

Abstract
MacroH2A1 is a histone variant harboring an ∼25-kDa carboxyl-terminal macrodomain. Due to its enrichment on the inactive X chromosome, macroH2A1 was thought to play a role in transcriptional repression. However, recent studies have shown that macroH2A1 occupies autosomal chromatin and regulates genes in a context-specific manner. The macrodomain may play a role in the modulation of gene expression outcomes via physical interactions with effector proteins, which may depend on the ability of the macrodomain to bind NAD+ metabolite ligands. Here, we identify proline, glutamic acid, and leucine-rich protein 1 (PELP1), a chromatin-associated factor and transcriptional coregulator, as a ligand-independent macrodomain-interacting factor. We used chromatin immunoprecipitation coupled with tiling microarrays (ChIP-chip) to determine the genomic localization of PELP1 in MCF-7 human breast cancer cells. We find that PELP1 genomic localization is highly correlated with that of macroH2A1. Additionally, PELP1 positively correlates with heterochromatic chromatin marks and negatively correlates with active transcription marks, much like macroH2A1. MacroH2A1 specifically recruits PELP1 to the promoters of macroH2A1 target genes, but macroH2A1 occupancy occurs independent of PELP1. This recruitment allows macroH2A1 and PELP1 to cooperatively regulate gene expression outcomes.
INTRODUCTION
The canonical nucleosome architecture, two copies each of histones H2A, H2B, H3, and H4, organizes the genomes of eukaryotes and is locally modified in a multitude of ways for various regulatory purposes. These modifications include the posttranslational modification (PTM) of histones, changes in nucleosome positioning, and the replacement of canonical histones with their histone variant counterparts (1, 2). MacroH2A1 is one such histone variant that can substitute for at least one copy of H2A in a subset of nucleosomes in vertebrates. At three times the size of histone H2A, macroH2A1 is made up of amino-terminal histone-like regions with 64% identity to H2A and a carboxyl-terminal ∼25-kDa macrodomain. Based largely on the observation that macroH2A1 is enriched on the transcriptionally silent inactive X (Xi) chromosome (3, 4), macroH2A1 was originally hypothesized to play a role in transcriptional repression (reviewed in reference 5). However, later studies demonstrated that macroH2A1 is not required for the initiation or maintenance of X-inactivation (6–9), casting doubt on a general role for macroH2A in transcriptional repression.
Data from several groups have demonstrated that macroH2A1 plays important roles in both tumor suppression and differentiation. Alterations in macroH2A1 splicing and expression occur in a variety of cancers (10–14). In addition, restoration of macroH2A1 expression in cancer cells suppresses both proliferation and anchorage-independent growth (11, 12). Perhaps relevant to macroH2A's role in tumor suppression, several reports have shown that macroH2A both promotes and maintains cellular differentiation (15–19). MacroH2A plays a key role blocking the reprogramming of differentiated cells back to a pluripotent state (15, 18, 20). Furthermore, embryonic stem cells lacking macroH2A1 are defective in their ability to differentiate (16, 17).
Recent work from our lab and others has determined that macroH2A1 is not only a component of chromatin on the Xi, it occupies approximately a quarter of the autosomal genome as well (21, 22). Additionally, macroH2A1 is generally associated with transcriptionally repressive heterochromatin across autosomes, where it colocalizes with other heterochromatin marks such as histone H3 lysine 27 trimethylation (H3K27me3) (21, 22). However, while macroH2A1 is a component of autosomal heterochromatin, it is not generally required for the repression of genes found in macroH2A1-containing domains (22), similar to the lack of a general requirement for macroH2A1 in the transcriptional repression of genes on the Xi chromosome. A growing body of evidence suggests that macroH2A1 can play either a positive or negative role in regulating the transcription of genes found in its domains in a context-specific manner (6, 21–24; reviewed in reference 25).
Macrodomains are ancient domains that have been identified in proteins from bacteria to humans (26). In macroH2A1-containing nucleosomes, this extra 25-kDa globular domain can recruit additional effector proteins to macroH2A1-containing chromatin in order to facilitate the regulation of gene expression (27–32). While certain macrodomains have been found to harbor enzymatic activity (reviewed in reference 33), work from Ladurner's group and others suggest that most macrodomains are ligand binding domains for NAD+ metabolites such as poly(ADP-ribose) (PAR), a posttranslational modification catalyzed by a family of PAR polymerases (PARPs), monomeric ADP-ribose, and O-acetyl-ADP-ribose, produced as a by-product of sirtuin family deacetylase reactions (32, 34–38). Ligand binding by macrodomains appears to have two functions that have been identified thus far. First, ligand binding can alter the affinity of some proteins to interact with macrodomains. For example, the macrodomain of macroH2A1.1 interacts specifically with automodified PARP-1 in a manner that requires the ability of the macrodomain to bind PAR (32). Second, macrodomains can mediate the recruitment of factors that contain these domains to genomic sites of PAR accumulation (32, 34, 35).
Here, we report the identification of the transcriptional coactivator proline-, glutamic acid-, and leucine-rich protein 1 (PELP1) as a novel factor that interacts with the macrodomain of macroH2A. PELP1, otherwise known as modulator of the nongenomic activities of estrogen receptor (MNAR), has been shown to promote estrogen receptor-dependent transcription as both a classical coactivator and through a controversial plasma membrane signaling mechanism (39, 40). PELP1 is also a chromatin-associated factor that has been shown to interact with a variety of transcription factors (e.g., androgen receptor [AR] and glucocorticoid receptor [GR]), covalently modified histone H3, and the linker histone H1 (41–45). Recently, PELP1 has been shown to stimulate transcription by displacing the linker histone H1 and/or by recruiting the lysine demethylase KDM1 to demethylate H3K9me2 (45). Taken together, these studies suggest that PELP1 is a multifunctional protein that can regulate chromatin-dependent transcriptional processes through a variety of mechanisms.
In the studies described herein, we document the physical and functional interactions of macroH2A1 with PELP1 using biochemical, genomic, and gene-specific analyses. Our results indicate that macroH2A1 and PELP1 function to cooperatively regulate the expression of a common set of target genes.
MATERIALS AND METHODS
Antibodies.
The rabbit polyclonal PELP1 and macroH2A1 antibodies used for Western blotting, chromatin immunoprecipitation coupled with tiling microarrays (ChIP-chip), and ChIP with quantitative PCR (ChIP-qPCR) were purchased from Bethyl Laboratories, Inc. (A300-180A), and Millipore (07-219), respectively. The antibodies were screened for (i) specificity by Western blotting MCF-7 cell extracts, (ii) the ability to immunoprecipitate their cognate antigens from formaldehyde cross-linked chromatin by a ChIP-Western blotting protocol, and (iii) a reduction in Western blot signal upon knockdown of PELP1 or macroH2A1. The rabbit polyclonal H3 antibody used for Western blotting, ChIP-chip, and ChIP-qPCR was purchased from Abcam (ab1791-100). The custom rabbit polyclonal antibody against PARP-1 used for Western blotting was generated by using a purified fragment of human PARP-1 (amino terminus, PARP-N; Pocono Rabbit Farm and Laboratory, Inc.) and previously characterized (46, 47). The rabbit polyclonal nucleolin (NCL; C-23) and SET antibodies used for Western blotting were purchased from Santa Cruz Biotechnology, Inc. (sc-13057 and sc-25564, respectively), and the mouse monoclonal β-actin antibody was purchased from Sigma-Aldrich (A5316).
Oligonucleotides.
Detailed information about the oligonucleotide sequences used for the short hairpin RNA (shRNA) constructs, reverse transcription-qPCR (RT-qPCR), and ChIP-qPCR can be found in the supplemental material.
GST-macro1.1 pulldown and protein identification.
The glutathione S-transferase (GST)–macro1.1 vector was made by cloning the nonhistone region of macroH2A1.1 (i.e., amino acids 123 to 368) into pGEX-2TK. The vector was induced in BL21(DE3), and the protein was purified using glutathione-agarose. GST alone or GST-macro1.1 (2 μg) was prebound to 15 μl of glutathione-agarose in a 150-μl final volume of buffer containing 20 mM Tris, pH 7.5, 0.1 M NaCl, 0.1 mM EDTA, 5% glycerol, and 0.1% Tween 20. After 30 min of prebinding, the beads were washed three times with the same buffer. The GST- or GST-macro1.1-bound beads were then incubated in a final volume of 150 μl with 250 μg of HeLa nuclear extract. Where indicated in Fig. 1C, 20 μM ADP-ribose (ADPR) or NAD+ was added to the reaction mixture. The binding reaction mixtures were incubated with agitation for 2 h at 4°C. The beads were then washed three times using the buffer conditions noted above. For Western blotting, the proteins were eluted directly with SDS and subjected to SDS-PAGE.
FIG 1.
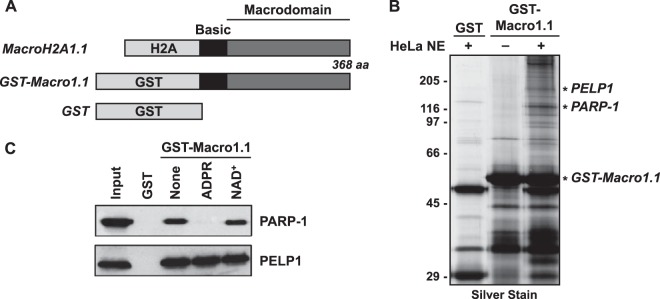
PELP1 interacts with the macrodomain of the histone variant macroH2A1.1 in an ADPR-independent manner. (A) Schematic diagram of the histone variant macroH2A1.1 and GST-tagged macrodomain. The histone H2A-like region (H2A), macrodomain, and the basic linker region are shown. The GST-macro1.1 fusion consists of GST fused to the nonhistone regions only. GST alone, used as a control, is shown for comparison. (B) Silver-stained image of GST-macro1.1-interacting proteins from a GST pulldown assay. HeLa nuclear extract (NE) was added as indicated. GST-macro1.1-specific protein bands were excised from the gel and identified by mass spectrometry. Macrodomain-interacting proteins, PELP1 and PARP-1, are shown. The full data set of identified proteins can be found in Fig. S3 and Table S1 in the supplemental material. (C) Western blots for PARP-1 and PELP1 from GST pulldown assay as described for panel B. ADPR or NAD+ at 20 μM was added as indicated.
For mass spectrometric identification the bound proteins were eluted with reduced glutathione (Sigma) and separated by SDS-PAGE. Proteins excised from gels were digested with trypsin, and the resulting peptide pools were analyzed by matrix-assisted laser desorption ionization–reflectron time of flight (MALDI-reTOF) mass spectrometry (MS) using a BRUKER UltraFlex two-stage TOF (TOF/TOF) instrument (Bruker Daltonics, Bremen, Germany) (48, 49). Selected experimental masses (m/z) were taken to search the human segment of a nonredundant (NR) protein database (accessed 8 April 2010; ∼233,131 entries; National Center for Biotechnology Information, Bethesda, MD), utilizing the Mascot Peptide Mass Fingerprint (PMF) program, version 2.3.01, for Windows (Matrix Science, Boston, MA), with a mass accuracy restriction better than 40 ppm and a maximum of one missed cleavage site allowed per peptide. To confirm PMF results with scores of ≤40, mass spectrometric sequencing of selected peptides was done by MALDI-TOF/TOF (tandem MS [MS/MS]) analysis on the same prepared samples, using the UltraFlex instrument in LIFT mode. Fragment ion spectra were taken to search the NR database using the Mascot MS/MS Ion Search program (Matrix Science, Boston, MA). Key identified proteins are listed in Table S1 in the supplemental material.
Luci and PELP1i inducible knockdown constructs.
For the Tet-on-inducible knockdown constructs (inducible luciferase [Luci] and PELP1i), the pSUPER.retro vector (puromycin resistant) was first modified by replacing the H1 promoter with one harboring a tetracycline (Tet) operator sequence from a pTER+ vector (50) using BglII and EcoRI restriction sites. The resulting vector is termed pSUPER.retro.TO (Tet on). Double-stranded oligonucleotides containing shRNA sequences targeting either luciferase (Luc control) or PELP1 were cloned into the pSUPER.retro.TO (puromycin resistant) vector using BglII and XhoI restriction sites, as described by the manufacturer. The shRNA sequences were based on sequences reported in the literature (51, 52) or designed using the Invitrogen BLOCK-iT RNAi Designer (Grand Island, NY). All constructs were confirmed by sequencing.
Generation, culture, and treatments of MCF-7-derived cell lines.
Parental MCF-7 human breast cancer cells, kindly provided by Benita Katzenellenbogen, were maintained in Eagle's minimal essential medium (MEM) containing Hanks' salts, l-glutamine, and nonessential amino acids (Sigma) supplemented with 5% bovine calf serum (CS; Sigma), 20 mM HEPES (pH 7.6), 100 units/ml penicillin, 100 μg/ml streptomycin, 25 μg/ml gentamicin, and 0.22% sodium bicarbonate. The Luc and macroH2A1 knockdown cell lines used in these studies were generated and cultured as described previously (22).
For the Tet-on-inducible knockdown system, parental MCF-7 cells were stably transfected with a Tet repressor (TetR) cDNA (pcDNA6/TR; kindly provided by Hans Clevers and H. T. Marc Timmers). Stable transfectants were clonally selected using hygromycin (200 μg/ml), expanded, and tested for TetR expression and activity by Western blotting and transient-transfection/luciferase reporter gene assays, respectively (data not shown). The resulting cell line, termed MCF-7 TetR, was maintained under conditions similar to those described above, with the exception of the serum (5% Tet-approved fetal bovine serum [Tet FBS]; Clontech) to reduce background shRNA expression in the absence of doxycycline (DOX). The MCF-7 TetR cell line was subsequently used to make the Luci and PELP1i knockdown cell lines by retroviral infection with the appropriate shRNA vectors.
Retroviruses were generated by transfection of the pSUPER.retro or pSUPER.retro.TO vector described above with an expression vector for the vesicular stomatitis virus G protein (VSV-G) envelope protein into Phoenix Ampho cells using GeneJuice transfection reagent (Novagen) according to the manufacturer's protocol. The resulting viruses were collected, filtered through a 0.45-μm-pore-size syringe filter to remove any remaining cells, and used to infect the MCF-7 TetR cells. Stably transduced cells were isolated under appropriate selection with puromycin (0.5 μg/ml; Sigma) or G418 sulfate (800 μg/ml; Gibco/BRL), expanded, and frozen in aliquots for future use. The cells were grown under subconfluent conditions for routine maintenance and most experimental procedures.
Luci and PELP1i knockdown cell lines were maintained as noted above and treated with doxycycline (2 μg/ml; Sigma-Aldrich) to induce the shRNA expression for a minimum of 9 days. For most experiments, one of the two shRNAs (number 2) for PELP1 was used as it gave a higher degree of PELP1 depletion. Both shRNAs were tested in gene-specific expression studies, however, to determine off-target effects (see Fig. S1 in the supplemental material). For ChIP-qPCR and RT-qPCR experiments, only the Luci and PELP1i conditions with doxycycline (+DOX) were compared due to the apparent leakiness of the system without doxycycline (−DOX condition).
Where indicated in Fig. 8, MCF-7 parental, Luc and macroH2A1 knockdown, or Luci and PELP1i knockdown cells were serum starved by washing the cells in phosphate-buffered saline (PBS) and replacing the medium with MEM without serum for 24 h or treating the cells with 12-O-tetradecanoylphorbol-13-acetate (TPA; 100 ng/ml) (Enzo Life Sciences) for 3 h (expression assay) or 1.5 h (ChIP assay).
FIG 8.

PELP1 acts to repress the transcriptional induction of the TPA-responsive gene CCL2. (A) RT-qPCR of CCL2 and AREG from Luc and macroH2A1 knockdown cells and Luci and PELP1i knockdown cells (+DOX) treated with vehicle (−) or with 100 nM TPA (+) for 3 h. Values represent the means plus standard errors of the means (error bars) from three or more independent determinations. Bars marked with asterisks are statistically different from the value for the corresponding TPA-treated luciferase knockdown control (Luc, in the case of macroH2A1; Luci in the case of PELP1i), as determined by Student's t test with a P value threshold of <0.05. The Luc and macroH2A1 knockdown data for AREG are from Gamble et al. (22). (B) MacroH2A1 and PELP1 ChIP assays were performed from parental MCF-7 cells treated with vehicle (−) or 100 nM TPA (+) for 1.5 h. Primers were designed to target regions upstream and downstream of the TSS. Values represent the means plus standard errors of the means (error bars) from three or more independent determinations. Bars marked with asterisks are statistically different from the value for the vehicle treated control, as determined by Student's t test with at P value threshold of <0.05. The macroH2A1 ChIP data for AREG are from Gamble et al. (22). Luc, luciferase; mH2A1, macroH2A1; i, inducible; U, upstream of the TSS; D, downstream of the TSS.
Chromatin immunoprecipitation assays.
ChIP assays were performed essentially as described previously (22, 53). The immunoprecipitations were performed from cross-linked parental or knockdown MCF-7 cells with antibodies against macroH2A1, PELP1, or histone H3, using a no-antibody control. The resulting input and ChIP DNA material was used for ChIP-chip or gene-specific ChIP-qPCR analyses. In all cases the DNA recovered from the PELP1 and macroH2A1 ChIPs was well above the signal seen in our no-antibody controls (see Fig. S2 in the supplemental material).
ChIP-chip.
The ChIP-chip sample processing and analyses were done essentially as described previously (22, 53). Briefly, PELP1-specific immunoprecipitated genomic DNA and reference DNA were blunted, amplified by ligation-mediated PCR (LM-PCR), labeled with Cy5 and Cy3, respectively, and used to probe a custom human oligonucleotide genomic array (Nimblegen) (22, 53). The PELP1 ChIP-chip was run in duplicate to ensure reproducibility.
Genomic data analyses.
The genomic data analyses for the PELP1 ChIP-chip were performed as described previously (22), using the statistical programming language R (R Development Core Team). All data processing scripts are available on request. Briefly, PELP1-bound regions (see Table S2 in the supplemental material) were defined as at least three consecutive windows with (i) positive means, (ii) at least six probes, and (iii) a P value of <0.016. PELP1-unbound regions were defined as at least three consecutive windows with (i) negative means, (ii) at least six probes, and (iii) a P value of <0.016.
For PELP1-macroH2A1 genomic comparisons, the macroH2A1 data set was accessed from the National Institutes of Health Gene Expression Omnibus (GEO) Database using accession number GSE9607. For expression-based classification of genes, MCF-7 expression microarray data were accessed from the National Institutes of Health GEO Database using accession number GSE9253, and expressed genes were divided into pentiles based on the degree of expression. The data were then compared to the genes represented on the ChIP-chip array, as described previously (22).
For gene ontology (GO) analyses, specific gene lists were entered into the Generic Gene Ontology Term Finder (http://go.princeton.edu/cgi-bin/GOTermFinder/GOTermFinder). A multiple-testing-corrected P value of 0.002 was used as the cutoff for significant enrichment. Specifically, we considered genes (i) bound by PELP1 (independent of macroH2A1 status), (ii) bound by macroH2A1 (independent of PELP1 status), (iii) bound by both PELP1 and macroH2A1, (iv) bound by PELP1 and not by macroH2A1, and (v) bound by macroH2A1 and not by PELP1. The gene lists can be found in Table S3 in the supplemental material.
For the multiple ChIP-chip correlation analysis, 362 ChIP-chip data sets were accessed from the National Institutes of Health GEO Database using criteria described previously (22). The data were processed with 1-kb windows identical to those of the macroH2A1 and PELP1 data sets, and Spearman correlations were determined between PELP1 and each factor. A correlation coefficient of ±0.20 and a P value of <10−100 were used as cutoffs for significant correlation. The full list of GEO accession numbers and analysis can be found in Table S4 in the supplemental material.
mRNA expression analyses by RT-qPCR.
For gene-specific mRNA expression analyses, total RNA was isolated using TRIzol reagent (Invitrogen), reverse transcribed, and subjected to real-time quantitative PCR using a set of gene-specific primers (primer sequences can be found in the supplemental material). All target gene transcripts were normalized to the β-actin transcript, which was unaffected by macroH2A1 or PELP1 knockdown (data not shown). All experiments were conducted a minimum of three times with independent RNA isolations to ensure reproducibility.
Quantitative PCR analyses (RT-qPCR and ChIP-qPCR).
Gene-specific mRNA expression and ChIP analyses were analyzed by quantitative PCR in a similar manner. Briefly, reaction mixtures containing DNA from either source, 1× SYBR green PCR master mix, and forward and reverse primers (250 nM for ChIP and 500 nM for expression assays) were used in 40 to 45 cycles of amplification (95°C for 15 s and 60°C for 1 min) using a Roche LightCycler 480 system (384-well) following an initial 10-min incubation at 95°C. Melting curve analysis was performed to ensure that only the targeted amplicon was amplified.
Statistical analyses.
For the ChIP-qPCR and RT-qPCR assays shown in Fig. 5 to 8 and in Fig. S1, S3, S4, S5, and S10 in the supplemental material, a paired Student t test with a P value threshold of <0.05 was used to determine the significance of differences between the control and experimental samples. The enrichment test shown in Fig. 3 was determined using a Fisher exact test.
FIG 5.
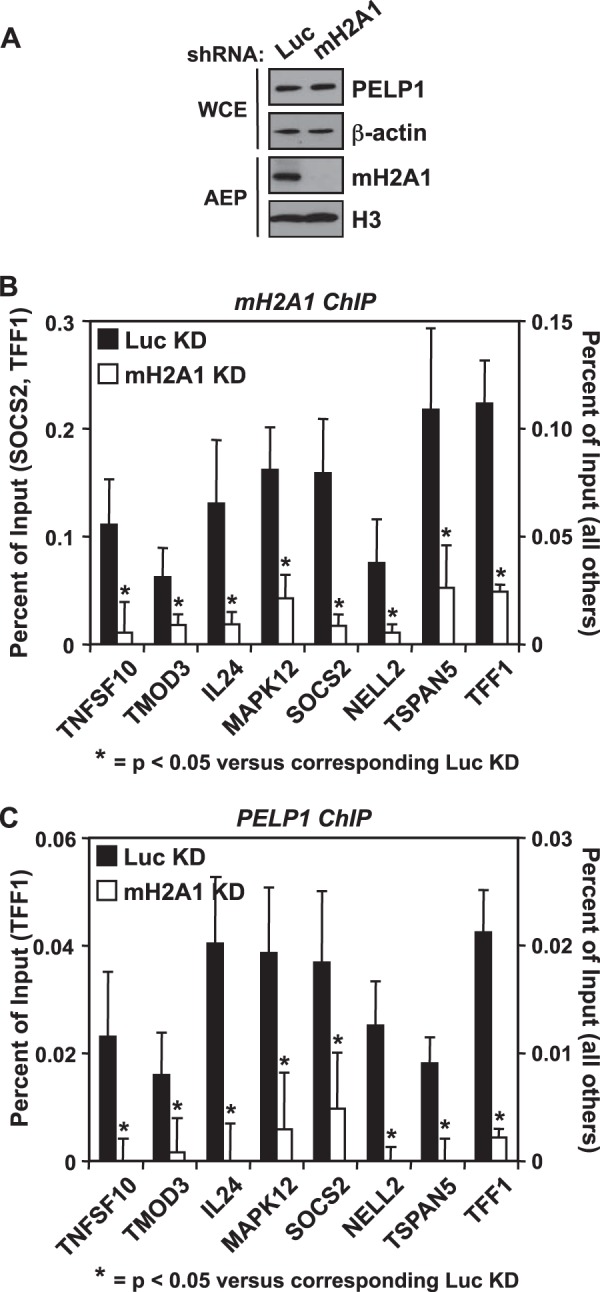
PELP1 is recruited to gene promoters by macroH2A1. (A) Western blot showing the shRNA-mediated depletion of macroH2A1 in MCF-7 cells compared to luciferase (Luc) knockdown cells. β-Actin and histone H3 were also analyzed as loading controls. (B and C) The occupancy of macroH2A1 (B) and PELP1 (C) was examined by ChIP analyses in the Luc and macroH2A1 knockdown cell lines. Values represent means plus standard errors of the means (error bars) from three or more independent determinations. All changes in occupancy for macroH2A1 and PELP1 upon macroH2A1 knockdown are statistically different from control cell line values, as determined by Student's t test with a P value threshold of <0.05, and are marked with an asterisk. WCE, whole-cell extract; AEP, acid-extracted pellet; Luc, luciferase; mH2A1, macroH2A1; KD, knockdown.
FIG 3.

PELP1 is significantly enriched in macroH2A1-bound regions and is also negatively correlated with gene expression. (A) Heat maps showing macroH2A1 and PELP1 ChIP-chip data across 2,149 promoters, tiled from kb −25 to +5 surrounding the transcription start sites. The promoters are ordered for average PELP1 intensity. (B) Averaging analysis of the log2 enrichment ratios for all windows from macroH2A1 (red) and PELP1 (blue) ChIP-chip data along the 30-kb regions represented on the array. Histone H3 (green) ChIP-chip data are shown for comparison. IP, immunoprecipitation. (C) Venn diagram representing the number of well-tiled windows bound by macroH2A1, PELP1, or both (Intersection). PELP1-bound regions, defined as consecutive windows bound by PELP1, can be found in Table S2 in the supplemental material. (D) Histogram depicting the fractions of macroH2A1 bound or unbound regions that are also bound by PELP1. The enrichment value (E) and P value from a Fisher exact test are shown. (E) Histogram depicting the average log2 ratios of PELP1 and macroH2A1 signal within 3 kb of the TSS at genes within each pentile of expression, as determined using expression microarray data in MCF-7 cells. The overall correlation coefficient (c.c.) and corresponding P value for PELP1 and macroH2A1 with gene expression are shown. mH2A1, macroH2A1.
Microarray data accession number.
Detailed information about the genomic regions included on the custom array and the data from the hybridizations described in this study can be accessed from the National Institutes of Health GEO Database (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE22254.
RESULTS
GST pulldown using the macrodomain of the histone variant macroH2A1.1 reveals interactions with a set of nuclear proteins.
Our previous work demonstrated that macroH2A1-containing chromatin can both positively and negatively regulate the expression of its target genes in a context-specific manner. The histone variant macroH2A1.1, depicted in Fig. 1A, consists of a canonical histone H2A region, followed by a basic linker and a globular macrodomain. The macrodomain of macroH2A is the most striking feature differentiating macroH2A1-containing nucleosomes from canonical nucleosomes. At ∼25 kDa, the globular macrodomain extends near the dyad axis of the nucleosome and makes minimal contact with DNA (54), suggesting that the macrodomain may make important protein-protein interactions that facilitate the regulation of transcription by macroH2A1. To identify the proteins interacting with this macrodomain, we performed a GST pulldown assay using the basic linker and macrodomain regions of macroH2A1.1 (GST-macro1.1) as bait (Fig. 1B). GST alone or GST-macro1.1 was immobilized using glutathione affinity resin and incubated with or without HeLa nuclear extract. After the resin was washed, the bound fraction was eluted from the resin using glutathione. The eluates were separated by SDS-PAGE and visualized by silver staining. GST-macro1.1-specific protein bands were excised from the gel and trypsinized, and protein fragments were identified by mass spectrometry.
The GST-macro1.1-interacting proteins we identified are chromatin associated and nucleolar components that participate in cellular activities such as transcription, ribosome biogenesis/export, RNA maturation, protein chaperoning, and maintenance of chromatin structure (see Fig. S3 and Table S1 in the supplemental material). Notably, both poly(ADP-ribose) polymerase 1 (PARP-1) and proline-, glutamic acid-, and leucine-rich protein 1 (PELP1) were identified as factors that interact with the macrodomain of macroH2A1.1. PARP-1 has previously been identified as a factor that interacts with the macroH2A macrodomain (30–32), validating our results. PELP1 is a nuclear protein (∼160 kDa) and has been shown to modulate both genomic and nongenomic activity of nuclear receptors through interactions with nuclear proteins (e.g., histone deacetylase 2 [HDAC2], SUMO2, estrogen receptor α [ERα], AR, GR, retinoid X receptor alpha [RXRA], and STAT3) (41–43, 55–58) and chromatin components (i.e., H1 and H3) (44, 45) involved in transcriptional regulation.
Previous findings indicate that the interaction of macroH2A1.1 and PARP-1 is modulated by the macrodomain ligand, ADP-ribose (ADPR) (32). We hypothesized that macroH2A1 and PELP1 may interact in a similar manner. To test this, the GST pulldown assay was repeated in the presence of the macrodomain ligand ADPR or NAD+ as a control (Fig. 1C). Using Western blot analyses, we were able to confirm the specific interactions of both PARP-1 and PELP1 with GST-macro1.1 compared to those with GST alone. As previously shown (32), PARP-1 binding was lost in the presence of ADPR but not in the presence of NAD+. However, we found that the PELP1 interaction with GST-macro1.1 was not interrupted in the presence of either ADPR or NAD+. These results demonstrate that PELP1 interacts with the macrodomain of macroH2A1 in an ADPR-independent manner.
The ability of PELP1 to bind to macroH2A1.1's non-histone-like region in an ADPR-independent manner suggests that PELP1 may be interacting with macroH2A1.1 in a region distinct from its macrodomain, such as the basic linker region (Fig. 1A). To test this, we used GST fused to the macrodomain of macroH2A1.1 without the linker region as bait in a PELP1 interaction assay. We found that the macrodomain of macroH2A1.1 alone is sufficient for binding to PELP1 (see Fig. S4A and B in the supplemental material). In addition, the ADPR-insensitive binding of PELP1 to macroH2A1.1 suggested that PELP1 may be able to interact with other macroH2A1 isoforms. Using GST pulldown assays similar to those described above, we determined that PELP1 binds to macroH2A1.2 and macroH2A2 even more strongly than it does to macroH2A1.1 (see Fig. S4A and B).
PELP1 occupies large chromatin domains in the MCF-7 genome and is enriched in macroH2A1-bound regions.
Recently, we reported the genomic localization patterns of macroH2A1 in MCF-7 cells using chromatin immunoprecipitation (ChIP) coupled to genomic tiling microarrays (i.e., ChIP-chip) (22). Those results indicated that macroH2A1 occupies large chromatin domains and that the boundaries of those occupied regions are near transcription start sites. For a subset of genes, macroH2A1-bound regions occupy the transcribed region. Since our in vitro assay suggested that macroH2A1 and PELP1 interact, we sought to determine if PELP1 associated with chromatin with a genomic localization pattern similar to that of macroH2A1.
We chose to study the interaction of macroH2A1 and PELP1 in MCF-7 cells for several reasons. First, our previous studies provided detailed information about the occupancy of macroH2A1 (predominantly macroH2A1.2) across a broad sampling of the genome (22). Second, compared to macroH2A1.1, PELP1 binds more strongly to macroH2A1.2 (see Fig. S4B in the supplemental material), which is the predominant macroH2A isoform in MCF-7 cells (22). Finally, the predominant expression of macroH2A1.2 in MCF-7 breast cancer cells recapitulates the loss of macroH2A1.1 seen in several types of cancer, including breast cancer (10, 12–14).
Using an antibody specific for PELP1, we performed ChIP-chip experiments in MCF-7 cells using a custom-designed Nimblegen platform, previously described (22, 53), representing 4.14 Mb of genomic DNA including 1,829 gene promoters typically tiled from kb −25 to +5 surrounding the transcription start sites (TSSs). Using our previously defined criteria (22), we determined that PELP1 significantly bound over 1,953 regions on the array, ranging in size from 1.5 to 7.5 kb (average, ∼1.9 kb) and covering ∼9% of the genome represented on the array. In total, PELP1 is found within 3 kb of the TSS for 257 (14.1%) genes on the array.
We compared the binding pattern of PELP1 to our previously published macroH2A1 ChIP-chip data set (National Institutes of Health GEO accession number GSE9607). Comparisons across several genomic loci, including all 44 ENCODE regions (59), revealed a striking correspondence between the PELP1 and macroH2A1 genomic occupancy (Fig. 2; see also Fig. S5 and S6 in the supplemental material). In order to determine if macroH2A1 and PELP1 associated with similar genomic regions on a global scale, we depicted each ChIP-chip data set as a heat map in which each row corresponds to the macroH2A1 or PELP1 ChIP-chip signal from kb −25 to +5 relative to the TSS for each TSS represented on the array. When the data from both heat maps are ordered for increasing average intensity of PELP1, an obvious correspondence between the two data sets can be observed (Fig. 3A). This pattern is also clearly evident by an averaging analysis of all TSSs represented on our array (Fig. 3B). As would be expected from these results, there is a significant correlation between the global localization pattern of macroH2A1 and PELP1 (Spearman correlation coefficient of ∼0.53; P value of <10−300). Further analysis indicated that 67.7% of the PELP1-bound regions overlap macroH2A1-containing domains (Fig. 3C). In fact, there is a greater than 27-fold enrichment of PELP1 binding in macroH2A1-containing regions of the genome (P value of <10−300) (Fig. 3D). Finally, of the 257 genes marked by PELP1 occupancy, 200 (78%) are cooccupied by macroH2A1. Overall, these data indicate that there is a tight statistical association between PELP1 and macroH2A1 binding across a broad sampling of the genome. Combined with the interaction data shown in Fig. 1, these data support the hypothesis that macroH2A1 and PELP1 are interacting with each other over many regions.
FIG 2.
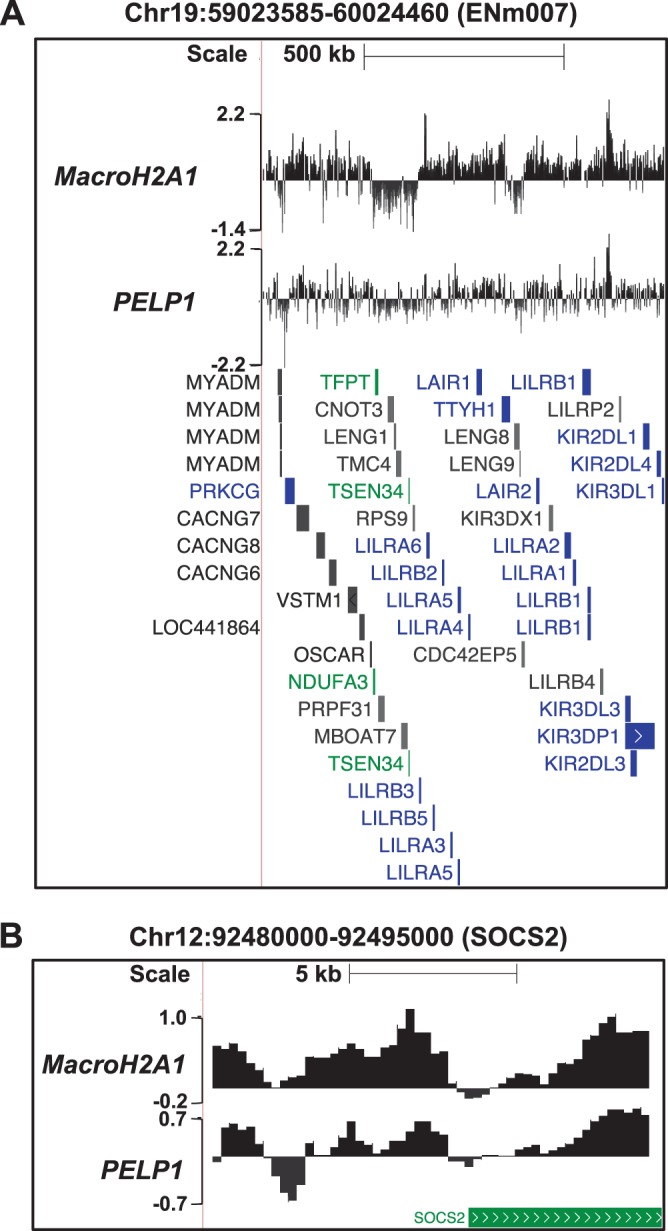
A comparison across several genomic loci reveals a striking correspondence between PELP1 and macroH2A1. Browser tracks show the log2 ratios of PELP1 and macroH2A1 ChIP-chip signals from MCF-7 cells across representative genomic regions represented on our custom-designed ChIP-chip array. The genomic locations are shown, and the position and orientation of NCBI Reference Sequence (RefSeq) genes are depicted below each track. The genes are color coded according to expression microarray data: green, expressed; blue, unexpressed; gray, ambiguous. (A) Data from a representative ENCODE region. (B) Data from a region centered at the SOCS2 locus. Chr, chromosome.
Despite the strong statistical enrichment of PELP1 in macroH2A1-containing domains, we were not able to detect PELP1 at all sites of macroH2A1 deposition in MCF-7 cells. In fact, PELP1 occupies only 29% of macroH2A1-bound regions (Fig. 3C). Additionally, 22% of PELP1-bound genes do not contain significant levels of macroH2A1 within 3 kb of the TSS. The lack of complete overlap of PELP1 and macroH2A1 binding may indicate that PELP1 binding functionally distinguishes a subset of macroH2A1-containing regions. Alternatively, these differences could be due to the reduced efficiency of the PELP1 ChIP compared to that of macroH2A1. To examine these possibilities, we set out to determine if PELP1 has a similar correlation to gene expression, histone marks, and gene ontology as previously observed for macroH2A1 (22).
MacroH2A1 is negatively correlated with gene expression in both IMR90 cells and MCF-7 cells (22). By calculating the average macroH2A1 and PELP1 ChIP-chip signal within 3 kb of all TSSs and comparing the result to previously published Affymetrix expression data from MCF-7 cells (60), we determined that, similar to macroH2A1 (correlation coefficient of −0.25, P value of 2 × 10−23), PELP1 (correlation coefficient of −0.17, P value of 7.7 × 10−12) is significantly negatively correlated with gene expression (Fig. 3E).
Previous gene ontology analysis determined that macroH2A1-containing domains are enriched for genes that are involved in developmental processes and signaling events. MacroH2A1-containing genes also often encode proteins that are secreted from the cell (22). We performed a similar gene ontology analysis for PELP1-bound genes, as well as for the genes that are bound by both macroH2A1 and PELP1. We found that PELP1-bound genes are enriched for a similar set of ontological processes (Table 1; see also Table S3 in the supplemental material).
TABLE 1.
Summary of the most significant gene ontology terms enriched in genes with macroH2A1, PELP1, or both within 3 kb surrounding the TSS
| Gene group and gene ontology terma | Aspectb | Corrected P value |
|---|---|---|
| MacroH2A1-bound genesc | ||
| Multicellular organismal process | BP | 4.46 × 10−5 |
| System development | BP | 7.44 × 10−5 |
| Multicellular organismal development | BP | 0.00011 |
| Anatomical structure development | BP | 0.00087 |
| Developmental process | BP | 0.00103 |
| Organ development | BP | 0.00288 |
| Regulation of localization | BP | 0.00342 |
| Signaling | BP | 0.00663 |
| Cell surface receptor linked signaling pathway | BP | 0.00948 |
| Extracellular region | CC | 2.60 × 10−7 |
| Extracellular region part | CC | 1.82 × 10−5 |
| Pattern binding | MF | 0.00072 |
| Polysaccharide binding | MF | 0.00072 |
| Glycosaminoglycan binding | MF | 0.00200 |
| PELP1-bound genesd | ||
| Multicellular organismal process | BP | 0.00020 |
| Cell surface receptor linked signaling pathway | BP | 0.00878 |
| Extracellular region part | CC | 0.00071 |
| Proteinaceous extracellular matrix | CC | 0.00128 |
| Extracellular matrix | CC | 0.00175 |
| MacroH2A1- and PELP1-bound genese | ||
| Multicellular organismal development | BP | 4.74 × 10−5 |
| Organ development | BP | 0.00023 |
| Multicellular organismal process | BP | 0.00024 |
| Developmental process | BP | 0.00026 |
| System development | BP | 0.00142 |
| Anatomical structure development | BP | 0.00216 |
| Extracellular region part | CC | 0.00011 |
| Proteinaceous extracellular matrix | CC | 0.00022 |
| Extracellular matrix | CC | 0.00023 |
| Extracellular region | CC | 0.00645 |
The enrichment of gene ontology terms in genes with macroH2A1-bound or PELP1-bound regions found within 3 kb of the TSS in MCF-7 cells was compared with all other genes represented on the ChIP-chip array for all three ontology aspects. Gene ontology terms were generated using the Generic Gene Ontology Term Finder (http://go.princeton.edu/cgi-bin/GOTermFinder/GOTermFinder), as described previously (22, 79).
The aspects covered are biological process (BP), cellular component (CC), and molecular function (MF).
The genes that correspond to macroH2A1-bound regions used for this analysis are irrespective of PELP1 status and can be found listed in Table S3 in the supplemental material.
The genes that correspond to PELP1-bound regions used for this analysis are irrespective of macroH2A1 status and can be found listed in Table S3 in the supplemental material.
The genes that correspond to both macroH2A1 and PELP1 bound regions used for this analysis can be found listed in Table S3 in the supplemental material.
Previously, we correlated macroH2A1 occupancy to 362 publicly available ChIP-chip data sets and determined that macroH2A1 positively correlates with heterochromatic histone marks, including histone H3 lysine 27 trimethylation (H3K27me3), while negatively correlating with active transcription marks, such as RNA polymerase II (Pol II) (22). By performing a similar analysis for PELP1 (Fig. 4A; see also Table S4 in the supplemental material), we found that, like macroH2A1, PELP1 positively correlates with the repressive H3K27me3 histone mark and negatively correlates with marks of active transcription (e.g., RNA Pol II) for ChIP-chip data derived from MCF-7 cells (Fig. 4B), suggesting that these two factors may be involved in similar functions. However, PELP1 also correlates with factors and histone marks independent of macroH2A1 (e.g., histone 3 lysine 9 trimethylation [H3K9me3]) (Fig. 4B).
FIG 4.
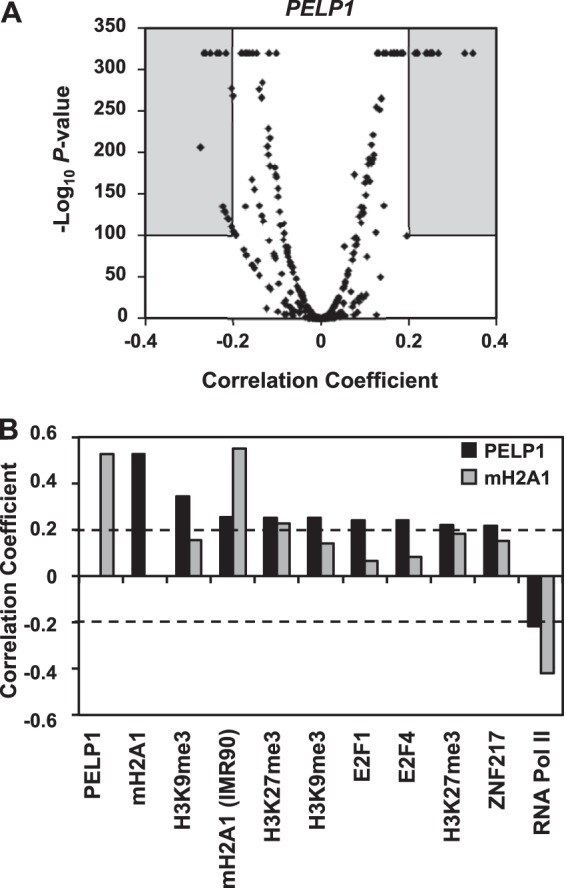
PELP1 occupancy correlates with heterochromatic chromatin marks and negatively correlates with active chromatin marks. (A) Volcano plot of Spearman's correlation coefficient for the PELP1 ChIP-chip data with each of 362 ChIP-chip data sets versus the corresponding significance score (−log2 P value). The gray boxes depict the data sets that positively or negatively correlate with PELP1 occupancy using a correlation coefficient of ±0.20 and P value of <10−100 as cutoffs of significance. The full analysis can be found in Table S4 in the supplemental material. (B) Correlation coefficients for factors positively or negatively correlating with PELP1 genomic localization in MCF-7 cells from the gray areas within the graph in panel A. The corresponding data for macroH2A1 are shown for comparison.
Overall, these analyses demonstrate that PELP1 and macroH2A1 have highly similar patterns of genomic occupancy. While bound to a subset of macroH2A1-containing regions, PELP1 genomic association correlates with gene expression, histone marks, and functional classes of genes in a manner that is highly similar to that of macroH2A1. Taken together, the results from our GST pulldown and ChIP-chip analyses suggest that PELP1 and macroH2A1 interact with each other and colocalize across a broad sampling of the genome of MCF-7 cells.
PELP1 is recruited to the genome by macroH2A1, but macroH2A1 occupancy is independent of PELP1.
The high degree of coincidence between macroH2A1 and PELP1 genomic occupancy led us to explore the hypothesis that macroH2A1 functions in recruiting PELP1 to the genome. In order to determine the effect of macroH2A1 knockdown on the localization of PELP1, we used an shRNA-mediated stable macroH2A1 knockdown cell line described previously (22). MacroH2A1 knockdown did not alter the overall levels of PELP1 in MCF-7 cells (Fig. 5A) compared to the luciferase (Luc) knockdown control. For our ChIP assays, we focused on gene promoters previously shown to contain and be regulated by macroH2A1 (22). At these promoters, knockdown of macroH2A1 significantly decreased macroH2A1 levels, as would be expected (Fig. 5B). We were able to confirm the occupancy of PELP1 at these gene promoters, as predicted from the ChIP-chip data. Interestingly, at all regions tested, we observed that PELP1 genomic occupancy decreased upon macroH2A1 knockdown (Fig. 5C), a finding independent of nucleosome loss (see Fig. S7 in the supplemental material), suggesting that macroH2A1 is required to recruit PELP1 to chromatin. Additionally, we observed that occupancy of PELP1 at macroH2A1-bound genes in A549 lung cancer cells was also reduced upon depletion of macroH2A1 (see Fig. S8), indicating that the interaction between macroH2A1 and PELP1 is a general feature and not specific to MCF-7 cells.
To determine whether macroH2A1 deposition was reciprocally dependent on PELP1, we used an analogous set of experiments. We developed a PELP1 knockdown cell line in MCF-7 cells to compare macroH2A1 promoter occupancy in control versus PELP1 knockdown cells. Our initial attempts to create a stable cell line using shRNA-mediated knockdown of PELP1, much like that shown above for macroH2A1, were unsuccessful as PELP1 expression reverted to wild-type levels after only a couple of passages (data not shown). In an alternative approach, we developed a Tet-on-inducible knockdown system where treatment of doxycycline (DOX) induced the expression of shRNA in MCF-7 cells via a Tet-responsive promoter. Using this system, we created MCF-7 cells stably expressing both the Tet repressor (TetR) and either a DOX-responsive Luc or PELP1 shRNA.
The resulting inducible knockdown cell lines, Luci and PELP1i, were treated with DOX or left untreated (see Materials and Methods) and tested for cellular PELP1 levels by Western blotting. As expected, DOX treatment induced shRNA expression, leading to a decrease in PELP1 protein by ∼80% compared to the Luc control (Fig. 6A). Notably, PELP1 knockdown did not have an effect on total cellular levels of macroH2A1 (Fig. 6A). We performed ChIP assays at the same promoter regions using our inducible knockdown cell lines to determine if macroH2A1 localization was altered in response to PELP1 depletion. Upon PELP1 knockdown, macroH2A1 recruitment remained constant even though PELP1 levels at the promoters were significantly reduced (Fig. 6B and C). These results demonstrate that while macroH2A1 genomic occupancy occurs independent of PELP1 recruitment, macroH2A1 is required in order to recruit PELP1 to the chromatin.
FIG 6.
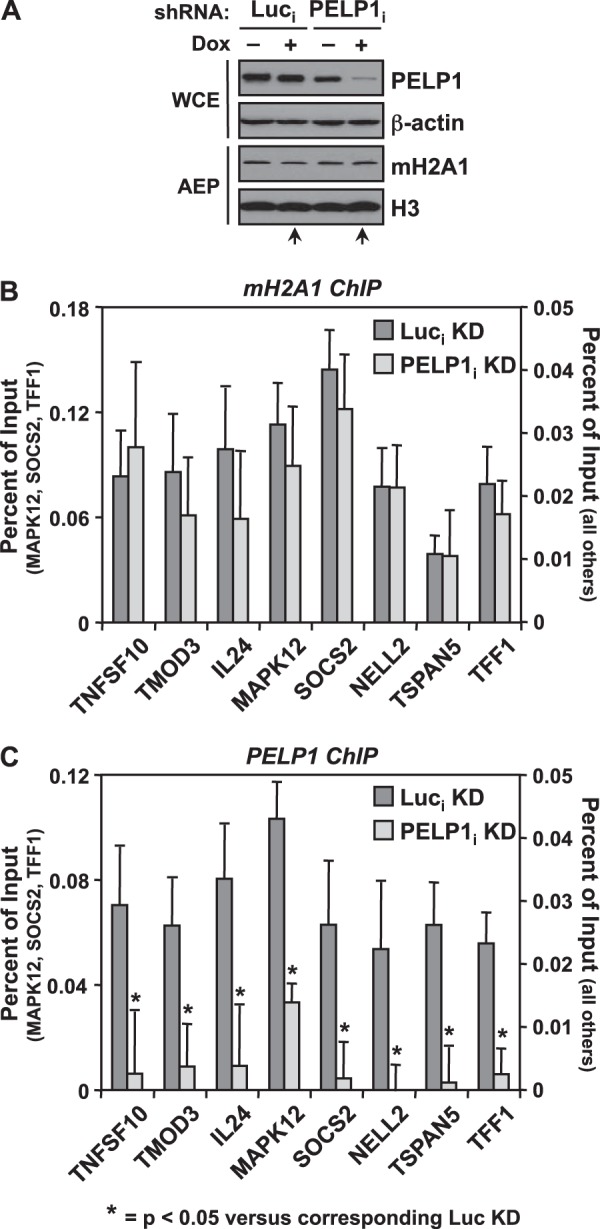
MacroH2A1 deposition is independent of PELP1. (A) Western blot showing the doxycycline-inducible shRNA-mediated depletion of PELP1 in MCF-7 cells compared to luciferase (Luc) knockdown cells. β-Actin and histone H3 were also analyzed as loading controls. Only the +DOX condition (lanes marked with arrows) for Luci and PELP1i was analyzed for gene-specific experiments. (B and C) The occupancy of macroH2A1 (B) and PELP1 (C) was examined by ChIP analyses in the Luci and PELP1i knockdown cell lines. Values represent the means plus standard errors of the means (error bars) from three or more independent determinations. All changes in occupancy for PELP1 upon PELP1 knockdown are statistically different from the control cell line values, as determined by Student's t test with a P value threshold of <0.05, and are marked with asterisks. Luc, luciferase; mH2A1, macroH2A1; KD, knockdown; DOX, doxycycline; i, inducible.
PELP1 regulates a subset of macroH2A1-regulated genes in a similar manner to macroH2A1 in both basal and signal-regulated transcription.
Thus far, our results demonstrate that macroH2A1 interacts with PELP1 and is responsible for the recruitment of PELP1 to target gene promoters. We hypothesized that these genes, previously shown to be regulated by macroH2A1 (22), would also be regulated by PELP1. We examined the expression levels of macroH2A1-regulated genes in Luci and PELP1i knockdown cell lines by RT-qPCR. Our analyses show that PELP1 knockdown alters the expression levels of a subset of macroH2A1-regulated genes (Fig. 7). Interestingly, the direction of regulation is similar to that of macroH2A1 knockdown, which is shown for comparison (data from Gamble et al. [22]). This suggests that both macroH2A1 and PELP1 are required for proper expression of a subset of genes.
FIG 7.
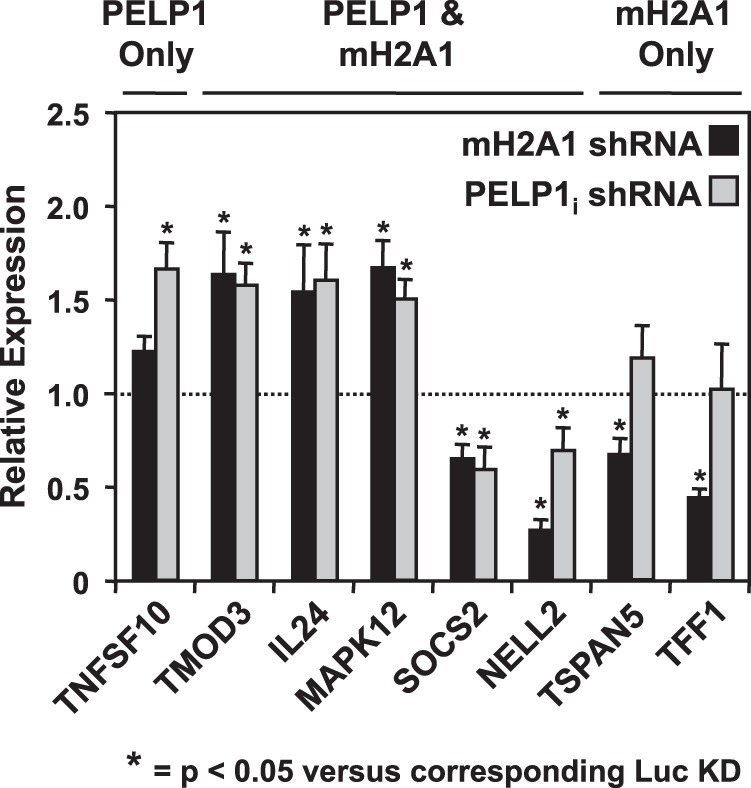
PELP1 knockdown alters the mRNA levels of a subset of macroH2A1-regulated genes in a similar manner to macroH2A1. Previously identified macroH2A1-regulated genes (22) were analyzed for changes in mRNA levels upon PELP1i knockdown. Total RNA from Luci or PELP1i knockdown cells (+DOX) was isolated, reverse transcribed, and subjected to qPCR using gene-specific primers. The effect of macroH2A1 knockdown (data from Gamble et al. [22]) is shown for comparison. Values represent the means plus standard errors of the means (error bars) from three or more independent determinations. Bars marked with asterisks are statistically different from the value of the luciferase knockdown control, as determined by Student's t test with at P value threshold of <0.05. mH2A1, macroH2A1.
We previously reported that macroH2A1 could also participate in signal-regulated transcription. As a model, we investigated both serum starvation and TPA signaling systems and showed that macroH2A1 can function both positively and negatively in regulated signal-induced transcription (22, 25). Specifically, macroH2A1 potentiates the response of serum starvation-induced genes found in macroH2A1 domains. Additionally, macroH2A1 abrogates the expression of TPA-responsive genes found in these domains. We asked whether PELP1 could modulate transcription in the same signaling pathways. To test this, we considered TPA- or serum starvation-induced genes whose expression patterns were altered upon macroH2A1 knockdown (Fig. 8; see also Fig. S9 in the supplemental material). We assayed the mRNA levels of these genes upon Luci and PELP1i knockdown. PELP1 involvement in signal-regulated transcription was limited to a subset of genes, as shown under resting conditions. For example, knockdown of PELP1 further enhanced the expression of the TPA-induced CCL2 gene similarly to macroH2A1 knockdown but had no effect on AREG expression (Fig. 8A). In addition, like macroH2A1, PELP1 occupancy upon TPA treatment was reduced downstream of the TSS at the CCL2 promoter (Fig. 8B) (25) but not at the AREG promoter. This result is not due to nucleosome loss as H3 levels remain constant (see Fig. S10A in the supplemental material). In the case of serum starvation, PELP1 acts similarly to macroH2A1 at the SOCS2 promoter but not at the ASCL1 promoter although the effects were less striking (see Fig. S9 and S10B) (22). Taken together, these results demonstrate that PELP1 modulates a subset of macroH2A1-regulated genes in a manner similar to macroH2A1 in both basal and signal-regulated systems.
DISCUSSION
The histone variant macroH2A1 can both positively and negatively regulate target genes within macroH2A1-containing chromatin in a context-specific manner (6, 21–23, 25). However, the mechanism by which macroH2A1 regulates the expression of its target genes has yet to be fully elucidated. As the distinguishing feature of macroH2A1, the large globular macrodomain plays a key role in determining the specific context in which an underlying gene will be modulated through specific recruitment and physical interaction of effector proteins. Here, we identify a chromatin-associated transcriptional coregulator, PELP1, as a novel interacting protein of the macrodomain of macroH2A1. Collectively, our biochemical, genomic, and gene-specific analyses suggest that macroH2A1 specifically recruits PELP1 to the genome. Together, they cooperatively regulate a subset of macroH2A1 target genes.
The macrodomain of macroH2A1 interacts with chromatin-associated and nucleolar components.
Our GST pulldown assay identified a host of novel macrodomain-interacting factors that can be classified into two categories: those that are chromatin-associated and those that are nucleolar components (Fig. 1B; see also Fig. S3 and Table S1 in the supplemental material). The chromatin-associated factors (PELP1, PARP-1, WDR18, and SET) are involved in cellular processes such as transcription and chromatin maintenance, while the nucleolar components (TCOF1, NCL, NPM1, MDN1, DDX46, TEX10, and Nol9) are generally involved in transcription, ribosome biogenesis/export, protein chaperoning, and RNA processing. The identification of these proteins fits well with emerging evidence that macroH2A1 is involved in regulating autosomal genes as many of these proteins are known to function in various aspects of transcriptional control. It is interesting that PELP1 and PARP-1 have also been shown to interact with proteins of the same family as those we identified interacting with the macrodomain of macroH2A1. For example, PELP1 can interact with WDR5 as part of the MLL1 complex (61, 62) or members of the DEAD box RNA helicase and AAA ATPase family members (63), which are involved in RNA processing. Additionally, PARP-1 has been shown to interact with nucleolar proteins and transcriptional coregulators (nucleolin and nucleophosmin) in a number of contexts (64–66). Recent studies have shown that PELP1 enhances ribosomal DNA (rDNA) transcription (67) and functions in a complex with WDR18, TEX10, and a nucleolar deSUMOylating enzyme to control rRNA processing and ribosome biogenesis (68). Most recently, macroH2A1 has been shown to repress rRNA gene transcription (69). Collectively, these results highlight the shared functions of macroH2A, PELP1, and PARP-1, which are supported by interactions with each other as well as with common binding partners.
PELP1 is recruited to specific chromatin environments.
Histone posttranslational modifications exert their effects by recruiting transcriptional coregulators that specifically recognize those marks. PELP1 has recently been shown to associate with H3K9me2 and H3K4me2 (45). Furthermore, PELP1 can also recruit additional chromatin-modifying enzymes to the sites where it is found. For example, PELP1 has been shown to recruit KDM1 (lysine demethylase 1) and HDAC2 to nucleosomes (41, 45). We show that PELP1-bound regions correlate with the histone modifications H3K27me3 and H3K9me3 and repressive transcription factors (e.g., ZNF217) (Fig. 4), all of which are known to generally mark heterochromatic chromatin and/or participate in the repression of gene expression (70, 71).
Similar to histone modifications, histone variants can also mark chromatin to specify positive or negative gene expression outcomes by recruiting specific effector proteins necessary to facilitate these outcomes. We demonstrate that PELP1 is broadly recruited to macroH2A1-containing chromatin across a broad sampling of the genome. These data suggest that PELP1 has alternative modes of interacting with chromatin. It is able to interact with specific histone PTMs (e.g., H3K9me2 and H3K4me2) and can also specifically recognize macroH2A1-containing nucleosomes. Furthermore, given the inclination of PELP1 to interact with histone-modifying enzymes (e.g., KDM1, HDAC2, p300/CBP [40, 41, 45]), it is possible that recruitment of PELP1 to macroH2A1-containing regions of the genome leads to further chromatin modifications. Further experiments are required to determine the role of PELP1 in further modifying macroH2A1-containing chromatin structure.
PELP1 and macroH2A1 are context-specific transcriptional coregulators.
For quite a while, macroH2A1 was exclusively considered a transcriptionally repressive histone mark. Recent work from our lab and from others has determined that macroH2A1 can function as a positive or negative regulator of transcription depending on the specific context (22, 25, 72). While originally identified as a coactivator for the estrogen receptor alpha (ERα) (40), PELP1 is now known to function as a coactivator for several transcription factors including ERα, ERβ, and RXR (40, 58), demonstrating that, like macroH2A1, PELP1 also modulates transcription in context-specific ways. Conversely, recent studies suggest that PELP1 can also function as a transcriptional corepressor in concert with several transcription factors, including GR, AP1, NF-κB, and serum response factor (SRF) (41, 73). Therefore, PELP1 can either positively or negatively regulate gene expression in a context-specific fashion. The coordinated regulation of PELP1 and macroH2A1 target gene expression described above demonstrates a connection between the determinants that specify these factors as transcriptionally permissive or repressive. Specifically, at genes coregulated by macroH2A1 and PELP1, if one factor supports target gene expression, so does the other and vice versa.
Macrodomains have been identified as ligand binding domains for NAD+ metabolites such as PAR, ADPR, and O-acetyl-ADPR (reviewed in reference 37; 32, 34–36, 38). This action may change (i) the affinity of macrodomain-interacting proteins or (ii) the recruitment of factors containing these domains to genomic sites of PAR accumulation (32, 34, 35). While PARP-1 has previously been shown to associate with the macrodomain of macroH2A1 in a manner dependent on macroH2A1's ability to bind NAD+ metabolite ligands (e.g., ADP-ribose), the interaction with PELP1 is unaffected by ADPR binding (Fig. 1C). So, what modulates the interaction of PELP1 with the macrodomain of macroH2A1? In addition, this finding brings up an interesting question of whether or not PELP1 interacts with other macrodomain-containing proteins. Interestingly, both PELP1 and macroH2A1 are subject to a variety of posttranslational modifications (57, 62, 74–77), which may modulate the ability of these factors to interact. Further studies are required to determine the mechanisms that regulate these interactions.
While there is much that remains to be elucidated about the concerted roles of macroH2A1 and PELP1 in transcriptional regulation of target genes, one major question centers around identifying the determinants that allow macroH2A1 and PELP1 to function as transcriptional activators and those specific for transcriptional repression. One possibility is that further histone modifications are required to specify direction of macroH2A1/PELP1-mediated transcriptional outcomes. In addition, macroH2A1 and PELP1 are themselves targets of various covalent modifications (reviewed in references 77 and 78), which may play a role in determining the direction of regulation. Alternatively, the specific transcription factors that bind near (or are specifically recruited to) macroH2A1/PELP1 positively and negatively regulated genes may be very different. Understanding the mechanisms that regulate the context-specific transcriptional outcomes mediated by PELP1 and macroH2A1 will lead to a greater understanding of how particular chromatin states regulate gene expression.
Supplementary Material
ACKNOWLEDGMENTS
We thank Shrikanth Gadad and Xin Luo for critical reading of the manuscript, Trevor Halle for assistance in making the MCF-7 TetR cell line, Lynne Lacomis for help with mass spectrometry and sample preparation, Rosemary Conry for preparing the GST-macrodomain fusions, and Hans Clevers and Marc Timmers for the Tet-inducible expression constructs.
This work was supported by grants from the NIH/NIDDK (DK069710), Cornell University's NanoBiotechnology Center (an STC Program of the NSF, agreement no. ECS-9876771), and the Endocrine Society to W.L.K., by a predoctoral fellowship from the American Heart Association to K.M.H., by postdoctoral fellowships from the American Heart Association and the NIH/NIDDK (F32DK079847) and a grant from the NIH/NCI (R01CA155232) to M.J.G., and by an NIH/NCI Cancer Center Support Grant (P30 CA08748) to P.T.
Footnotes
Published ahead of print 21 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01315-13.
REFERENCES
- 1.Turner BM. 2012. The adjustable nucleosome: an epigenetic signaling module. Trends Genet. 28:436–444. 10.1016/j.tig.2012.04.003 [DOI] [PubMed] [Google Scholar]
- 2.Volle C, Dalal Y. 2014. Histone variants: the tricksters of the chromatin world. Curr. Opin. Genet. Dev. 25C:8–14. 10.1016/j.gde.2013.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costanzi C, Pehrson JR. 1998. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 393:599–601. 10.1038/31275 [DOI] [PubMed] [Google Scholar]
- 4.Mietton F, Sengupta AK, Molla A, Picchi G, Barral S, Heliot L, Grange T, Wutz A, Dimitrov S. 2009. Weak but uniform enrichment of the histone variant macroH2A1 along the inactive X chromosome. Mol. Cell. Biol. 29:150–156. 10.1128/MCB.00997-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ladurner AG. 2003. Inactivating chromosomes: a macro domain that minimizes transcription. Mol. Cell 12:1–3. 10.1016/S1097-2765(03)00284-3 [DOI] [PubMed] [Google Scholar]
- 6.Changolkar LN, Costanzi C, Leu NA, Chen D, McLaughlin KJ, Pehrson JR. 2007. Developmental changes in histone macroH2A1-mediated gene regulation. Mol. Cell. Biol. 27:2758–2764. 10.1128/MCB.02334-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Csankovszki G, Panning B, Bates B, Pehrson JR, Jaenisch R. 1999. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet. 22:323–324. 10.1038/11887 [DOI] [PubMed] [Google Scholar]
- 8.Rasmussen TP, Wutz AP, Pehrson JR, Jaenisch RR. 2001. Expression of Xist RNA is sufficient to initiate macrochromatin body formation. Chromosoma 110:411–420. 10.1007/s004120100158 [DOI] [PubMed] [Google Scholar]
- 9.Wutz A, Rasmussen TP, Jaenisch R. 2002. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 30:167–174. 10.1038/ng820 [DOI] [PubMed] [Google Scholar]
- 10.Dardenne E, Pierredon S, Driouch K, Gratadou L, Lacroix-Triki M, Espinoza MP, Zonta E, Germann S, Mortada H, Villemin JP, Dutertre M, Lidereau R, Vagner S, Auboeuf D. 2012. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 19:1139–1146. 10.1038/nsmb.2390 [DOI] [PubMed] [Google Scholar]
- 11.Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, Polsky D, Osman I, Garcia BA, Hernando E, Bernstein E. 2010. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468:1105–1109. 10.1038/nature09590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novikov L, Park JW, Chen H, Klerman H, Jalloh AS, Gamble MJ. 2011. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol. Cell. Biol. 31:4244–4255. 10.1128/MCB.05244-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sporn JC, Jung B. 2012. Differential regulation and predictive potential of MacroH2A1 isoforms in colon cancer. Am. J. Pathol. 180:2516–2526. 10.1016/j.ajpath.2012.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sporn JC, Kustatscher G, Hothorn T, Collado M, Serrano M, Muley T, Schnabel P, Ladurner AG. 2009. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 28:3423–3428. 10.1038/onc.2009.26 [DOI] [PubMed] [Google Scholar]
- 15.Barrero MJ, Sese B, Kuebler B, Bilic J, Boue S, Marti M, Izpisua Belmonte JC. 2013. Macrohistone variants preserve cell identity by preventing the gain of H3K4me2 during reprogramming to pluripotency. Cell Rep. 3:1005–1011. 10.1016/j.celrep.2013.02.029 [DOI] [PubMed] [Google Scholar]
- 16.Barrero MJ, Sese B, Marti M, Izpisua Belmonte JC. 2013. Macro histone variants are critical for the differentiation of human pluripotent cells. J. Biol. Chem. 288:16110–16116. 10.1074/jbc.M113.466144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Creppe C, Janich P, Cantarino N, Noguera M, Valero V, Musulen E, Douet J, Posavec M, Martin-Caballero J, Sumoy L, Di Croce L, Benitah SA, Buschbeck M. 2012. MacroH2A1 regulates the balance between self-renewal and differentiation commitment in embryonic and adult stem cells. Mol. Cell. Biol. 32:1442–1452. 10.1128/MCB.06323-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaspar-Maia A, Qadeer ZA, Hasson D, Ratnakumar K, Leu NA, Leroy G, Liu S, Costanzi C, Valle-Garcia D, Schaniel C, Lemischka I, Garcia B, Pehrson JR, Bernstein E. 2013. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun. 4:1565. 10.1038/ncomms2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasque V, Radzisheuskaya A, Gillich A, Halley-Stott RP, Panamarova M, Zernicka-Goetz M, Surani MA, Silva JC. 2012. Histone variant macroH2A marks embryonic differentiation in vivo and acts as an epigenetic barrier to induced pluripotency. J. Cell Sci. 125:6094–6104. 10.1242/jcs.113019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pasque V, Gillich A, Garrett N, Gurdon JB. 2011. Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J. 30:2373–2387. 10.1038/emboj.2011.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buschbeck M, Uribesalgo I, Wibowo I, Rue P, Martin D, Gutierrez A, Morey L, Guigo R, Lopez-Schier H, Di Croce L. 2009. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat. Struct. Mol. Biol. 16:1074–1079. 10.1038/nsmb.1665 [DOI] [PubMed] [Google Scholar]
- 22.Gamble MJ, Frizzell KM, Yang C, Krishnakumar R, Kraus WL. 2010. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 24:21–32. 10.1101/gad.1876110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agelopoulos M, Thanos D. 2006. Epigenetic determination of a cell-specific gene expression program by ATF-2 and the histone variant macroH2A. EMBO J. 25:4843–4853. 10.1038/sj.emboj.7601364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Changolkar LN, Singh G, Pehrson JR. 2008. macroH2A1-dependent silencing of endogenous murine leukemia viruses. Mol. Cell. Biol. 28:2059–2065. 10.1128/MCB.01362-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gamble MJ, Kraus WL. 2010. Multiple facets of the unique histone variant macroH2A. Cell Cycle 9:2568–2574. 10.4161/cc.9.13.12144 [DOI] [PubMed] [Google Scholar]
- 26.Pehrson JR, Fuji RN. 1998. Evolutionary conservation of histone macroH2A subtypes and domains. Nucleic Acids Res. 26:2837–2842. 10.1093/nar/26.12.2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chakravarthy S, Gundimella SK, Caron C, Perche PY, Pehrson JR, Khochbin S, Luger K. 2005. Structural characterization of the histone variant macroH2A. Mol. Cell. Biol. 25:7616–7624. 10.1128/MCB.25.17.7616-7624.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez-Munoz I, Lund AH, van der Stoop P, Boutsma E, Muijrers I, Verhoeven E, Nusinow DA, Panning B, Marahrens Y, van Lohuizen M. 2005. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc. Natl. Acad. Sci. U. S. A. 102:7635–7640. 10.1073/pnas.0408918102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nusinow DA, Hernandez-Munoz I, Fazzio TG, Shah GM, Kraus WL, Panning B. 2007. Poly(ADP-ribose) polymerase 1 is inhibited by a histone H2A variant, MacroH2A, and contributes to silencing of the inactive X chromosome. J. Biol. Chem. 282:12851–12859. 10.1074/jbc.M610502200 [DOI] [PubMed] [Google Scholar]
- 30.Ouararhni K, Hadj-Slimane R, Ait-Si-Ali S, Robin P, Mietton F, Harel-Bellan A, Dimitrov S, Hamiche A. 2006. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev. 20:3324–3336. 10.1101/gad.396106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratnakumar K, Duarte LF, LeRoy G, Hasson D, Smeets D, Vardabasso C, Bonisch C, Zeng T, Xiang B, Zhang DY, Li H, Wang X, Hake SB, Schermelleh L, Garcia BA, Bernstein E. 2012. ATRX-mediated chromatin association of histone variant macroH2A1 regulates alpha-globin expression. Genes Dev. 26:433–438. 10.1101/gad.179416.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, Colombelli J, Altmeyer M, Stelzer EH, Scheffzek K, Hottiger MO, Ladurner AG. 2009. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 16:923–929. 10.1038/nsmb.1664 [DOI] [PubMed] [Google Scholar]
- 33.Gamble MJ. 2013. Expanding the functional repertoire of macrodomains. Nat. Struct. Mol. Biol. 20:407–408. 10.1038/nsmb.2552 [DOI] [PubMed] [Google Scholar]
- 34.Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, Owen-Hughes T, Boulton SJ. 2009. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325:1240–1243. 10.1126/science.1177321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, Washburn MP, Florens L, Ladurner AG, Conaway JW, Conaway RC. 2009. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. U. S. A. 106:13770–13774. 10.1073/pnas.0906920106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, Bycroft M, Ladurner AG. 2005. The macro domain is an ADP-ribose binding module. EMBO J. 24:1911–1920. 10.1038/sj.emboj.7600664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kraus WL. 2009. New functions for an ancient domain. Nat. Struct. Mol. Biol. 16:904–907. 10.1038/nsmb0909-904 [DOI] [PubMed] [Google Scholar]
- 38.Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG. 2005. Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 12:624–625. 10.1038/nsmb956 [DOI] [PubMed] [Google Scholar]
- 39.Cheskis BJ, Greger J, Cooch N, McNally C, McLarney S, Lam HS, Rutledge S, Mekonnen B, Hauze D, Nagpal S, Freedman LP. 2008. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids 73:901–905. 10.1016/j.steroids.2007.12.028 [DOI] [PubMed] [Google Scholar]
- 40.Vadlamudi RK, Wang RA, Mazumdar A, Kim Y, Shin J, Sahin A, Kumar R. 2001. Molecular cloning and characterization of PELP1, a novel human coregulator of estrogen receptor alpha. J. Biol. Chem. 276:38272–38279. 10.1074/jbc.M103783200 [DOI] [PubMed] [Google Scholar]
- 41.Choi YB, Ko JK, Shin J. 2004. The transcriptional corepressor, PELP1, recruits HDAC2 and masks histones using two separate domains. J. Biol. Chem. 279:50930–50941. 10.1074/jbc.M406831200 [DOI] [PubMed] [Google Scholar]
- 42.Khan MM, Hadman M, De Sevilla LM, Mahesh VB, Buccafusco J, Hill WD, Brann DW. 2006. Cloning, distribution, and colocalization of MNAR/PELP1 with glucocorticoid receptors in primate and nonprimate brain. Neuroendocrinology 84:317–329. 10.1159/000097746 [DOI] [PubMed] [Google Scholar]
- 43.Nair SS, Guo Z, Mueller JM, Koochekpour S, Qiu Y, Tekmal RR, Schule R, Kung HJ, Kumar R, Vadlamudi RK. 2007. Proline-, glutamic acid-, and leucine-rich protein-1/modulator of nongenomic activity of estrogen receptor enhances androgen receptor functions through LIM-only coactivator, four-and-a-half LIM-only protein 2. Mol. Endocrinol. 21:613–624. 10.1210/me.2006-0269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nair SS, Mishra SK, Yang Z, Balasenthil S, Kumar R, Vadlamudi RK. 2004. Potential role of a novel transcriptional coactivator PELP1 in histone H1 displacement in cancer cells. Cancer Res. 64:6416–6423. 10.1158/0008-5472.CAN-04-1786 [DOI] [PubMed] [Google Scholar]
- 45.Nair SS, Nair BC, Cortez V, Chakravarty D, Metzger E, Schule R, Brann DW, Tekmal RR, Vadlamudi RK. 2010. PELP1 is a reader of histone H3 methylation that facilitates oestrogen receptor-alpha target gene activation by regulating lysine demethylase 1 specificity. EMBO Rep. 11:438–444. 10.1038/embor.2010.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frizzell KM, Gamble MJ, Berrocal JG, Zhang T, Krishnakumar R, Cen Y, Sauve AA, Kraus WL. 2009. Global analysis of transcriptional regulation by poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in MCF-7 human breast cancer cells. J. Biol. Chem. 284:33926–33938. 10.1074/jbc.M109.023879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL. 2004. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119:803–814. 10.1016/j.cell.2004.11.002 [DOI] [PubMed] [Google Scholar]
- 48.Erdjument-Bromage H, Lui M, Lacomis L, Grewal A, Annan RS, McNulty DE, Carr SA, Tempst P. 1998. Examination of micro-tip reversed-phase liquid chromatographic extraction of peptide pools for mass spectrometric analysis. J. Chromatogr. A 826:167–181. 10.1016/S0021-9673(98)00705-5 [DOI] [PubMed] [Google Scholar]
- 49.Sebastiaan Winkler G, Lacomis L, Philip J, Erdjument-Bromage H, Svejstrup JQ, Tempst P. 2002. Isolation and mass spectrometry of transcription factor complexes. Methods 26:260–269. 10.1016/S1046-2023(02)00030-0 [DOI] [PubMed] [Google Scholar]
- 50.van de Wetering M, Oving I, Muncan V, Pon Fong MT, Brantjes H, van Leenen D, Holstege FC, Brummelkamp TR, Agami R, Clevers H. 2003. Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 4:609–615. 10.1038/sj.embor.embor865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dimple C, Nair SS, Rajhans R, Pitcheswara PR, Liu J, Balasenthil S, Le XF, Burow ME, Auersperg N, Tekmal RR, Broaddus RR, Vadlamudi RK. 2008. Role of PELP1/MNAR signaling in ovarian tumorigenesis. Cancer Res. 68:4902–4909. 10.1158/0008-5472.CAN-07-5698 [DOI] [PubMed] [Google Scholar]
- 52.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. 2004. Rational siRNA design for RNA interference. Nat. Biotechnol. 22:326–330. 10.1038/nbt936 [DOI] [PubMed] [Google Scholar]
- 53.Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. 2008. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 319:819–821. 10.1126/science.1149250 [DOI] [PubMed] [Google Scholar]
- 54.Changolkar LN, Pehrson JR. 2002. Reconstitution of nucleosomes with histone macroH2A1.2. Biochemistry 41:179–184. 10.1021/bi0157417 [DOI] [PubMed] [Google Scholar]
- 55.Brann DW, Zhang QG, Wang RM, Mahesh VB, Vadlamudi RK. 2008. PELP1—a novel estrogen receptor-interacting protein. Mol. Cell. Endocrinol. 290:2–7. 10.1016/j.mce.2008.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manavathi B, Nair SS, Wang RA, Kumar R, Vadlamudi RK. 2005. Proline-, glutamic acid-, and leucine-rich protein-1 is essential in growth factor regulation of signal transducers and activators of transcription 3 activation. Cancer Res. 65:5571–5577. 10.1158/0008-5472.CAN-04-4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosendorff A, Sakakibara S, Lu S, Kieff E, Xuan Y, DiBacco A, Shi Y, Shi Y, Gill G. 2006. NXP-2 association with SUMO-2 depends on lysines required for transcriptional repression. Proc. Natl. Acad. Sci. U. S. A. 103:5308–5313. 10.1073/pnas.0601066103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh RR, Gururaj AE, Vadlamudi RK, Kumar R. 2006. 9-cis-retinoic acid up-regulates expression of transcriptional coregulator PELP1, a novel coactivator of the retinoid X receptor alpha pathway. J. Biol. Chem. 281:15394–15404. 10.1074/jbc.M601593200 [DOI] [PubMed] [Google Scholar]
- 59.ENCODE Project Consortium. 2004. The ENCODE (ENCyclopedia Of DNA Elements) project. Science 306:636–640. 10.1126/science.1105136 [DOI] [PubMed] [Google Scholar]
- 60.Kininis M, Chen BS, Diehl AG, Isaacs GD, Zhang T, Siepel AC, Clark AG, Kraus WL. 2007. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Mol. Cell. Biol. 27:5090–5104. 10.1128/MCB.00083-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG. 2005. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121:873–885. 10.1016/j.cell.2005.04.031 [DOI] [PubMed] [Google Scholar]
- 62.Kashiwaya K, Nakagawa H, Hosokawa M, Mochizuki Y, Ueda K, Piao L, Chung S, Hamamoto R, Eguchi H, Ohigashi H, Ishikawa O, Janke C, Shinomura Y, Nakamura Y. 2010. Involvement of the tubulin tyrosine ligase-like family member 4 polyglutamylase in PELP1 polyglutamylation and chromatin remodeling in pancreatic cancer cells. Cancer Res. 70:4024–4033. 10.1158/0008-5472.CAN-09-4444 [DOI] [PubMed] [Google Scholar]
- 63.Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, Lamond AI. 2002. Directed proteomic analysis of the human nucleolus. Curr. Biol. 12:1–11. 10.1016/S0960-9822(01)00650-9 [DOI] [PubMed] [Google Scholar]
- 64.Fu Z, Fenselau C. 2005. Proteomic evidence for roles for nucleolin and poly[ADP-ribosyl] transferase in drug resistance. J. Proteome Res. 4:1583–1591. 10.1021/pr0501158 [DOI] [PubMed] [Google Scholar]
- 65.Isabelle M, Moreel X, Gagne JP, Rouleau M, Ethier C, Gagne P, Hendzel MJ, Poirier GG. 2010. Investigation of PARP-1, PARP-2, and PARG interactomes by affinity-purification mass spectrometry. Proteome Sci. 8:22. 10.1186/1477-5956-8-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meder VS, Boeglin M, de Murcia G, Schreiber V. 2005. PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J. Cell Sci. 118:211–222. 10.1242/jcs.01606 [DOI] [PubMed] [Google Scholar]
- 67.Gonugunta VK, Nair BC, Rajhans R, Sareddy GR, Nair SS, Vadlamudi RK. 2011. Regulation of rDNA transcription by proto-oncogene PELP1. PLoS One 6:e21095. 10.1371/journal.pone.0021095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Finkbeiner E, Haindl M, Raman N, Muller S. 2011. SUMO routes ribosome maturation. Nucleus 2:527–532. 10.4161/nucl.2.6.17604 [DOI] [PubMed] [Google Scholar]
- 69.Cong R, Das S, Douet J, Wong J, Buschbeck M, Mongelard F, Bouvet P. 2014. macroH2A1 histone variant represses rDNA transcription. Nucleic Acids Res. 42:181–192. 10.1093/nar/gkt863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Banck MS, Li S, Nishio H, Wang C, Beutler AS, Walsh MJ. 2009. The ZNF217 oncogene is a candidate organizer of repressive histone modifiers. Epigenetics 4:100–106. 10.4161/epi.4.2.7953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reid G, Gallais R, Metivier R. 2009. Marking time: the dynamic role of chromatin and covalent modification in transcription. Int. J. Biochem. Cell Biol. 41:155–163. 10.1016/j.biocel.2008.08.028 [DOI] [PubMed] [Google Scholar]
- 72.Choo JH, Kim JD, Kim J. 2007. MacroH2A1 knockdown effects on the Peg3 imprinted domain. BMC Genomics 8:479. 10.1186/1471-2164-8-479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kayahara M, Ohanian J, Ohanian V, Berry A, Vadlamudi R, Ray DW. 2008. MNAR functionally interacts with both NH2- and COOH-terminal GR domains to modulate transactivation. Am. J. Physiol. Endocrinol. Metab. 295:E1047–E1055. 10.1152/ajpendo.90429.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abbott DW, Chadwick BP, Thambirajah AA, Ausio J. 2005. Beyond the Xi: macroH2A chromatin distribution and post-translational modification in an avian system. J. Biol. Chem. 280:16437–16445. 10.1074/jbc.M500170200 [DOI] [PubMed] [Google Scholar]
- 75.Bernstein E, Muratore-Schroeder TL, Diaz RL, Chow JC, Changolkar LN, Shabanowitz J, Heard E, Pehrson JR, Hunt DF, Allis CD. 2008. A phosphorylated subpopulation of the histone variant macroH2A1 is excluded from the inactive X chromosome and enriched during mitosis. Proc. Natl. Acad. Sci. U. S. A. 105:1533–1538. 10.1073/pnas.0711632105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chu F, Nusinow DA, Chalkley RJ, Plath K, Panning B, Burlingame AL. 2006. Mapping post-translational modifications of the histone variant MacroH2A1 using tandem mass spectrometry. Mol. Cell. Proteomics 5:194–203. 10.1074/mcp.M500285-MCP200 [DOI] [PubMed] [Google Scholar]
- 77.Nagpal JK, Nair S, Chakravarty D, Rajhans R, Pothana S, Brann DW, Tekmal RR, Vadlamudi RK. 2008. Growth factor regulation of estrogen receptor coregulator PELP1 functions via protein kinase A pathway. Mol. Cancer Res. 6:851–861. 10.1158/1541-7786.MCR-07-2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thambirajah AA, Li A, Ishibashi T, Ausio J. 2009. New developments in post-translational modifications and functions of histone H2A variants. Biochem. Cell Biol. 87:7–17. 10.1139/O08-103 [DOI] [PubMed] [Google Scholar]
- 79.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. 2004. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20:3710–3715. 10.1093/bioinformatics/bth456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.