

ABSTRACT
The emergence of drug-resistant hepatitis B virus (HBV) is a major problem for antiviral treatment in chronic hepatitis B infection. In this study, we analyzed the evolution of drug-resistant mutations and characterized the effects of the rtA181T and rtI233V mutations on viral replication and drug resistance. We performed a clonal analysis of the HBV polymerase gene from serum samples during viral breakthrough treated with antiviral agents. A series of mutant clones containing rtA181T and/or rtI233V mutations were constructed and determined the effect of these mutations on the replication ability and drug resistance. An in vitro study revealed that the effect of the rtA181T mutation on viral replication and drug resistance is dependent on the mutations in the overlapping surface gene. Compared to the rtA181T surface missense mutation (rtA181T/sW172S), the introduction of rtA181T surface nonsense mutation (rtA181T/sW172*) resulted in decreased viral replication and increased drug resistance. Complementation assay revealed that the truncated PreS1 is responsible for reduced replication of rtA181T/sW172* mutant. Moreover, the rtA181T/sW172* mutant exhibited a defect in viral particle secretion. The rtI233V mutation that emerged during adefovir therapy reduced viral replication and conferred resistance to adefovir. Our data suggest that the impact of the rtA181T mutation on replication and drug resistance differs based on the mutation status of the corresponding surface gene. The rtI233V mutation also affects replication ability and drug resistance. This observation suggests the need for genotypic analysis of overlapping surface genes to manage antiviral drug resistance if clinical isolates harbor the rtA181T mutation.
IMPORTANCE The emergence of drug-resistant HBV that are no longer susceptible to nucleos(t)ide analogues is a major problem for antiviral treatment in chronic hepatitis B infection. Among drug-resistant mutations, the single rtA181T mutation is known to confer cross-resistance to antiviral drugs. This mutation causes intermediate or reduced susceptibility to tenofovir. Moreover, the clinical occurrence of the rtA181T mutation during antiviral therapy is also high. Our study revealed that the effect of the rtA181T mutation on viral replication and drug resistance is dependent on the mutations in the overlapping surface gene. This observation suggests the need for genotypic analysis of overlapping surface genes to manage antiviral drug resistance if clinical isolates harbor the rtA181T mutation. We believe that our study will not only extend the understanding of the drug resistance mechanism, but it will also ultimately provide new treatment options for patients with multidrug resistant HBV.
INTRODUCTION
The hepatitis B virus (HBV) is a hepatotropic DNA virus causing chronic hepatitis B. More than 400 million people suffer from chronic hepatitis B worldwide, leading to cirrhosis and hepatocellular carcinoma (HCC) (1, 2). Thus, effectively controlling HBV infection is considered a major issue in chronic hepatitis B management.
Currently, several nucleos(t)ide analogues, including lamivudine (LMV), adefovir (ADV), entecavir (ETV), telbivudine (LdT), and tenofovir (TDF) are approved for treatment in patients with hepatitis B. These drugs are used to suppress viral replication and support liver disease remission and subsequently prevent the progression to cirrhosis and HCC (3). The first approved anti-HBV agent LMV showed a good response to the initial treatment; however, genotypic resistance increased up to 70% after 5 years of treatment (4). ADV, still used worldwide, was effective against wild type (WT) and LMV-resistant HBV mutants (5–7). Thus, patients with LMV-resistant HBV were switched to ADV or a ADV-LMV combination therapy (8). It is reported that ADV alone or ADV-LMV combination therapy produced effective antiviral effects in patients with LMV-resistant chronic hepatitis B (9, 10). However, ADV monotherapy in patients with LMV-resistant chronic hepatitis B frequently experienced ADV resistance, compared to untreated patients (11). Therefore, ADV and LMV combination therapy was recommended for LMV-resistant HBV (12). Recently, ETV was found to be effective in treating both LMV- and ADV-resistant HBV (13).
Among several drug resistance-conferring mutations, the single rtA181T mutation can confer cross-resistance by removing or reducing the genetic barrier to drugs (14). The rtA181T mutation has been reported in patients undergoing LMV, ADV, clevudine, or LdT therapy (14–16). According to the 2012 EASL Guidelines, the rtA181T mutation leads to LMV, ADV, and LdT resistance. The mutation causes intermediate or reduced susceptibility to TDF, and mutant strains are only sensitive to ETV treatment (13). In vitro phenotypic studies showed that the rtA181T mutant showed decreased susceptibility to LMV, ADV, and TDF (17).
Due to the overlapping nature of the open reading frames coding for the polymerase and the surface protein, the rtA181T nucleotide substitution in the polymerase results in either an sW172* (stop codon) or missense mutation such as sW172S in the mRNA encoding the surface protein. Very recently, the sequence dynamics of ADV-resistant HBV variants in patients with chronic HBV infection were analyzed using ultradeep pyrosequencing techniques to determine the population of minor viral variants (18). The authors of that study found that even though the dynamics of ADV-resistant variants were highly complex, the rtA181T substitution was mainly associated with an sW172* substitution (80 to 90% of variants compared to other missense mutations) during therapy. Furthermore, the rtA181T/sW172* mutant was reportedly associated with tumorigenesis (19, 20) and HBsAg and virion secretion defects (15, 21). The frequency of HBV encoding rtA181T/sW172* was as high as 42% in patients receiving LMV and ADV combination therapy (15). These reports emphasized the importance of finely characterizing rtA181T mutants and raised the question whether the effect of the evolutionally dominant rtA181T/sW172* mutation on replication and drug resistance differs from that of the missense surface protein mutation.
On the other hand, the influence of the I233V mutation on ADV resistance is controversial. Schildgen et al. reported that the I233V mutation confers primary resistance to ADV (22), whereas another group showed that it results in susceptibility (23). Therefore, in the present study, we investigated the evolution of drug resistance mutations in serial samples obtained from a patient with sequential LMV, ADV, and combination therapy and the effect of rtI233V and rtA181T/sW172* mutations on viral replication and drug resistance; our data show that both the rtI233V and the rtA181T/sW172* mutations have an effect.
MATERIALS AND METHODS
Patient.
A 47-year-old man with chronic hepatitis B was treated with LMV as a first-line HBV therapy. HBV DNA was reduced to undetectable levels (<300 copies/ml) in real-time PCR analysis after LMV therapy. However, viral breakthrough occurred after 36 months of LMV treatment. After switching to ADV, HBV DNA was decreased to ∼40,000 copies/ml; however, viral breakthrough with the ADV-resistant mutation was observed after 28 months of ADV treatment. LMV was added to the ADV, and the HBV DNA level gradually decreased after the combination treatment. An increased HBV DNA level was observed after 15 months of combination therapy (Fig. 1A).
FIG 1.


Clinical characteristics and in vitro susceptibility assay of RT mutants isolated at the time of treatment failure. (A) Clinical course during chronic hepatitis B infection in a patient. Arrowheads indicate the time points of serum sampling used in the present study. Clone series denoted by “#” were selected for further study. Analysis of coexisting HBV quasispecies at the time of viral breakthrough is summarized. (B to D) The HBV RT genes from each serum sample were converted into HBV1.2mer replicons, and the substitutions were compared to the WT. Cloned HBV DNAs were transfected into Huh7 cells, and cells were treated for 4 days with or without 20 μM LMV or ADV. The intracellular HBV DNA was analyzed by Southern blotting. The relative replication ability and drug susceptibility compared to the WT were quantified by using a PhosphorImager. Aliquots of same cell lysates were analyzed by Southern blotting to detect HBV core and actin proteins to verify the transfection yield and loading amount.
Construction of the HBV replicon and direct sequencing of the polymerase RT domain.
To analyze the complete sequence of the HBV reverse transcriptase (RT) gene that evolved during the antiviral treatment, HBV DNA was isolated from serially obtained patient serum samples, which were collected before the treatment and at the time of viral breakthrough (Fig. 1), by using the QIAamp MinElute virus spin kit (Qiagen) according to the manufacturer's manual. After the RT gene PCR, HBV1.2mer replicons harboring the patient-derived RT gene were constructed using the pGEM-4z vector (Promega) and sequenced as previously described (24). All artificial HBV1.2mer clones, including rtI233V, rtA181T/sW172S, rtA181T/sW172*, rtN236T, rtA181T/sW172*+rtN236T, rtA181T/sW172S+rtI233V, and rtA181T/sW172*+rtI233V, were generated by site-directed mutagenesis using WT HBV1.2mer. Plasmids for the expression of truncated PreS1, PreS2, and S were cloned into pCMV-tag2A by using the rtA181T/sW172* HBV1.2mer as the template. The polymerase constructs, including Pol-WT, Pol-rtA181T/sW172S, and Pol-rtA181T/sW172*, were cloned into pCMV-tag2A using WT, rtA181T/sW172S, and rtA181T/sW172* HBV1.2mer as the templates, respectively. The polymerase-null HBV replicon [HBV Pol(−)] was kindly provided by W. S. Ryu (Yonsei University).
Antiviral drug treatment.
LMV and ADV were provided by GlaxoSmithKline (Brentford, United Kingdom) and Gilead Sciences (Foster City, CA), respectively. Huh7 human hepatoma cells were cultured in Dulbecco modified Eagle medium (Gibco-BRL) supplemented with 10% heat-inactivated fetal bovine serum (Gibco-BRL) and 1% penicillin-streptomycin (Gibco-BRL). Then, 2-μg portions of WT and mutant HBV replicons were transfected into Huh7 cells using Lipofectamine 2000 (Invitrogen). At 4 h after transfection, the medium was replaced with a fresh medium containing the indicated concentrations of LMV, ADV, TDF, and ETV, as previously described (24). A fresh drug-containing medium was replaced every day for 4 days, and the cells were harvested.
Drug susceptibility and replication assay.
In vitro drug susceptibility and replication assays were performed using Southern blotting as described previously (24, 25). Briefly, cell pellets were lysed with 100 μl of HEPES buffer containing 1% NP-40. Transfected plasmid DNA was removed from the cell lysate by nuclease treatment for 15 min at 37°C in reaction buffer. Core particles were precipitated with a 26% polyethylene glycol (PEG) solution. After incubation for 2 h on ice, the capsid was digested with proteinase K at 37°C for 2 h in the presence of sodium dodecyl sulfate (SDS). HBV DNA was extracted with phenol and precipitated with ethanol. Purified DNA was separated on a 0.8% agarose gel. After transfer to a Hybond-N+ membrane (Amersham), HBV DNA was detected using a highly pure and randomized HBV probe labeled with [α-32P]dCTP (500 μCi/μmol; Perkin-Elmer). HBV replication was quantified using a phosphorimager and Multi-Gauge v3.2 software (Fujifilm, Tokyo, Japan).
To analyze the virion secretion, the culture supernatant was loaded on top of 20% sucrose in TEN buffer and subjected to ultracentrifugation at 39,000 rpm for 18 h to separate extracellular HBV particles. HBV DNA was separated as described above and subject to Southern blot analysis.
Northern blot analysis.
Total RNA was extracted by using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. A 20-μg portion of total RNA was separated on a 1% formaldehyde-agarose gel and transferred to a nitrocellulose membrane (GE Healthcare). To detect HBV RNA, the membrane was hybridized with purified 32P-labeled HBV probe used for Southern blot analysis. One microgram of total RNA was used as a loading control.
Western blot assay, HBsAg and HBeAg enzyme-linked immunosorbent assay (ELISA).
Cell lysates were resolved on a 12% SDS-polyacrylamide gel and transferred onto polyvinylidene fluoride membranes (Millipore). The membranes were blocked in TBS containing 5% skim milk (BD) and 0.1% Tween 20 and then probed with the following: a rabbit polyclonal anti-HBV core (Dako), a horse polyclonal HBs (Novus, Abcam, and Santa Cruz), or a mouse monoclonal anti-β-actin (Sigma) antibody. To analyze the HBsAg and HBeAg secretion, culture supernatants were collected and analyzed using the Enzygnost HBsAg 6.0 and Enzygnost HBe monoclonal kit (Siemens) according to the manufacturer's protocols, respectively.
RESULTS
Sequence evolution of HBV polymerase RT isolated from a patient with chronic hepatitis B during sequential antiviral therapy.
Clinical characteristics of the patient are presented in Fig. 1. Five sequential serum samples from the patient were collected before the treatment and at the time of viral breakthrough during the indicated antiviral therapy. Serum HBV DNA was purified and the patient-derived RT genes were converted into HBV1.2mer replicons. Approximately 10 clones per each serum were obtained, and all clones were sequenced. The complete sequence data of the HBV RT domain are summarized in Table 1. The amino acid substitutions were determined by comparing RT domain sequences with the WT genotype C sequences, as previously reported (24).
TABLE 1.
Analysis of HBV RT mutations isolated from a patient receiving sequential antiviral therapya

Position numbers and wild-type (GQ872210) amino acids are indicated in the upper and lower column headings, respectively; the specific individual mutations are indicated in the body of the table.
At baseline, when no antiviral agent was used, the rtI233V mutation was predominantly detected. Interestingly, most of the rtI233V clones harbored an rtV191I substitution on the same HBV genome. The corresponding change in rtV191I in the overlapping surface gene was sW182* (which introduces a stop codon). Therefore, the dominant HBV clone at baseline (the “20 series”) was the rtV191I/sW182*+rtI233V mutant.
At viral breakthrough during LMV treatment, three conserved mutations (L80I, L199V, and M204I) were predominantly detected and colocalized on the same genome (the clone 22 series). Mutations L80I and M204I were well-known LMV resistance mutations. Intriguingly, the rtI233V mutation, the ADV resistance of which is still under debate (22, 23), could not be detected after LMV treatment, suggesting no beneficial effect of this mutation on LMV resistance.
After LMV viral breakthrough, the antiviral drug was switched to ADV. Clonal analyses revealed that the mutants containing rtH55R+I233V+N236T emerged and were dominantly selected during ADV treatment failure (the “10 series”). The reemergence of rtI233V, which disappeared during LMV therapy, suggests that this mutation may have some beneficial role in ADV resistance for the HBV mutants harboring rtN236T.
The most notable mutational emergence after LMV and ADV combination therapy was the rtA181T mutation, implying that this mutation confers cross-resistant to both drugs, as suggested previously (17). Clonal analyses revealed that in most of the rtA181T mutants, this mutation colocalizes with rtI233V and rtN236T on the same HBV genome following ADV or LMV+ADV breakthrough (Table 1).
Since the HBV surface gene sequence overlaps with that of the polymerase, we analyzed the corresponding changes with respect to the rtA181T mutation in the surface gene. Early during LMV and ADV treatment, the surface gene mutations to rtA181T/sW172* (clone 13) were dominantly selected; however, most clones evolved to rtA181T/sW172S (clone 51) late during the combination therapy with a slight virus titer increase in patient serum. These data suggest that the replication ability of the rtA181T/sW172S mutant is superior to that of the rtA181T/sW172* mutant under LMV and ADV treatment-induced selection pressure (Fig. 1, bottom).
Taken together, the HBV RT domain sequence evolution study revealed that rtI233V is associated with ADV resistance and the role of rtA181T in replication and drug resistance may be different by the corresponding mutations in the overlapping surface gene.
In vitro drug susceptibility of RT mutants obtained during sequential antiviral therapy.
To analyze whether the serial emergence of clones was phenotypically concordant with the clinical information, we performed an in vitro drug susceptibility assay using representative clones obtained from each time point of the indicated viral breakthrough.
The replication capacity of clones (the “20 series”) obtained at baseline did not differ greatly from the WT (NCBI GQ872210), while those clones had several substitutions in the RT domain (Fig. 1B). Replication after LMV treatment decreased to WT levels, indicating that those substitutions were not involved in LMV resistance. Notably, mutants harboring rtI233V were sensitive to LMV treatment. These results suggest that mutations in the quasispecies obtained at baseline have no role in LMV resistance.
Series 22 clones obtained from serum samples after viral breakthrough during LMV therapy revealed that the replication level was reduced up to 80% of that in WT (Fig. 1C). As expected, the rtL80I+M204I or rtM204I mutations conferred resistance to LMV as previously described (26–28). These mutant clones exhibited a typical drug resistance pattern by showing a low replication capacity with the least drug treatment suppression.
All clones obtained during ADV resistance (the 10 series) harbored the rtI233V+N236T mutation and showed resistance to ADV consistent with previous reports (Fig. 1D) (17, 29, 30). Notably, the replication capacity of clone 10-16 was extremely low compared to that of clone 10-12. Furthermore, clone 10-16 showed more resistance to ADV (Fig. 1D). The only difference in mutation profile between these two clones was the emergence of the rtA181T mutation (Table 1). These results suggest that the rtA181T mutation is responsible for the reduction in replication ability and the enhanced ADV resistance.
Clones 13 and 51 were obtained during the LMV and ADV combination therapy (Fig. 2A and B). Although all clones lost the LMV-resistant mutations (rtL80I and rtM204I), the rtA181T mutation was de novo in most of the clones harboring rtA181T+I233V+N236T on the same HBV genome (Table 1). Southern blot analysis showed that the rtI233V mutation dramatically decreased the viral replication (clone 13-13 and 13-19 in Fig. 2A). However, the emergence of the rtI233V mutation in the rtA181T+N236T mutant did not affect the resistance to LMV or ADV (Fig. 2A). A drug susceptibility assay revealed that these clones were only partially resistant to LMV and fully resistant to ADV (Fig. 2A and B).
FIG 2.

In vitro drug susceptibility assay of HBV RT mutants derived from a patient during combination therapy. (A and B) Cloned HBV DNAs obtained during LMV and ADV combination therapy were transfected into Huh7 cells, and cells were treated for 4 days with 20 μM LMV, ADV, or both drugs. The intracellular HBV DNA level was analyzed by Southern blotting. The relative replication ability and drug susceptibility compared to that of WT were quantified by using a PhosphorImager. (C) Summary of HBV clones that have substitutions at the rt181 and rt233 positions. The relative replication abilities of each clone compared to the WT are presented as the mean value of three independent experiments.
As shown in Fig. 2A, the rtI233V mutation reduced viral replication. Similarly, as suggested in Fig. 1D, the rtA181T mutation seemed to be responsible for reducing genomic replication. However, as summarized in Fig. 2C, some clinical isolates harboring the rtA181T mutation showed a relatively high replication ability (ca. 70% of WT), irrespective of the rtI233V mutation. Unexpectedly, the replication ability of clone 51-14, which harbors both the rtA181T and the rtI233V mutations on the same HBV genome, did not considerably differ from that of the WT or clone 13-19, which does not bear the rtI233V mutation (Fig. 2B and C). These somewhat discrepant results indicate that the decreasing effect of rtA181T and rtI233V mutations on replication may be the result of unknown mechanisms or factors.
Collectively, these results suggest that the effect of the rtA181T and rtI233V mutations on viral replication and drug resistance may be regulated through an unidentified mechanism.
Impact of rtA181T and rtI233V mutants derived from clinical isolates on HBV replication and drug resistance.
First, we investigated whether the decrease in the replication capacity of clone 13-13 in the cytoplasm resulted from an enhanced virion secretion. As shown in Fig. 3A, clone 13-13 showed rather a secretion defect of virions and naked capsids. Interestingly, comparison of Western blot analysis the clone 13-13 surface antigens (HBsAgs) revealed a completely different band pattern compared to that of WT and clone 13-19, which also harbors the rtA181T mutation. WT HBV expresses three HBsAgs with six bands owing to alternative protein glycosylation, as shown in a previous study (25). However, clone 13-13 showed only one band corresponding to a lower molecular weight than the intact small HBsAg, suggesting the expression of a truncated HBsAgs (Fig. 3A). To confirm this, we compared the nucleotide sequences and found that the rtA181T mutation in the polymerase was associated with two different mutations in the corresponding surface gene, sW172* or sW172S (Fig. 3B). An HBV rtA181T/sW172* mutant (clone 13-13) showed both decreased replication and secretion defects while the A181T/sW172S mutant (clone 13-19) did not (Fig. 3A). The polymerase proteins expressed from the two mutants were identical except for the region encoded by the rtI233V mutation. Therefore, we questioned whether the decreased replication was the result of the surface stop codon at this specific site. To answer this, we analyzed the sequence of all 42 clinical isolates (Table 1) obtained in the present study and found several mutants harboring nonsense mutations in the surface gene at another position, rtV191I/sW182* (Fig. 3B and Fig. 4A). We also found other mutants harboring the rtA181T/sW172S mutation.
FIG 3.

Effect of rtA181T and rtI233V mutations on virion secretion and expression of surface proteins. (A) After transfection of the WT and the 13-13 and 13-19 clones in Huh7 cells, the intracellular and secreted HBV DNA were analyzed by Southern blotting. Intracellular HBsAg was detected by using an anti-HBs antibody (Novus). (B) Schematic representation of HBV polymerase sequences overlapping with the surface gene sequences. The nucleotide changes at position rt181 and corresponding substitutions in the overlapping surface gene for the WT and the 13-13 and 13-19 clones are indicated. XhoI and NcoI sites used for RT gene cloning for the HBV1.2mer. The corresponding substitutions in the overlapping surface gene are indicated by arrows.
FIG 4.

Analysis of replication ability and ADV resistance of HBV mutants derived from clinical isolates harboring the rtA181T, rtV191I, and rtI233V mutations. (A) Sequence analysis of clones harboring the rtA181T, rtV191I, and rtI233V mutations. The substitutions in the overlapping surface protein and the relative replication levels compared to WT are indicated. (B and C) Ten selected clones were transfected into Huh7 cells, and the replication ability and ADV susceptibility were analyzed by Southern blotting. ADV treatment (20 μM) lasted for 4 days. Intracellular HBsAg was detected by Western blotting with an anti-HBs antibody (Abcam).
With these mutants, we determined the relative replication ability of each clone compared to the WT (Fig. 4A). Surprisingly, all of the mutants with rtA181T/sW172* (clones 13-11, 13-13, 13-17, and 13-18) showed replication defects, whereas the mutants with rtV191I/sW182* or rtA181T/sW172S exhibited relatively high replication levels. The HBV mutants harboring a surface stop codon at another position, rtV191I/sW182*, had enhanced replication. The effect of the rtI233V mutation on replication in these clones did not seem to be significant. All mutants showed resistance to ADV (Fig. 4C).
Western blot analysis revealed that all mutants harboring a surface stop codon expressed the truncated HBsAg proteins. The detection level of truncated HBsAg proteins was varied probably due to mutation-induced alterations in protein stability or antigenicity (Fig. 4B). Transfection efficiency was confirmed by HBeAg ELISA and all clones showed similar levels of HBeAg (data not shown).
Collectively, these data suggest that the effect of the rtA181T mutation on viral replication is dependent on the mutations in the overlapping surface gene and the clinical isolates harboring a surface stop codon only at the rt181 position greatly reduced genomic replication and led to secretion defects.
Impact of artificial mutants containing rtA181T and/or rtI233V on HBV replication and drug resistance.
We demonstrated that the clinically isolated mutants harboring rtA181T/sW172* and I233V mutations exhibited reduced replication. To investigate the effect of the point mutations, including rtA181T/sW172*, rtA181T/sW172S, and rtI233V, on genomic replication and drug resistance, we constructed several artificial mutants using a WT HBV template (Fig. 5A). Another mutation at the rt181 position, rtA181V/sL173P, which is known to confer ADV resistance (31, 32) was used for comparison.
FIG 5.


Characterization of artificial mutants containing rtA181T/sW172*, rtA181T/sW172S, and rtI233V and functional analysis of mutant polymerase and surface proteins. (A) Artificial mutant clones (2 μg) were transfected into Huh7 cells. Cells were harvested at 4 days after transfection. Culture supernatants were used to measure the secreted HBsAg. Intracellular HBV DNA and surface antigens were detected by Southern and Western blotting, respectively. Two different types of anti-HBs antibodies were used to detect the intracellular surface antigens. Relative replications (compared to WT) were calculated by Southern blotting and PhosphorImager analysis. (B) Transfected cells were treated with 20 μM ADV for 4 days and subjected to Southern blot analysis. (C) Relative replication and ADV susceptibility of artificial mutants, compared to the corresponding WT values. Data were obtained by using at least three independent Southern blot analyses (*, P < 0.05). (D) Portions (2 μg) of the indicated clones were transfected into Huh7 cells. HBV RNA was detected by Northern blotting by using 20 μg of total RNA. The relative HBV RNA represents the mean value from two independent experiments. The total RNA (1 μg) was separated on 0.8% agarose gel as a loading control. (E) Effect of WT and mutant polymerases on the replication of polymerase-null HBV. The HBV1.2 Pol(−) (1 μg) and CMV-Pol constructs (1 μg) were cotransfected into Huh7 cells. The levels of HBV replication and polymerase expression were detected by Southern and Western blotting, respectively. (F) Dominant-negative effect of truncated PreS1 proteins on WT HBV replication. After cotransfection of the indicated plasmids into Huh7 cells, the intracellular HBV DNA was analyzed by Southern blotting. Quantification of replication was obtained by at least three independent analyses. Expression levels of surface proteins were detected by indicated antibodies. The transfection efficiency was determined from the level of secreted HBeAg. Asterisks indicate the truncated surface proteins.
As shown in Fig. 5A and C, the rtI233V mutation reduced the genomic replication alone or in combination with the rtA181T mutation (lane 2 versus lane 3, lane 4 versus lane 9, and lane 5 versus lane 10). The replication ability of the rtA181T/sW172* mutant was approximately half of the WT, whereas that of the rtA181T/sW172S mutant was similar to the WT, indicating that the attenuated replication of the rtA181T mutant was the result of surface truncation. The HBsAg expression and secretion profile was determined by different antibodies and by ELISA to avoid the possible difference of antigenicity to mutant HBsAgs (Fig. 5A). The expression of truncated HBsAg and relative levels of mutant HBsAgs were confirmed by Western blot and ELISA. Similar transfection efficiency of each mutant was also confirmed by HBeAg ELISA (data not shown).
To examine the effect of rtA181T and I233V mutations on ADV resistance, we performed a drug susceptibility assay (Fig. 5B), and the results are summarized in Fig. 5C. The results demonstrated that the rtI233V mutation, which emerged during ADV therapy, reduced viral replication and conferred resistance to ADV. Most importantly, the rtA181T/sW172*+I233V mutant, which was dominant during LMV+ADV therapy (Fig. 1), showed the lowest replication level with the highest ADV resistance (Fig. 5C).
It is possible that the observed difference in replication between rtA181T/sW172* and rtA181T/sW172S mutants is exerted at the nucleotide level, i.e., a cis effect on the pgRNA, even though the mutant pgRNAs produce the polymerases with the same amino acid sequences. To clarify this, we performed complementation experiments with the cytomegalovirus (CMV)-driven WT and mutant polymerases after cotransfection of polymerase-null HBV [HBV1.2 Pol(−)]. First, we found that the transcription levels of viral RNAs between WT and rtA181T mutants are not changed (Fig. 5D). In addition, the CMV-driven WT and mutant polymerases exerted similar polymerase activity on the replication of HBV1.2 Pol(−) (Fig. 5E). These data suggest that the cis effect on the pgRNA is not involved in replication difference between rtA181T/sW172* and rtA181T/sW172S mutants.
Finally, we assessed which of the truncated surface proteins are responsible for reduced replication of rtA181T/sW172* mutant. To do this, we cloned the CMV-driven mutant surface constructs (truncated PreS1, PreS2, and S) and tested the effect on the replication of WT HBV. As shown in Fig. 5F, only the truncated PreS1 protein exerted the strong dominant-negative effect on viral replication. These data indicate that the truncated PreS1 is responsible for reduced replication of rtA181T/sW172* mutant.
The rtA181T/sW172* mutants seemed to exhibit increased drug resistance compared to the rtA181T/sW172S mutants at single dose of drug (Fig. 5C). Therefore, to determine the 50% inhibitory concentration (IC50) of rtA181T/sW172* and rtA181T/sW172S mutants against antiviral drugs, we performed a drug susceptibility assay using recently approved drugs (Fig. 6). The level of genomic replication of the rtA181T/sW172S mutant sharply decreased as drug concentration increased, which was not the case for the rtA181T/sW172* mutant (Fig. 6A and C). Notably, the replication levels of both mutants at the final drug concentration were similar, although the rtA181T/sW172* mutant replication ability was approximately half that of the rtA181T/sW172S mutant when not ADV treated (Fig. 6A and B). Conclusively, the fold change in resistance of the rtA181T/sW172* mutant to ADV was significantly increased compared to that of the rtA181T/sW172S mutant. Similarly, the fold change in resistance of the rtA181T/sW172* mutant to TDF was slightly increased, likely due to similarity in the TDF and ADV chemical structures. However, both mutants were sensitive to TDF and ETV (Fig. 6C).
FIG 6.
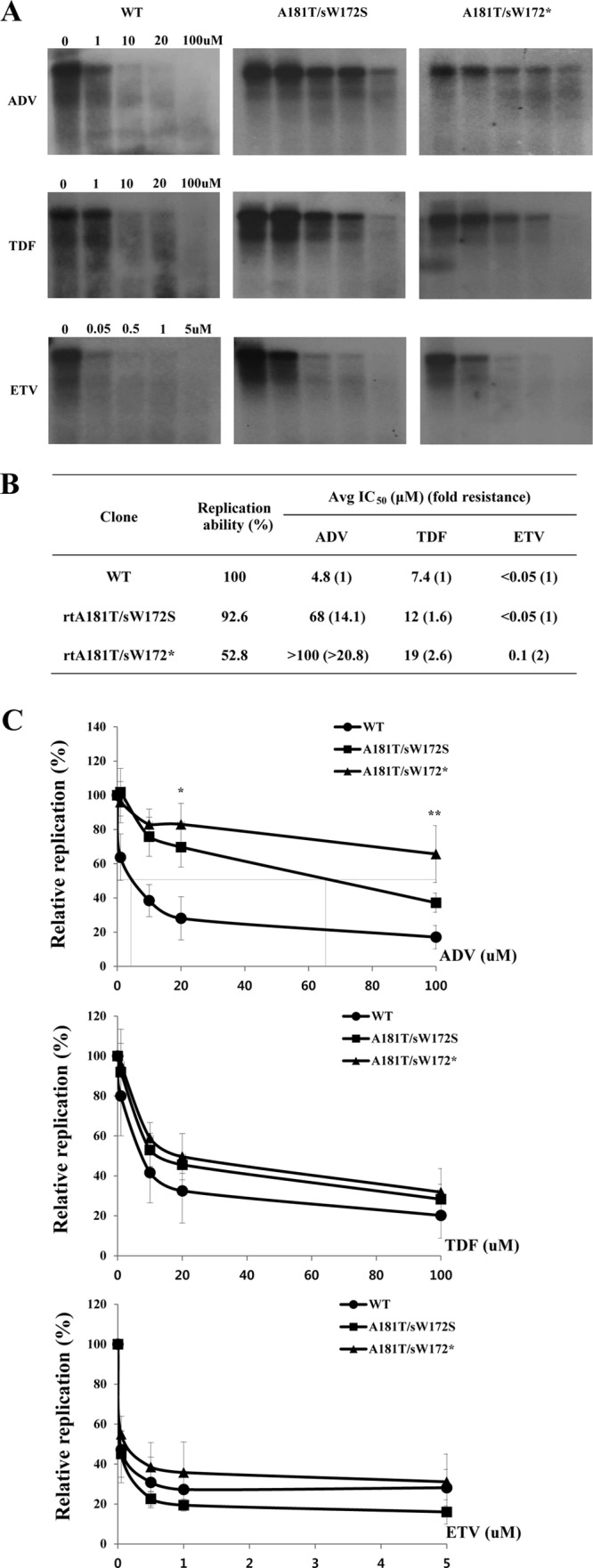
In vitro susceptibility assay of WT, rtA181T/sW172S mutant, and rtA181T/sW172* mutant to recently approved antiviral drugs. (A) Cells were transfected with indicated plasmids and treated for 4 days with increasing concentrations of ADV, TDF, and ETV. Then, the intracellular HBV DNA was subjected to Southern blot analysis. (B and C) Determination of average IC50 and fold change in resistance of WT, rtA181T/sW172* mutant, and rtA181T/sW172S mutant against ADV, TDF, and ETV treatment. Data were obtained by using at least three independent Southern blot analyses (*, P < 0.05; **, P < 0.01).
These data demonstrate that the rtA181T/sW172* mutation reduces viral replication and increases drug resistance compared to the rtA181T/sW172S mutation. Our observations imply that the rtA181T mutation impact on HBV replication and drug resistance can be affected by the overlapping changes in surface gene.
DISCUSSION
In this study, we performed clonal analyses of the drug-resistant mutants evolved during sequential mono- and multi-antiviral therapy. We identified several conserved mutations, including rtA181T, rtM204I, rtI233V, and rtN236T, which are associated with treatment failure. Our data showed that the rtA181T/sW172* and rtI233V mutations affected viral replication and drug resistance. The rtI233V mutation, which emerged during ADV therapy, reduced viral replication and conferred resistance to ADV. Intriguingly, the rtA181T/sW172* mutant exhibited a decrease in viral replication and an increase in drug resistance compared to the rtA181T/sW172S missense mutant. Importantly, we found that the rtA181T/sW172*+I233V mutant, which was dominantly selected during the LMV-ADV combination therapy (Fig. 1), exhibited the lowest level of replication with the highest level of ADV resistance.
Recent studies revealed that the clinical occurrence of the rtA181T/sW172* mutation during antiviral therapy is high (15, 18). The ultradeep pyrosequencing of HBV quasispecies isolated from patients with ADV resistance demonstrated that 80 to 90% of rtA181T mutations are associated with an rtA181T/sW172* substitution in the surface gene (18). Wagner and Locarnini reported that rtA181T/sW172* was detected in 2% of total LMV resistance cases, 18% of total ADV resistance cases, and 42% of LMV and ADV combination therapy cases, whereas HBV encoding rtA181T/sW172* was observed in only 0.2% of isolates from untreated patients (15). The high frequency of the rtA181T/sW172* mutation, not the other missense mutations, in the surface gene during drug treatment may be associated with increased drug resistance, as shown in the present study (Fig. 6).
Many studies suggested that the rtA181T mutation is continuously detected during ADV or LMV-ADV combination therapy (15, 17, 18, 33, 34). Currently, the AASLD, EASL, and KASL Guidelines recommend TDF for ADV resistance (13, 35, 36). The patient enrolled in the present study is now undergoing treatment with TDF. Therefore, it was worth testing whether the rtA181T/sW172* mutant was sensitive to TDF. Similar to the data from the above guidelines, the rtA181T mutant was resistant to ADV and sensitive to ETV and TDF regardless of its overlapping mutations in the surface gene. However, the fold resistance of the rtA181T mutant to all three drugs showed a meaningful difference depending on the surface gene change. The rtA181T/sW172* mutant had an ∼2-fold increase in resistance against all drugs compared to the rtA181T/sW172S missense mutant (Fig. 6).
As shown in Fig. 1, most of the HBV clones isolated from the patient before the antiviral therapy already had the rtI233V mutation. Although the effect of the rtI233V mutation on ADV resistance has been controversial (22, 23), we demonstrated here that the rtI233V mutant was resistant to ADV (Fig. 5C). It is well known that ADV monotherapy for LMV resistance confers a higher risk for ADV resistance (11, 31). Therefore, the existence of the rtI233V mutation in the patient before therapy, as observed in our patient, allowed for one possible explanation for the increased risk of ADV resistance in patients with LMV-resistant hepatitis B.
The rtA181T/sW172* mutant showed a viral particle secretion defect (Fig. 3). Recently, similar to our genotype C result, it was reported that the rtA181T/sW172* mutant showed a secretory defect with a dominant-negative effect in genotype D (15). That study reported that the truncated S protein is responsible for the defect of virion secretion but not viral replication. In the present study, we demonstrate that the truncated PreS1 protein dominantly inhibits the replication of WT HBV (Fig. 5F). The rtA181T/sW172* mutant obtained here always coexisted with the WT or missense mutant in the corresponding surface gene in the patient's serum at the time of viral breakthrough. This suggests that the secretion defect mutant required the trans complementation of the surface protein by other viral quasispecies for the emergence of this mutant.
It is well characterized that the retention of foreign proteins in the cytoplasm induces endoplasmic reticulum (ER) stress that leads to the overall inhibition of gene transcription and is also associated with liver disease (37). Another surface truncation mutant known as pre-S2 mutation is also associated with hepatocarcinogenesis via accumulation in the ER and induction of ER stress (38). In this regard, the emergence of the rtA181T/sW172* mutant in patients with chronic hepatitis B was reportedly associated with tumorigenesis (19, 20). We, along with another group (15), demonstrated that the rtA181T/sW172* mutant had a HBsAg and virion secretion defect. Therefore, it is possible that the reduced secretion of virions and HBsAgs is caused by accumulation in the ER, which leads to ER stress and eventually results in liver disease.
How does the rtA181T/sW172* mutant increase drug resistance? Nucleotide analogues such as ADV are taken up into the liver cells by both nonspecific and receptor-mediated endocytosis, followed by cleavage into adefovir by intracellular esterases. Adefovir is subsequently phosphorylated by adenylate kinases and nucleoside diphosphate kinases and competes with dATP for binding to the HBV polymerase. One possible explanation for the increased drug resistance is that the truncated HBsAgs induce sustained ER stress, which eventually leads to the downregulation of several proteins involved in ADV pharmacokinetics.
In summary, we have shown that the effect of the rtA181T mutation on viral replication and drug resistance is dependent on the mutations in the overlapping surface gene. Compared to the rtA181T/sW172S mutant, the rtA181T/sW172* mutant showed decreased viral replication and increased drug resistance with a secretion defect. The rtI233V mutation also affected replication ability and drug resistance. Based on our findings, it may be necessary to perform genotypic analyses of the overlapping surface gene to manage antiviral drug resistance if clinical isolates harbor the rtA181T mutation.
ACKNOWLEDGMENT
This study was supported by Konkuk University.
Footnotes
Published ahead of print 2 April 2014
REFERENCES
- 1.Lai CL, Ratziu V, Yuen MF, Poynard T. 2003. Viral hepatitis B. Lancet 362:2089–2094. 10.1016/S0140-6736(03)15108-2 [DOI] [PubMed] [Google Scholar]
- 2.Lee WM. 1997. Hepatitis B virus infection. N. Engl. J. Med. 337:1733–1745. 10.1056/NEJM199712113372406 [DOI] [PubMed] [Google Scholar]
- 3.Zoulim F, Locarnini S. 2009. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 137:1593–1608. 10.1053/j.gastro.2009.08.063 [DOI] [PubMed] [Google Scholar]
- 4.Hoofnagle JH, Doo E, Liang TJ, Fleischer R, Lok AS. 2007. Management of hepatitis B: summary of a clinical research workshop. Hepatology 45:1056–1075. 10.1002/hep.21627 [DOI] [PubMed] [Google Scholar]
- 5.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, Chang TT, Kitis G, Rizzetto M, Marcellin P, Lim SG, Goodman Z, Wulfsohn MS, Xiong S, Fry J, Brosgart CL, Adefovir Dipivoxil 438 Study Group 2003. Adefovir dipivoxil for the treatment of hepatitis B e antigen-negative chronic hepatitis B. N. Engl. J. Med. 348:800–807. 10.1056/NEJMoa021812 [DOI] [PubMed] [Google Scholar]
- 6.Marcellin P, Chang TT, Lim SG, Tong MJ, Sievert W, Shiffman ML, Jeffers L, Goodman Z, Wulfsohn MS, Xiong S, Fry J, Brosgart CL, Adefovir Dipivoxil 437 Study Group 2003. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N. Engl. J. Med. 348:808–816. 10.1056/NEJMoa020681 [DOI] [PubMed] [Google Scholar]
- 7.Perrillo R, Hann HW, Mutimer D, Willems B, Leung N, Lee WM, Moorat A, Gardner S, Woessner M, Bourne E, Brosgart CL, Schiff E. 2004. Adefovir dipivoxil added to ongoing lamivudine in chronic hepatitis B with YMDD mutant hepatitis B virus. Gastroenterology 126:81–90. 10.1053/j.gastro.2003.10.050 [DOI] [PubMed] [Google Scholar]
- 8.Lok AS, McMahon BJ, Practice Guidelines Committee American Association for the Study of Liver Diseases (AASLD) 2004. Chronic hepatitis B: update of recommendations. Hepatology 39:857–861. 10.1002/hep.20110 [DOI] [PubMed] [Google Scholar]
- 9.Pellicelli AM, Barbaro G, Francavilla R, Romano M, Barbarini G, Mazzoni E, Mecenate F, Paffetti A, Barlattani A, Struglia C, Villani R, Nauri L, Nosotti L, Armignacco O, Ferri F, Camporiondo MP, Soccorsi F, Club Epatologi Ospedalieri (CLEO) Group 2008. Adefovir and lamivudine in combination compared with adefovir monotherapy in HBeAg-negative adults with chronic hepatitis B virus infection and clinical or virologic resistance to lamivudine: a retrospective, multicenter, nonrandomized, open-label study. Clin. Ther. 30:317–323. 10.1016/j.clinthera.2008.02.012 [DOI] [PubMed] [Google Scholar]
- 10.Peters MG, Hann Hw Hw, Martin P, Heathcote EJ, Buggisch P, Rubin R, Bourliere M, Kowdley K, Trepo C, Gray Df Df, Sullivan M, Kleber K, Ebrahimi R, Xiong S, Brosgart CL. 2004. Adefovir dipivoxil alone or in combination with lamivudine in patients with lamivudine-resistant chronic hepatitis B. Gastroenterology 126:91–101. 10.1053/j.gastro.2003.10.051 [DOI] [PubMed] [Google Scholar]
- 11.Lee YS, Suh DJ, Lim YS, Jung SW, Kim KM, Lee HC, Chung YH, Lee YS, Yoo W, Kim SO. 2006. Increased risk of adefovir resistance in patients with lamivudine-resistant chronic hepatitis B after 48 weeks of adefovir dipivoxil monotherapy. Hepatology 43:1385–1391. 10.1002/hep.21189 [DOI] [PubMed] [Google Scholar]
- 12.Lok AS, McMahon BJ. 2007. Chronic hepatitis B. Hepatology 45:507–539. 10.1002/hep.21513 [DOI] [PubMed] [Google Scholar]
- 13.European Association for the Study of the Liver. 2012. EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J. Hepatol. 57:167–185 [DOI] [PubMed] [Google Scholar]
- 14.Gish R, Jia JD, Locarnini S, Zoulim F. 2012. Selection of chronic hepatitis B therapy with high barrier to resistance. Lancet Infect. Dis. 12:341–353. 10.1016/S1473-3099(11)70314-0 [DOI] [PubMed] [Google Scholar]
- 15.Warner N, Locarnini S. 2008. The antiviral drug selected hepatitis B virus rtA181T/sW172* mutant has a dominant negative secretion defect and alters the typical profile of viral rebound. Hepatology 48:88–98. 10.1002/hep.22295 [DOI] [PubMed] [Google Scholar]
- 16.Yoo BC, Kim JH, Chung YH, Lee KS, Paik SW, Ryu SH, Han BH, Han JY, Byun KS, Cho M, Lee HJ, Kim TH, Cho SH, Park JW, Um SH, Hwang SG, Kim YS, Lee YJ, Chon CY, Kim BI, Lee YS, Yang JM, Kim HC, Hwang JS, Choi SK, Kweon YO, Jeong SH, Lee MS, Choi JY, Kim DG, Kim YS, Lee HY, Yoo K, Yoo HW, Lee HS. 2007. Twenty-four-week clevudine therapy showed potent and sustained antiviral activity in HBeAg-positive chronic hepatitis B. Hepatology 45:1172–1178. 10.1002/hep.21629 [DOI] [PubMed] [Google Scholar]
- 17.Villet S, Pichoud C, Billioud G, Barraud L, Durantel S, Trépo C, Zoulim F. 2008. Impact of hepatitis B virus rtA181V/T mutants on hepatitis B treatment failure. J. Hepatol. 48:747–755. 10.1016/j.jhep.2008.01.027 [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez C, Chevaliez S, Bensadoun P, Pawlotsky JM. 2013. Characterization of the dynamics of hepatitis B virus resistance to adefovir by ultra-deep pyrosequencing. Hepatology 58:890–901. 10.1002/hep.26383 [DOI] [PubMed] [Google Scholar]
- 19.Lai MW, Yeh CT. 2008. The oncogenic potential of hepatitis B virus rtA181T/surface truncation mutant. Antivir. Ther. 13:875–879 [PubMed] [Google Scholar]
- 20.Yeh CT, Chen T, Hsu CW, Chen YC, Lai MW, Liang KH, Chen TC. 2011. Emergence of the rtA181T/sW172* mutant increased the risk of hepatoma occurrence in patients with lamivudine-resistant chronic hepatitis B. BMC Cancer 11:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai J, Chen EQ, Bai L, Gong DY, Zhou QL, Cheng X, Huang FJ, Tang H. 2012. Biological characteristics of the rtA181T/sW172* mutant strain of Hepatitis B virus in animal model. Virol. J. 9:280. 10.1186/1743-422X-9-280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schildgen O, Sirma H, Funk A, Olotu C, Wend UC, Hartmann H, Helm M, Rockstroh JK, Willems WR, Will H, Gerlich WH. 2006. Variant of hepatitis B virus with primary resistance to adefovir. N. Engl. J. Med. 354:1807–1812. 10.1056/NEJMoa051214 [DOI] [PubMed] [Google Scholar]
- 23.Curtis M, Zhu Y, Borroto-Esoda K. 2007. Hepatitis B virus containing the I233V mutation in the polymerase reverse-transcriptase domain remains sensitive to inhibition by adefovir. J. Infect. Dis. 196:1483–1486. 10.1086/522521 [DOI] [PubMed] [Google Scholar]
- 24.Kwon SY, Park YK, Ahn SH, Cho ES, Choe WH, Lee CH, Kim BK, Ko SY, Choi HS, Park ES, Shin GC, Kim KH. 2010. Identification and characterization of clevudine-resistant mutants of hepatitis B virus isolated from chronic hepatitis B patients. J. Virol. 84:4494–4503. 10.1128/JVI.02066-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin GC, Ahn SH, Choi HS, Lim KH, Choi DY, Kim KP, Kim KH. 2013. Hepatocystin/80K-H inhibits replication of hepatitis B virus through interaction with HBx protein in hepatoma cell. Biochim. Biophys. Acta 1832:1569–1581. 10.1016/j.bbadis.2013.04.026 [DOI] [PubMed] [Google Scholar]
- 26.Hashimoto Y, Suzuki F, Hirakawa M, Kawamura Y, Yatsuji H, Sezaki H, Hosaka T, Akuta N, Kobayashi M, Saito S, Suzuki Y, Kobayashi M, Arase Y, Ikeda K, Kumada H. 2010. Clinical and virological effects of long-term (over 5 years) lamivudine therapy. J. Med. Virol. 82:684–691. 10.1002/jmv.21681 [DOI] [PubMed] [Google Scholar]
- 27.Natsuizaka M, Hige S, Ono Y, Ogawa K, Nakanishi M, Chuma M, Yoshida S, Asaka M. 2005. Long-term follow-up of chronic hepatitis B after the emergence of mutations in the hepatitis B virus polymerase region. J. Viral Hepat. 12:154–159. 10.1111/j.1365-2893.2005.00559.x [DOI] [PubMed] [Google Scholar]
- 28.Warner N, Locarnini S, Kuiper M, Bartholomeusz A, Ayres A, Yuen L, Shaw T. 2007. The L80I substitution in the reverse transcriptase domain of the hepatitis B virus polymerase is associated with lamivudine resistance and enhanced viral replication in vitro. Antimicrob. Agents Chemother. 51:2285–2292. 10.1128/AAC.01499-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Locarnini S, Mason WS. 2006. Cellular and virological mechanisms of HBV drug resistance. J. Hepatol. 44:422–431. 10.1016/j.jhep.2005.11.036 [DOI] [PubMed] [Google Scholar]
- 30.Zaaijer HL, Takkenberg RB, Weegink CJ, Rebers SP, Menting S, Reesink HW, Schinkel J, Molenkamp R. 2009. Susceptibility of hepatitis B virus to lamivudine restored by resistance to adefovir. J. Med. Virol. 81:413–416. 10.1002/jmv.21401 [DOI] [PubMed] [Google Scholar]
- 31.Fung SK, Chae HB, Fontana RJ, Conjeevaram H, Marrero J, Oberhelman K, Hussain M, Lok AS. 2006. Virologic response and resistance to adefovir in patients with chronic hepatitis B. J. Hepatol. 44:283–290. 10.1016/j.jhep.2005.10.018 [DOI] [PubMed] [Google Scholar]
- 32.Yim HJ, Hussain M, Liu Y, Wong SN, Fung SK, Lok AS. 2006. Evolution of multidrug resistant hepatitis B virus during sequential therapy. Hepatology 44:703–712. 10.1002/hep.21290 [DOI] [PubMed] [Google Scholar]
- 33.Gaia S, Barbon V, Smedile A, Olivero A, Carenzi S, Lagget M, Alessandria C, Rizzetto M, Marzano A. 2008. Lamivudine-resistant chronic hepatitis B: an observational study on adefovir in monotherapy or in combination with lamivudine. J. Hepatol. 48:540–547. 10.1016/j.jhep.2007.12.018 [DOI] [PubMed] [Google Scholar]
- 34.Lampertico P, Viganò M, Manenti E, Iavarone M, Sablon E, Colombo M. 2007. Low resistance to adefovir combined with lamivudine: a 3-year study of 145 lamivudine-resistant hepatitis B patients. Gastroenterology 133:1445–1451. 10.1053/j.gastro.2007.08.079 [DOI] [PubMed] [Google Scholar]
- 35.Korean Association for the Study of the Liver. 2012. KASL Clinical Practice Guidelines: management of chronic hepatitis B. Clin. Mol. Hepatol. 18:109–162. 10.3350/cmh.2012.18.2.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lok AS, McMahon BJ. 2009. Chronic hepatitis B: update 2009. Hepatology 50:661–662. 10.1002/hep.23190 [DOI] [PubMed] [Google Scholar]
- 37.Malhi H, Kaufman RJ. 2011. Endoplasmic reticulum stress in liver disease. J. Hepatol. 54:795–809. 10.1016/j.jhep.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang LH, Huang W, Lai MD, Su IJ. 2012. Aberrant cyclin A expression and centrosome overduplication induced by hepatitis B virus pre-S2 mutants and its implication in hepatocarcinogenesis. Carcinogenesis 33:466–472. 10.1093/carcin/bgr296 [DOI] [PubMed] [Google Scholar]