrasts with previous observations that inhibition of CDK9 resulted in only slight changes of RNAP II density on examined genes (33), and an accumulation of RNAP II around the TSS (58, 59). This apparent discrepancy could be explained by the existence of additional Ser 2 kinases, e.g., CDK12 and -13 (10, 11), which may not be inhibited by the CDK9 inhibitors (60), and thus could perhaps partially compensate for CDK9. In any event, our results provide strong evidence for the importance of Ser 2 phosphorylation in transcription. It is notable that an S2A derivative analogous to the one we analyzed here was found to confer full viability, under normal growth conditions, in S. pombe (29), and a full-length S2A-containing CTD was viable albeit slow growing in S. cerevisiae (48). These findings are somewhat surprising, given the important functions attributed to Ser 2 phosphorylation, and additional work is required to understand the basis for this.
Perhaps the most unexpected of our findings was the dispensability of Ser 7. Previous studies provided evidence that Ser7 phosphorylation plays an important role in snRNA expression and 3′ processing (34, 49). However, DT40 cells expressing Rpb1-S7A were fully viable and displayed no defects in any aspect of gene expression, including expression of U1 and U2 snRNA genes. What might be the basis for this discrepancy? One possibility is a difference between chicken and human cells in the mechanism of U snRNA gene expression. However, the snRNA genes and factors involved in their expression are all highly conserved, and we are unaware of any evidence suggesting evolutionarily based differences in the basic steps of gene expression among vertebrate organisms. A second possibility stems from the usage of α-amanitin in the previous studies. For example, α-amanitin treatment accelerates the degradation of several proteins, including the transcription elongation factor DSIF (61), known to be important for U gene expression (62). Thus, we suggest that the S7A mutation coupled with reduced accumulation of a required factor such as DSIF in the presence of α-amanitin results in a “synthetic” phenotype reflected in defective snRNA expression. This model nonetheless envisions a role for Ser 7 phosphorylation; indeed, our results provided considerable albeit indirect support for this. Specifically, we suggest that the strong and specific defects in snRNA 3′ processing we observed in Rpb1(27–52) cells reflected changes in Ser 7. As mentioned above, the heptads in the C-terminal half of the CTD display considerable divergence from the consensus, and this divergence is by far the greatest at Ser 7. Only 6 of the 26 C-terminal heptads contain Ser at this position, and 9 contain a basic residue (8 Lys residues and 1 Arg; indeed, the Arg residue, via methylation, exerts a negative effect on snRNA gene expression [63]). Deviations from the consensus occur at other positions and could contribute to the snRNA 3′-processing defect. However, these changes are scattered and mostly conservative and therefore perhaps unlikely to play a role. As we detected no other defects in the Rpb1(27–52) cells, it may be that defective snRNA gene expression underlies the inviability of these cells.
We have also provided new insight into how phosphorylation of another CTD residue, Thr 4, is controlled. It was previously shown that Thr 4 phosphorylation was inhibited in vivo by the specific CDK9 inhibitors DRB and flavopiridol (14, 15). However, Hintermair et al. found that CDK9 was unable to phosphorylate Thr 4 in vitro, and instead those authors provided evidence that Plk3 could do so (15). In addition, those authors were unable to detect Thr 4 phosphorylation in vivo in the context of an S2A Rpb1 derivative, perhaps suggesting that the apparent CDK 9 requirement may instead indicate that Ser 2 phosphorylation is a prerequisite for Thr 4 phosphorylation. In contrast, though, our own prior experiments also examined Thr 4 phosphorylation in an S2A derivative (the one anaylzed here), and we found that Thr 4 phosphorylation was modestly affected (14). One explanation for these discrepancies is likely the antibodies employed. A negative result when analyzing mutant CTD derivatives may be difficult to interpret, depending on the sensitivity of the antibody to alterations in its epitope. Indeed, Hintermair et al. (15) noted that the antibody they used was unable to recognize P-Thr 4 when Ser 2 or Ser 5 was also phosphorylated. In any event, the in vitro and in vivo assays presented here strengthen the case for CDK9 being a Thr 4 kinase. In keeping with this, it would be somewhat surprising if Plk3 were the principal Thr 4 kinase, as it has well-established roles in other cellular processes, e.g., the cell cycle and stress response, and has been reported to be localized primarily in the nucleolus (64). None of this, however, rules out the possibility that Plk3 indeed phosphorylates Thr 4 under some conditions. This may be analogous to the case with Ser 5, which we showed previously can be modified during the M phase by the cell cycle kinase Cdc2/cyclin B (65). Finally, our data providing evidence that Fcp1 dephosphorylates Thr4 in vitro and in vivo both explain how Thr 4 is dephosphorylated and also extend the roles of this well-studied CTD phosphatase. While Fcp1 can dephosphorylate both Ser 2 and Ser 5, its primary function is thought to be the dephosphorylation of Ser 2 at the 3′ ends of genes (reviewed in reference 3). Thus, an attractive model would include CDK9 phosphorylation of both Ser 2 and Thr 4 during transcription elongation and with the same two residues dephosphorylated by Fcp1 at the ends of genes.
Supplementary Material
ACKNOWLEDGMENTS
We thank our lab members, especially Emanuel Rosonina, for discussion and critical reading of the manuscript. We thank Stephane Larochelle and Robert Fisher for CDK9 baculoviruses and Patrick Cramer for the Fcp1 expression vector.
This work was supported by NIH grant R01GM097174.
Footnotes
Published ahead of print 21 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00181-14.
REFERENCES
- 1.Buratowski S. 2009. Progression through the RNA polymerase II CTD cycle. Mol. Cell 36:541–546. 10.1016/j.molcel.2009.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore MJ, Proudfoot NJ. 2009. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136:688–700. 10.1016/j.cell.2009.02.001 [DOI] [PubMed] [Google Scholar]
- 3.Hsin JP, Manley JL. 2012. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 26:2119–2137. 10.1101/gad.200303.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heidemann M, Hintermair C, Voss K, Eick D. 2013. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta 1829:55–62. 10.1016/j.bbagrm.2012.08.013 [DOI] [PubMed] [Google Scholar]
- 5.Egloff S, Dienstbier M, Murphy S. 2012. Updating the RNA polymerase CTD code: adding gene-specific layers. Trends Genet. 28:333–341. 10.1016/j.tig.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 6.Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL. 2009. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol. Cell. Biol. 29:5455–5464. 10.1128/MCB.00637-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, Ansari AZ. 2009. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol. Cell 34:387–393. 10.1016/j.molcel.2009.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho EJ, Kobor MS, Kim M, Greenblatt J, Buratowski S. 2001. Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev. 15:3319–3329. 10.1101/gad.935901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marshall NF, Peng J, Xie Z, Price DH. 1996. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 271:27176–27183. 10.1074/jbc.271.43.27176 [DOI] [PubMed] [Google Scholar]
- 10.Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, Adelman K, Lis JT, Greenleaf AL. 2010. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 24:2303–2316. 10.1101/gad.1968210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J, Peterlin BM. 2011. The cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 25:2158–2172. 10.1101/gad.16962311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baskaran R, Dahmus ME, Wang JYJ. 1993. Tyrosine phosphorylation of mammalian RNA polymerase II carboxyl-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 90:11167–11171. 10.1073/pnas.90.23.11167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hintermair C, Kremmer E, Eick D, Cramer P. 2012. CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science 336:1723–1725. 10.1126/science.1219651 [DOI] [PubMed] [Google Scholar]
- 14.Hsin JP, Sheth A, Manley JL. 2011. RNAP II CTD phosphorylated on threonine-4 is required for histone mRNA 3′-end processing. Science 334:683–686. 10.1126/science.1206034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hintermair C, Heidemann M, Koch F, Descostes N, Gut M, Gut I, Fenouil R, Ferrier P, Flatley A, Kremmer E, Chapman RD, Andrau JC, Eick D. 2012. Threonine-4 of mammalian RNA polymerase II CTD is targeted by Polo-like kinase 3 and required for transcriptional elongation. EMBO J. 31:2784–2797. 10.1038/emboj.2012.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hausmann S, Shuman S. 2002. Characterization of the CTD phosphatase Fcp1 from fission yeast. Preferential dephosphorylation of serine 2 versus serine 5. J. Biol. Chem. 277:21213–21220. 10.1074/jbc.M202056200 [DOI] [PubMed] [Google Scholar]
- 17.Bataille AR, Jeronimo C, Jacques PE, Laramee L, Fortin ME, Forest A, Bergeron M, Hanes SD, Robert F. 2012. A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol. Cell 45:158–170. 10.1016/j.molcel.2011.11.024 [DOI] [PubMed] [Google Scholar]
- 18.Zhang DW, Mosley AL, Ramisetty SR, Rodriguez-Molina JB, Washburn MP, Ansari AZ. 2012. Ssu72 phosphatase-dependent erasure of phospho-Ser7 marks on the RNA polymerase II C-terminal domain is essential for viability and transcription termination. J. Biol. Chem. 287:8541–8551. 10.1074/jbc.M111.335687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiang K, Nagaike T, Xiang S, Kilic T, Beh MM, Manley JL, Tong L. 2010. Crystal structure of the human symplekin-Ssu72-CTD phosphopeptide complex. Nature 467:729–733. 10.1038/nature09391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiang K, Manley JL, Tong L. 2012. An unexpected binding mode for a Pol II CTD peptide phosphorylated at Ser7 in the active site of the CTD phosphatase Ssu72. Genes Dev. 26:2265–2270. 10.1101/gad.198853.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosley AL, Pattenden SG, Carey M, Venkatesh S, Gilmore JM, Florens L, Workman JL, Washburn MP. 2009. Rtr1 is a CTD phosphatase that regulates RNA polymerase II during the transition from serine 5 to serine 2 phosphorylation. Mol. Cell 34:168–178. 10.1016/j.molcel.2009.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egloff S, Zaborowska J, Laitem C, Kiss T, Murphy S. 2012. Ser7 phosphorylation of the CTD recruits the RPAP2 Ser5 phosphatase to snRNA genes. Mol. Cell 45:111–122. 10.1016/j.molcel.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiang K, Manley JL, Tong L. 2012. The yeast regulator of transcription protein Rtr1 lacks an active site and phosphatase activity. Nat. Commun. 3:946. 10.1038/ncomms1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tietjen JR, Zhang DW, Rodriguez-Molina JB, White BE, Akhtar MS, Heidemann M, Li X, Chapman RD, Shokat K, Keles S, Eick D, Ansari AZ. 2010. Chemical-genomic dissection of the CTD code. Nat. Struct. Mol. Biol. 17:1154–1161. 10.1038/nsmb.1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim H, Erickson B, Luo W, Seward D, Graber JH, Pollock DD, Megee PC, Bentley DL. 2010. Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat. Struct. Mol. Biol. 17:1279–1286. 10.1038/nsmb.1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayer A, Lidschreiber M, Siebert M, Leike K, Soding J, Cramer P. 2010. Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 17:1272–1278. 10.1038/nsmb.1903 [DOI] [PubMed] [Google Scholar]
- 27.Ho CK, Shuman S. 1999. Distinct roles for CTD Ser-2 and Ser-5 phosphorylation in the recruitment and allosteric activation of mammalian mRNA capping enzyme. Mol. Cell 3:405–411. 10.1016/S1097-2765(00)80468-2 [DOI] [PubMed] [Google Scholar]
- 28.Fabrega C, Shen V, Shuman S, Lima CD. 2003. Structure of an mRNA capping enzyme bound to the phosphorylated carboxy-terminal domain of RNA polymerase II. Mol. Cell 11:1549–1561. 10.1016/S1097-2765(03)00187-4 [DOI] [PubMed] [Google Scholar]
- 29.Schwer B, Shuman S. 2011. Deciphering the RNA polymerase II CTD code in fission yeast. Mol. Cell 43:311–318. 10.1016/j.molcel.2011.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris DP, Greenleaf AL. 2000. The splicing factor, Prp40, binds the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 275:39935–39943. 10.1074/jbc.M004118200 [DOI] [PubMed] [Google Scholar]
- 31.David CJ, Boyne AR, Millhouse SR, Manley JL. 2011. The RNA polymerase II C-terminal domain promotes splicing activation through recruitment of a U2AF65-Prp19 complex. Genes Dev. 25:972–983. 10.1101/gad.2038011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fong N, Bentley DL. 2001. Capping, splicing, and 3′-processing are independently stimulated by RNA polymerase II: different functions for different segments of the CTD. Genes Dev. 15:1783–1795. 10.1101/gad.889101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni Z, Schwartz BE, Werner J, Suarez JR, Lis JT. 2004. Coordination of transcription, RNA processing, and surveillance by P-TEFb kinase on heat shock genes. Mol. Cell 13:55–65. 10.1016/S1097-2765(03)00526-4 [DOI] [PubMed] [Google Scholar]
- 34.Egloff S, O'Reilly D, Chapman RD, Taylor A, Tanzhaus K, Pitts L, Eick D, Murphy S. 2007. Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science 318:1777–1779. 10.1126/science.1145989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.West ML, Gorden JL. 1995. Construction and analysis of yeast RNA polymerase II CTD deletion and substitution mutations. Genetics 140:1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stiller JW, McConaughy BL, Hall BD. 2000. Evolutionary complementation for polymerase II CTD function. Yeast 16:57–64. [DOI] [PubMed] [Google Scholar]
- 37.Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, Eick D. 2007. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 318:1780–1782. 10.1126/science.1145977 [DOI] [PubMed] [Google Scholar]
- 38.Pirngruber J, Shchebet A, Schreiber L, Shema E, Minsky N, Chapman RD, Eick D, Aylon Y, Oren M, Johnsen SA. 2009. CDK9 directs H2B monoubiquitination and controls replication-dependent histone mRNA 3′-end processing. EMBO Rep. 10:894–900. 10.1038/embor.2009.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilman M. 2001. Ribonuclease protection assay. Curr. Protoc. Mol. Biol. Chapter 4:Unit4.7. 10.1002/0471142727.mb0407s24 [DOI] [PubMed] [Google Scholar]
- 40.Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, Blethrow JD, Shokat KM, Fisher RP. 2006. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat. Struct. Mol. Biol. 13:55–62. 10.1038/nsmb1028 [DOI] [PubMed] [Google Scholar]
- 41.Hirose Y, Manley JL. 1998. RNA polymerase II is an essential mRNA polyadenylation factor. Nature 395:93–96. 10.1038/25786 [DOI] [PubMed] [Google Scholar]
- 42.Kamenski T, Heilmeier S, Meinhart A, Cramer P. 2004. Structure and mechanism of RNA polymerase II CTD phosphatases. Mol. Cell 15:399–407. 10.1016/j.molcel.2004.06.035 [DOI] [PubMed] [Google Scholar]
- 43.Ghosh A, Shuman S, Lima CD. 2008. The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol. Cell 32:478–490. 10.1016/j.molcel.2008.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eick D, Geyer M. 2013. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem. Rev. 113:8456–8490. 10.1021/cr400071f [DOI] [PubMed] [Google Scholar]
- 45.Chapman RD, Conrad M, Eick D. 2005. Role of the mammalian RNA polymerase II C-terminal domain (CTD) nonconsensus repeats in CTD stability and cell proliferation. Mol. Cell. Biol. 25:7665–7674. 10.1128/MCB.25.17.7665-7674.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chapman RD, Palancade B, Lang A, Bendaude O, Eick D. 2004. The last CTD repeat of the mammalian RNA polymerase II large subunit is important for its stability. Nucleic Acids Res. 32:35–44. 10.1093/nar/gkh172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartolomei MS, Halden NF, Cullen CR, Corden JL. 1988. Genetic analysis of the repetitive carboxyl-terminal domain of the largest subunit of mouse RNA polymerase II. Mol. Cell. Biol. 8:330–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cassart C, Drogat J, Migeot V, Hermand D. 2012. Distinct requirement of RNA polymerase II CTD phosphorylations in budding and fission yeast. Transcription 3:231–234. 10.4161/trns.21066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Egloff S, Szczepaniak SA, Dienstbier M, Taylor A, Knight S, Murphy S. 2010. The integrator complex recognizes a new double mark on the RNA polymerase II carboxyl-terminal domain. J. Biol. Chem. 285:20564–20569. 10.1074/jbc.M110.132530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosonina E, Blencowe BJ. 2004. Analysis of the requirement for RNA polymerase II CTD heptapeptide repeats in pre-mRNA splicing and 3′-end cleavage. RNA 10:581–589. 10.1261/rna.5207204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Medlin JE, Uguen P, Taylor A, Bentley DL, Murphy S. 2003. The C-terminal domain of pol II and a DRB-sensitive kinase are required for 3′-processing of U2 snRNA. EMBO J. 22:925–934. 10.1093/emboj/cdg077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fong N, Bird G, Vigneron M, Bentley DL. 2003. A 10 residue motif at the C-terminus of the RNA pol II CTD is required for transcription, splicing and 3′-end processing. EMBO J. 22:4274–4282. 10.1093/emboj/cdg396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP. 2012. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 19:1108–1115. 10.1038/nsmb.2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahn SH, Kim M, Buratowski S. 2004. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′-end processing. Mol. Cell 13:67–76. 10.1016/S1097-2765(03)00492-1 [DOI] [PubMed] [Google Scholar]
- 55.Bird G, Zorio DA, Bentley DL. 2004. RNA polymerase II carboxy-terminal domain phosphorylation is required for cotranscriptional pre-mRNA splicing and 3′-end formation. Mol. Cell. Biol. 24:8963–8969. 10.1128/MCB.24.20.8963-8969.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gu B, Eick D, Bensaude O. 2013. CTD serine-2 plays a critical role in splicing and termination factor recruitment to RNA polymerase II in vivo. Nucleic Acids Res. 41:1591–1603. 10.1093/nar/gks1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gornemann J, Kotovic KM, Hujer K, Neugebauer KM. 2005. Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol. Cell 19:53–63. 10.1016/j.molcel.2005.05.007 [DOI] [PubMed] [Google Scholar]
- 58.Glover-Cutter K, Kim S, Espinosa J, Bentley DL. 2008. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat. Struct. Mol. Biol. 15:71–78. 10.1038/nsmb1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng B, Li T, Rahl PB, Adamson TE, Loudas NB, Guo J, Varzavand K, Cooper JJ, Hu X, Gnatt A, Young RA, Price DH. 2012. Functional association of Gdown1 with RNA polymerase II poised on human genes. Mol. Cell 45:38–50. 10.1016/j.molcel.2011.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bartkowiak B, Greenleaf AL. 2011. Phosphorylation of RNAPII: To P-TEFb or not to P-TEFb? Transcription 2:115–119. 10.4161/trns.2.3.15004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsao DC, Park NJ, Nag A, Martinson HG. 2012. Prolonged alpha-amanitin treatment of cells for studying mutated polymerases causes degradation of DSIF160 and other proteins. RNA 18:222–229. 10.1261/rna.030452.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mandal SS, Chu C, Wada T, Handa H, Shatkin AJ, Reinberg D. 2004. Functional interactions of RNA-capping enzyme with factors that positively and negatively regulate promoter escape by RNA polymerase II. Proc. Natl. Acad. Sci. U. S. A. 101:7572–7577. 10.1073/pnas.0401493101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sims RJ, III, Rojas LA, Beck D, Bonasio R, Schuller R, Drury WJ, III, Eick D, Reinberg D. 2011. The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science 332:99–103. 10.1126/science.1202663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zimmerman WC, Erikson RL. 2007. Finding Plk3. Cell Cycle 6:1314–1318. 10.4161/cc.6.11.4275 [DOI] [PubMed] [Google Scholar]
- 65.Xu YX, Hirose Y, Zhou XZ, Lu KP, Manley JL. 2003. Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 17:2765–2776. 10.1101/gad.1135503 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.