

ABSTRACT
In February 2013, very severe acute clinical symptoms were observed in calves, heifers, and dairy cattle in several farms in North Rhine Westphalia and Lower Saxony, Germany. Deep sequencing revealed the coexistence of three distinct genome variants within recent highly virulent bovine viral diarrhea virus type 2 (BVDV-2) isolates. While the major portion (ca. 95%) of the population harbored a duplication of a 222-nucleotide (nt) segment within the p7-NS2-encoding region, the minority reflected the standard structure of a BVDV-2 genome. Additionally, unusual mutations were found in both variants, within the highly conserved p7 protein and close to the p7-NS2 cleavage site. Using a reverse genetic system with a BVDV-2a strain harboring a similar duplication, it could be demonstrated that during replication, genomes without duplication are generated de novo from genomes with duplication. The major variant with duplication is compulsorily escorted by the minor variant without duplication. RNA secondary structure prediction allowed the analysis of the unique but stable mixture of three BVDV variants and also provided the explanation for their generation. Finally, our results suggest that the variant with duplication plays the major role in the highly virulent phenotype.
IMPORTANCE This study emphasizes the importance of full-genome deep sequencing in combination with manual in-depth data analysis for the investigation of viruses in basic research and diagnostics. Here we investigated recent highly virulent bovine viral diarrhea virus isolates from a 2013 series of outbreaks. We discovered a unique special feature of the viral genome, an unstable duplication of 222 nucleotides which is eventually deleted by viral polymerase activity, leading to an unexpectedly mixed population of viral genomes for all investigated isolates. Our study is of high importance to the field because we demonstrate that these insertion/deletion events allow another level of genome plasticity of plus-strand RNA viruses, in addition to the well-known polymerase-induced single nucleotide variations which are generally considered the main basis for viral adaptation and evolution.
INTRODUCTION
In February 2013, severe clinical symptoms, including bloody diarrhea, hemorrhages, and mucosal lesions, and a very high mortality rate were observed in calves, heifers, and dairy cattle in several farms in North Rhine Westphalia (NRW) and Lower Saxony, Germany. The first investigations revealed infections with a bovine viral diarrhea virus type 2 (BVDV-2) strain of the noncytopathogenic biotype, according to cell culture experiments (1). Subsequently, BVDV infections with a mortality rate of approximately 40% were reported from the Netherlands (2).
BVDV-1 and BVDV-2 belong to the genus Pestivirus within the family Flaviviridae and comprise a single-stranded positive-sense RNA genome of approximately 12.5 kb (3). The genome encodes a polyprotein which is co- and posttranslationally processed by viral and host proteases into 12 proteins (NH2-Npro-C-Erns-E1–E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B-COOH) (4). BVDV strains are further subdivided genetically into at least 16 subtypes (a to p) for BVDV-1 and three subtypes (a to c) for BVDV-2 (5). Phenotypically, BVDV strains can be distinguished into two biotypes (cytopathogenic [cp] and noncytopathogenic [ncp]) depending on their effect on cell culture (6–10), with the cytopathic phenotype being evoked by various mutations (reviewed in reference 5). BVDV is contagious and has the unique ability to produce persistently infected animals when infecting fetuses in the first gestation phase (11, 12). These persistently infected animals constantly shed very large amounts of virus (13). Cytopathogenic strains usually emerge de novo in persistently infected animals and cause a characteristic and severe clinical picture known as “mucosal disease” (14, 15).
In Germany, about 95% of the circulating BVDV strains belong to BVDV-1, and only 5% represent BVDV-2 (1, 16). In the past, severe acute infections seem to have been caused particularly by BVDV-2 (17). Correspondingly, the only reported German case of severe acute BVDV infection was associated with a BVDV-2a strain (18).
Here we analyzed isolates from a recent BVDV outbreak series by using next-generation sequencing. Furthermore, we used an available BVDV-2 reverse genetic system with a strain carrying a genome resembling the genomes of the recent highly virulent strains to provide more insights into the genetics of the observed unique virus mixture.
MATERIALS AND METHODS
Cells, viruses, and RNA extraction.
Bovine esophageal cells (KOP-R cells; also called RIE244 cells) were obtained from the Collection of Cell Lines in Veterinary Medicine (CCLV) at the Friedrich-Loeffler-Institut, Germany. Cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% BVDV-free fetal calf serum (FCS) and were used for virus isolation and propagation and for transfection of in vitro-transcribed RNA. The isolated viruses from six calves from different farms analyzed in this study are listed in Table 1. BVDV-2 wild-type strain 890 (v890WT) (19) was kindly provided by H. Hehnen (Bayer AG, Monheim, Germany). The recombinant virus v890FL was reconstituted after transfection of RNA transcribed from the infectious cDNA clone p890FL into KOP-R cells (20). RNAs from cell culture supernatant and body fluids were extracted using TRIzol LS (Invitrogen, Life Technologies, Darmstadt, Germany) in combination with Direct-zol RNA columns (Zymo Research, Freiburg, Germany) or in combination with RNeasy minikit columns (Qiagen, Hilden, Germany).
TABLE 1.
Viruses analyzed in this study
| Sample | Collection date (mo/yr) | Regiona | Virulence level | Presence of duplicationb | INSDC accession no. |
|---|---|---|---|---|---|
| NRW 12-13 | 02/2013 | NRW | High | − | HG426483 |
| + | HG426484 | ||||
| NRW 14-13 | 02/2013 | NRW | High | − | HG426485 |
| + | HG426486 | ||||
| NRW 19-13-1 | 03/2013 | NRW | High | − | HG426487 |
| + | HG426488 | ||||
| NRW 19-13-8c | 03/2013 | NRW | High | − | HG426489 |
| + | HG426490 | ||||
| D37-13-2 | 06/2013 | NRW | High | − | HG426479 |
| + | HG426480 | ||||
| D75-13-609 | 06/2013 | NRW | High | − | HG426481 |
| + | HG426482 | ||||
| VOE 4007 | 2007 | BW | Low/moderate | − | HG426495 |
| SH 2210-23 | 03/2010 | SH | Low/moderate | − | HG426494 |
| Potsdam 1600 | 2000 | BR | Low/moderate | − | HG426491 |
| SH 2210-14 | 03/2010 | SH | Low/moderate | − | HG426492 |
| SH 2210-17 | 03/2010 | SH | Low/moderate | − | HG426493 |
Federal state of Germany in which the samples were collected. NRW, North Rhine-Westphalia; BW, Baden-Württemberg; SH, Schleswig-Holstein; BR, Brandenburg.
Presence (+) or absence (−) of partial duplication of p7-NS2 detected by PCR and sequence analyses.
Virulence of the isolate was confirmed by an animal experiment (Fig. 1).
Animal experiment.
The animal trial was approved by the ethics committee of the responsible federal state, Mecklenburg Western Pomerania (approval no. LALLF, Az 7221.3-2.5-009/13). Eight calves were inoculated intranasally with 106 50% tissue culture infective doses (TCID50) of the NRW 19-13-8 isolate. In order to simulate an emergency vaccination scenario, four animals were vaccinated at day 5 postinfection with a single dose of a BVDV live vaccine (Vacoviron; Merial, Lyon, France). Blood and organ samples were tested by reverse transcription-quantitative PCR (RT-qPCR), using a standardized protocol (21).
Sequencing and sequence analysis.
Deep sequencing was applied by using both 454 (FLX genome sequencer; Roche, Mannheim, Germany) and Illumina (MiSeq; Illumina, San Diego, CA) technologies. cDNA synthesis, library preparation, and pyrosequencing were performed as previously described (22). For Illumina sequencing, the 454 libraries were transformed into Illumina libraries by using a Nextera XT kit (Illumina) according to the manufacturer's recommendations. Raw sequence data were analyzed and assembled using the Genome Sequencer software suite (v. 2.6; Roche). Phylogenetic analysis of the open reading frames of different BVDV strains was performed with MEGA 5 (23). The TMHMM tool (24) in Geneious 6.1.2 (Biomatters Ltd., Auckland, New Zealand) was used for transmembrane prediction. For signal peptide prediction of potential cleavage sites, the Emboss tool sigcleave (25) in Geneious 6.1.2 was used. PCR amplicons of the p7-NS2 region were sequenced by classical Sanger sequencing with a BigDye Terminator v1.1 cycle sequencing kit (Applied Biosystems, Darmstadt, Germany). Subsequently, nucleotide (nt) sequences were determined with a model 3130 genetic analyzer (Applied Biosystems).
Variant quantification.
For calculation of the variant distributions within isolates, RT-PCR of the p7-NS2 region was performed using primers 5′-TAC ATG ATA CTG TCT GAG CAG-3′ and 5′-GCA GTT AAT ACT ACT GCT ACA G-3′ for BVDV-2c and primers 5′-GCT GAC ACA CAG TGA TAT TGA GGT TGT GGT C-3′ and 5′-GTC ACG TAG CTA GCC AGC AAA AG-3′ for the BVDV-2a strain. The SuperScript III One-Step RT-PCR system with Platinum Taq High Fidelity polymerase (Life Technologies, Darmstadt, Germany) was used for reverse transcription-PCR per the manufacturer's recommendations. In vitro-transcribed RNA was digested with DNase I (Invitrogen), and subsequently, RNA was purified with Agencourt RNAClean XP beads (Beckman Coulter, Krefeld, Germany) prior to PCR. PCR products were analyzed with a Bioanalyzer 2100 instrument (Agilent Technologies, Böblingen, Germany), using an Agilent DNA 7500 chip. Quantification of the PCR products was done using 2100 Expert software (version B.02.08.SI648 [SR2]; Agilent).
BVDV-2a 890 clone and transfection.
BVDV-2a 890 (v890WT), a virulent noncytopathogenic strain, was isolated from a cow which died from acute infection. The BVDV-2a 890 open reading frame has a length of 11,922 nucleotides corresponding to 3,973 amino acids. The open reading frame is larger due to a 228-nucleotide insertion (19). The BVDV-2a 890 cDNA clone p890FL (here designated p890FL dup+) was described by Mischkale et al. (20). The cDNA clone p890FL dup− was generated by deleting one copy of the duplicated p7-NS2 region from p890FL dup+ by restriction-free cloning (26). In vitro transcription of the cDNA was performed using the T7 RiboMax large-scale RNA production system (Promega, Mannheim, Germany) according to the manufacturer's instructions, after linearizing the plasmids with SmaI. The amount of RNA was estimated by ethidium bromide staining after agarose gel electrophoresis. For RNA transfection, bovine cells were detached using a trypsin solution, washed twice with phosphate-buffered saline without Ca2+ and Mg2+ (PBS−), and mixed with 1 to 5 μg of in vitro-synthesized RNA. Electroporation was done by using a GenePulser transfection unit (Bio-Rad Laboratories GmbH, Munich, Germany) (two pulses at 850 V, 25 μF, and 156 Ω). Supernatants of transfected cells were harvested at 72 h posttransfection, and serial passages of the recombinant virus v890FL were performed on KOP-R cells.
Nucleotide sequence accession numbers.
All genome sequences generated in this study are available from the International Nucleotide Sequence Database Collaboration (INSDC) databases under accession numbers HG426479 to HG426495.
RESULTS
As the observed clinical picture was rather untypical for most BVDV strains, we conducted an animal experiment in order to clearly establish the isolated BVDV strain as the causative agent of disease. Interestingly, the postinoculation application of a live BVDV vaccine did not influence the clinical course, and all animals died acutely or had to be euthanized within 11 days, resulting in a mortality rate of 100%. All animals showed very high viral loads in blood and organs, with quantification cycle (Cq) values of <20 in the RT-qPCR, corresponding to about 105 genome copies per μl blood. For comparison, the viral loads in blood of animals acutely infected with BVDV are usually approximately 101 to 102 genome copies per μl. Hence, this experiment unequivocally confirmed the unusual phenotype for a representative recent German isolate (Fig. 1). For detailed genetic characterization, complete viral genomes were deep sequenced directly from sample material from one euthanized calf and from the previously isolated field strains.
FIG 1.
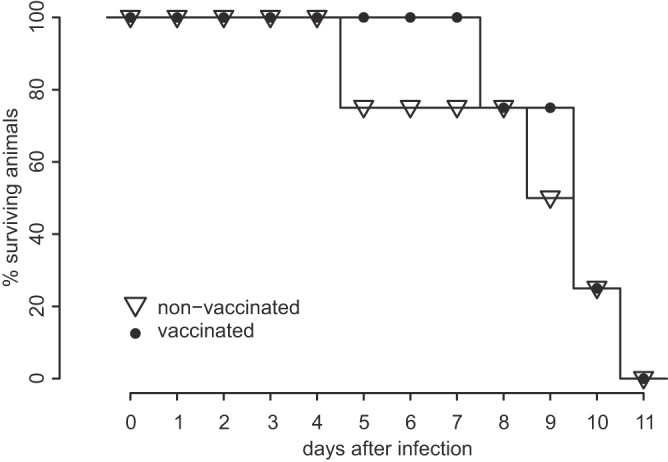
Survival of 8-week-old calves infected with the German BVDV-2c isolate NRW 19-13-8, with (n = 4) or without (n = 4) emergency vaccination at 5 days postinfection.
Recent BVDV-2c strains carry a genomic duplication giving rise to a new protein.
In order to correlate the observed phenotype with a specific genotype, we applied deep sequencing of the complete genomes of four isolates, using both 454 and Illumina technologies. Sequence assemblies resulted in genome variants with (dup+) or without (dup−) a 222-nt duplication within the p7-NS2 coding region. Figure 2 schematically depicts the dup− and dup+ variants at the nucleotide and amino acid levels, in comparison with a German prototypic BVDV-2c strain (Potsdam 1600) and a virulent noncytopathogenic BVDV-2a strain (BVDV-2a 890) (19). Interestingly, both BVDV-2c dup+ and BVDV-2a 890 showed very similar duplications within the p7-NS2 coding region. Heretofore, BVDV-2a 890 was the only known strain with such a duplication (19). Prediction of transmembrane helices (Geneious, v. 6.1.2) revealed a striking structural similarity of the resulting proteins of the two p7-NS2-duplicated strains (Fig. 2B). However, both mutants showed different adaptations in the primary structure (Fig. 2B). Comparison of the dup+ variant and the prototype BVDV-2c strain Potsdam 1600 unveiled a number of adaptive mutations within the second copy of the duplicated region, close to the p7-NS2 cleavage site (Fig. 2), while the first copy, with a single exception, perfectly fitted the prototype genome. Signal peptides were predicted (Emboss sigcleave [25]) for all genomes for both the prototypic and adapted p7-NS2 cleavage sites. Strikingly, comparison of the absolute scores calculated by sigcleave for the predicted signal peptides showed that in both BVDV-2c and BVDV-2a 890, the adapted signal sequences received higher scores than the prototypic copies. Figure 2 displays the locations of the predicted signal peptides by use of vertical black arrows sized relative to the Potsdam 1600 score (arrow sizes were calculated by determining the scoreStrainX/scorePotsdam1600 ratio). According to the cleavage site prediction, the duplication putatively results in either an extended p7 or NS2 protein or an additional short protein, of 74 amino acids (NS21–37-p734–70), with a hydrophobicity pattern very similar to that of the p7 protein (Fig. 2B).
FIG 2.

Schematic representation of p7- and NS2-encoding regions of the genomes of a prototypic BVDV-2c strain (Potsdam 1600), dup− and dup+ variants from the recent BVDV infections in NRW, Germany, and the previously described highly virulent BVDV-2a strain 890. Corresponding parts of the genomes and the deduced proteins are depicted in corresponding colors (light and dark). (A) Structural variations at the genomic level, with the observed nonsynonymous mutations located within the duplicated region highlighted. (B) Implications of the genomic variations at the amino acid level, with the amino acid substitutions located within the duplication highlighted. The predicted transmembrane helices (TM) are shown as red arrows. Predicted cleavage sites are indicated by vertical arrows sized relative to the prototype cleavage site according to scores obtained with Emboss sigcleave.
BVDV-2c isolates are a mix of three different genomic variants.
In order to confirm the existence of the duplication within the p7-NS2 region, PCR followed by Sanger sequencing of the PCR products was done. Analyses of PCR amplicons confirmed the existence of the dup+ genome and clearly showed that both the dup+ and dup− variants coexisted in all samples from the recent outbreak (Fig. 3A; Table 1). Detailed analyses of the obtained deep-sequencing reads shed further light on the composition of the population. Due to specific sequence signatures, it was possible to sort the reads into different subpopulations. The first subpopulation comprised reads spanning the newly generated NS2–p7 fusion site plus at least 20 nt at either side. These were extracted from the complete data set and could be assembled into a single contig (a single contiguous sequence built from multiple short reads) representing nucleotides 3290 to 3721 of the dup+ open reading frame, thereby proving the existence of the dup+ genomes. The second subpopulation contained reads representing the duplicated p7-NS2 region, but without the newly generated NS2–p7 fusion site. Assembly of the reads representing the second subpopulation resulted in dup− full-genome sequences with either an adapted or nonadapted p7 cleavage site. Accordingly, reads of the second subpopulation could be subdivided further by the existence of adaptive mutations. In-depth analysis of the sequencing reads clearly showed that the mutations at positions 143, 149, and 155 in p7 (Fig. 2A) were strictly linked. The mutations at positions 4 and 14 in the NS2-encoding region (Fig. 2A) were likewise strictly coupled. In contrast, mutations at positions 61 and 64 in NS2 (Fig. 2A) could not be found together in a single read but, rather, were mutually exclusive.
FIG 3.

Agilent Bioanalyzer 2100 analyses (with a DNA 7500 chip) of PCR products. (Upper panels) Gel view of Agilent traces. Bands representing dup+ and dup− genomes are marked by arrows labeled accordingly. (Lower panels) Bar plots of relative quantities of PCR products determined by Agilent software, representing dup+ and dup− genomes. (A) Analysis of recent BVDV-2c strains. NRW 19-13-8a (lane 7) depicts results for the NRW 19-13-8 isolate, reisolated from the animal trial. (B) Analysis of BVDV-2a 890 wild-type strain (v890WT) and clones. Results obtained for plasmids of the variants with (p890FL dup+) and without (p890FL dup−) duplication and the p890FL dup+-derived in vitro transcript are shown. In addition, results for virus rescued from in vitro-transcribed RNA and passaged in cell culture 2 (v890FL dup+ P2), 3 (v890FL dup+ P3), and 4 (v890FL dup+ P4) times are displayed.
The dup− genomes could be distinguished based on the different patterns of mutations found in the different copies of the duplicated region (Fig. 4A). As mentioned above, reads representing the dup+ genome can be identified by the unique NS2–p7 fusion site. By analogy, reads representing either of the possible dup− genomes must have certain combinations of nucleotide variations arising from combinations of the different modules (p7100–210 copies 1 and 2 and NS21–111 copies 1 and 2). As can be seen in Fig. 4A, the dup−1 variant comprised a G64A mutation within its NS21–111 module as the single variation and was flanked by the original context at its 3′ end, i.e., flanked by NS2112–453 instead of the NS2–p7 fusion site. The dup−2 variant (Fig. 4A) contained six mutations scattered across the p7100-NS111 region in the dup+ second copy and was flanked by the original p71–100 module, instead of the newly arisen fusion site, at its 5′ end. Finally, the dup−3 variant (Fig. 4A) was reflected by reads with A4G, C14T, and T61G mutations in the NS21–111 module but was flanked by a completely prototypic p7100–210 module, i.e., without the T143C, T149C, and G155T mutations within that region. However, none of the reads encompassing p7100–210 and NS21–111 represented the first, i.e., unmutated, copy of the p7100–210 module and, at the same time, the A4G and C14T mutations within NS21–111. Therefore, analysis of the sequence reads obtained from deep sequencing enabled us to rule out the existence of the dup−3 variant.
FIG 4.

Schema of theoretically possible dup− variants and potential RNA secondary structures facilitating their generation. (A) Schematic representation of theoretically possible dup− variants fitting the BVDV-2c prototype Potsdam 1600 that could be generated from dup+ genomes by deletions. Different shades of blue indicate different copies of the p7-NS2 region. (B) Circular graphs of RNA secondary structure predictions of the p7-NS2 region for the positive and negative strands. The outer circle represents the 5′-to-3′ sequence (clockwise). Arcs within the circle show base interactions (red, G-C; blue, A-U; and green, G-U). Along the circle, the modules of the duplicated region are highlighted. The sections marked in red represent interacting RNA stretches which bring corresponding regions of the two copies into close proximity, thereby providing an opportunity for the polymerase to circumvent exactly one copy. (C) Schematic straight representations of the circular parts of panel B, without base interactions.
Altogether, from our in-depth analysis, it was concluded that three different genome variants existed in the populations: the genome with a duplication (dup+), in which adapted and nonadapted p7-NS2 cleavage sites coexisted in a single genome in a fixed order, as well as a mixed population of two different genomes without duplication (dup−), with either an adapted or nonadapted p7-NS2 cleavage site (Fig. 4A).
dup− genomes are generated de novo from dup+ genomes during replication.
The observation that the dup− genomes comprised either the adapted or nonadapted p7-NS2 cleavage site implied that the dup− genomes were generated de novo during replication by deletion of either of the copies. To confirm this theory, we used the cloned BVDV-2a 890 strain (p890FL) as a model system. For this purpose, PCR products were generated from RNA derived from v890FL dup+ after 2, 3, and 4 cell culture passages. RNA from v890FL after the first passage was not analyzed because the results would not be reliable due to in vitro-transcribed RNA from transfection remaining in the preparation. In parallel, we generated PCR products from v890WT, p890FL dup+, and in vitro-transcribed RNAs thereof, with p890FL dup− as a control. These PCR products were analyzed using an Agilent Bioanalyzer 2100 and by sequencing. As expected, both p890FL dup− and p890FL dup+ contained only one variant each (Fig. 3B, lanes 3 and 4), i.e., the variant encoded by the plasmid. Not surprisingly, v890WT showed PCR products for both dup+ and dup− genomes (Fig. 3B, lane 2), confirming the general mechanism of the generation of dup− from dup+ genomes. Most importantly, this analysis revealed that during replication, v890FL dup+ constantly generated dup− genomes (Fig. 3B, lanes 6 to 8). Surprisingly, in vitro transcription of p890FL dup+ by T7 DNA-dependent RNA polymerase also generated dup− variants (Fig. 3B, lane 5). Therefore, it can be concluded that a single mechanism enables the de novo generation of dup− genomes both in vitro and during viral replication.
The dup+ genome is obligatorily accompanied by a dup− genome.
Interestingly, neither a dup+ nor dup− genome alone could be found within any of the recent highly virulent BVDV-2c isolates, but both genomes were obligatorily accompanied by each other (Fig. 3A). Despite all efforts, we did not succeed in isolating a pure dup− variant (data not shown). The first attempts to separate dup− from dup+ genomes were limiting-dilution experiments using 10-fold serial dilutions in 96-well microtiter plates with KOP-R cells. We then tried molecular cloning with the model system, i.e., generation of a plasmid-borne complete genome (p890FL dup−) from which virus might be rescued in cell culture after transfection of in vitro-transcribed RNA. However, in contrast to efficient rescue of the v890FL dup+ variant, we were not able to rescue v890FL dup− from several independent clones and transfection experiments. A reason for the failure to isolate a dup− strain might possibly be replication incompetence of the dup− variants caused by the observed adaptive mutations, i.e., dup− strains might need a helper virus for their replication. In addition, as dup+ genomes constantly generate dup− genomes de novo during replication (Fig. 3), it was not possible to isolate dup+ genomes from dup− genomes.
RNA secondary structure enables copy deletions.
We hypothesized that the deletions might be facilitated by genomic secondary structures. In order to test this hypothesis, we used mfold RNA folding (27) for secondary structure prediction within the duplicated p7-NS2 region, flanked by 111 nt at each side. Figure 4B displays the predicted secondary structures in circle graphs. In these graphs, the bases are arranged clockwise along the outer circle, and each arc within the circle represents an interaction between two bases. The color of each arc indicates the predicted base pairing (red = G-C, blue = A-U, and green = G-U). For convenience, the interacting regions that enable the deletion of either of the copies are highlighted in red on the outer circle (Fig. 4B). All predicted secondary structures enable the generation of dup−1 and dup−2 genomes, but not dup−3 genomes, by bringing corresponding regions of the two copies into close proximity, thereby providing an opportunity for the polymerase to circumvent one of the copies during replication. Although base interactions are not found exactly at the start positions of the duplicated modules (Fig. 4B and C), elimination of the bases between interacting sites results in the observed mutation patterns of dup− genomes. During synthesis of the negative strand, generation of dup−2 genomes would occur, while dup−1 genomes would be generated during positive-strand synthesis.
dup+ genomes are the major component of the virus population.
As shown above, dup− genomes are probably generated from dup+ genomes during replication of the virus. Therefore, we were interested in the proportions of dup+ and dup− genomes. Quantification of the portions of the different genomes (dup+, dup−1, and dup−2 genomes) was possible based on the analysis of reads from deep sequencing, representing the different combinations of the modules. According to in-depth sequence analysis (see above), the portion of dup− genomes within BVDV-2c isolate NRW 19-13-8 was estimated to be approximately 1%. Moreover, we were interested in the portions of dup−1 and dup−2 genomes within isolate NRW 19-13-8. Therefore, we sequenced the dup− PCR product excised from an agarose gel with Illumina MiSeq sequencing to a sequence depth of roughly 68,000. Within this data set, we identified approximately 33% dup−2 and 66% dup−1 genomes based on the mutation patterns described above.
Because the estimation of the dup+ and dup− proportions was not possible for all isolates due to insufficient sequence depth, as an alternative measure, we quantified the dup+ and dup− genomes of the different isolates based on PCR products in combination with Agilent Bioanalyzer 2100 analysis (Fig. 3; Tables 2 and 3). Qualitative and semiquantitative PCR analyses of the isolates of the recent outbreak (Fig. 3A, lanes 3 to 6), virus reisolated from an experimentally infected animal (Fig. 3A, lane 7), and the reference strain Potsdam 1600 (Fig. 3A, lane 1) showed that in all isolates except for Potsdam 1600 (only dup− genomes), both dup+ and dup− genomes were present. Agilent quantification of PCR products revealed similar and surprisingly stable proportions (approximately 5% dup− and 95% dup+ genomes) compared to sequence read analysis of BVDV-2c. For v890WT in comparison with BVDV-2c, we found a clearly larger proportion of dup− genomes (33%) (Fig. 3B, lane 2). In the rescued v890FL dup+ strain, the mix of dup+ and dup− genomes was as stable as the mix for BVDV-2c through different passages, with about 75% dup+ and 25% dup− genomes (Fig. 3B, lanes 6 to 8). As expected, 100% of the respective PCR products were detected for the controls, p890FL dup− and p890FL dup+ (Fig. 3B, lanes 3 and 4).
TABLE 2.
Variant quantification for BVDV-2c
| Sample | % dup− genomes | % dup+ genomes |
|---|---|---|
| Potsdam 1600 | 100 | NAc |
| NRW 12-13 | 3.3 | 96.7 |
| NRW 14-13 | 4.8 | 95.2 |
| NRW 19-13-1 | 4.4 | 95.6 |
| NRW 19-13-8 | 5.1 | 94.9 |
| NRW 19-13-8a | 4.7 | 95.3 |
| Avgb | 4.5 | 95.5 |
Reisolated after animal experiment.
Average without Potsdam 1600 data.
NA, not applicable.
TABLE 3.
Variant quantification for BVDV-2a 890
| Sample | % dup− genomes | % dup+ genomes |
|---|---|---|
| v890WT | 32.7 | 67.3 |
| p890FL dup− | 100 | NAb |
| p890FL dup+ | NAb | 100 |
| p890FL dup+ in vitro transcript | 24.1 | 75.9 |
| v890FL dup+ 2nd passage | 21.2 | 78.8 |
| v890FL dup+ 3rd passage | 23.9 | 76.1 |
| v890FL dup+ 4th passage | 21.5 | 78.5 |
| Avga | 22.2 | 77.8 |
Average for passaged v890FL dup+.
NA, not applicable.
Recent BVDV-2c strains are closely related to one another.
Finally, phylogenetic analysis of the full-length dup+ and dup− genome sequences (MEGA 5) (20) (Fig. 5) confirmed the preliminary results for the 5′-untranslated region (1). Sequences of recent highly virulent isolates clustered closely with various prototypic German BVDV-2c strains (Fig. 5, “References, Germany”; also see Table 1) but formed their own subcluster within this group. Sequence identities of dup− genomes and German BVDV-2c references ranged between 97.4% and 98%. For dup+ genomes, the identity decreased to 95.6% to 96.2% due to the duplication.
FIG 5.

Phylogenetic analysis (neighbor-joining tree with Tamura-Nei parameter [43]; gamma distribution = 3.71; 1,000 bootstrap replicates) of the open reading frames of different BVDV strains. Recent highly virulent isolates form a distinct subcluster within genotype 2c (highlighted in bold). Strain BVDV-2a 890 clusters within genotype 2a (highlighted in bold italics). Bootstrap values of >50 are indicated at the branches.
DISCUSSION
Severe BVDV outbreaks with type 2 strains have been described before (28), but detailed sequence analysis by deep sequencing, to our knowledge, was not used before to characterize the full-length genomes of the causative strains. Therefore, we applied next-generation sequencing to define the genetic properties of the isolated BVDV strain. Interestingly, deep sequencing resulted in several novel findings and revealed two major important points: (i) the majority of BVDV-2c strains have a characteristic duplication of 222 nt within the p7-NS2 region (dup+ variant), and the minority (less than 5%) resembles the genome of related BVDV-2 strains which are normally not highly virulent (dup− variants); and (ii) the dup+ variant is the source for the de novo generation of the dup− variants during virus replication. This could be proven by using the available infectious clone system (20) for BVDV-2a 890 (p890FL dup+), which constantly generates the dup− variant during virus replication. As a consequence of the constant de novo generation of dup− genomes by deletion of the duplication from dup+ genomes, the virus variants cannot be separated. A separate investigation of the dup+ variant, e.g., concerning virulence, is therefore impossible. Although individual characterization of dup+ or dup− genomes is not feasible, it is very unlikely that the increased virulence of the mixed population is caused by the dup− variant, for two important reasons. (i) The observed constantly low percentage of 5% or even less of the virus population for all tested highly virulent isolates does not provide an indication for dup− genomes as the cause of high virulence. (ii) The genome structure of dup− genomes fits the well-known prototypic structure of BVDV-2 strains, for which, in the vast majority of cases, no high virulence was reported (28). Our results also clearly show that the existence of the dup+ and dup− variants is always linked. The dup− variants are therefore not ancestors of the duplicated variant, as we initially expected, but rather the duplicated genome is the source of the versions without duplication. The fact that the percentage of dup− genomes remains constant for different isolates, both after animal infection and cell culture passaging and in the different field isolates, argues for an equilibration between dup+ and dup− genomes and for a marked replication advantage of the dup+ virus. We therefore concluded that the dup+ variant alone or the dup+ variant together with the less prominent dup− variant is responsible for the observed phenotype.
A duplication resembling that of recent BVDV-2c strains was reported previously (19), but only the dup+ variant was detected. Interestingly, we were also able to detect a dup− variant in the available BVDV-2a 890 wild-type virus isolate (v890WT) by PCR analysis and subsequent sequencing of the PCR products. Ridpath and Bolin (19) concluded that the duplication was not a prerequisite for virulence, since five other virulent BVDV-2 isolates were negative when tested for the presence of the insertion. However, it could not be excluded that the duplication is a crucial factor for the respective strain or strain composition and is therefore directly linked with high virulence. The pathogenesis of BVDV-2c behind the observed clinical picture in both experimental and field infections might be caused by mechanisms similar to those discussed for highly virulent classical swine fever viruses, which are related pestiviruses (29). As a main factor, a higher replication efficacy together with immunopathogenic mechanisms, including cytokine dysregulation, are assumed (30). Additional studies looking for immunotolerant persistently infected calves in the outbreak region, as well as molecular attenuation of the original isolates, will allow further insights.
For members of other virus families, duplications have been reported before, either for complete genes (31–33) or for only parts within open reading frames (34–36), and in some cases were also connected to very high virulence. For example, Perdue and colleagues (35) reported a highly pathogenic influenza virus in which the polybasic cleavage site was generated by duplication of part of a purine-rich region.
In most cases, BVDV variants with insertions, point mutations, or duplications in the nonstructural proteins are observed in cytopathic (cp) strains isolated from animals suffering from the so-called mucosal disease (15). In these cases, a cp strain is generated de novo from the persisting noncytopathic (ncp) virus, resulting in a “matching pair” of cp and ncp viruses which finally kill the host. Several different cp variants have been described, including viruses with sequence duplications, and the p7-to-NS3 genome region is the region affected most often (37, 38). A similar mechanism probably led to the generation of a noncytopathogenic highly virulent virus variant by duplication within the p7-NS2 region. Similar BVDV-2c strains without duplication were isolated in Germany (e.g., the Potsdam 1600 strain), and most likely, an inconspicuous persistently infected calf was the source of the novel variant, which subsequently spread to other farms.
Interestingly, the duplication occurred upstream of the known “hot spot” region for cp BVDV (37). The observed unique adaptive mutations in the p7-NS2 region are remarkable, since the p7-encoding genome part, in general, appears to be highly conserved in all pestiviruses (39). Only these very specific single nucleotide polymorphisms (SNPs) allowed the detection and differentiation of the duplicated genome parts by deep sequencing. In addition, these changes are unique, and these combinations of SNPs cannot be found in any other pestivirus sequence in the databases, proving that they are most likely the consequence of an adaptation to the duplicated sequences. We therefore cannot exclude that besides the duplication, these adaptive SNPs especially are important for the very virulent phenotype. Furthermore, our results fuel the implication of the p7-NS2 duplication in the high-virulence phenotype, which was controversially discussed before (19).
We showed that de novo generation of two of the three theoretically possible dup− variants indeed occurs during viral replication of the dup+ genome. dup− genomes are also produced by T7 DNA-dependent RNA polymerase in vitro during transcription of viral genomes from the cloned virus. In addition, we have a clear indication that RNA secondary structures possibly easily enable the generation of the two deleted variants, during minus- and plus-strand synthesis. The observed proportions of 33% dup−2 genomes, generated during negative-strand synthesis, and 66% dup−1 genomes, generated during positive-strand synthesis, are in accordance with the data of Gong and colleagues (40), who reported that for BVDV, synthesis and accumulation of positive-strand viral RNA are 2- to 10-fold higher than those of negative-strand viral RNA. The mechanism of RNA polymerase “jumping” at RNA secondary structures was also suggested for the generation of the above-mentioned highly pathogenic avian influenza virus (35). By analogy, the mechanism underlying the initial duplication could be an intermolecular interaction between similar genomic regions leading to recombination of two genome copies during replication. This would be the reverse of what is shown in Fig. 4 for RNA secondary structures that enable the deletion of either of the copies. The introduction of the adaptive mutations after the insertion of the duplication would then be accomplished easily during genome replication by the error-prone viral RNA-dependent RNA polymerase. These different lines of argument indicate a general mechanism for the introduction of variations into viral genomes that is dependent on the viral RNA polymerases and their immanent errors in combination with nucleic acid secondary structures. Unfortunately, the de novo-generated BVDV-2c dup− variants could not be separated from the dup+ variant, and it could therefore not be proven if autonomous replication and virus growth are supported by these genomes. This might be caused by the adaptive SNPs in the duplicated region or elsewhere in the genome. The constant but minor presence of the dup− variants in all tested isolates of both BVDV-2 strains could also be an indication of a reduced replication activity of the dup− genome variants.
At the amino acid level, the duplication can possibly lead to an extended p7 or NS2 protein. The extended p7 or NS2 protein would occur if the nascent polyprotein were cleaved at only one of the two cleavage sites indicated in Fig. 2B. Alternatively, the duplication could give rise to an additional short protein (NS21–37-p734–70), which would be generated by cleavage of the polyprotein at both predicted cleavage sites (Fig. 2B). According to transmembrane segment prediction, this potentially newly arising NS21–37-p734–70 fusion protein clearly resembles the p7 protein. Thus, it can be speculated that an additional protein (NS21–37-p734–70) is processed which structurally fits the p7 protein. As p7 is assumed to act as a so-called “viroporin” and is involved in virus assembly and release (41, 42), a new p7-like protein would be a hint for more effective assembly and release and could therefore cause a higher level of virulence. However, future investigations are especially necessary in order to elucidate the functional consequences of the duplication for protein processing and functionality.
In conclusion, we describe here a very detailed deep-sequencing analysis of a unique pestivirus strain composition connected with extreme virulence for cattle, demonstrating the importance of next-generation sequencing for the analysis of novel and unexpected genomes. Our study emphasizes the importance of full-genome deep sequencing in combination with manual in-depth data analysis for the investigation of viruses in basic research and diagnostics.
ACKNOWLEDGMENTS
We thank Moctezuma Reimann, Bianka Hillmann, Doreen Reichelt, and Christian Korthase for excellent technical assistance.
This work was supported by the EMIDA ERA-NET project “Molecular Epidemiology of Epizootic Diseases using Next Generation Sequencing Technology” (Epi-SEQ) (grant 2811ERA094) and by the FP7 project “Rapid Field Diagnostics and Screening in Veterinary Medicine” (RAPIDIA-FIELD) (grant 289364).
Footnotes
Published ahead of print 9 April 2014
REFERENCES
- 1.Schirrmeier H. 2013. Auftreten von akuten Infektionen mit BVDV-2c in Deutschland. Mitt. Ganzen Nord. 2:17–21 [Google Scholar]
- 2.ProMED-Mail. 26 March 2013. Bovine viral diarrhea—Netherlands: (GE), BVDV type II. Archive number 20130326.1603817. http://www.promedmail.org/
- 3.Colett MS, Larson R, Gold C, Strick D, Anderson DK, Purchio AF. 1988. Molecular cloning and nucleotide sequence of the pestivirus bovine viral diarrhea virus. Virology 165:191–199. 10.1016/0042-6822(88)90672-1 [DOI] [PubMed] [Google Scholar]
- 4.Collett MS, Larson R, Belzer SK, Retzel E. 1988. Proteins encoded by bovine viral diarrhea virus: the genomic organization of a pestivirus. Virology 165:200–208. 10.1016/0042-6822(88)90673-3 [DOI] [PubMed] [Google Scholar]
- 5.Peterhans E, Bachofen C, Stalder H, Schweizer M. 2010. Cytopathic bovine viral diarrhea viruses (BVDV): emerging pestiviruses doomed to extinction. Vet. Res. 41:44. 10.1051/vetres/2010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ridpath JF, Bolin SR. 1998. Differentiation of types 1a, 1b and 2 bovine viral diarrhoea virus (BVDV) by PCR. Mol. Cell. Probes 12:101–106. 10.1006/mcpr.1998.0158 [DOI] [PubMed] [Google Scholar]
- 7.Fulton RW, Saliki JT, Confer AW, Burge LJ, d'Offay JM, Helman RG, Bolin SR, Ridpath JF, Payton ME. 2000. Bovine viral diarrhea virus cytopathic and noncytopathic biotypes and type 1 and 2 genotypes in diagnostic laboratory accessions: clinical and necropsy samples from cattle. J. Vet. Diagn. Invest. 12:33–38. 10.1177/104063870001200106 [DOI] [PubMed] [Google Scholar]
- 8.Gillespie JH, Baker JA, McEntee K. 1960. A cytopathogenic strain of virus diarrhea virus. Cornell Vet. 50:73–79 [PubMed] [Google Scholar]
- 9.Lee KM, Gillespie JH. 1957. Propagation of virus diarrhea virus of cattle in tissue culture. Am. J. Vet. Res. 18:952–953 [PubMed] [Google Scholar]
- 10.Underdahl NR, Grace OD, Hoerlein AB. 1957. Cultivation in tissue-culture of cytopathogenic agent from bovine mucosal disease. Proc. Soc. Exp. Biol. Med. 94:795–797. 10.3181/00379727-94-23091 [DOI] [PubMed] [Google Scholar]
- 11.Bielefeldt Ohmann H. 1988. BVD virus antigens in tissues of persistently viraemic, clinically normal cattle: implications for the pathogenesis of clinically fatal disease. Acta Vet. Scand. 29:77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyling A, Houe H, Jensen AM. 1990. Epidemiology of bovine virus diarrhoea virus. Rev. Sci. Tech. 9:75–93 [DOI] [PubMed] [Google Scholar]
- 13.Baker JC. 1987. Bovine viral diarrhea virus: a review. J. Am. Vet. Med. Assoc. 190:1449–1458 [PubMed] [Google Scholar]
- 14.Donis RO, Dubovi EJ. 1987. Differences in virus-induced polypeptides in cells infected by cytopathic and noncytopathic biotypes of bovine virus diarrhea-mucosal disease virus. Virology 158:168–173. 10.1016/0042-6822(87)90250-9 [DOI] [PubMed] [Google Scholar]
- 15.Brownlie J, Clarke MC, Howard CJ. 1984. Experimental production of fatal mucosal disease in cattle. Vet. Rec. 114:535–536. 10.1136/vr.114.22.535 [DOI] [PubMed] [Google Scholar]
- 16.Beer M, Wolf G, Kaaden OR. 2002. Phylogenetic analysis of the 5′-untranslated region of German BVDV type II isolates. J. Vet. Med. B Infect. Dis. Vet. Public Health 49:43–47. 10.1046/j.1439-0450.2002.00536.x [DOI] [PubMed] [Google Scholar]
- 17.Carman S, van Dreumel T, Ridpath J, Hazlett M, Alves D, Dubovi E, Tremblay R, Bolin S, Godkin A, Anderson N. 1998. Severe acute bovine viral diarrhea in Ontario, 1993–1995. J. Vet. Diagn. Invest. 10:27–35. 10.1177/104063879801000106 [DOI] [PubMed] [Google Scholar]
- 18.Martin R, Kühne S. 2005. Verlauf einer Herdeninfektion mit BVDV-2 Ein Fallbericht. Tierarztl. Praxis Grosstiere 33:224–231 [Google Scholar]
- 19.Ridpath JF, Bolin SR. 1995. The genomic sequence of a virulent bovine viral diarrhea virus (BVDV) from the type 2 genotype: detection of a large genomic insertion in a noncytopathic BVDV. Virology 212:39–46. 10.1006/viro.1995.1451 [DOI] [PubMed] [Google Scholar]
- 20.Mischkale K, Reimann I, Zemke J, König P, Beer M. 2010. Characterisation of a new infectious full-length cDNA clone of BVDV genotype 2 and generation of virus mutants. Vet. Microbiol. 142:3–12. 10.1016/j.vetmic.2009.09.036 [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann B, Depner K, Schirrmeier H, Beer M. 2006. A universal heterologous internal control system for duplex real-time RT-PCR assays used in a detection system for pestiviruses. J. Virol. Methods 136:200–209. 10.1016/j.jviromet.2006.05.020 [DOI] [PubMed] [Google Scholar]
- 22.Becker N, Jöst Ziegler HU, Eiden M, Höper D, Emmerich P, Fichet-Calvet E, Ehichioya DU, Czajka C, Gabriel M, Hoffmann B, Beer M, Tenner-Racz K, Racz P, Günther S, Wink M, Bosch S, Konrad A, Pfeffer M, Groschup MH, Schmidt-Chanasit J. 2012. Epizootic emergence of Usutu virus in wild and captive birds in Germany. PLoS One 7:e32604. 10.1371/journal.pone.0032604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonnhammer EL, von Heijne G, Krogh A. 1998. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6:175–182 [PubMed] [Google Scholar]
- 25.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. 10.1016/S0168-9525(00)02024-2 [DOI] [PubMed] [Google Scholar]
- 26.Geiser M, Cebe R, Drewello D, Schmitz R. 2001. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques 31:88–90, 92 [DOI] [PubMed] [Google Scholar]
- 27.Mathews DH, Turner DH, Zuker M. 2007. RNA secondary structure prediction. Curr. Protoc. Nucleic Acid Chem. Chapter 11:Unit 11.2. 10.1002/0471142700.nc1102s28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ridpath JF, Neill JD, Frey M, Landgraf JG. 2000. Phylogenetic, antigenic and clinical characterization of type 2 BVDV from North America. Vet. Microbiol. 77:145–155. 10.1016/S0378-1135(00)00271-6 [DOI] [PubMed] [Google Scholar]
- 29.Lange A, Blome S, Moennig V, Greiser-Wilke I. 2011. Pathogenesis of classical swine fever—similarities to viral haemorrhagic fevers: a review. Berl. Munch. Tierarztl. Wochenschr. 124:36–47. 10.2376/0005-9366-124-36 [DOI] [PubMed] [Google Scholar]
- 30.Graham SP, Everett HE, Johns HL, Haines FJ, La Rocca SA, Khatri M, Wright IK, Drew T, Crooke HR. 2010. Characterisation of virus-specific peripheral blood cell cytokine responses following vaccination or infection with classical swine fever viruses. Vet. Microbiol. 142:34–40. 10.1016/j.vetmic.2009.09.040 [DOI] [PubMed] [Google Scholar]
- 31.Pickup DJ, Ink BS, Parsons BL, Hu W, Joklik WK. 1984. Spontaneous deletions and duplications of sequences in the genome of cowpox virus. Proc. Natl. Acad. Sci. U. S. A. 81:6817–6821. 10.1073/pnas.81.21.6817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suhre K. 2005. Gene and genome duplication in Acanthamoeba polyphaga mimivirus. J. Virol. 79:14095–14101. 10.1128/JVI.79.22.14095-14101.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyko VP, Karasev AV, Agranovsky AA, Koonin EV, Dolja VV. 1992. Coat protein gene duplication in a filamentous RNA virus of plants. Proc. Natl. Acad. Sci. U. S. A. 89:9156–9160. 10.1073/pnas.89.19.9156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qi F, Ridpath JF, Berry ES. 1998. Insertion of a bovine SMT3B gene in NS4B and duplication of NS3 in a bovine viral diarrhea virus genome correlate with the cytopathogenicity of the virus. Virus Res. 57:1–9. 10.1016/S0168-1702(98)00073-2 [DOI] [PubMed] [Google Scholar]
- 35.Perdue ML, García M, Senne D, Fraire M. 1997. Virulence-associated sequence duplication at the hemagglutinin cleavage site of avian influenza viruses. Virus Res. 49:173–186. 10.1016/S0168-1702(97)01468-8 [DOI] [PubMed] [Google Scholar]
- 36.Trento A, Galiano M, Videla C, Carballal G, Garcia-Barreno B, Melero JA, Palomo C. 2003. Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J. Gen. Virol. 84:3115–3120. 10.1099/vir.0.19357-0 [DOI] [PubMed] [Google Scholar]
- 37.Meyers G, Tautz N, Stark R, Brownlie J, Dubovi EJ, Collett MS, Thiel H-J. 1992. Rearrangement of viral sequences in cytopathogenic pestiviruses. Virology 191:368–386. 10.1016/0042-6822(92)90199-Y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyers G, Thiel HJ. 1996. Molecular characterization of pestiviruses. Adv. Virus Res. 47:53–118. 10.1016/S0065-3527(08)60734-4 [DOI] [PubMed] [Google Scholar]
- 39.Leifer I, Hoffmann B, Höper D, Bruun Rasmussen T, Blome S, Strebelow G, Höreth-Böntgen D, Staubach C, Beer M. 2010. Molecular epidemiology of current classical swine fever virus isolates of wild boar in Germany. J. Gen. Virol. 91:2687–2697. 10.1099/vir.0.023200-0 [DOI] [PubMed] [Google Scholar]
- 40.Gong Y, Trowbridge R, Macnaughton TB, Westaway EG, Shannon AD, Gowans EJ. 1996. Characterization of RNA synthesis during a one-step growth curve and of the replication mechanism of bovine viral diarrhoea virus. J. Gen. Virol. 77:2729–2736. 10.1099/0022-1317-77-11-2729 [DOI] [PubMed] [Google Scholar]
- 41.Elbers K, Tautz N, Becher P, Stoll D, Rumenapf T, Thiel HJ. 1996. Processing in the pestivirus E2-NS2 region: identification of proteins p7 and E2p7. J. Virol. 70:4131–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Largo E, Gladue DP, Huarte N, Borca MV, Nieva JL. 2014. Pore-forming activity of pestivirus p7 in a minimal model system supports genus-specific viroporin function. Antiviral Res. 101:30–36. 10.1016/j.antiviral.2013.10.015 [DOI] [PubMed] [Google Scholar]
- 43.Tamura K, Nei M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10:512–526 [DOI] [PubMed] [Google Scholar]