

ABSTRACT
Agnoprotein is a small multifunctional regulatory protein required for sustaining the productive replication of JC virus (JCV). It is a mostly cytoplasmic protein localizing in the perinuclear area and forms highly stable dimers/oligomers through a Leu/Ile/Phe-rich domain. There have been no three-dimensional structural data available for agnoprotein due to difficulties associated with the dynamic conversion from monomers to oligomers. Here, we report the first nuclear magnetic resonance (NMR) structure of a synthetic agnoprotein peptide spanning amino acids Thr17 to Glu55 where Lys23 to Phe39 encompassing the Leu/Ile/Phe-rich domain forms an amphipathic α-helix. On the basis of these structural data, a number of Ala substitution mutations were made to investigate the role of the α-helix in the structure and function of agnoprotein. Single L29A and L36A mutations exhibited a significant negative effect on both protein stability and viral replication, whereas the L32A mutation did not. In addition, the L29A mutant displayed a highly nuclear localization pattern, in contrast to the pattern for the wild type (WT). Interestingly, a triple mutant, the L29A+L32A+L36A mutant, yielded no detectable agnoprotein expression, and the replication of this JCV mutant was significantly reduced, suggesting that Leu29 and Leu36 are located at the dimer interface, contributing to the structure and stability of agnoprotein. Two other single mutations, L33A and E34A, did not perturb agnoprotein stability as drastically as that observed with the L29A and L36A mutations, but they negatively affected viral replication, suggesting that the role of these residues is functional rather than structural. Thus, the agnoprotein dimerization domain can be targeted for the development of novel drugs active against JCV infection.
IMPORTANCE Agnoprotein is a small regulatory protein of JC virus (JCV) and is required for the successful completion of the viral replication cycle. It forms highly stable dimers and oligomers through its hydrophobic (Leu/Ile/Phe-rich) domain, which has been shown to play essential roles in the stability and function of the protein. In this work, the Leu/Ile/Phe-rich domain has been further characterized by NMR studies using an agnoprotein peptide spanning amino acids T17 to Q54. Those studies revealed that the dimerization domain of the protein forms an amphipathic α-helix. Subsequent NMR structure-based mutational analysis of the region highlighted the critical importance of certain amino acids within the α-helix for the stability and function of agnoprotein. In conclusion, this study provides a solid foundation for developing effective therapeutic approaches against the dimerization domain of the protein to inhibit its critical roles in JCV infection.
INTRODUCTION
Determining the three-dimensional (3D) structures of proteins is essential for not only studying their regulatory roles but also developing therapeutic strategies against them. The human polyomavirus JC virus (JCV) encodes a limited number of regulatory proteins, one of which is a 71-amino-acid long agnoprotein. This protein was shown to play important regulatory roles in the JCV replication cycle (1, 2). A counterpart of this protein is also encoded by the human polyomavirus BK virus (BKV) and simian virus 40 (SV40). These three polyomaviruses undergo a productive life cycle in the presence of agnoprotein. Interestingly, the remaining human polyomaviruses, including Merkel cell polyomavirus, human polyomavirus 9 (HPyV9), trichodysplasia spinulosa-associated polyomavirus, HPyV10, HPyV6, HPyV7, KI polyomavirus, and WU polyomavirus (3), do not contain an open reading frame for this protein, nor do they have a productive replication cycle in tissue culture, suggesting that, if provided in trans, agnoprotein may stimulate the growth properties of these viruses. Agnoprotein possesses a highly basic amino acid composition with dominant Arg (R) and Lys (K) residues located at the amino- and carboxy-terminal halves of the protein. The middle portion of the protein is, however, composed of mostly uncharged residues, creating a hydrophobic region rich in Leu/Ile/Phe residues. The two negatively charged residues Glu34 (E34) and Asp38 (D38) are also dispersed within this hydrophobic region. Although Glu34 is highly conserved in the agnoprotein of polyomaviruses, Asp38 is replaced by Glu38 and Gln38 in BKV and SV40, respectively. It is also interesting to note that all three Phe residues of agnoprotein (Phe31, Phe35, and Phe39) localize to its hydrophobic region. The functional importance of these Phe residues has recently been evaluated by genetic and biochemical approaches, and it was demonstrated that they positively influence viral DNA replication (1, 4) and play regulatory roles in the perinuclear distribution of agnoprotein in infected cells (4, 5). Agnoprotein possesses only one highly conserved Cys residue, which is located right after the hydrophobic region of the protein (Cys40).
The 3D structure of full-length agnoprotein has yet to be resolved. However, previous computer modeling studies revealed that the Leu/Ile/Phe-rich domain of the protein is involved in forming an amphipathic α-helix (1, 6, 7) and is highly conserved in the agnoproteins of JCV, SV40, and BKV (1). These 3D modeling studies also suggest that a small portion of the amino-terminal region and carboxy-terminal half of the protein may adopt an intrinsically disordered conformation. Such disordered regions have been shown to provide significant flexibility to a protein to interact with various binding partners and, thus, to diversify and perhaps amplify protein functions (8, 9). For example, recent work by Ou et al. (10) demonstrated that E4-ORF3, a small adenovirus protein, forms a multivalent functional matrix through a dimerization/oligomerization process in infected cells, leading to the inactivation of several tumor suppressor proteins, including p53, TIM24, MRE11/Rad50/NBS1 (MRN), and promyelocytic leukemia factor (10). Similarly, the intrinsically unstructured regions of agnoprotein could play various roles in the function of agnoprotein and thereby contribute to the overall JCV replication cycle (1).
JCV agnoprotein was previously shown to interact with a number of cellular and viral proteins, including YB-1 (11), p53 (12), FEZ1 and HP1-α (13), adaptor protein complex 3 (14), JCV small t antigen (Sm t-Ag) (15), and JCV large T antigen (LT-Ag) (16), and has been implicated in various aspects of the JCV life cycle, including viral replication (4, 17), viral transcription (11), functioning as a viroporin (14, 18), encapsidation (19), and interfering with exocytosis (20). In addition, this protein deregulates cell cycle progression, where cells stably expressing agnoprotein largely accumulate at the G2/M phase of the cell cycle (12). Agnoprotein was also shown to induce apoptosis in differentiating oligodendrocytes (21, 22). Despite all this intense research, the exact regulatory function of agnoprotein in viral replication is not fully understood.
Replication studies with null mutants of agnoprotein showed that this protein is required for sustaining successful propagation of orthopolyomaviruses, including JCV, BKV, and SV40 (2, 23, 24). Even the constitutive expression of LT-Ag is not fully capable of compensating for the loss of agnoprotein function in infected cells. In other words, in the absence of agnoprotein, LT-Ag alone is not able to sustain an efficient viral replication cycle (24). Agnoprotein is a cytoplasmic protein, with high concentrations accumulating in the perinuclear region of infected cells, but a small portion of the protein is also consistently detected in the nucleus, indicating a possible nuclear function for agnoprotein. To support this notion in functional assays, we demonstrated that agnoprotein enhances the DNA binding activity of LT-Ag to the viral origin (Ori) without itself directly interacting with DNA (4). We also demonstrated that agnoproteins of BKV, SV40, and JCV form highly stable, SDS-resistant homodimers and oligomers (7, 18). In particular, the Leu/Ile/Phe-rich region of the protein plays a direct role in forming such structures and is required for the stable expression of the protein (1).
All these studies strongly suggest a critical role for agnoprotein in the life cycle of JCV. JCV lytically infects oligodendrocytes in the central nervous system and causes a white matter disease known as progressive multifocal leukoencephalopathy (PML) in a subset of patients with immunosuppressive conditions, including those with cancer, organ transplantation, and human immunodeficiency virus type 1 (HIV-1) infection (25–28). Little is known about how JCV enters the cells and traffics into the nucleus, although recent studies suggest that certain polysaccharides may be important for cell entry (29, 30). JCV asymptomatically infects a majority of the human population worldwide (70 to 80%) during childhood and establishes a latent asymptomatic infection in the body but can reactivate under a subset of conditions of immune dysfunction. However, the mechanism(s) of reactivation is currently unknown. In recent years, PML has also been encountered in a certain subpopulation of autoimmune disorder patients, including patients with multiple sclerosis and Crohn's disease, who are treated with immunomodulatory drugs (such as natalizumab and efalizumab) targeted against certain cell surface receptors on T cells (31–33). This makes JCV a serious risk factor for those groups of patients.
In this study, we have determined the 3D structure of a synthetic JCV agnoprotein peptide encompassing amino acids Thr17 to Glu54 by nuclear magnetic resonance (NMR) and demonstrate that amino acids Lys23 to Phe39 form an amphipathic α-helix. To assess the contribution of a specific amino acid or groups of amino acids present within the α-helix region to the structure and function of the agnoprotein, NMR structure-based mutational analysis of three groups of mutations (groups I, II, and III) in the region was performed. The mutations were grouped on the basis of their special orientation in the α-helix, each of which represents one of the faces of the helix. In group I, single (E34A and D38A) or combined (E34A+D38A) mutations at the charged residues were found to have a limited effect on both agnoprotein expression and viral DNA replication. In group II, a single mutation (L33A) caused a substantial negative effect on viral DNA replication, while the protein expression profile for the mutant with this mutation remained similar to that of the wild type (WT). A double mutation in this group (I30A+L33A), however, did not have more of an effect than that of the single mutant on either process, suggesting that the I30A mutation has a compensatory effect on the functional loss resulting from the L33A mutation. In group III, the individual mutations L29A and L36A, but not L32A, had a substantial negative effect on both agnoprotein expression and viral DNA replication, while a combination of those mutations (L29A+L32A+L36A) caused even more dramatic effects on both processes. For example, the agnoprotein expression level dropped to undetectable levels, and this effect appeared to be at the level of protein degradation rather than agnoprotein gene transcription, suggesting that these three amino acids are highly critical for the structural integrity and the stability of agnoprotein. Thus, the interface formed between these three residues can primarily be targeted for the development of novel therapeutics against agnoprotein to destabilize its whole protein structure and, therefore, to prevent its activity during the JCV replication cycle in PML patients.
MATERIALS AND METHODS
Prediction of secondary structures and hydrophobicity of agnoprotein.
The primary sequence of the agnoprotein peptide from T17 to Q52 was analyzed to predict the secondary structure present in the peptide. Two programs were used, University College London's PSIPRED server (v3.3; http://bioinf.cs.ucl.ac.uk/psipred/) (34) and the NPS@ (network protein sequence analysis) web server (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sspred.html) available from Pole Bioinformatique Lyonnais (33). A 36-amino-acid region of agnoprotein from T17 to Q52 was predicted to contain a hydrophobic 19-amino-acid α-helix encompassing R24 to G42.
CD.
Circular dichroism (CD) spectra were recorded at room temperature (20°C) in a 0.1-cm-path-length quartz cell on a Jobin-Yvon model C8 spectropolarimeter calibrated with d-camphor-10-sulfonate. The experiments were performed at pH 3.0 either in 100% H2O or in an H2O and 50% 2,2,2-trifluoroethanol (TFE) mixture (1:1, vol/vol). The peptide concentration in each sample was 50 μM. Three averaged scans were collected at 0.5-nm-interval steps using an integration time of 2 s and a light band-pass or slit width of 2 nm. Curves were smoothed from 200 to 260 nm using 25 data points. Respective intensities are expressed as the mean residue molar ellipticity [θ], calculated from the equation [θ]obs/(10 · C · n · L), where [θ]obs is the observed ellipticity)in millidegrees), L is the optical path length (in centimeters [0.1 cm]), C is the final concentration of the peptide (in molar), and n represents the number of amino acid residues.
Synthesis and preparation of the agnoprotein peptide for NMR.
Synthesis of a full-length agnoprotein (>98% pure) is highly challenging and costly for NMR studies. Therefore, a relatively short region of agnoprotein encompassing the dimerization domain of the protein from T17 to Q52 (hereafter referred to as the “agnoprotein peptide”) was used for the present studies (AnaSpect EGT, Fremont, CA). The peptide was synthesized by automated solid-phase synthesis using standard 9-fluorenylmethoxy carbonyl (Fmoc) chemistry and purified by reverse-phase high-performance liquid chromatography (HPLC), using procedures reported previously for the production of retroviral proteins (35). The peptide was analyzed by use of a mass spectrometer and HPLC and found to have a purity of greater than 98%. The agnoprotein peptide showed a strong propensity to aggregate in pure water, and thus, different solvent conditions (H2O, methanol-CDCl3 [50/50], TFE [30%], and dodecylphosphatidylcholine [100 eq]) were examined to determine the optimal conditions for the NMR study.
Analysis of the agnoprotein peptide by NMR.
The results of all NMR experiments were recorded at a sample concentration of 0.5 mM in water with 30% (vol/vol) TFE, pH 3.0, at 293 K, 303 K, and 313 K on an Avance Bruker spectrometer operating at 600.13 MHz and equipped with a cryoprobe. Two-dimensional spectra were obtained with 2,048 real points in the direct dimension t2 and 512 increments in the indirect dimension t1 of acquisition, and a spectral width of 7,183.91 Hz in both dimensions. The transmitter frequency was set to the water signal. The solvent resonance was suppressed by using a 3-9-19 pulse sequence (36) with gradients during the relaxation delay of 1.6 s between free induction decays or by using excitation sculpting with gradients (37). A mixing time of 250 ms was used in the nuclear Overhauser effect spectroscopy (NOESY) experiments (38), and the temperature was controlled externally using a special temperature control system (BCU 05; Bruker) for all experiments. All data were processed using Bruker Topspin (v1.3) software (Biospin; Bruker). A π/6 phase-shifted sine bell window function was applied, and data were zero filled once prior to Fourier transformation in both dimensions. The final size of the frequency domain matrices was 2,048 real points in both dimensions. For all experiments, a 1H frequency scale was directly referenced to water. The data were then analyzed with the spectrum analysis program CcpNmr Analysis (39).
Structure determination for the agnoprotein peptide.
The calculation of the tertiary structure of the peptide was performed iteratively with the software programs ARIA (40) and CNS (41). ARIA was used for the initial CcpNmr Analysis-derived data (39), which generated a table of chemical shifts and a list of nuclear Overhauser effect (NOE) peaks from assigned NMR spectra. A set of distance restraints was generated from NOE cross-signal volumes measured on a 250-ms-mixing-time NOESY spectrometer at 313 K by automatic integration of the peaks in the spectra. Eight iterations were performed by ARIA. For each iteration, 200 structures were calculated and the NOE distance restraints were recalibrated by ARIA after each iteration. No dihedral torsion angle restraints were included in the calculations. The constraint violation threshold was set to 0.2 Å, and systematically violated constraints were eliminated. During the 9th iteration, the structure was analyzed and refined by performing a molecular dynamics step in a water box. This process was repeated until the introduction of all the experimental data was consistent with the lowest-energy structures obtained. The structures were evaluated with the PROCHECK program (42), and PyMOL software (43) was used to visualize the structures. Calculations were performed on a Dell Precision 690 workstation.
Theoretical model of a dimeric structure of the agnoprotein peptide.
The experimental distance restraints obtained for the calculation of the monomeric structure of the agnoprotein peptide were duplicated and used to build a theoretical model of a dimeric structure of the dimerization domain based on the primary sequence and structure homology observed with the C-terminal domain from D52 to S96 of HIV-1 Vpr (44). The Protein Data Bank (PDB; http://www.rcsb.org/pdb/home/home.do) accession number for Vpr is 1X9V (44). The X-PLOR program (45–47) was used to build the dimeric structure with a distance geometry simulated annealing protocol. Energy minimization with X-PLOR was performed in a vacuum; i.e., the dielectric constant was set to 1, and surface charges were removed. All distance restraints were specified with the R-6 averaging option, and the intermolecular NOEs used to build the theoretical dimeric model were determined by homology with the dimeric structure of the C-terminal domain from D52 to S96 of Vpr (44). Distance geometry and simulated annealing regularization metric matrix distance geometry calculations were performed to embed and optimize the initial structures. The 10 best optimized structures (out of 100) were selected on the basis of the criteria of an acceptable covalent geometry, low distance restraint violations, and favorable nonbonded energy. The structures were further optimized with restrained molecular dynamics simulations. Noncrystallographic symmetry constraints were maintained throughout the computations because of the inherent symmetry of the system. The quality of the structures was evaluated with PROCHECK (42) and with the iCing valuation package (48). PyMOL software was used to visualize the structures (43).
Cell lines.
SVG-A is a human cell line established by transformation of primary human fetal glial cells with an origin-defective SV40 mutant (49), and SVG-A cells do not express either SV40 capsid proteins (VPs) or agnoprotein but express SV40 LT-Ag. SVG-A cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (penicillin-streptomycin [100 μg/ml], ciprofloxacin [10 μg/ml]). They were maintained at 37°C in a humidified atmosphere supplemented with 7% CO2.
Plasmid constructs.
The construction of the pMAL-c5x-Agno(1-71) and Bluescript KS(+)-JCV Mad-1 WT plasmids was previously described (7). Various alanine (Ala) substitution mutations on agnoprotein (Agno) in a viral background (JCV Mad-1) were created using a QuikChange site-directed mutagenesis kit (catalog no. 200521; Agilent) and appropriate synthetic oligonucleotides. The resulting plasmids were designated Bluescript KS(+)-JCV Mad-1 Agno(L29A), Bluescript KS(+)-JCV Mad-1 Agno(I30A), Bluescript KS(+)-JCV Mad-1 Agno(L32A), Bluescript KS(+)-JCV Mad-1 Agno(L33A), Bluescript KS(+)-JCV Mad-1 Agno(E34A), Bluescript KS(+)-JCV Mad-1 Agno(L36A), Bluescript KS(+)-JCV Mad-1 Agno(L32A+L36A), Bluescript KS(+)-JCV Mad-1 Agno(I30A+L33A), Bluescript KS(+)-JCV Mad-1 Agno(E34A+D38A), and Bluescript KS(+)-JCV Mad-1 Agno(L29A+L32A+L36A). Each mutant agnoprotein gene was then subcloned into the NcoI/EcoRI sites of the pMAL-c5x vector as described previously (7), and the resulting plasmids were designated pMAL-c5x-JCV Agno(L29A), pMAL-c5x-JCV Agno(I30A), pMAL-c5x-JCV Agno(L32A), pMAL-c5x-JCV Agno(L33A), pMAL-c5x-JCV Agno(E34A), pMAL-c5x-JCV Agno(L36A), pMAL-c5x-JCV Agno(L32A+L36A), pMAL-c5x-JCV Agno(I30A+L33A), pMAL-c5x-JCV Agno(E34A+D38A), and pMAL-c5x-JCV Agno(L29A+L32A+L36A) to produce maltose binding protein (MBP)–agnoprotein (MBP-Agno) fusion proteins as described previously (1). The integrities of the whole viral genome in the viral backgrounds and the mutations made in each construct were verified by DNA sequencing.
Expression and purification of recombinant WT agnoprotein and its mutants.
Bacterial expression plasmids [pMAL-pMAL-c5x alone, pMAL-c5x-JCV Agno(L29A), pMAL-c5x-JCV Agno(I30A), pMAL-c5x-JCV Agno(L32A), pMAL-c5x-JCV Agno(L33A), pMAL-c5x-JCV Agno(E34A), pMAL-c5x-JCV Agno(L36A), pMAL-c5x-JCV Agno(L32A+L36A), pMAL-c5x-JCV Agno(I30A+L33A), pMAL-c5x-JCV Agno(E34A+D38A), and pMAL-c5x-JCV Agno(L29A+L32A+L36A)] were transformed into the Escherichia coli DH5α strain; and MBP-Agno fusion proteins were produced and purified as previously described (1). Briefly, bacterial cultures were grown in 100 ml Luria-Bertani broth (LB) supplemented with ampicillin (100 μg/ml) at 37°C overnight. On the next day, cultures were first diluted 1:15 in 0.5 liter fresh LB supplemented with ampicillin (100 μg/ml) and glucose (2 g/liter) and grown at 37°C to an optical density at 600 nm of 0.5, induced with 0.3 mM isopropyl-β-thiogalactopyranoside (IPTG), and incubated for an additional 2 h at 28°C. Subsequently, bacterial cultures were harvested by centrifugation, and the resulting pellet was resuspended in amylose column buffer (20 mM Tris-HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA). The cell suspension was treated with lysozyme (0.5 mg/ml) for 30 min on ice in the presence of protease inhibitors (catalog no. P8465; Sigma), followed by sonication. Clear lysates were obtained after centrifugation (15,000 rpm; F21-8 × 50y rotor; Thermo Scientific) and were incubated with 200 μl of amylose fast-flow resin (1:1, buffer/beads; catalog no. E8021S; New England BioLabs) prepared in amylose column buffer overnight at 4°C. MBP-Agno fusion protein-bound amylose resins were washed with five column volumes of amylose column buffer, affinity purified, stored on the beads at +4°C until use. The quality of the proteins was analyzed by SDS-PAGE with Coomassie staining as described previously (7).
Indirect immunofluorescence microscopy.
Indirect immunofluorescence microscopy studies were performed as previously described (24). Briefly, SVG-A cells were separately transfected/infected with Bluescript KS(+)-JCV Mad-1 WT and various Bluescript KS(+)-JCV Mad-1 Agno mutant DNAs which had already been linearized by BamHI. At the 5th day posttransfection/infection, cells at subconfluence were seeded on polylysine-coated glass chamber slides (catalog no. 177380; Thermo Scientific), and on the next day, the cells were washed twice with 1× phosphate-buffered saline (PBS), fixed in ice-cold acetone, blocked with 5% bovine serum albumin (prepared in 1× PBS) for 2 h, and incubated overnight with a combination of two different antibodies (anti-JCV Agno primary polyclonal rabbit antibody [1:200 dilution]) [50] plus anti-VP1 monoclonal mouse antibody [pAB597; 1:200 dilution]). Antibody dilutions were made in 1× PBS–0.01% Tween 20 (PBST) incubation buffer, and the samples were incubated in the same buffer. Following incubation of the samples with the primary antibodies, cells were washed with PBST buffer (3 times for 10 min each time) and incubated with a rhodamine-conjugated goat anti-mouse antibody (catalog no.12-509; EMD Millipore) plus fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit secondary antibody (catalog no. AP307F; EMD Millipore) for 45 min. Cells were finally washed with incubation buffer three times for 10 min each time, mounted using Vectashield mounting medium (catalog no. H-1200; Vector Laboratories Inc.), and examined under a fluorescence microscope (Nikon Eclipse TE300; objectives, ×40/×1.3 oil and ×10 eyepiece; Slidebook [v5.0] operating software) for detection of JCV agnoprotein and JCV VP1.
Preparation of whole-cell extracts and Western blotting.
Whole-cell extracts were prepared as described previously (17). Briefly, in parallel with the isolation of viral DNA from transfectants, SVG-A cells (uninfected and infected; 1 × 106 cells/sample) were lysed in a lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.5% NP-40 on a rocking platform at 4°C for 30 min. The cell lysate was then cleared by centrifugation at 15,000 × g for 10 min at 4°C and stored at −80°C until use. For Western blot analysis of the protein samples, 40 μg of the whole-cell extract prepared from uninfected and infected cells was separated on an SDS-15% polyacrylamide gel, transferred onto a nitrocellulose membrane (catalog no. 162-0112; Bio-Rad) for 16 h at 40 mA, and analyzed using antiagnoprotein polyclonal antibody (11). Western blots were developed using a Pierce ECL 2 Western blotting substrate kit (catalog no. PI80196; Thermo Scientific) according to the manufacturer's recommendations.
RT-PCR.
Total RNA was isolated from untransfected cells (SVG-A cells alone) and transfected/infected cells (2.2 × 107 cells) at the 5th day posttransfection/infection, as indicated in the respective figure legends, using an RNeasy Plus minikit (catalog no. 74132; Qiagen), and 15 μg of total RNA was fractionated on a 1% agarose–RNA gel to monitor the integrity of the isolated total RNA. cDNA synthesis was carried out using a SuperScript III one-step reverse transcription (RT)-PCR system (catalog no. 12574-026; Invitrogen). Briefly, 300 ng total RNA (DNase I treated) isolated from different samples was mixed with 1 μl of oligo(dT)12-18 (0.5 μg/μl) and 1 μl of a deoxynucleoside triphosphate mix (10 nM each dATP, dCTP, dGTP, and dTTP), and the reaction volume was brought up to 13 μl. The samples were then heated at 65°C for 5 min and cooled on ice for 1 min. After a brief centrifugation, 4 μl of 5× first-strand buffer (catalog no. 12574-026; Invitrogen), 1 μl dithiothreitol (0.1 M), and 1 μl SuperScript III reverse transcriptase (200 units/μl; Invitrogen) were added to each reaction mixture, and the components were gently mixed by pipetting. The reaction mixtures were then incubated at 55°C for 1 h to synthesize cDNA. Reverse transcriptase was inactivated by heating of the reaction mixture samples at 70°C for 15 min. Finally, RNA complementary to the cDNA was eliminated by incubating the reaction mixture with RNase H (1 U/μl/reaction mixture; Invitrogen) at 37°C for 30 min. The RT products were then PCR amplified as follows. Two microliters of the RT reaction mixture for each sample was subjected to PCR amplification using a Fail Safe DNA polymerase mix (PCR premix F; catalog no. SF99060; Epicentre) according to the recommendations of the manufacturer. The following primers were used in the PCR: Forward-(JCV Mad-1) (covering the region from bp 277 to 303), Reverse-(JCV Mad-1) (covering the region from bp 1338 to 1318), and Reverse-(JCV Mad-1) (covering the region from bp 2533 to 2513). PCR conditions were as follows: 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min for 35 cycles. Amplified gene products were analyzed by agarose gel electrophoresis (1.5%). Each RT product was also used to PCR amplify GAPDH (glyceraldehyde-3-phosphate dehydrogenase) cDNA as a control using the following primers: GAPDH-Forward (5′-TTCTCCCCATTCCGTCTTCC-3′) and GAPDH-Reverse (5′-GTACATGGTATTCACCACCC-3′). The PCR conditions for GAPDH amplification were the same as those described above for JCV transcripts. The DNA bands of interest for each data point were gel purified using a QIAquick gel extraction kit (catalog no. 28704; Qiagen) and were sequenced commercially (Genewiz) to verify the presence of the expected PCR products.
Replication assays.
Replication assays were carried out as previously described (1). Briefly, SVG-A cells were separately transfected/infected with Bluescript KS(+)-JCV Mad-1 WT and the various Bluescript KS(+)-JCV Mad-1 Agno mutant DNAs which had already been linearized by BamHI digestion. Briefly, the linearized viral DNA (8 μg each) was transfected/infected into SVG-A cells (2 × 106 cells/75-cm2 flask) using the Lipofectamine 2000 reagent according to the manufacturer's recommendations (catalog no. 11668027; Invitrogen,). After 5 h of incubation, the transfectants were washed with PBS and refed with DMEM supplemented with 10% FBS and 1% ampicillin-streptomycin. The next day, transfected cells were transferred into 162-cm2 flasks. Half of the volume of the medium was replenished every 3 days posttransfection, and low-molecular-weight DNA containing both input and replicated viral DNA was isolated at the time points indicated below using Qiagen spin columns as described previously (51), digested with the BamHI and DpnI enzymes, separated on a 1% agarose gel, and analyzed by Southern blotting as described previously (24). The DpnI enzyme specifically digests transfected and bacterially methylated DNA and leaves the newly replicated DNA intact. The probes for the Southern blots were prepared from the whole genome of JCV Mad-1 by utilizing a Ready-Prime random labeling kit (catalog no. 300385; Agilent Technologies Inc.).
Accession numbers.
We have deposited the NMR structural data for the agnoprotein peptide in the Biological Magnetic Resonance Bank (BMRB) under accession number 19702. The NMR structural data for the agnoprotein peptide have also been deposited in the Protein Data Bank under accession number 2MJ2.
RESULTS
Agnoprotein is predicted to contain a hydrophobic α-helix.
Analysis of the primary sequence of agnoprotein using the NPS@ web server (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sspred.html) (52) and the PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/) (34) allowed prediction of the presence of 2 α-helices in the full-length sequence of agnoprotein (Fig. 1A). These two predicted helices, from V2 to A10 and G20 to C40, were located in the N-terminal domain and in the central region of the protein, respectively. The rest of the protein, from S11 to S19 and T41 to T71, was predicted to possess an unstructured conformation (Fig. 1A). Analysis of the potential hydrophobic domains in the agnoprotein was performed with the program MPEx (53), which allowed prediction of the presence of a hydrophobic domain spanning residues R24 to G42 (Fig. 1B) which corresponds, more or less, to the second predicted α-helix (G20 to C40).
FIG 1.

Secondary structure prediction for agnoprotein. (A) The full-length sequence of agnoprotein was analyzed by use of the PSIPRED server (34) and the NPS@ web server (52) for secondary structure predictions. These showed the presence of two α-helices (pink cylinders) encompassing residues V2 to A10 and G20 to C40. Other regions of the agnoprotein, S11 to S19 and T41 to T71, are predicted to have an unstructured conformation. Conf., confidence of the prediction; Pred., the prediction of the secondary structure; C, H, and AA, coil, helix, and amino acid, respectively. (B) Prediction of the hydrophobic domains of agnoprotein. The program MPEx is an interactive tool that facilitates the characterization and the prediction of favorable hydrophobic regions in proteins using experiment-based physical and biological hydrophobicity scales (53). One hydrophobic domain spanning residues R24 to G42 approximately corresponding to the second predicted α-helix from G20 to C40 was identified. (C) The circular dichroism spectra of the agnoprotein peptide in 100% H2O and in 50% TFE (TFE-dH2O [1:1, vol/vol]) at pH 3.0 were determined as described in Materials and Methods.
Analysis of the agnoprotein peptide by CD.
CD experiments on the agnoprotein peptide were performed in 100% H2O and in 50% TFE at pH 3.0 (Fig. 1C). The spectra of the agnoprotein peptide in both solutions displayed the characteristic negative bands at 209 nm (parallel π → π* transitions) and at 222 nm and indicate the formation of an α-helix. The part of the spectrum containing the maximum at 190 nm was not completely represented because the signal was saturated. The results showed that the agnoprotein peptide adopts an α-helical structure in both distilled H2O (dH2O) and 50% TFE and the helical content in the spectrum for agnoprotein in 50% TFE seems to be higher than that in the spectrum for agnoprotein in 100% H2O (Fig. 1C).
Optimization of NMR spectroscopy conditions for determining agnoprotein structure.
One-dimensional (1D) proton NMR experiments were recorded either in pure water or in the presence of 30% TFE (vol/vol) at two different temperatures, 293 K and 313 K (Fig. 2). The spectrum in H2O (Fig. 2A) showed very broad peaks due the tendency of the peptide to oligomerize or aggregate. The aggregation of the peptide was prevented by using TFE, as evidenced by a narrowing of the line width, a spreading of the resonances, and a stabilization of the secondary structures of the peptide. Increasing the temperature from 293 K (Fig. 2B) to 313 K (Fig. 2C) resulted in a reduction of the line width. Similar results were obtained when TFE was used to solubilize HIV-1 Vpr (54). The hydrophobic solvent (30% TFE) prevented protein oligomerization but forced the structure to adopt an open U-shaped form by disrupting the intramolecular hydrophobic interactions. Previously, the use of acetonitrile (CH3CN), which is less hydrophobic than TFE, prevented oligomerization of Vpr but allowed the tertiary structure of the protein to be maintained due to hydrophobic interactions between the three helices (43, 47, 54). However, in this study, we used 30% TFE to prevent aggregation of the agnoprotein peptide.
FIG 2.

Comparison of the 1D 1H NMR spectra of the agnoprotein peptide determined under different conditions. The spectra were acquired on an Avance Bruker spectrometer operating at 600.13 MHz and equipped with a cryoprobe. Experiments were carried out to assess the ability of various conditions to increase the solubility of the agnoprotein peptide and to prevent its aggregation. (A) In pure water at pH 3.0, the peptide appears to be aggregated. (B) In the presence of 30% (vol/vol) TFE, pH 3.0, at 293 K, the peptide appears to be structured with a large spreading of the chemical shifts, but the line width remains relatively broad. (C) The assay was performed under the same conditions used for the assay whose results are presented in B but at a temperature of 313 K, which allows spectra with a narrower line width to be obtained. All experiments were conducted using a 0.5 mM concentration of the agnoprotein peptide. (D) A representative NOESY spectrum of the agnoprotein peptide. The spectrum was recorded with a mixing time of 200 ms in the presence of 30% (vol/vol) TFE at pH 3.0 and a temperature of 313 K. Only the region of the spectrum showing the interresidual correlations between the α proton (i) and the amide proton (i + 1) is shown. The intraresidual correlations for each residue (i) are labeled.
NMR analysis of the agnoprotein peptide.
Double-quantum-filtered correlated spectroscopy (DQF-COSY) (55, 56) and total correlated spectroscopy (TOCSY) (57) experiments were used to identify the spin systems, and NOESY experiments (38, 58) were used for sequential and medium-range distance assignment (59). Many overlaps occur in the NH/H-α region of the NOESY spectra (Fig. 2D), and characteristic connectivities dαN (i, i + 3) and dαβ (i, i + 3) and strong NOEs dNN (i, i + 1) and dβN (i, i + 1) (where i and i + 1 indicate the α proton and the amide proton, respectively) were identified, indicating the presence of an α-helix spanning residues K23 to F39 (Fig. 2D). The chemical shift index (CSI) presented in Fig. 3C is consistent with these data. The N- and C-terminal moieties from T17 to K22 and C40 to Q52 of the peptide do not present any particular secondary structure element (Fig. 3). The full 1H resonance assignment was performed and is shown in Table 1.
FIG 3.

Summary of sequential and short-range NOE connectivities for the agnoprotein peptide. (A) Amino acid sequence of the agnoprotein peptide. (B) Characteristic strong dNN (i, i + 1), dαN (i, i + 1), medium-range dαN (i, i + 2; i, i + 3; i, i + 4), and dαβ (i, i + 3) signals identified by TOCSY, DQF-COSY, and NOESY experiments. α, β, and N are the alpha, beta, and amide protons of each amino acid, respectively. dαβ, dNN, and dβN are the distances between alpha and beta, between amide and amide, and between beta and amide protons of two distinct residues, respectively. The thickness of the lines is related to the estimated intensity of the NOE (strong, medium, and weak). (C) The Δδ and CSI represented indicate the secondary structure formation according to the H-α chemical shift. (D) Schematic organization of the agnoprotein peptide, showing the secondary structures deduced from the NOESY spectra. Δδ is the chemical shift index (CSI) used to display and identify the location and the type of protein secondary structures using 1Hα chemical shift data, which are shifted upfield in helices and downfield in beta strands relative to their random coil values.
TABLE 1.
Proton chemical shifts in the presence of 30% TFE calibrated to H2O
| Position and residue | Proton chemical shift(s) (ppm) |
|||||||
|---|---|---|---|---|---|---|---|---|
| NH | H-α | H-β | H-γ | H-δ | H-ε | H-ζ | H-η | |
| 2 Trp | 8.78 | 4.83 | 3.35, 3.35 | 7.34 | 10.04, 7.66 | 7.50, 7.15 | 7.23 | |
| 3 Ser | 8.17 | 4.31 | 3.78, 3.87 | |||||
| 4 Gly | 7.50 | 3.68, 3.77 | ||||||
| 5 Thr | 7.82 | 4.14 | 4.26 | 1.29 | ||||
| 6 Lys | 8.04 | 4.14 | 1.80, 1.86 | 1.48, 1.48 | 1.65, 1.65 | |||
| 7 Lys | 7.90 | 4.18 | 1.86, 1.86 | 1.46, 170 | ||||
| 8 Arg | 7.88 | 4.12 | 1.79, 1.91 | 1.64, 1.65 | 3.21, 3.21 | 7.15 | ||
| 9 Ala | 8.03 | 4.12 | 1.48 | |||||
| 10 Gln | 8.04 | 4.04 | 2.18, 2.18 | 2.37, 2.48 | 6.95, 6.62 | |||
| 11 Arg | 7.77 | 4.08 | 1.98, 1.98 | 1.72, 1.76 | 3.20, 3.20 | 7.19 | ||
| 12 Ile | 7.78 | 3.91 | 2.00 | 0.97; 1.71, 1.20 | 0.86 | |||
| 13 Leu | 7.89 | 4.14 | 1.76, 1.76 | 0.93, 0.93 | ||||
| 14 Ile | 7.83 | 3.80 | 1.92 | 0.90; 1.72, 1.28 | 0.86 | |||
| 15 Phe | 7.69 | 4.36 | 3.36, 3.25 | 7.26 | ||||
| 16 Leu | 8.43 | 4.10 | 1.93, 1.93 | 1.74 | 0.91, 0.95 | |||
| 17 Leu | 8.49 | 4.11 | 1.90, 1.88 | 1.63 | 0.88, 0.93 | |||
| 18 Glu | 8.29 | 3.94 | 2.06, 2.24 | 2.67, 2.39 | ||||
| 19 Phe | 8.24 | 4.23 | 3.24, 2.97 | 7.15 | 7.25 | 7.09 | ||
| 20 Leu | 8.61 | 4.04 | 1.97, 1.97 | 1.63 | 0.87, 0.93 | |||
| 21 Leu | 8.57 | 4.11 | 1.84, 1.84 | 1.53 | 0.93, 0.86 | |||
| 22 Asp | 8.18 | 4.44 | 2.68, 2.93 | |||||
| 23 Phe | 8.33 | 4.33 | 2.85, 3.11 | 7.17 | 7.25 | |||
| 24 Cys | 8.41 | 4.30 | 2.97, 3.06 | |||||
| 25 Thr | 7.87 | 4.30 | 4.34 | 1.28 | ||||
| 26 Gly | 7.95 | 3.95, 3.95 | ||||||
| 27 Glu | 8.04 | 4.24 | 2.08, 1.97 | 2.37, 2.37 | ||||
| 28 Asp | 8.20 | 4.69 | 2.89, 2.83 | |||||
| 29 Ser | 7.99 | 4.48 | 3.97, 3.88 | |||||
| 30 Val | 7.95 | 4.10 | 2.17 | 0.97, 0.97 | ||||
| 31 Asp | 8.18 | 4.66 | 2.85, 2.85 | |||||
| 32 Gly | 8.12 | 3.95, 3.95 | ||||||
| 33 Lys | 7.89 | 4.35 | 1.83, 1.83 | 1.46, 1.46 | 1.70, 1.70 | 3.01, 3.01 | ||
| 34 Lys | 8.05 | 4.35 | 1.76, 1.91 | 1.49, 1.49 | 1.65,1.65 | 3.20, 3.20 | ||
| 35 Arg | 8.15 | 4.36 | 1.90, 1.79 | 1.68, 1.68 | 3.21,3.21 | |||
| 36 Gln | 7.92 | 4.26 | 2.15, 1.99 | 2.33, 2.33 | 7.41, 6.65 | |||
Structural calculations of the monomeric α-helical domain of the agnoprotein peptide.
A total of 452 interproton distance restraints were identified on the 250-ms-mixing-time NOESY spectra recorded at pH 3.0 and 313 K in the presence of 30% (vol/vol) TFE, and all were used for calculation of the structure of the JCV agnoprotein domain using the software ARIA, which facilitates assignment of the NOE constraints and manages the resulting ambiguities. The ambiguous distance restraint method, first introduced by Nilges (40), allows the use of ambiguous distance constraints iteratively in molecular modeling. Among these distance restraints, 293 were intraresidual, 100 were sequential, and 59 were medium range. The structures were calculated by restrained simulated annealing and energy minimization, and 17 structures were selected according to their lowest total energy and number of NOE restraint violations for structural analysis (Table 2) and for generation of the average structure. The 17 best structures of the protein were superimposed on the backbone atoms of the domain from K23 to F39 (Fig. 4B).
TABLE 2.
Structural statistics for the monomeric structure of JCV agnoprotein peptide
| Parametera | Value for JCV agnoprotein peptide monomer |
|---|---|
| No. of restraints for calculation | 452 |
| Unambiguous | 369 |
| Ambiguous | 83 |
| Total no. of NOE restraints | 452 |
| Intraresidue | 293 |
| Sequential (|i − j| = 1)a | 100 |
| Medium range (|i − j| ≤ 4) | 59 |
| RMSD structure statistics | |
| Bonds (Å) | 2.7 × 10−3-2.93 × 10−3 |
| Bond angles (°) | 0.44–0.50 |
| Improper torsions (°) | 0.88–1.23 |
| NOE restraints (Å) | 1.34 × 10−2-1.63 × 10−2 |
| Final energies (kcal/mol) | |
| Total | −1,146 to −998.6 |
| Bonds | 4.42–5.21 |
| Angles | 32.43–41.91 |
| Improper angles | 9.78–17.38 |
| Dihedrals | 164.5–167.86 |
| van der Waals | −98.55 to −55.67 |
| Electrostatic | −1,270.34 to −1,173.67 |
| NOE | 4.08–5.98 |
| Ramachandran plot of residues (%) in: | |
| Most favored regions | 80.3 |
| Additional allowed regions | 18.0 |
| Generously allowed regions | 1.7 |
| Disallowed regions | 0 |
| Atomic RMSD (Å) on backbone atoms (K23 to F29) | |
| Pairwise | 0.66 ± 0.22 |
| To mean structure | 1.04 ± 0.15 |
j represents an amino acid different from amino acid i. Residues represented by i and j may be consecutive in the sequence or separated by 1, 2, or 3 residues.
FIG 4.

NMR structure of the agnoprotein peptide in the presence of 30% TFE (vol/vol) at pH 3.0 and a temperature of 313 K calculated using ARIA (40) and CNS (41). (A) The NMR structure-based α-helical region and the Leu/Ile/Phe-rich domain of agnoprotein within the α-helix are indicated. (B) Superimposition of the 17 energetically most favorable structures of the peptide on the backbone atoms of the domain from K23 to F39 of the averaged structure. The α-helix and unstructured regions are in red and green, respectively. The structure is characterized by two unstructured regions from residues T17 to K22 and C40 to Q52, which are colored green. In some structures, a short α-helix from E43 to D47 is formed in the C-terminal domain. (C) Helical wheel representation of the α-helix (K23 to F39) of the agnoprotein peptide. Hydrophilic and hydrophobic residues are colored blue and red, respectively. The distribution of the residues on the two sides of the helix suggests amphipathic properties for this structure. The α-helical domain from residues K23 to F39 shows amphipathic properties: hydrophobic residues (I28, L29, I30, F31, L32, L33, F35, L36, L37, and F39) form a large surface of interaction, while hydrophilic charged residues (K23, E34, R27, and D38) form a very narrow surface.
NMR structure analysis of the agnoprotein peptide reveals the presence of an α-helix.
Analysis of the agnoprotein peptide shows that the K23 to F39 domain assumes an α-helical structure which is surrounded by two unstructured domains in its N- and C-terminal regions (Fig. 4B). In some structures, the E43 to D47 region near the C terminus also contains a short α-helix (Fig. 4B). The root mean square deviation (RMSD) calculated on the backbone atoms of the helix from K23 to F39 and for all nonhydrogen atoms was evaluated to be 0.66 ± 0.22 Å with respect to the mean coordinates, and the pairwise RMSD was calculated to be 1.04 ± 0.15 Å (Table 2). The domain from K23 to F39 has been represented in a helical wheel projection and shows hydrophobic properties with the structural characteristics of an amphipathic helix (Fig. 4C). The agnoprotein peptide sequence used in the NMR study is shown in Fig. 4A. The space-filling demonstration of the NMR structure of agnoprotein reveals two faces of opposite polarity, a large hydrophobic surface (Fig. 5A and B) containing residues L29, L36, A25, L32, F39, I28, and F35 and a smaller hydrophilic surface on the opposite side of the helix composed of residues K23, E34, R27, and D38 (Fig. 5A and B). The amphipathicity of this helical domain may explain the propensity of the JCV agnoprotein to aggregate and to dimerize/oligomerize.
FIG 5.

Repartition of the hydrophobic and hydrophilic residues in the α-helix from residues K23 to F39 of the agnoprotein peptide. (A) A surface representation shows the large surface occupied by the hydrophobic residues (red, left) and the hydrophilic residues (blue, right). (B) Cartoon representation of the α-helical domain from residues K23 to F39 (gray), with hydrophobic residues (I28, L29, I30, F31, L32, L33, F35, L36, L37, and F39) shown in red and hydrophilic charged residues (K22, K23, E34, R27, and D38) shown in blue.
Theoretical model for the dimeric structure of the agnoprotein peptide.
We previously reported that agnoprotein forms dimers and oligomers through its Leu/Ile/Phe-rich domain (1). Our goal for this study was to obtain a possible theoretical homodimeric structure of agnoprotein using the NMR data to support our previous findings. Future goals include obtaining various theoretical oligomeric structures for agnoprotein once its full-length NMR structure is resolved. To obtain the theoretical structure of a homodimer of the agnoprotein helical domain, the NOE constraints identified by NMR for the monomeric structure of the peptide were duplicated and applied to each monomer within the dimer. The constraints involving the hydrophobic leucine residues L29, L32, and L36 in the agnoprotein were assigned by homology with those experimentally determined in the leucine zipper domain of the C-terminal region of Vpr (Fig. 6E). These constraints were used unambiguously (as defined for NMR constraints in the ARIA program) to build a theoretical dimeric structure of the agnoprotein dimerization domain using the program X-PLOR (60). The structure of the dimer of the JCV agnoprotein domain was calculated by restrained simulated annealing, molecular dynamics, and energy minimization implemented with a total of 368 unambiguous intramolecular NOE-derived distances for each monomer and 40 intermolecular constraints unambiguously defined. Out of 100 calculated structures, 10 were selected according to their low overall energy and their low number of distance restraint violations. None of the structures exhibited NOE violations greater than 0.2 Å, and all of them presented good covalent geometry, with no bond or angle violations. The distribution of the φ and ψ angles revealed that 62.3% of the residues were found in the most favorable regions, 30.6% were located in additional allowed regions, and 6.5% were located in the generously allowed regions. Only 0.6% of the residues were found in disallowed regions (Table 3). The superimposition of the 10 lowest-energy structures on the backbone atoms of residues R24 to C40 of the average structure showed a good convergence of the structures with an average RMSD of 0.60 ± 0.15 Å to the mean structure represented in Table 3. A pairwise RMSD between structures was evaluated to be 0.89 ± 0.24 Å (Table 3). Input data for structural calculations of the theoretical model of the dimer and statistics are given in Table 3. Each domain within the dimeric structure displays the same characteristics seen in the monomer structure. The two amphipathic α-helices associate in an antiparallel manner through interactions between hydrophobic residues L29, L32, and L36 from each monomer (Fig. 6A). The starting point of the refined model of agnoprotein was based on the Vpr NMR structure. To further support the validity of the theoretical dimer formation, we superimposed the 17 best calculated dimer structures on top of the average structure and have demonstrated that all 17 structures form homodimers through their α-helical agnoprotein domains (Fig. 6B). The relative orientation of the residues essential for the formation of the interface (L29, L32, and L36) is represented on a helical wheel and marked with arrows in Fig. 4C. It is of interest to note that an extra hydrophobic surface constituted by residues I30 and L33 is also possible for intra- and intermolecular interactions and dimer/oligomer formation (Fig. 6C). Another interface made up of the three phenylalanines (F31, F35, and F39) is also available for inter- or intramolecular interactions as shown in Fig. 4C. It is also important to note that the calculated dimeric models are only theoretical structures and may represent a few of the multiple potential conformational variants that are present in the infected cells. In addition, since agnoprotein is found in the nucleus, cytoplasm, and, perhaps, cellular membranes, it is possible that agnoprotein exists in different conformations in different regions of infected cells to allow functional flexibility at each location.
FIG 6.

Theoretical model of the dimeric structure of agnoprotein. (A) The energetically most favorable structure of agnoprotein calculated with X-PLOR (45, 46) is shown with the backbone in a ribbon representation and the side chains of the important hydrophobic and aromatic residues shown in stick representations. The structure is characterized by two antiparallel α-helical domains from residues K23 to F39 with an interface composed of residues, L29, L32, and L36 (orange). Residues I30 and L33 (dark orange) are perpendicular to this interface, and aromatic residues (green) are on the opposite face of the α-helix. The domain from residues K23 to F39 is well structured with good convergence, while the N- and C-terminal domains (not shown) are unstructured. (B) Seventeen different dimeric structures of the agnoprotein peptide were superimposed on the average structure. Hydrophobic amino acids (L29, L32, and L36) at the interface of the protein peptide are colored red. (C) An alternative theoretical model of the dimeric structure of agnoprotein. The energetically most favorable structure of agnoprotein was calculated using X-PLOR (45, 46), as described for panel A. (D) Surface-filling representation of the NMR structure of agnoprotein. The positions of the various amino acids in the α-helix are indicated. (E) Sequence alignment of Vpr and JCV agnoprotein within their dimerization domains. Primary sequences for both Vpr and JCV agnoprotein are shown. Sequences labeled in red for both proteins were aligned using the Multalin (77) and CLUSTALW (78) programs. The best correspondence in the alignment was found using Multalin when L29, I30, L33, L36, and L37 in the JCV agnoprotein corresponded, respectively, to L60, I61, L64, L67, and L68 in Vpr.
TABLE 3.
Structural statistics for theoretical model of JCV agnoprotein peptide dimer
| Parameter | Value for JCV agnoprotein peptide dimer |
|---|---|
| Total no. of NOE restraints | |
| Intraresidue | 229 |
| Sequential (|i − j| = 1)a | 90 |
| Medium range (|i − j| ≤ 4) | 49 |
| Intermolecular | 40 |
| RMSD structure statistics | |
| Bonds (Å) | 8.48 × 10−4–9.48 × 10−4 |
| Bond angles (°) | 0.424–0.426 |
| Improper torsions (°) | 0.317–0.320 |
| NOE restraints (Å) | 2.90 × 10−3–5.93 × 10−3 |
| Final energies (kcal/mol) | |
| Total | 70.97–72.98 |
| Bonds | 0.873–1.090 |
| Angles | 60.05–60.69 |
| Improper angles | 9.58–9.75 |
| van der Waals | 0.03–0.79 |
| NOE | 0.37–1.58 |
| Ramachandran plot of residues (%) in: | |
| Most favored regions | 62.3 |
| Additional allowed regions | 30.6 |
| Generously allowed regions | 6.5 |
| Disallowed regions | 0.6 |
| Atomic RMSD (Å) on the backbone atoms (K23 to F39) | |
| Pairwise | 0.89 ± 0.24 |
| To mean structure | 0.60 ± 0.15 |
j represents an amino acid different from amino acid i. Residues represented by i and j may be consecutive in the sequence or separated by 1, 2, or 3 residues.
Effect of NMR-based mutations on dimer/oligomer formation by agnoprotein.
We have recently reported the formation of highly stable dimers/oligomers for the agnoproteins of JCV, BKV, and SV40 (7) and also demonstrated that the Leu/Ile/Phe-rich region of the protein plays an essential role in this phenomenon (1). Our NMR studies confirmed that the Leu/Ile/Phe-rich region is a part of an α-helix (Fig. 4A and B). In addition, modeling studies using NMR structural data suggested the possibility that residues Leu29, Leu32, and Leu36 (Fig. 6A) or Ile30 and Leu33 (Fig. 6C) may facilitate homodimer formation by JCV agnoprotein. Surface representation of the α-helical region highlighted the spatial orientation of residues on each face of the α-helical region (Fig. 6D). On the basis of the NMR structural data and the spatial orientation of the amino acid within the different faces of the α-helix, we made single or multiple Ala substitution mutations and grouped them into three groups: group I, I30A, L33A, and I30A+L33A; group II, E34A and E34A+D38A; and group III, L29A, L32A, L36A, L32A+L36, and L29A+L32A+L36A. We then analyzed them for their ability to form dimers and oligomers relative to that of the WT. For this purpose, these agnoprotein mutants were fused to maltose binding protein (MBP) as described in Materials and Methods, expressed in E. coli as MBP-Agno fusion proteins, and affinity purified. As we previously demonstrated, the stable dimer and oligomer formation by agnoprotein can be monitored by employing SDS-PAGE under reducing conditions, followed by Coomassie blue staining in vitro (1, 7). As shown in Fig. 7A, MBP-WT agnoprotein readily formed stable dimers (lane 3) and oligomers, while MBP alone did not (lane 1) (1). Similarly, we also analyzed the ability of all mutants to form dimers/oligomers. Neither a single mutation (I30A) nor double mutations (I30A+L33A) had a drastic effect on the ability of the mutants to form dimers or oligomers of agnoprotein. However, these mutants showed an extra banding pattern, migrating faster than agnoprotein monomers on SDS-polyacrylamide gels than the WT (Fig. 7B, lanes 3 to 5, arrow). Such a banding pattern was not present for either the E34A or the E34A+D38A mutant (Fig. 7C; compare lane 2 to lanes 3 and 4). In parallel, dimer/oligomer formation for single (L29A, L32A and L36A), double (L32A+L36A), and triple (L29A+L32A+L36A) mutants was also evaluated. Results showed that, similar to the observations for the I30A and L33A mutants, no significant change in dimer/oligomer formation was observed compared to that for the WT, but an extra and faster-migrating band for monomers of agnoprotein still persisted for the single mutant (Fig. 7D, lanes 3 to 5) and double mutant (Fig. 7D, lane 6). However, the triple (L29A+L32A+L36A) mutant showed noticeable degradation products resulting from agnoprotein monomers (Fig. 7D, bracket), suggesting that Leu29, Leu32, and Leu36 may play important roles in the stability and function of agnoprotein. Another interesting point about this triple mutant was that it was expressed at considerably lower levels than the WT during the expression of this mutant protein in E. coli. These dimerization findings are consistent with our theoretical dimerization studies where these three residues (L29A, L32A, and L36A) were found to play a role in dimer formation (Fig. 6A). Thus, it appears that these residues play critical roles in the stability of agnoprotein. The relative level of dimer/oligomer formation by all three groups of mutants (groups I, II, and III) is summarized in Table 4.
FIG 7.

Dimer/oligomer formation by α-helix domain mutants of agnoprotein. (A) MBP and MBP-Agno were expressed in E. coli, purified, and analyzed by SDS-8% PAGE, followed by Coomassie blue staining, as described in Materials and Methods. (B to D) Alanine (Ala, A) substitution mutations (I30A, L33A, I30+L33A, E34A, E34+D38A, L29A, L32A, L36A, L32A+L36A, L29A+L32A+L36A) were expressed in E. coli, purified, and analyzed by SDS-8% PAGE, followed by Coomassie blue staining, as described in Materials and Methods. Arrows, a distinct band migrating faster than the WT monomer; bracket (in panel D), degraded agnoprotein monomers; lanes Markers and M, molecular size markers (in kilodaltons).
TABLE 4.
Dimer/oligomer formation and protein expression by JCV agnoprotein mutants
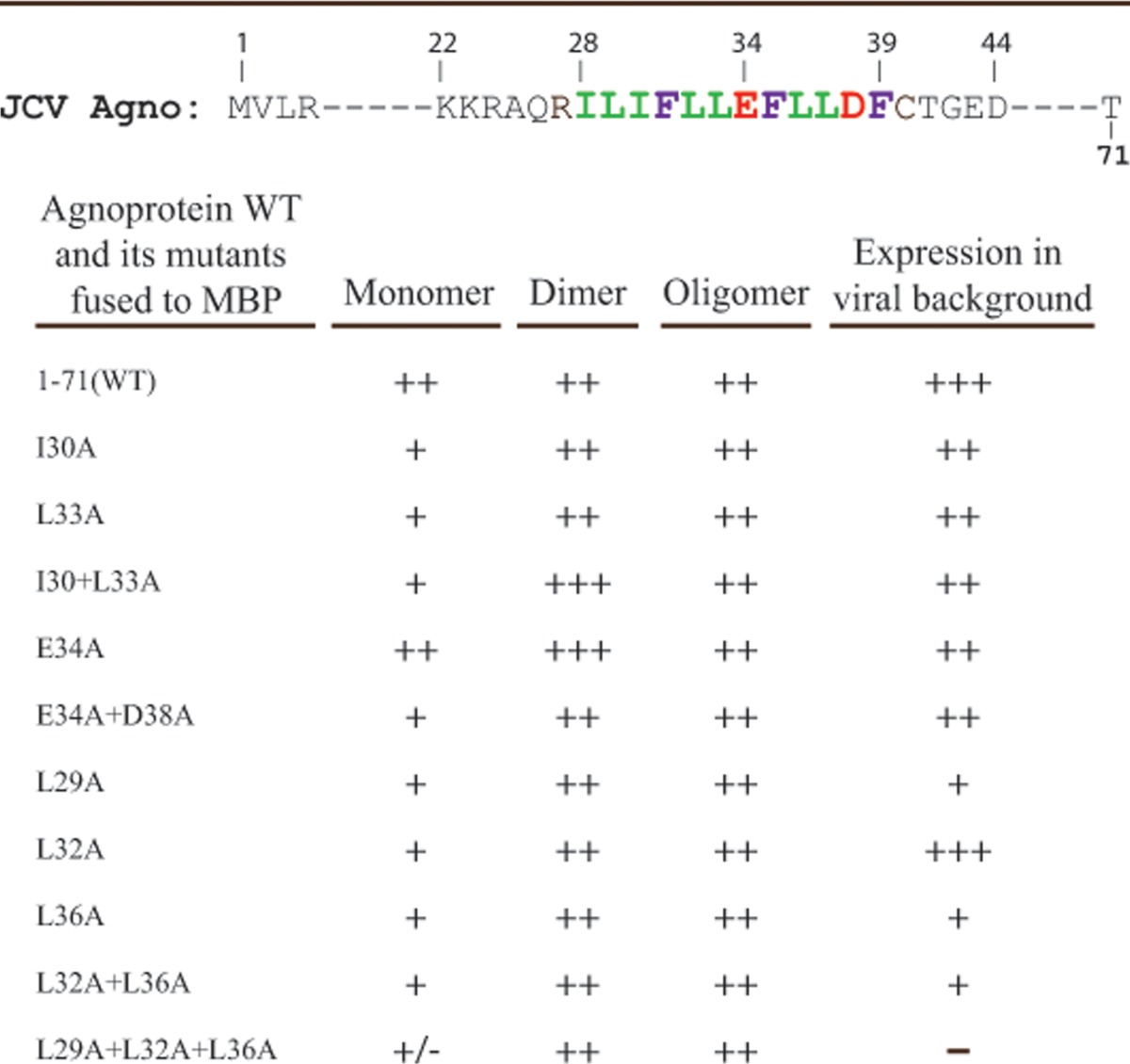
Expression properties of the α-helix domain mutants in the viral background.
JCV grows more slowly in tissue culture systems than its counterparts, BKV and SV40. It takes 6 to 7 days to complete its first full life cycle in SVG-A cells, whereas it takes SV40 and BKV 2 to 3 days to complete the first full life cycle in appropriate cell lines. The expression profile of various NMR-based agnoprotein mutants in transfected/infected cells was analyzed by Western blotting. For this, whole-cell extracts were prepared from SVG-A cells transfected/infected with either the JCV Mad-1 WT genome or mutant genomes carrying Ala mutations in the dimerization domain of agnoprotein and analyzed by Western blotting using antiagnoprotein antibody. As shown in Fig. 8A, the level of agnoprotein expression was readily detectable both for WT agnoprotein and for a single E34A or double E34A+E38A mutant, although the expression levels of both mutants were found to be lower than that of the WT for both the 5th and 15th day posttransfection/infection data points. Note that, as previously reported (7), a faster-migrating band for agnoprotein WT that was absent for both mutants was also detected at the 15th day data point. It is believed that it is either a monomer or a cleavage product of agnoprotein in JCV-infected cells.
FIG 8.

Analysis of the expression levels of the α-helix domain mutants of agnoprotein by Western blotting. (A to D) SVG-A cells (2 × 106 cells/75-cm2 flask) were transfected/infected with Mad-1 Agno WT or Mad-1 Agno Ala substitution mutant genomes (8 μg each) separately. Whole-cell extracts were prepared at the time points indicated and evaluated for the expression level of agnoprotein (WT and mutant) by Western blotting (40 μg protein/lane) using antiagnoprotein antibody (50), as described in Materials and Methods. Blots were also stripped and reprobed for anti-β actin antibody (clone EP1123Y, rabbit monoclonal antibody; Millipore) to monitor for the equal loading of each lane. − Cont., whole-cell extracts prepared from untransfected SVG-A cells loaded as a negative control. Numbers on the left are molecular mass markers (in kilodaltons).
In parallel, the expression levels of the Ala substitution mutants with mutations at two hydrophobic residues, Ile30 and Leu33, which are located on the same face of the helix (the L33A single mutation or the I30+L33A double mutation) were also analyzed (Fig. 8B). The levels of protein expression for both mutants were found to be relatively reduced compared to that for the WT at both data points (5th and 15th days). A faster-migrating band associated with WT and L33A was not detectable for the double (I30A+L33A) mutant under these conditions.
A third group of mutants with mutations which were also located at the same face of the α-helix (Fig. 6D) was also analyzed for agnoprotein expression in the viral background by Western blotting. The expression profile of a single L32A mutant was identical to that of the WT (Fig. 8C, lanes 3 and 7). However, when both Leu32 and Leu36 were together mutated to Ala (L32A+L36A), protein expression was drastically reduced (98% and 89% reductions for the 5th and 15th day data points, respectively) compared to that for the WT, suggesting a critical contribution of Leu36 to agnoprotein stability, because the L32A mutation alone did not show a noticeable effect on protein expression. Moreover, we were unable to detect agnoprotein expression at all when all three residues (Leu29, Leu32, and Leu36) were mutated to Ala (Fig. 8C, lanes 5 and 9). Finally, we also investigated the individual contributions of Leu29 and Leu36 to the stability of agnoprotein in transfected/infected cells by Western blotting. The results showed that the expression levels of both proteins were significantly reduced compared to those for the WT (Fig. 8D; compare lane 2 to lanes 3 and 4). These results collectively suggest that residues L29 and L36, individually and in combination, significantly contribute to the stable expression of agnoprotein in infected cells.
The L29A+L32A+L36A triple mutant expresses viral late transcripts.
The JCV late genome produces a polycistronic message which undergoes alternative splicing (1) and produces two primary messages (M1 and M2) following the removal of intron 1 and intron 2 (Fig. 9A). We have recently demonstrated that these splicing events occur for the WT and for some other dimerization domain mutants previously characterized by RT-PCR followed by DNA sequencing (1).
FIG 9.

Analysis of the expression levels of various agnoprotein mutants by RT-PCR. (A) Graphical presentation of JCV Mad-1 WT late messages M1 and M2 prior to splicing. The positions of intron 1 and intron 2 (according to the JCV Mad-1 strain numbering) are indicated. (B) RT-PCR amplification of the JCV late transcripts using different primers. Total RNA was isolated from SVG-A cells either untransfected (lane 2, negative control [− Cont.]) or transfected/infected with JCV WT or various agnoprotein mutant genomes, as indicated (lanes 3 to 6), and was subjected to RT-PCR using specific primers, as described in Materials and Methods. DNA bands of interest for each data point were gel purified and sequenced to verify the presence of the spliced forms of the M1 and M2 products. In lane 7, the JCV Mad-1 WT genome (10 ng) was PCR amplified using the specific primers described for the experimental samples and used as a positive control (+ Cont.) in gel analysis. Transf./Inf., transfection/infection. (C) (Top) Each RT product was also subjected to PCR to amplify the GAPDH cDNA as a control for RT-PCRs using specific primers, as described in Materials and Methods; (bottom) total RNA was also analyzed on an agarose gel to verify the integrity of the isolated RNA that was used for RT-PCRs. Numbers on the left in panels B and C are molecular mass markers (in kilodaltons).
An undetectable level of agnoprotein expression from a triple (L29A+L32A+L36A) mutant by Western blotting (Fig. 8C) suggested the possibility that the observed effect would be at the level of transcription rather than at the level of translation. To distinguish between these two possibilities, an RT-PCR analysis was performed with total RNA isolated from SVG-A cells transfected/infected either with the JCV WT genome (control) or with various mutants with mutations in the JCV agnoprotein genome, followed by DNA sequencing of the RT-PCR products as described in Materials and Methods.
As shown in Fig. 9B, results from RT-PCR studies clearly demonstrated that a splice product of the JCV late transcript was produced for both the triple (L29A+L32A+L36A) mutant and the single (L29A and L36A) mutants (Fig. 9B, top and bottom), suggesting that the lack of agnoprotein detection in Western blots (Fig. 8C, lanes 5 and 9) is most likely due to the rapid degradation of the protein rather than the absence of its mRNA in infected cells. In Fig. 9C, the total RNA was also analyzed by RT-PCR for the amplification of GAPDH (top) as a control and by agarose gel electrophoresis (bottom) to demonstrate the integrity of the total RNA. In addition, RT-PCR products were sequenced to further verify the splicing products.
Immunocytochemistry analysis of the agnoprotein dimerization domain mutants.
In parallel with the Western blot analyses of the agnoprotein mutants described in Fig. 8, the expression and cellular distribution properties of the various α-helix domain mutants were also analyzed by immunocytochemistry (Fig. 10). As expected, WT agnoprotein exhibited a predominantly cytoplasmic distribution, with high concentrations detected in the perinuclear area (11), and a small amount of the protein was also localized to the nucleus (4). Three single mutants (the L32A, L33A, and L36A mutants) and a double mutant (the I30A+L33A mutant) displayed a distribution similar to that shown by the WT. However, the I30A+L33A double mutant exhibited a punctate distribution in infected cells distinct from that of the WT. Another double mutant, the L32A+L36A mutant, not only showed a low level of expression, which is consistent with the Western blot data shown in Fig. 8C, but also exhibited a mostly perinuclear distribution. In addition, the expression and distribution properties of either a single (E34A) or a double (E34A+D38A) mutant were found to be not significantly different from those of the WT (data not shown).
FIG 10.

Analysis of the subcellular localization of the α-helix domain mutants by immunocytochemistry. In parallel with the Western blot studies whose results are shown in shown in Fig. 8, samples were also prepared and evaluated for agnoprotein expression (WT and agnoprotein mutants) by immunocytochemistry using primary antibodies (antiagnoprotein polyclonal antibody [49] and anti-VP1 monoclonal antibody [pAB597]) and secondary antibodies (goat anti-rabbit FITC-conjugated [green] and goat anti-mouse, rhodamine-conjugated [red] antibodies), as described in Materials and Methods.
Interestingly, in contrast to all other amino acid substitution mutants, a single amino acid substitution mutant, the L29A mutant, showed a drastic change in its cellular distribution profile in infected cells and accumulated heavily in the nucleus, suggesting that agnoprotein may, in fact, shuttle back and forth between the nucleus and the cytoplasm to fulfill its functions. In addition, consistent with our findings from Western blotting studies (Fig. 8C), a triple (L29+L32A+L36A) mutant showed no detectable level of expression. The three mutated amino acids in the triple mutant are perhaps localized at the dimer interface of agnoprotein monomers, as suggested from the dimer modeling studies (Fig. 6A), and therefore, alterations in these three hydrophobic residues greatly affect the stability of agnoprotein.
Analysis of the effect of NMR-based mutations made within the α-helical region of agnoprotein on viral DNA replication.
In parallel with the expression studies, we also examined the replication properties of agnoprotein mutants relative to those of the WT by DpnI assays, followed by Southern blotting (1). JCV Mad-1 WT and mutant viral genomes were individually transfected/infected into SVG-A cells, and low-molecular-mass DNA was isolated at the 5th and 15th days posttransfection/infection, as described in Materials and Methods. After isolation, DNA samples were digested with the BamHI enzyme, which digests the JCV genome once and thereby linearizes the circular genome. DNA samples were also treated with the DpnI enzyme to eliminate the input DNA (bacterially produced and methylated), while keeping the newly replicated viral DNA intact (61). DNA samples were then analyzed by Southern blotting using a probe prepared from the JCV Mad-1 genome as the template. As previously observed (1), the replication efficiency of each mutant was almost indistinguishable from that of the WT (1) if the data were taken during the first replication cycle (on the 5th day posttransfection/infection) (Fig. 11A, C, E, and G). However, significant deviations in the replication efficiency of various mutants compared to that of the WT were observed by the second replication cycle (15th day). For example, replacement of two negatively charged residues (E34 and D38, singly or in combination) with Ala noticeably affected the replication efficiency of JCV. We observed ∼53% and ∼39% reductions in the viral replication efficiency for single (E34A) and double (E34A+D38A) mutants, respectively (Fig. 11A, lanes 6 and 7, and B). Single (L33A) and double (I30A+L33A) Ala substitutions in two hydrophobic residues (I30 and L33) also resulted in similar reductions (∼58% and ∼43%, respectively) in replication efficiency (Fig. 11C, lanes 6 and 7, and D). Another series of single (L29A, L32A, and L36A), double (L32A+L36A), and triple (L29A+L32A+L36A) mutants was also evaluated by the replication assays (Fig. 11E to H). While the L32A mutation did not have a noticeable effect on viral replication (Fig. 11E, lane 7), the mutant with the L32A+L36A double mutation exhibited a moderate reduction in replication (∼26%) (Fig. 11E, lane 8). However, two individual single mutations alone, L29A and L36A, showed a more substantial effect on replication (∼32% and ∼42% reductions, respectively) (Fig. 11G, lanes 7 and 8) than the L32A and L36A combination of mutations (Fig. 11E, lane 8), suggesting that the L32A mutation plays a compensatory role in the L36A-mediated reduction. Note that all the mutants with these mutations exhibited a detectable level of expression in infected cells by Western blotting (Fig. 8A and B) and immunocytochemistry analysis (Fig. 10). The greatest decline in viral replication was observed when three hydrophobic residues (L29, L32, and L36) were simultaneously mutated to Ala (∼66% decline; Fig. 11E, lane 9, and F). The behavior of this triple mutant in replication assays is consistent with our findings from our in vitro dimerization (Fig. 7D, lane 7) and in vivo protein expression (Western blotting and immunocytochemistry) studies. That is, in in vitro dimerization studies, agnoprotein monomers with triple mutations were found to be degraded, and in in vivo protein expression studies, this mutant protein was not detectable by Western blotting (Fig. 8C, lanes 4 and 9) or immunocytochemistry (Fig. 10). Taken together, results from the replication studies suggest that, although several individual mutations or combinations of various mutations were found to have significant effects on viral DNA replication in vivo, the observed effect was more pronounced for the triple (L29A+L32A+L36A) mutant.
FIG 11.

Analysis of the replication efficiency of the agnoprotein dimerization domain mutants by DpnI/Southern blotting. (A, C, E, G) In parallel with the Western blots of the mutants shown in Fig. 8, low-molecular-mass DNA containing both input and replicated viral DNA was also isolated from untransfected or transfected cells, as indicated, using Qiagen spin columns (51) and digested with the BamHI and DpnI restriction enzymes. Digested DNA was separated on a 1% agarose gel, transferred onto a nitrocellulose membrane (catalog no. 162-0112; Bio-Rad), and probed for detection of newly replicated DNA using a probe prepared from the JCV Mad-1 WT. In lane 1 of each blot, 2 ng of JCV Mad-1 WT linearized by BamHI digestion was loaded as a positive control (+ Contr.). Replication assays were repeated several times, and representative data are shown here. (B, D, F, H). Quantitative analysis of Southern blots was performed using semiquantitative densitometry with the NIH ImageJ program, and the results are given in arbitrary units. The replication efficiency of each data point relative to that of the WT is presented.
DISCUSSION
Agnoprotein is an important regulatory protein of several polyomaviruses, including JCV, BKV, and SV40. In the absence of its expression, these viruses are unable to sustain their productive life cycle (24). The amino acid composition of this protein shows that it is highly basic and is composed of various positively charged residues (Lys and Arg). However, the central portion of the protein possesses a Leu/Ile/Phe-rich hydrophobic region. In this study, we have demonstrated the involvement of this hydrophobic region in the formation of an amphipathic α-helix by NMR. Previously, we had attempted to reveal the crystal structure of this protein using various expression systems, including bacterial and baculovirus expression systems, with no success due to difficulties associated with purification and a dynamic interconversion of monomers to dimers or oligomers. Hence, in this study, NMR was employed to obtain the 3D structure of agnoprotein using a synthetic agnoprotein peptide from T17 to Q52. We are also in the process of obtaining the NMR structure for the full-length agnoprotein, despite the difficulties associated with its synthesis. NMR studies revealed that the agnoprotein peptide contains an amphipathic α-helix, as had previously been predicted by various computer programs (1, 5), and that surrounding regions adopt an unstructured conformation. The critical roles of the α-helical domain in stable expression and dimer/oligomer formation by agnoprotein were recently demonstrated (1). However, we have further analyzed this region by NMR structure-based alanine substitution mutagenesis. Functional studies with these mutants demonstrated that the simultaneous conversion of Leu29, Leu32, and Leu36 to Ala showed the most negative effect compared to the effects of all other mutations on the stable expression of the protein and replication cycle of the virus. That is, we were unable to detect the expression of this mutant protein in infected cells by either Western blotting (Fig. 8C) or immunocytochemistry (Fig. 10), suggesting that this mutant protein is highly unstable and the observed effect does not appear to be at the level of transcription (Fig. 9B). These observations are consistent with our theoretical (Fig. 6A) and in vitro dimerization (Fig. 7B) studies. Theoretical dimer formation studies suggest an interaction between the Leu29 and Leu36 residues, which are located at the same interface of the α-helical domain, if a dimer occurs in an antiparallel manner (Fig. 6A). In vitro dimerization studies, on the other hand, consistently revealed that the triple (L29A+L32A+L36A) mutant containing both mutant residues is unable to form stable monomers. In contrast, this mutant showed the ability to form stable dimers and oligomers (Fig. 7D). One possible explanation for this discrepancy is as follows: the amphipathic α-helical domain of agnoprotein possesses a wide-range hydrophobic surface (Fig. 4C), and this may provide an opportunity for the α-helical region to create alternative interaction interfaces. Therefore, even though experimental evidence suggests that this triple (L29A+L32A+L36A) mutant creates a primary dimer interface between two monomers, similar possible alternative dimer interfaces may also exist on the other two hydrophobic surfaces (Fig. 4C). In fact, an alternative theoretical dimer study suggests that such a possibility can possibly occur through the interaction of the I30 and L33 residues if a dimer forms in an antiparallel orientation (Fig. 6C). Another important point about the triple mutant is that its expression level in a bacterial expression system is highly reduced compared to that of the WT. This observation also supports our findings from the protein expression studies in infected cells by Western blotting (Fig. 8C) and immunocytochemistry (Fig. 10). In addition, we made an unexpected observation when analyzing the subcellular distribution of the α-helix domain mutants. In particular, the L29A mutant showed a predominantly nuclear localization pattern, in contrast to the conventional cytoplasmic distribution of WT agnoprotein (Fig. 10). This type of behavior by the L29A mutant suggests that agnoprotein may shuttle between the nucleus and cytoplasm to fulfill its regulatory functions during the viral replication cycle. Consistent with this concept, we have recently demonstrated that agnoprotein enhances the binding activity of JCV large T antigen to the origin of viral DNA replication (4). Moreover, consistent with the nuclear localization of the L29A mutant, we have recently demonstrated that various α-helix domain mutants of agnoprotein also showed defects in splicing of the viral late transcripts, suggesting that agnoprotein may be directly or indirectly involved in the regulation of splicing of JVC transcripts (1).
In addition to possessing an α-helical domain (Fig. 4B), our prediction (Fig. 1A) and NMR structure (Fig. 4B) data indicate that agnoprotein has intrinsically disordered structural regions, and the functional significance of these regions is unknown. There are several other examples of intrinsically disordered regions from viral proteins in the literature. These types of proteins are known to have a high net charge and low overall hydrophobicity in these regions (8, 9, 62, 63). It is suggested that these regions may provide considerable flexibility to a protein to interact with multiple targets in the host cells during the viral infection cycle. One prime example of such proteins is the HIV-1 Vif protein, which plays important roles in viral pathogenesis by facilitating the degradation of APOBEC3G, an endogenous cellular inhibitor (cytosine deaminase) of HIV-1 replication (64). Our NMR structural studies of the agnoprotein peptide (Fig. 4B), as well as predictions by modeling of the full-length agnoprotein (Fig. 1A), show that small portions of the N-terminal and C-terminal domains of agnoprotein possess such intrinsic disordered structures, implying that these regions may indeed play important regulatory roles in interacting with a variety of partners of agnoprotein in infected cells and contribute to the function of the protein.
Moreover, the analysis of the primary sequence of the JCV agnoprotein is predicted to have an additional α-helix structure located toward the N-terminal end of the protein spanning residues V2 to A10 (Fig. 1A). In contrast to the structural features of the second α-helix, such as having a cluster of hydrophobic residues forming a wide surface on one side of the helix and a narrow hydrophilic face on the other side (Fig. 4C), which may sufficiently explain the propensity of agnoprotein to form homodimers and oligomers, the first α-helix from V2 to A10 does not exhibit such amphipathic properties (Fig. 1A).
Like agnoprotein, a variety of viral proteins reported in the literature form dimers and oligomers, including hepatitis C virus nonstructural protein 4B (65), Ebola virus VP40 (66), polyomavirus LT-Ag (67, 68), and HIV-1 proteins (Rev, Vpr, Vif, and Vpu) (44, 69–72). For instance, Rev binds to HIV RNA and regulates the transport of unspliced or partially spliced RNA molecules from the nucleus to the cytoplasm. Rev initially binds to a highly structured ∼350-nucleotide (nt) Rev response element (RRE) RNA in dimeric forms. The recently resolved crystal structure of the Rev-RNA complex revealed that dimeric forms of Rev cooperatively bind to viral RNA to form a hexameric unit complexed with RNA. This hexameric complex has a 500-fold higher affinity for the RRE than the monomeric Rev molecule. The Rev-RRE nucleoprotein complex then targets the host export factor Crm1 (XpoI) (72, 73) to shuttle the Rev-RNA complex to the cytoplasm. In the cytoplasm, Rev unloads its cargo and free Rev is reimported into the nucleus for further rounds of nuclear export of HIV-1 RNA (73, 74). Our recent RNA chromatin immunoprecipitation assays suggest that agnoprotein is an RNA binding protein (data not shown) rather than a DNA binding protein (75). As mentioned above, we believe that dimeric and oligomeric forms of agnoprotein may play important roles in the transport of JCV transcripts from the nucleus to the cytoplasm in a Crm1-dependent (72, 73) or -independent (76) manner. This possibility will be further investigated.
The sequences of agnoprotein and HIV-1 Vpr were compared (Fig. 6E). Interestingly, Vpr contains an Leu/Ile-rich domain from residues 60 to 81 in its C-terminal portion which is critical for its dimerization. We have previously demonstrated by NMR spectroscopy the ability of this peptide to form an α-helix spanning from residues 53 to 75 and to dimerize in an antiparallel orientation through hydrophobic interactions between leucine and isoleucine residues (44, 47, 54). We now also propose a theoretical homodimeric structure for the α-helical domain of agnoprotein from residues K23 to F39, by analogy with the domain from residues 53 to 75 of Vpr. The alignment of the two domains of Vpr and the agnoprotein shows a good consensus primary sequence and a high degree of homology for the hydrophobic residues, leucines and isoleucines L29, I30, L33, L36, and L37 in agnoprotein and L60, I61, L64, L67, and L68 in the C-terminal domain of Vpr (77, 78) (Fig. 6E). The putative homodimeric structure of agnoprotein was built by homology with the experimental dimeric structure of Vpr and shows the formation of a hydrophobic interface between the two monomers (Fig. 6A). Interestingly, an extra hydrophobic surface constituted by residues A25, I28, L32, F35, and F39 is available for intermolecular interactions (Fig. 4C).
Accumulating experimental evidence suggests that agnoprotein is a multifunctional protein, despite its small size. Then, the question of how agnoprotein achieves multiple functions, despite its small size, arises. It seems that different regions of agnoprotein are involved in different functions. For instance, our recent protein-protein interaction studies with agnoprotein and LT-Ag showed that agnoprotein stimulates LT-Ag binding to the viral origin of replication (Ori), through which it contributes to the viral replication cycle (4). We have also previously reported that alterations in the protein kinase C (PKC) phosphorylation sites of JCV agnoprotein (conversion of Ser7, Ser11, and Thr21 to Ala) resulted in phenotypes that are unable to sustain the viral replication cycle (19). Deletion of either the intrinsically disordered C-terminal region (the sequence from amino acids 51 to 71) (2, 79) or internal sequences containing the α-helix region (amino acids 17 to 42) (7) resulted in a replication-incompetent virus. Recent fine mapping studies of the multimerization (α-helix) domain of agnoprotein also revealed the involvement of this domain in determining the stability of agnoprotein as well as in regulation of the splicing of the viral late transcripts (1).
Without the 3D structure of the proteins, it is difficult to study their regulatory roles. We have attempted to obtain the 3D crystal structure of bacterially expressed JCV agnoprotein (MBP-Agno) several times, with no success, due to the dynamic process of conversion of monomers to dimers or to oligomers (7). We have now obtained the NMR structure of a synthetic peptide of agnoprotein. On the basis of the NMR structure, the contribution of specific individual amino acids and groups of amino acids located within the α-helical domain of agnoprotein to multimerization, the stability of the protein, and viral DNA replication has been analyzed. Our genetic and biochemical characterization of the specific α-helix domain mutants revealed that certain amino acids within the α-helix are critically important for the function of agnoprotein, including L29 and L36, both individually and in combination. Thus, overall these comprehensive studies provide us new opportunities to develop effective inhibitors against the unique dimerization/oligomerization domain of JCV agnoprotein to interfere with its functions during the viral replication cycle.
ACKNOWLEDGMENTS
We thank past and present members of the Department of Neuroscience for their insightful discussion and sharing of ideas and reagents.
This work was made possible by grants awarded to M.S. (RO1NS43108) by the NIH and by the Temple University School of Medicine Drug Discovery Initiative (161107).
Footnotes
Published ahead of print 26 March 2014
REFERENCES
- 1.Sami Saribas A, Abou-Gharbia M, Childers W, Sariyer IK, White MK, Safak M. 2013. Essential roles of Leu/Ile/Phe-rich domain of JC virus agnoprotein in dimer/oligomer formation, protein stability and splicing of viral transcripts. Virology 443:161–176. 10.1016/j.virol.2013.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellis LC, Norton E, Dang X, Koralnik IJ. 2013. Agnogene deletion in a novel pathogenic JC virus isolate impairs VP1 expression and virion production. PLoS One 8:e80840. 10.1371/journal.pone.0080840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Gascun CF, Carr MJ. 2013. Human polyomavirus reactivation: disease pathogenesis and treatment approaches. Clin. Dev. Immunol. 2013:373579. 10.1155/2013/373579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saribas AS, White MK, Safak M. 2012. JC virus agnoprotein enhances large T antigen binding to the origin of viral DNA replication: evidence for its involvement in viral DNA replication. Virology 433:12–26. 10.1016/j.virol.2012.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Unterstab G, Gosert R, Leuenberger D, Lorentz P, Rinaldo CH, Hirsch HH. 2010. The polyomavirus BK agnoprotein co-localizes with lipid droplets. Virology 399:322–331. 10.1016/j.virol.2010.01.011 [DOI] [PubMed] [Google Scholar]
- 6.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738. 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saribas AS, Arachea BT, White MK, Viola RE, Safak M. 2011. Human polyomavirus JC small regulatory agnoprotein forms highly stable dimers and oligomers: implications for their roles in agnoprotein function. Virology 420:51–65. 10.1016/j.virol.2011.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dyson HJ, Wright PE. 2005. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6:197–208. 10.1038/nrm1589 [DOI] [PubMed] [Google Scholar]
- 9.Fink AL. 2005. Natively unfolded proteins. Curr. Opin. Struct. Biol. 15:35–41. 10.1016/j.sbi.2005.01.002 [DOI] [PubMed] [Google Scholar]
- 10.Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, Land HS, Soria C, Powers CJ, May AP, Shu X, Tsien RY, Fitzpatrick JA, Long JA, Ellisman MH, Choe S, O'Shea CC. 2012. A structural basis for the assembly and functions of a viral polymer that inactivates multiple tumor suppressors. Cell 151:304–319. 10.1016/j.cell.2012.08.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Safak M, Sadowska B, Barrucco R, Khalili K. 2002. Functional interaction between JC virus late regulatory agnoprotein and cellular Y-box binding transcription factor, YB-1. J. Virol. 76:3828–3838. 10.1128/JVI.76.8.3828-3838.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darbinyan A, Darbinian N, Safak M, Radhakrishnan S, Giordano A, Khalili K. 2002. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene 21:5574–5581. 10.1038/sj.onc.1205744 [DOI] [PubMed] [Google Scholar]
- 13.Okada Y, Suzuki T, Sunden Y, Orba Y, Kose S, Imamoto N, Takahashi H, Tanaka S, Hall WW, Nagashima K, Sawa H. 2005. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: role in nuclear egress of viral particles. EMBO Rep. 6:452–457. 10.1038/sj.embor.7400406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki T, Orba Y, Makino Y, Okada Y, Sunden Y, Hasegawa H, Hall WW, Sawa H. 2013. Viroporin activity of the JC polyomavirus is regulated by interactions with the adaptor protein complex 3. Proc. Natl. Acad. Sci. U. S. A. 110:18668–18673. 10.1073/pnas.1311457110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sariyer IK, Khalili K, Safak M. 2008. Dephosphorylation of JC virus agnoprotein by protein phosphatase 2A: inhibition by small t antigen. Virology 375:464–479. 10.1016/j.virol.2008.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Safak M, Khalili K. 2001. Physical and functional interaction between viral and cellular proteins modulate JCV gene transcription. J. Neurovirol. 7:288–292. 10.1080/13550280152537111 [DOI] [PubMed] [Google Scholar]
- 17.Safak M, Barrucco R, Darbinyan A, Okada Y, Nagashima K, Khalili K. 2001. Interaction of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J. Virol. 75:1476–1486. 10.1128/JVI.75.3.1476-1486.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki T, Orba Y, Okada Y, Sunden Y, Kimura T, Tanaka S, Nagashima K, Hall WW, Sawa H. 2010. The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathog. 6:e1000801. 10.1371/journal.ppat.1000801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sariyer IK, Akan I, Palermo V, Gordon J, Khalili K, Safak M. 2006. Phosphorylation mutants of JC virus agnoprotein are unable to sustain the viral infection cycle. J. Virol. 80:3893–3903. 10.1128/JVI.80.8.3893-3903.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johannessen M, Walquist M, Gerits N, Dragset M, Spang A, Moens U. 2011. BKV agnoprotein interacts with alpha-soluble N-ethylmaleimide-sensitive fusion attachment protein, and negatively influences transport of VSVG-EGFP. PLoS One 6:e24489. 10.1371/journal.pone.0024489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merabova N, Kaminski R, Krynska B, Amini S, Khalili K, Darbinyan A. 2012. JCV agnoprotein-induced reduction in CXCL5/LIX secretion by oligodendrocytes is associated with activation of apoptotic signaling in neurons. J. Cell. Physiol. 227:3119–3127. 10.1002/jcp.23065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merabova N, Kaniowska D, Kaminski R, Deshmane SL, White MK, Amini S, Darbinyan A, Khalili K. 2008. JC virus agnoprotein inhibits in vitro differentiation of oligodendrocytes and promotes apoptosis. J. Virol. 82:1558–1569. 10.1128/JVI.01680-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myhre MR, Olsen GH, Gosert R, Hirsch HH, Rinaldo CH. 2010. Clinical polyomavirus BK variants with agnogene deletion are non-functional but rescued by trans-complementation. Virology 398:12–20. 10.1016/j.virol.2009.11.029 [DOI] [PubMed] [Google Scholar]
- 24.Sariyer IK, Saribas AS, White MK, Safak M. 2011. Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol. J. 8:255. 10.1186/1743-422X-8-255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Major EO. 2010. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu. Rev. Med. 61:35–47. 10.1146/annurev.med.080708.082655 [DOI] [PubMed] [Google Scholar]
- 26.Berger JR, Concha M. 1995. Progressive multifocal leukoencephalopathy: the evolution of a disease once considered rare. J. Neurovirol. 1:5–18. 10.3109/13550289509111006 [DOI] [PubMed] [Google Scholar]
- 27.Berger JR. 2011. The basis for modeling progressive multifocal leukoencephalopathy pathogenesis. Curr. Opin. Neurol. 24:262–267. 10.1097/WCO.0b013e328346d2a3 [DOI] [PubMed] [Google Scholar]
- 28.Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. 1992. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin. Microbiol. Rev. 5:49–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Assetta B, Maginnis MS, Gracia Ahufinger I, Haley SA, Gee GV, Nelson CD, O'Hara BA, Allen Ramdial SA, Atwood WJ. 2013. 5-HT2 receptors facilitate JC polyomavirus entry. J. Virol. 87:13490–13498. 10.1128/JVI.02252-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neu U, Maginnis MS, Palma AS, Stroh LJ, Nelson CD, Feizi T, Atwood WJ, Stehle T. 2010. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 8:309–319. 10.1016/j.chom.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. 2005. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 353:375–381. 10.1056/NEJMoa051847 [DOI] [PubMed] [Google Scholar]
- 32.Kleinschmidt-DeMasters BK, Tyler KL. 2005. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N. Engl. J. Med. 353:369–374. 10.1056/NEJMoa051782 [DOI] [PubMed] [Google Scholar]
- 33.Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. 2005. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N. Engl. J. Med. 353:362–368. 10.1056/NEJMoa051586 [DOI] [PubMed] [Google Scholar]
- 34.McGuffin LJ, Bryson K, Jones DT. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16:404–405. 10.1093/bioinformatics/16.4.404 [DOI] [PubMed] [Google Scholar]
- 35.Cornille F, Wecker K, Loffet A, Genet Roques RB. 1999. Efficient solid-phase synthesis of Vpr from HIV-1 using low quantities of uniformly 13C-, 15N-labeled amino acids for NMR structural studies. J. Pept. Res. 54:427–435. 10.1034/j.1399-3011.1999.00129.x [DOI] [PubMed] [Google Scholar]
- 36.Piotto M, Saudek V, Sklenar V. 1992. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2:661–665. 10.1007/BF02192855 [DOI] [PubMed] [Google Scholar]
- 37.Hwang TL, Shaka AJ. 1995. Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. J. Magn. Reson. A 112:275–279 [Google Scholar]
- 38.Jeener J, Meier BH, Bachmann P, Ernst RR. 1979. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 71:5246–5253. 10.1063/1.438333 [DOI] [Google Scholar]
- 39.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. 2005. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696. 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 40.Nilges M. 1995. Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J. Mol. Biol. 245:645–660 [DOI] [PubMed] [Google Scholar]
- 41.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921. 10.1107/S0907444998003254 [DOI] [PubMed] [Google Scholar]
- 42.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. 1996. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8:477–486 [DOI] [PubMed] [Google Scholar]
- 43.DeLano W. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA [Google Scholar]
- 44.Bourbigot S, Beltz H, Denis J, Morellet N, Roques BP, Mely Y, Bouaziz S. 2005. The C-terminal domain of the HIV-1 regulatory protein Vpr adopts an antiparallel dimeric structure in solution via its leucine-zipper-like domain. Biochem. J. 387:333–341. 10.1042/BJ20041759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arkin IT, Sukharev SI, Blount P, Kung C, Brunger AT. 1998. Helicity, membrane incorporation, orientation and thermal stability of the large conductance mechanosensitive ion channel from E. coli. Biochim. Biophys. Acta 1369:131–140. 10.1016/S0005-2736(97)00219-8 [DOI] [PubMed] [Google Scholar]
- 46.Fleming KG, Hohl TM, Yu RC, Muller SA, Wolpensinger B, Engel A, Engelhardt H, Brunger AT, Sollner TH, Hanson PI. 1998. A revised model for the oligomeric state of the N-ethylmaleimide-sensitive fusion protein, NSF. J. Biol. Chem. 273:15675–15681. 10.1074/jbc.273.25.15675 [DOI] [PubMed] [Google Scholar]
- 47.Morellet N, Bouaziz S, Petitjean P, Roques BP. 2003. NMR structure of the HIV-1 regulatory protein VPR. J. Mol. Biol. 327:215–227. 10.1016/S0022-2836(03)00060-3 [DOI] [PubMed] [Google Scholar]
- 48.Doreleijers JF, Sousa da Silva AW, Krieger E, Nabuurs SB, Spronk CA, Stevens TJ, Vranken WF, Vriend G, Vuister GW. 2012. CING: an integrated residue-based structure validation program suite. J. Biomol. NMR 54:267–283. 10.1007/s10858-012-9669-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. 1985. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc. Natl. Acad. Sci. U. S. A. 82:1257–1261. 10.1073/pnas.82.4.1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Del Valle L, Gordon J, Enam S, Delbue S, Croul S, Abraham S, Radhakrishnan S, Assimakopoulou M, Katsetos CD, Khalili K. 2002. Expression of human neurotropic polyomavirus JCV late gene product agnoprotein in human medulloblastoma. J. Natl. Cancer Inst. 94:267–273. 10.1093/jnci/94.4.267 [DOI] [PubMed] [Google Scholar]
- 51.Ziegler K, Bui T, Frisque RJ, Grandinetti A, Nerurkar VR. 2004. A rapid in vitro polyomavirus DNA replication assay. J. Virol. Methods 122:123–127. 10.1016/j.jviromet.2004.08.012 [DOI] [PubMed] [Google Scholar]
- 52.Combet C, Blanchet C, Geourjon C, Deleage G. 2000. NPS@: network protein sequence analysis. Trends Biochem. Sci. 25:147–150. 10.1016/S0968-0004(99)01540-6 [DOI] [PubMed] [Google Scholar]
- 53.Snider C, Jayasinghe S, Hristova K, White SH. 2009. MPEx: a tool for exploring membrane proteins. Protein Sci. 18:2624–2628. 10.1002/pro.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wecker K, Morellet N, Bouaziz S, Roques BP. 2002. NMR structure of the HIV-1 regulatory protein Vpr in H2O/trifluoroethanol. Comparison with the Vpr N-terminal (1–51) and C-terminal (52–96) domains. Eur. J. Biochem. 269:3779–3788. 10.1046/j.1432-1033.2002.03067.x [DOI] [PubMed] [Google Scholar]
- 55.Rance M, Sorensen OW, Bodenhausen G, Wagner G, Ernst RR, Wuthrich K. 1983. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 117:479–485. 10.1016/0006-291X(83)91225-1 [DOI] [PubMed] [Google Scholar]
- 56.Marion D, Wuthrich K. 1983. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 113:967–974. 10.1016/0006-291X(83)91093-8 [DOI] [PubMed] [Google Scholar]
- 57.Griesinger C, Otting G, Wuthrich K, Ernst RR. 1988. Clear TOCSY for 1H spin system identification in macromolecules. J. Am. Chem. Soc. 110:7870–7872. 10.1021/ja00231a044 [DOI] [Google Scholar]
- 58.Kumar A, Ernst RR, Wuthrich K. 1980. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 95:1–6. 10.1016/0006-291X(80)90695-6 [DOI] [PubMed] [Google Scholar]
- 59.Wuthrich K. 1986. NMR of proteins and nucleic acids. Wiley Interscience, Hoboken, NJ [Google Scholar]
- 60.Brunger AT. 1992. A system for X-ray crystallography and NMR. Yale University Press, New Haven, CT [Google Scholar]
- 61.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365–369. 10.1016/0022-2836(67)90307-5 [DOI] [PubMed] [Google Scholar]
- 62.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. 2005. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 272:5129–5148. 10.1111/j.1742-4658.2005.04948.x [DOI] [PubMed] [Google Scholar]
- 63.Uversky VN, Oldfield CJ, Dunker AK. 2005. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 18:343–384. 10.1002/jmr.747 [DOI] [PubMed] [Google Scholar]
- 64.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650. 10.1038/nature00939 [DOI] [PubMed] [Google Scholar]
- 65.Gouttenoire J, Penin F, Moradpour D. 2010. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev. Med. Virol. 20:117–129. 10.1002/rmv.640 [DOI] [PubMed] [Google Scholar]
- 66.Hoenen T, Biedenkopf N, Zielecki F, Jung S, Groseth A, Feldmann H, Becker S. 2010. Oligomerization of Ebola virus VP40 is essential for particle morphogenesis and regulation of viral transcription. J. Virol. 84:7053–7063. 10.1128/JVI.00737-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cuesta I, Nunez-Ramirez R, Scheres SH, Gai D, Chen XS, Fanning E, Carazo JM. 2010. Conformational rearrangements of SV40 large T antigen during early replication events. J. Mol. Biol. 397:1276–1286. 10.1016/j.jmb.2010.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Foster EC, Simmons DT. 2010. The SV40 large T-antigen origin binding domain directly participates in DNA unwinding. Biochemistry 49:2087–2096. 10.1021/bi901827k [DOI] [PubMed] [Google Scholar]
- 69.Bernacchi S, Mercenne G, Tournaire C, Marquet R, Paillart JC. 2011. Importance of the proline-rich multimerization domain on the oligomerization and nucleic acid binding properties of HIV-1 Vif. Nucleic Acids Res. 39:2404–2415. 10.1093/nar/gkq979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu JX, Sharpe S, Ghirlando R, Yau WM, Tycko R. 2010. Oligomerization state and supramolecular structure of the HIV-1 Vpu protein transmembrane segment in phospholipid bilayers. Protein Sci. 19:1877–1896. 10.1002/pro.474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. 2010. HIV Rev response element (RRE) directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc. Natl. Acad. Sci. U. S. A. 107:12481–12486. 10.1073/pnas.1007022107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Daugherty MD, Liu B, Frankel AD. 2010. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat. Struct. Mol. Biol. 17:1337–1342. 10.1038/nsmb.1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cullen BR. 2003. Nuclear mRNA export: insights from virology. Trends Biochem. Sci. 28:419–424. 10.1016/S0968-0004(03)00142-7 [DOI] [PubMed] [Google Scholar]
- 74.Cullen BR. 2003. Nuclear RNA export. J. Cell Sci. 116:587–597. 10.1242/jcs.00268 [DOI] [PubMed] [Google Scholar]
- 75.Saribas SA, Mun S, Johnson J, El-Hajmoussa M, White MK, Safak S. 2014. Human polyoma JC virus minor capsid proteins, VP2 and VP3, enhance large T antigen binding to the origin of viral DNA replication: evidence for their involvement in regulation of the viral DNA replication. Virology 449:1–16. 10.1016/j.virol.2013.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yatherajam G, Huang W, Flint SJ. 2011. Export of adenoviral late mRNA from the nucleus requires the Nxf1/Tap export receptor. J. Virol. 85:1429–1438. 10.1128/JVI.02108-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Corpet F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16:10881–10890. 10.1093/nar/16.22.10881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Akan I, Sariyer IK, Biffi R, Palermo V, Woolridge S, White MK, Amini S, Khalili K, Safak M. 2006. Human polyomavirus JCV late leader peptide region contains important regulatory elements. Virology 349:66–78. 10.1016/j.virol.2006.01.025 [DOI] [PubMed] [Google Scholar]