

ABSTRACT
Intrinsic immune mechanisms mediated by constitutively expressed proteins termed “restriction factors” provide frontline antiviral defense. We recently demonstrated that the DNA sensor IFI16 restricts human cytomegalovirus (HCMV) replication by downregulating viral early and late but not immediate-early mRNAs and their protein expression. We show here that at an early time point during the in vitro infection of low-passage-number human embryonic lung fibroblasts, IFI16 binds to HCMV DNA. However, during a later phase following infection, IFI16 is mislocalized to the cytoplasmic virus assembly complex (AC), where it colocalizes with viral structural proteins. Indeed, upon its binding to pUL97, IFI16 undergoes phosphorylation and relocalizes to the cytoplasm of HCMV-infected cells. ESCRT (endosomal sorting complex required for transport) machinery regulates the translocation of IFI16 into the virus AC by sorting and trafficking IFI16 into multivesicular bodies (MVB), as demonstrated by the interaction of IFI16 with two MVB markers: Vps4 and TGN46. Finally, IFI16 becomes incorporated into the newly assembled virions as demonstrated by Western blotting of purified virions and electron microscopy. Together, these results suggest that HCMV has evolved mechanisms to mislocalize and hijack IFI16, trapping it within mature virions. However, the significance of this IFI16 trapping following nuclear mislocalization remains to be established.
IMPORTANCE Intracellular viral DNA sensors and restriction factors are critical components of host defense, which alarm and sensitize immune system against intruding pathogens. We have recently demonstrated that the DNA sensor IFI16 restricts human cytomegalovirus (HCMV) replication by downregulating viral early and late but not immediate-early mRNAs and their protein expression. However, viruses are known to evolve numerous strategies to cope and counteract such restriction factors and neutralize the first line of host defense mechanisms. Our findings describe that during early stages of infection, IFI16 successfully recognizes HCMV DNA. However, in late stages HCMV mislocalizes IFI16 into the cytoplasmic viral assembly complex and finally entraps the protein into mature virions. We clarify here the mechanisms HCMV relies to overcome intracellular viral restriction, which provides new insights about the relevance of DNA sensors during HCMV infection.
INTRODUCTION
Intrinsic immunity constitutes a frontline antiviral defense system mediated by constitutively expressed proteins, termed restriction factors (RFs), that are already present and active before a virus enters a cell (1, 2). The term “restriction factor” was originally adopted by investigators studying retroviruses. In the case of primate lentiviruses, the proteins TRIM5α and tetherin (CD317, BST/HMI), as well as members of the APOBEC family of cytidine deaminases, are prominent examples of host cell factors that can restrict the replication of human immunodeficiency virus type 1 (HIV-1) at distinct steps of the viral life cycle. However, HIV-1 has evolved evasion strategies to counter all of these factors. One evasion strategy that viruses may use is to exploit the effects of an RF for its own purposes or to generate an interfering protein that neutralizes the effect of an RF. Another strategy involves the virus hijacking an RF during its phase of maturation to guarantee protection (reviewed in references 3 and 4). While the interference of retroviral replication by cellular RFs and retroviral evasion strategies have been studied in great detail, research into the ways through which RFs restrict other viral infections, such as rhabdoviruses, filoviruses, influenza viruses, hepatitis C virus, and herpesviruses, is still in its infancy (reviewed in reference 5). In particular, in the case of the human cytomegalovirus (HCMV), a betaherpesvirus, the cellular components of nuclear domains 10 (ND10s) (i.e., promyelocytic leukemia protein [PML], hDaxx, and Sp100) have been identified as restriction factors that are involved in mediating intrinsic immunity against this virus (6–8).
The IFI16 protein, a member of the p200 family of proteins, now designated the PYHIN family, contains an N-terminal PYRIN domain and two partially conserved 200-amino-acid domains (HIN domains). IFI16 displays multifaceted activity due to its ability to bind to various target proteins (i.e., transcription factors, signaling proteins, and tumor suppressor proteins) and to modulate various cell functions (9). In addition, IFI16 has been shown to bind to and function as a pattern recognition receptor (PRR) of virus-derived intracellular DNA and trigger the expression of antiviral cytokines via the STING/TBK1/IRF3 signaling pathway (10–20). Although many different functions have been ascribed to IFI16 (and to other proteins of the PYHIN family), its role as an antiviral restriction factor has not yet been fully described. Recent studies from our laboratory implicate the involvement of IFI16 in host defense against HCMV (21). The evidence supporting such a role of IFI16 is as follows: (i) small interfering RNA (siRNA)-mediated depletion of IFI16 in primary human embryonic lung fibroblasts (HELFs) significantly increases HCMV replication efficiency as a result of augmented viral DNA synthesis; (ii) similarly, viral plaque formation is enhanced in the presence of an exogenous dominant-negative IFI16 mutant that competes with the endogenous IFI16; (iii) overexpression of functional IFI16 in HCMV-infected HELFs decreases both virus yield and viral DNA copy number; and (iv) early and late, but not immediate-early, viral mRNAs and proteins are strongly downregulated under these same conditions, suggesting that IFI16 exerts its main antiviral effect at the level of viral genome synthesis. This unique defense mechanism distinguishes the activity of IFI16 from that described for ND10.
In more general terms, human viruses have to face powerful RF responses and thus have evolved a number of strategies to overcome RF attack. Viral antagonists can act through highly specialized mechanisms, such as coupling RFs to protein degradation pathways, causing their relocalization and thus downregulating their functionality, or even by mimicking RF substrates (5). In the case of HCMV, viral regulatory proteins (such as IE1p72, pp71, and others) mediate an efficient evasion from the antiviral state instituted by ND10, either by means of proteasomal degradation or by disrupting the host's subnuclear structure (6, 22).
In this study, we investigated the mechanisms used by HCMV to evade IFI16 restriction activity. We observed that starting from 72 to 96 h postinfection (hpi), nuclear levels of IFI16 protein started to decrease in the nucleus and gradually increased in the cytoplasm of infected cells, where it relocalized to the virus assembly complex (AC), as shown by its colocalization with the viral structural proteins gB and pp65. Finally, through the use of immunogold electron microscopy and coprecipitation experiments, we provide evidence indicating that IFI16 eventually transits into the maturing virions embedded in the outer tegument layer. In conclusion, these data suggest that in order to overcome the restriction activity of IFI16, HCMV may stimulate its subcellular relocalization from the nucleus to the viral AC, followed by its inclusion into mature virions.
MATERIALS AND METHODS
Cells, viruses, and DNA constructs.
Low-passage HELFs and human embryo kidney 293 cells (HEK 293; Microbix Biosystems, Inc.) were cultured in Eagle minimal essential medium (Life Technologies Italia) supplemented with 10% fetal calf serum (Sigma-Aldrich). Low-passage human umbilical vein endothelial cells (HUVECs) were grown in endothelial cell growth medium 2 (Lonza) supplemented with 2% fetal bovine serum and 1% penicillin-streptomycin solution (Sigma-Aldrich) as previously described (23). The HCMV laboratory strain AD169 (ATCC-VR538) and the HCMV clinical isolate derivative VR1814 were propagated and titrated as previously described (21, 24). UV-inactivated AD169 was prepared using a double pulse of UV-B light (1.2 J/cm2). The mutant HCMV (AD169) BAC213 (ΔUL97/GFP+) was produced as previously described (25, 26). siRNA UL97 and siRNA CTRL were purchased from Sigma and electroporated at a final concentration of 300 nM. Plasmids expressing wild-type (WT) and dominant-negative forms of Vps4A (pBJ-Vps4AWT and pBJ-Vps4AE228Q, respectively) were obtained as previously described (27, 28).
Antibodies and reagents.
Primary antibodies were obtained from various sources, as shown in Table S1 in the supplemental material. Conjugated antibodies included fluorescein isothiocyanate-labeled anti-rabbit antibodies (Sigma-Aldrich), Texas Red-labeled anti-mouse and anti-rabbit antibodies (Invitrogen), and horseradish peroxidase-labeled anti-mouse and anti-rabbit antibodies (GE Healthcare). The chemicals used were Gö6976 (inhibitor of serine/threonine protein kinases, particularly pUL97 and protein kinase C [PKC]; Calbiochem) (29), phosphonoformic acid (PFA; Foscarnet), and ganciclovir (HCMV inhibitors; Sigma-Aldrich).
Cell viability assay.
Cells were seeded at a density of 104/well in a 96-well culture plate. After 24 h, the cells were treated with different doses (from 0.5 to 5 μM) of Gö6976. At 72 h after treatment, cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich) method.
Quantitative real-time RT-PCR.
Real-time quantitative reverse transcription-PCR (RT-PCR) analysis was performed on an Mx 3000 P apparatus (Stratagene). Total RNA was extracted with the NucleoSpin RNA kit (Macherey-Nagel), and 1 μg was retrotranscribed using a Revert-Aid H-Minus FirstStrand cDNA synthesis kit (Fermentas). Reverse-transcribed cDNAs were amplified in duplicate using Brilliant Sybr green QPCR master mix (Fermentas) for IFI16. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GADPH) was used to normalize for variations in cDNA levels (primers IFI16 forward [ACTGAGTACAACAAAGCCATTTGA], IFI16 reverse [TTGTGACATTGTCCTGTCCCCAC], GADPH forward [TCGGAGTCAACGGATTTGGTC], and GADPH reverse [CGTTCTCAGCCTTGACGGTG]).
ChIP assay.
Chromatin immunoprecipitation (ChIP) assays were conducted similar to the methods published by Cristea et al. (11) and Li et al. (18). HELFs were infected with HCMV at an MOI of 5. At 6 hpi, cells were cross-linked with 1% paraformaldehyde for 15 min and then processed for ChIP assay using an EpiTect ChIP OneDay kit (Qiagen) according to the manufacturer's instructions. Anti-IFI16 antibody (5 μg) was used to pull down the protein-chromatin complexes. Rabbit IgG was used as a negative control. The immunoprecipitated DNA was recovered by column purification and analyzed by PCR using HCMV or human specific primers (the primer sequences are available on request).
Immunofluorescence microscopy.
Immunofluorescence analysis was performed as previously described (30) using the appropriate dilution of primary antibodies (see Table S1 in the supplemental material) for 1 h at room temperature, followed by 1 h with secondary antibodies in the dark at room temperature. Nuclei were counterstained with DAPI (4′,6′-diamidino-2-phenylindole) where indicated. Finally, coverslips were mounted with Vectashield mounting medium (Vector Laboratories, Ltd.), and cells were visualized with a Leica TCS SP2 confocal microscope equipped with a UV laser (351–364 nm) and argon-krypton laser (457 to 675 nm; Leica Microsystems, S.r.l.), using a ×63 oil immersion objective NA 1.4 lens. Fluorescence in situ hybridization (FISH) was combined with immunofluorescence by performing the hybridization first as described below and then incubating the coverslips with primary and secondary antibodies.
FISH.
HCMV-infected HELFs were grown on glass slides, fixed as described above, and permeabilized with 0.5% Triton X-100 for 20 min at 4°C. The probe used for FISH was a bacterial artificial chromosome (BAC) DNA containing the entire HCMV genome (a gift from Jay Nelson, Oregon Health and Science University), labeled using the biotin-nick translation system (Roche Diagnostics GmbH) according to the manufacturer's protocol. The probe was added to the hybridization buffer (0.2 ng/μl yeast t-RNA, 50% formamide, 15% SSC [(1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 0.1% Tween 20) at a concentration of 2 ng/μl and then incubated at 72°C for 5 min in order to denature the probe and the sample. Hybridization was continued overnight at 37°C in a humidified chamber. After stringent washing, the cells were blocked with 10% normal goat serum. HCMV probes were then detected using the tyramide signal amplification procedure according to manufacturer's instructions (Perkin-Elmer Life and Analytical Sciences, Inc.). Images were analyzed using a confocal laser scanning microscope.
Immunoprecipitation assay.
Uninfected cells or cells infected with HCMV (multiplicity of infection [MOI] of 1) for different times were washed and lysed in radioimmunoprecipitation assay (RIPA) buffer. A total of 200 μg of protein was incubated with 2 μg of immunoprecipitating or control antibody for 1 h at room temperature with rotation, and the immune complexes were collected using protein G-Sepharose (Sigma-Aldrich). The Sepharose beads were pelleted and washed three times with RIPA buffer, boiled with sample buffer, and resolved on an 8% SDS-PAGE gel to assess the protein binding by Western blotting.
Western blot analysis.
Nuclear and cytoplasmic extracts were collected using a nuclear extract kit (Active Motif) according to the manufacturer's instructions and subjected to immunoblot analysis as previously described (31). Briefly, an equal amount of cell extracts were fractionated by electrophoresis on SDS-polyacrylamide gels and transferred to Immobilon-P membranes (Millipore). After blocking, membranes were incubated overnight at 4°C with the appropriate primary antibodies. Membranes were then washed and incubated for 1 h at room temperature with secondary antibodies. Proteins were detected using an enhanced chemiluminescence detection kit (Thermo Scientific).
Scanning densitometry of the bands was performed using Quantity One software (version 4.6.9; Bio-Rad Laboratories S.r.l.). Background values were subtracted from each calculated value.
Virion purification and viral protein extraction.
Virus containing media was collected at 192 hpi (MOI of 1), centrifuged at 3,000 rpm for 10 min to remove large cellular debris and then filtered using a Filtropur 0.45 (Sarstedt). To pellet viral particles, the media was then centrifuged at 13,000 × g for 2 h at 4°C in a Beckman SW32 Ti rotor. The viral pellet was resuspended in 1× phosphate-buffered saline (PBS) and centrifuged in a 20/41/70% discontinuous sucrose gradient composed of the following steps: 0.5 ml of 60% (wt/wt) sucrose, 1.5 ml of 41% sucrose, and 1 ml of 20% sucrose. Sucrose solutions were made in 1× PBS. After centrifugation overnight at 130,000 × g at 4°C, using a Beckman SW40 Ti rotor, the virus containing band was removed from the gradient and lysed with 50 mM Tris-HCl (pH 6.8)–2% SDS for 30 min at 4°C. After heating for 10 min at 95°C and clarification, the viral protein extract was collected.
In vitro kinase assay.
The kinase activity of FLAG-tagged pUL97 was determined in vitro after immunoprecipitation of the kinase from whole-cell lysates of HEK 293 cells, previously electroporated using a MicroPorator MP-100 (Digital Biotechnology) according to the manufacturer's instructions (a single 1,200-V pulse, 30-ms pulse width). The following UL97 expression constructs were used: pcDNA-UL97–M2, pcDNA-UL97(181-707)-M2, and pcDNA-UL97(1-595)-M2 (25). Immunoprecipitates were subsequently pelleted, washed, and subjected to in vitro kinase assay reaction (2.5 μCi of [γ-32P]ATP [Amersham Biosciences]) at 30°C for 30 min, as previously described (26), in the presence of the recombinant full-length IFI16 as the substrate (5 μg). After incubation, samples were separated by SDS-PAGE, transferred to Immobilon-P membranes (Millipore), and processed for autoradiography and immunoblotting.
Immunogold labeling of isolated viral particles.
For transmission electron microscopic analysis, samples were allowed to adsorb onto carbon and Formvar-coated grids and fixed in 4% paraformaldehyde. Grids were then washed with PBS and water and, when appropriate, the samples were permeabilized with 0.2% Triton. The grids were stained with primary antibodies, followed by gold-labeled secondary antibodies in the presence of 10% human serum (32). Grids were then negatively stained using 0.5% uranyl acetate for 1 min. Images were captured using a CM10 electron microscope (Philips).
RESULTS
IFI16 colocalizes with HCMV genome early during infection.
IFI16 has previously been shown to interact with HSV-1 as well as HCMV DNA early during infection (13, 15, 17, 18, 33, 34). To confirm that endogenous IFI16 interacts with viral DNA during natural HCMV infection, HELFs were mock infected or infected with HCMV for 12 h. Combined immunofluorescence and FISH analysis was performed using IFI16 antibodies and a probe for the HCMV genome. In mock-infected cells, IFI16 was distributed into defined spots, which appear to reorganize following HCMV infection and colocalize with the HCMV-DNA (Fig. 1A). The colocalization of IFI16 with the viral genome is reinforced by Z-stack images, generated by three-dimensional (3D) reconstruction of confocal images to improve colocalization analysis (Fig. 1A, far right pictures). Consistent with FISH analysis and in accord with the results reported by Li et al. (18), ChIP assays at 6 hpi demonstrated that endogenous IFI16 specifically recognizes virus DNA loci, but not host chromosomal DNA (Fig. 1B). Altogether, these results are compatible with the proposed role of IFI16 as a nuclear sensor for HCMV DNA.
FIG 1.
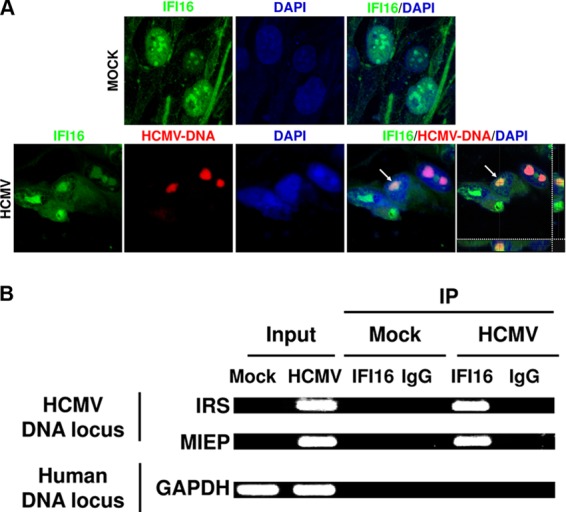
The HCMV genome is recognized by IFI16 at early time points following infection. (A) HELFs were mock infected (MOCK) or infected with HCMV (strain AD169, MOI of 2 PFU/cell), fixed in 1% paraformaldehyde at 12 hpi, and subjected to combined FISH with a BAC DNA probe containing the entire HCMV genome (red) and immunofluorescence analysis with anti-IFI16 antibodies (green). Cell nuclei are visualized in blue. Images were taken by confocal microscopy, and the far right hand picture shows 3D image reconstruction of stacks of confocal images. At least five fields were digitally reconstructed to generate the 3D images for each condition; representative images are shown. (B) HELFs were infected at an MOI of 5 and processed for ChIP assays 6 h later to test the association of endogenous IFI16 with HCMV and host DNA.
HCMV relocalizes IFI16 nuclear protein into the cytoplasm.
IFI16 is typically located within the nucleus (11, 15, 35, 36), but it translocates to the cytoplasm following infection with HSV-1 (12, 13, 15, 17, 34), Kaposi's sarcoma-associated herpesvirus (KSHV) (14, 16), or Epstein-Barr virus (EBV) (10). To determine the subcellular localization of IFI16 during early and late infection with HCMV, two different approaches were adopted: Western blot analysis and immunofluorescence (IF). In the first case, HELFs, synchronized by serum starvation to increase infection efficiency (37, 38), were mock infected or infected with HCMV at an MOI of 1; they were then fractionated into nuclear and cytoplasmic components. The purity of the nuclear and cytoplasmic fractions was monitored by Western blotting for the presence of TATA-binding protein (TBP) and tubulin, respectively (Fig. 2A). Immediate early antigen (IEA) protein labeling was used to assess HCMV infection by Western blotting (Fig. 2A, lower panel). In mock-infected cells, IFI16 was exclusively nuclear (Fig. 2A, lane 1). Notably, at 24 hpi, HCMV-infected cells showed an increase in nuclear IFI16 that peaked at 48 h, decreased by 96 hpi, and almost disappeared by 144 hpi (Fig. 2A, lanes 2 to 6). However, at 48 hpi IFI16 was also detected at appreciable levels in the cytoplasm of HCMV-infected cells and gradually increased at later time points (Fig. 2A, lane 10). Consistent with the Western blot results, RT-PCR analysis confirmed that HCMV infection upregulates IFI16 also at the mRNA level (∼2-fold between 12 and 24 h of HCMV versus mock-infected cells) (Fig. 2B).
FIG 2.

IFI16 accumulates in the cytoplasm of HCMV-infected cells at late time points postinfection. (A) HELFs were infected with HCMV at an MOI of 1 PFU/cell. Nuclear and cytoplasmic fractions were prepared at the indicated time points and subjected to Western blotting and subsequent densitometry for IFI16. The results were normalized to TBP and tubulin, respectively (*, P < 0.05; **, P < 0.01; ***, P < 0.001 [one-way analysis of variance, followed by Bonferroni's post test]). Nuclear fractions were stained for HCMV IEA as a positive control for viral infection (lower panel). (B) HELFs were infected with HCMV (MOI of 1 PFU/cell) or left untreated. Total RNA was isolated at the indicated time postinfection and assayed by quantitative real-time PCR to determine the relative levels of IFI16, normalized to the levels of cellular GADPH. The data shown are the average of three experiments ± the SD (*, P < 0.05; **, P < 0.01 [one-way analysis of variance, followed by Bonferroni's post test]). (C) Kinetics of IFI16 subcellular localization upon HCMV infection. HELFs were infected with HCMV at an MOI of 1 PFU/cell for the indicated time points and subjected to confocal microscopy analysis. IFI16 (green) and viral proteins (red) were visualized using primary antibodies, followed by secondary antibody staining, in the presence of 10% human serum. Nuclei are visualized in blue. The far right-hand picture of each panel shows a Z stack of confocal images, generating a 3D reconstruction, obtained as described in Fig. 1A (right panel). A graph shows the IFI16 delocalization levels. The bars indicate the percentages of cells positive for cytoplasmic IFI16 immunofluorescence over the course of infection. Images were acquired by using a microscope with a ×20 objective lens, and three random fields from two slides of each time point were counted using ImageJ software to calculate the ratio of cytoplasmic-IFI16 expressing cells to infected cells. The data represent means ± the SD (left panel). (D) IFI16 is mislocalized in HUVECs by the HCMV clinical isolate derivative VR1814. HUVECs were infected with HCMV (MOI of 1 PFU/cell), fixed in 1% paraformaldehyde at the indicated time points, and subjected to confocal immunofluorescence analysis as described in panel C. Images were acquired at ×63 magnification, and representative images are shown.
These results demonstrate that HCMV induces the cytoplasmic translocation of IFI16 early on during infection. In apparent contrast to our results, Cristea et al. (11) and Li et al. (18) found that endogenous IFI16 remained nuclear during early HCMV infection. In our study, nuclear delocalization started from 96 hpi, a time point not examined by Cristea et al. (11) and Li et al. (18). Moreover, IFI16 nuclear egress into the cytoplasm was only observed when synchronized cell cultures were used. This condition was created to increase virus infection efficiency. Thus, considering the different postinfection time points analyzed and the virus MOI used, the discrepancies between our results the results reported by the other investigators can be easily explained.
To gain deeper insight into the nuclear disappearance of IFI16, a detailed analysis using confocal microscopy at time points ranging from 12 to 144 hpi was performed. As shown in Fig. 2C, all infected cells positive for IEA staining showed the nuclear presence of IFI16 during the first 12 hpi. In contrast, between 72 and 96 hpi, IFI16 became undetectable in the nucleus of the majority of the cells, accompanied by its appearance in the cytoplasm. Consistent with the Western blot outcome, the IFI16 nuclear decline in infected cells was accompanied by the appearance of IFI16 in the cytoplasm, specifically in the compartment that overlapped with the virus AC, as shown by its colocalization with the viral glycoprotein gB. To verify that the signal observed in the AC was specific for IFI16 and not due to rabbit IgG binding to HCMV-encoded Fc receptor-like proteins (39, 40), the staining was performed after blocking the Fc receptors using 10% HCMV-negative human serum prior to the addition of the specific rabbit IgG, as described in Buchkovich et al. (41). Similar results were obtained using monoclonal anti-IFI16 antibodies (Santa Cruz) (data not shown), which have also been used in other studies to demonstrate the nuclear export of IFI16 (14, 15).
Finally, to exclude the possibility that IFI16 nuclear egression was limited to HELFs infected by the AD169 strain, IF analysis was performed on HUVECs infected with the endotheliotropic VR1814 strain. A similar pattern of IFI16 relocalization from the nucleus accompanied by its appearance in the AC compartment was observed, demonstrating that IFI16 nuclear delocalization is related to HCMV infection (Fig. 2D).
HCMV early/late proteins induce the nucleocytoplasmic relocalization of IFI16.
The observation that the relocalization of IFI16 protein into the cytoplasm sharply increases from 48 hpi, accompanied by its gradual nuclear disappearance, suggests that an early or late viral protein(s) is(are) responsible for driving IFI16 into the cytoplasm. To test this possibility, HELFs were infected with UVB-inactivated HCMV, or with wild-type HCMV in the presence of 100 μM phosphonoformic acid (PFA) or 100 μM ganciclovir (GCV). To confirm that the infection was successfully established, FISH staining was performed (Fig. 3, right panel). As shown in Fig. 3 (left panel), all treatments blocked IFI16 nucleocytoplasmic translocation. Since treatment with PFA or GCV inhibits viral DNA synthesis and the accumulation of early-late and late viral proteins to different extents, an early-late or late HCMV gene product is likely to be responsible for IFI16 subcellular relocalization.
FIG 3.

HCMV inhibition blocks the mislocalization of IFI16. HELFs were infected with wild-type or UV-inactivated HCMV (1 PFU/cell; 1.2 J/cm2 for two pulses) and treated with phosphonoformic acid (PFA; 100 μM) or ganciclovir (GCV; 100 μM) as indicated. Cells were fixed 72 h later in 1% paraformaldehyde and processed by immunofluorescence analysis for IFI16 (green) and gB (red) (left panel) or subjected to combined FISH with a BAC DNA probe containing the entire HCMV genome (red) and immunofluorescence analysis with anti-IFI16 antibodies (green); cell nuclei were visualized in blue (right panel). Images were taken by confocal microscopy (×63 magnification).
HCMV pUL97 contributes to the nucleocytoplasmic translocation of IFI16.
To gain insight into the mechanism responsible for the HCMV-induced nuclear reduction and relocalization of IFI16 into the cytoplasm, we focused on the recently described HCMV nuclear egress complex (NEC) composed of viral and cellular proteins (25, 26, 42). In this context, the viral protein kinase pUL97 is known to play an important role by phosphorylating and reorganizing nuclear lamins A/C, a step required for the nuclear egress of viral capsids (26, 43). We therefore hypothesized that pUL97 might also be involved in the regulation of IFI16 nucleocytoplasmic relocalization. To test this, HELFs were infected with a green fluorescent protein (GFP)-tagged recombinant UL97 deletion mutant BAC (BAC ΔUL97) or AD169 (AD169 UL97+) as a control (25, 43) and used for immunostaining at 32 days postinfection or 96 hpi, respectively. The lack of production of pUL97 by BAC ΔUL97 was confirmed by immunofluorescence staining (data not shown). The choice to perform the experiments at different time points was due to the delayed replication kinetics of viruses lacking a functional pUL97 kinase compared to the wild-type strain, as previously reported (25, 44, 45). As shown in Fig. 4A, many AD169 UL97+-infected cells showed IFI16 relocalization into the cytoplasm. In contrast, cells infected with a UL97 deletion mutant BAC (BAC ΔUL97) displayed IFI16 nuclear accumulation (Fig. 4A, row 1). To gain further supporting evidence of the specific involvement of pUL97 in the cytoplasmic relocalization of IFI16, HELFs were electroporated with a mixture of three different small interfering RNAs targeting the UL97 gene (siRNA UL97) or with scrambled control siRNA (siRNA CTRL). After 24 h, the cells were infected with HCMV for 72 h. The siRNA-mediated knockdown reduced the expression of pUL97 protein by ca. 90 to 95%, as indicated by Western blotting (see Fig. S1A in the supplemental material). As shown in Fig. 4A (row 2) and consistent with the results of the BAC mutant experiments, inhibition of pUL97 expression prevented IFI16 nuclear egress compared to infected cells pretreated with control siRNA. Moreover, treating infected cells with the pUL97 inhibitor Gö6976 (2 μM [26]) strongly suppressed IFI16 relocalization (Fig. 4A, row 3), in agreement with the results of previous studies (26, 29). No such effect was observed when cells were treated with vehicle control (DMSO). To exclude the possibility that the Gö6976 inhibitor might influence the observed effect independent of viral infection, an MTT assay was used to examine and quantify its effect on HELF survival (see Fig. S1B in the supplemental material). Finally, to investigate whether pUL97 alone is sufficient to induce IFI16 nuclear egress in the absence of other viral gene products, the protein was transfected into HELFs. In contrast to what we observed following virus infection, IFI16 retained its nuclear localization 72 h after protein electroporation, indicating that pUL97 alone, in the absence of viable and functional virus, is not sufficient to trigger the relocalization of IFI16 into the cytoplasm (see Fig. S1C in the supplemental material).
FIG 4.

pUL97 mediates HCMV-induced IFI16 mislocalization. (A) pUL97 inhibition impairs IFI16 nuclear egress. HELFs were treated as described in detail below and in Results, fixed at the time points indicated below, and double stained with the appropriated antibodies. HELFs infected with a UL97 deletion mutant BAC (BAC ΔUL97) or with AD169 UL97+ as a control at an MOI of 1 PFU/ml were fixed and immunostained at 32 days or 96 hpi, respectively (row 1); HELFs were electroporated with a mixture of three different small interfering RNAs targeting UL97 (siRNA UL97) or with scrambled control siRNA (siRNA CTRL) and 24 h later infected with HCMV at an MOI of 1 PFU/cell for 72 h (row 2); HCMV-infected HELFs were treated with the pUL97 inhibitor Gö6976 (2 μM) or with an equal volume of vehicle control (DMSO) and immunostained after 72 h (row 3). (B) IFI16 interacts with pUL97. Total cell protein extracts from HELFs infected with HCMV at an MOI of 1 for 96 h were immunoprecipitated with polyclonal antibodies against IFI16 (left panel) or monoclonal antibodies against UL97 (right panel), and control antibody. Samples were then immunoblotted with antibodies for pUL97 and IFI16, respectively. Nonimmunoprecipitated whole-cell extract (Input) obtained from HCMV-infected cells was used to normalize the proteins subjected to immunoprecipitation. (C) Phosphorylation of IFI16 by pUL97 in vitro. HEK 293 cells were transfected with wild-type pUL97 (lane 1), catalytically active pUL97 (N-terminally truncated pUL97-181-707) (lane 2), or inactive C-terminally truncated pUL97 (pUL97-1-595, lane 3). At 48 h posttransfection, cells were lysed and subjected to immunoprecipitation (IP) with monoclonal antibodies for pUL97, followed by in vitro kinase reaction with recombinant IFI16 (rIFI16) as the substrate. Labeled phosphorylation products were separated by SDS-PAGE and visualized by exposing the blots to autoradiography film (upper panel). Lysate control samples taken prior to immunoprecipitation were used for Western blot analysis with the monoclonal antibodies for pUL97 to monitor the levels of expressed proteins (lower panel).
To investigate the interplay between IFI16 and pUL97, total protein extracts from HELFs infected with HCMV for 96 h were used for coimmunoprecipitation with anti-IFI16 or the appropriate control antibodies. Precipitates were then immunostained with a monoclonal anti-pUL97 antibody. As shown in Fig. 4B (left panel), virus pUL97 indeed binds to IFI16. This interaction is specific as no migrating bands were present when coprecipitation was performed using control antibodies. The presence of pUL97 in all protein extracts was monitored by the staining of INPUT (nonimmunoprecipitated whole-cell extract) control samples (Fig. 4B). To confirm further the specificity of the interaction, coimmunoprecipitation and immunoblotting experiments were performed in reverse order, i.e., anti-UL97 was used for immunoprecipitation and anti-IFI16 for immunoblotting (Fig. 4B, right panel). In line with previous results, a band corresponding to IFI16 was detectable when protein extracts were immunoprecipitated with an antibody against virus pUL97.
The interaction between IFI16 and pUL97 suggested that IFI16 might be directly phosphorylated by pUL97 kinase. To address this hypothesis, we performed an in vitro kinase assay (26) using wild-type and mutant pUL97, immunoprecipitated from lysates of transiently transfected HEK 293 cells, and incubated with highly purified recombinant IFI16 protein as the substrate. IFI16 phosphorylation was exclusively detectable for wild-type pUL97 and catalytically active pUL97 [i.e., N-terminally truncated pUL97(181-707) (pUL97-181-707; Fig. 4C, lanes 1 and 2, respectively)], whereas an inactive C-terminally truncated version (pUL97-1-595) (lane 3) did not produce a phosphorylation signal.
IFI16 colocalizes with the viral AC following nuclear egress.
It has been proposed that HCMV acquires its final envelope from the trans-Golgi network (TGN) or from TGN-derived particles (28, 42). In addition, many cellular markers, like those of early, recycling, and late endosomes, as well as the endosomal sorting complex required for transport (ESCRT) and several viral tegument and envelope proteins, including gB, all localize to the AC (28). Since IFI16 staining appears to overlap the AC, as shown by confocal analysis, we wanted to gain insights into the fate of IFI16 following nuclear egression. We performed an immunofluorescence assay at 96 hpi using anti-IFI16 antibodies, together with antibodies recognizing the virion envelope protein gB, vacuolar protein sorting-4A (Vps4A, a component of the ESCRT machinery), or TGN46 (a marker of the trans-Golgi network) (Fig. 5A). As shown in Fig. 2C and 5A (confocal Z-stack images), a high level of IFI16 colocalization could be detected with viral gB, Vps4A, and TGN46. These results strongly suggest that IFI16 mislocalizes out of the nucleus and associates with AC-containing virion particles.
FIG 5.

HCMV infection induces IFI16 sorting into multivesicular bodies (MVBs). (A) IFI16 colocalizes with components of MVBs and the ESCRT pathway in HCMV-infected cells. HELFs were infected with HCMV at an MOI of 1 PFU/cell; 96 h later cells were fixed, permeabilized, and costained with anti-IFI16, anti-TGN46, anti-Vps4A, and anti-HCMV gB antibodies. Nuclei were visualized in blue. Images were taken by confocal microscopy, and the far right hand panel shows a 3D image reconstruction of stacks of confocal images. At least five fields were digitally reconstructed for each condition, and a representative image is shown. (B) IFI16 interacts with Vps4A in HCMV-infected cells. Total cell protein extracts obtained by HELFs treated as described above were immunoprecipitated with antibodies against Vps4A (upper panel) or IFI16 (lower panel) and the appropriate control antibody (CTRL). Immunoprecipitated samples and whole-cell extracts (INPUT) were then immunoblotted using antibodies against Vps4A or IFI16. (C) Effect of blocking MVB biogenesis on IFI16 localization. HELFs cotransfected with a construct expressing Vps4A (Vps4WT) or the mutated form Vps4AE228Q were infected with HCMV (MOI of 1 PFU/cell) at 24 h posttransfection. Cells were fixed and photographed at 72 hpi. Vps4A was detected by anti-FLAG primary antibody (red) and IFI16 by the polyclonal anti-IFI16 antibody (green). Representative images were taken using ×63 magnification.
To better define the relationship between IFI16 and ESCRT components, total protein extracts from HELFs infected with HCMV for 96 h were immunoprecipitated with anti-Vps4A or control antibodies and immunoblotted with polyclonal anti-IFI16 antibodies. As shown in Fig. 5B (upper panel), no interaction between Vps4A and IFI16 was observed in mock-infected cells. In contrast, HCMV infection induced a strong interaction between Vps4A and IFI16, as shown by the co-IP reactions. The same results were obtained in reverse order (Fig. 5B, lower panel). Vps4A induction by HCMV was also evident in the very same total protein extracts. Finally, no bands were detected when cell extracts were immunoprecipitated with control antibodies. Overall, these results indicate that there might be a connection between the egress of viral proteins from the nucleus and the mislocalization of IFI16 protein into the AC.
Functional significance of the IFI16-Vps4 interaction.
To evaluate the impact of dysfunctional MVBs on IFI16 localization, we used a previously described construct expressing a dominant-negative Vps4A (27, 28). HELFs were transfected with FLAG-tagged pBJ5-Vps4AE228Q or the corresponding wild-type form pBJ5-Vps4AWT and 24 h later infected with HCMV. The transfection efficiency of HELFs is low (∼20%), such that only the FLAG-expressing subpopulation of cells was studied. Confocal microscopy at 72 hpi (96% HCMV-positive cells [data not shown]) demonstrated that in the pBJ5-Vps4E228Q-transfected cells IFI16 remains strictly nuclear (Fig. 5C, lower panel) in all of the infected cells. In contrast, in cells transfected with the wild-type vector, we observed that IFI16 mislocalizes to the cytoplasm (Fig. 5C, upper panel), confirming the dependence of IFI16 mislocalization on functional MVB biogenesis, which serves as a platform for HCMV envelopment/egress.
Immunogold labeling of IFI16 in purified HCMV particles.
The colocalization of IFI16 and viral gB in the AC opened up the possibility that IFI16 may be incorporated into viral particles during maturation. To investigate this possibility, HCMV virions were fractionated by sucrose gradient from supernatants of HELFs infected at an MOI of 1 for 192 h and analyzed by Western blotting for the viral proteins IEA, UL44, and pp65, and cellular proteins IFI16 and p53 (the latter is translocated into the cytoplasm during HCMV infection) (46). As shown in Fig. 6A, pp65 and IFI16 were detected in highly purified virions, indicating their incorporation. The specificity of IFI16 incorporation into viral particles was supported by the finding that none of the nonstructural viral proteins, such as IEA, UL44, or cellular p53, was identified by Western blotting. Importantly, total cell extracts from mock- or HCMV-infected HELFs were included in order to estimate the levels of IFI16 induction by HCMV infection. The presence of IFI16 in purified virions was confirmed by coimmunoprecipitation experiments with anti-pp65 antibodies. As shown in Fig. 6B, IFI16 indeed interacted with the HCMV tegument protein pp65. Consistent with our results, Li et al. (18) have recently demonstrated that early during infection, pp65 associates with IFI16 by interacting with its pyrin domain, inhibiting its subsequent immune signaling (11, 18).
FIG 6.

IFI16 is associated with purified HCMV particles. (A) HCMV particles (indicated as virions) were purified by sucrose gradient from supernatants of infected HELFs (192 hpi, MOI of 1) and analyzed by immunoblotting for the viral proteins IEA, UL44, and pp65, and the cellular proteins IFI16 or p53. Total cell extract from mock- or HCMV-infected cells were included as controls. (B) IFI16 interacts with pp65 in purified virions. HELFs were infected as described for panel A. Protein extracts were obtained from purified virions and immunoprecipitated with anti-pp65 (upper panel) or anti-IFI16 (lower panel) and the appropriate control antibodies (CTRL) and then immunoblotted with anti-IFI16 or anti-pp65 antibodies, respectively. Nonimmunoprecipitated whole-cell extracts (INPUT) were immunoblotted with anti-IFI16 or anti-pp65 antibodies and used to normalize the proteins subjected to immunoprecipitation. (C) Immunoelectron microscopy analysis of purified virions stained, in the presence of 10% human serum, for IFI16 and HCMV gB and pp65, or left unstained (SecAb). A 15-nm gold-conjugated secondary antibody was used to detect proteins. Scale bar, 100 nm.
To investigate further the incorporation of IFI16 into mature virions, we labeled purified virus particles with IFI16-specific antibodies, followed by gold-conjugated secondary antibodies, and analyzed them by using electron microscopy. The integrity of the purified virions was further verified by negative staining that showed two classes of spherical enveloped particles: 200-nm-diameter HCMV virions and larger structures corresponding to dense bodies (DBs). The ratio of virions to DBs was about 1:2. The specificity of the immunogold labeling was assessed by omitting the primary antibody. The virus preparation was permeabilized or left unmasked, so that antibodies could recognize within the inner layers of the viral particles or on their surface, respectively. As shown in Fig. 6C, gB, used as a control, was observed in the outer envelope of the purified virions, while pp65 was present inside the viral particles. Altogether, these results demonstrate that a percentage of IFI16 protein becomes trapped within mature virions.
DISCUSSION
The importance of the role played by restriction factors in controlling viral infection is substantiated by the diverse mechanisms the viruses have evolved to antagonize it (47–49). We recently demonstrated that the IFN-inducible protein IFI16 may act as a restriction factor for HCMV replication by downregulating viral early and late mRNA and protein expression (21). In the present study, we examined how HCMV can overcome the antiviral activity of the nuclear restriction factor IFI16. Consistent with its property as a pathogenic DNA sensor (10–14, 17–20, 33, 50), detailed kinetics studies exploiting immunofluorescence show that in the early phases of infection, IFI16 binds to viral DNA, also confirmed by FISH combined with Western blot analysis. These results are in line with previous studies showing that following HCMV infection IFI16 binds viral DNA and triggers the expression of antiviral cytokines via the STING/TBK1/IRF3 signaling pathway (18).
During a late phase postinfection, however, IFI16 levels decreases inside the nucleus and this is accompanied by a parallel increase in its presence in the cytoplasmic AC, as shown by Western blotting and confocal microscopy analysis. This nucleocytoplasmic egress of IFI16 in HCMV-infected cells is driven, at least in part, by the viral protein kinase pUL97, which binds and phosphorylates nuclear IFI16. Subsequently, the IFI16-AC complex mediates its incorporation into newly assembled virions. IFI16 mislocalization and assembly into mature virions appears to be regulated by the ESCRT machinery through its sorting and trafficking into multivesicular bodies.
Other studies have examined the effects of herpesvirus infection on IFI16 degradation. Orzalli et al. (15, 51) demonstrated that during alphaherpesvirus HSV-1 infection, the viral nuclear ICP0 protein leads to IFI16 degradation. Similarly, Johnson et al. (13) showed that alphaherpesvirus HSV-1 specifically targets IFI16 for rapid proteasomal degradation later on postinfection. Interestingly, two other herpesviruses, namely, gammaherpesvirus 1 EBV and gammaherpesvirus 2 KSHV, which undergo latency in endothelial cells or in B and epithelial cells, respectively, were not found to cause IFI16 degradation early on during infection, suggesting a relationship between virus replication/latency and IFI16 fate (10, 13, 14).
A key point that could actually complete this scenario is to understand whether IFI16 relocalizes and interacts with inflammasome- and STING-related pathway components upon HCMV infection, especially to promote the release of the proinflammatory cytokines interleukin-1β or beta interferon, respectively, thereby enhancing the antiviral response. Recent studies explored these issues in other herpesvirus models, such as HSV-1 (12, 13, 15, 17, 51), KHSV (14, 16), and EBV (10). Orzalli et al. (15, 51) found that during HSV-1 infection IFI16, which is required for induction of IRF-3 signaling in these cells, remains nuclear. An unknown factor must be exported from the nucleus to activate IRF-3 through cytoplasmic STING, which is required for IRF-3 activation and signaling (15, 51). In contrast, Johnson et al. (13) showed that early during in vitro infection of HFF, HSV-1 induced the activation of IFI16 inflammasome and maturation of IL-1β. For such activity, IFI16 recognized the HSV-1 genome in infected cell nuclei and colocalized with ASC in the cytoplasm. Consistent with these findings, evidence of inflammasome activation, such as the activation of caspase-1 and cleavage of pro-IL-1β, -IL-18, and -IL-33 in EBV- or KHSV-infected cells, respectively, has been accumulated (10, 14, 16). Interaction of ASC with IFI16 but not with AIM2 or NOD-like receptor P3 (NLRP3) was detected. During HCMV infection, IFI16 acts as a nuclear DNA sensor, binding viral DNA and triggering the expression of antiviral cytokines via the STING/TBK1/IRF3 signaling pathway (18). No data have been provided thus far, demonstrating the ability of IFI16 to activate inflammasome through ASC interaction. Therefore, further investigations, should be addressed to pursue the interplay between IFI16 and these critical components of innate immunity.
Thus, although the egression of IFI16 from the nucleus into the cytoplasm following pathogenic or damaged DNA sensing has now been widely demonstrated, the mechanisms it relies on have not been clarified. Therefore, in the present study we sought to exploit the HCMV model in order to gain some insight into the mechanisms underlying IFI16 mislocalization. The finding that IFI16 egress from the nucleus was first detected at 48 hpi and the fact that it could be blocked by pretreating cells with inhibitors of viral L gene expression suggest that HCMV L genes may be responsible for IFI16 mislocalization. During HCMV replication, DNA-filled capsids bud through the inner nuclear membrane (INM) and transit from the nucleus direct to the AC located in the cytoplasm close to the nuclear membrane (42). The HCMV-specific nuclear egress complex (NEC) is composed of both viral and cellular proteins, in particular protein kinases with the capacity to induce the destabilization of the nuclear lamina (43). The viral protein kinase pUL97, along with cellular protein kinase C (PKC), plays an important role by phosphorylating several types of nuclear lamins, events that lead to the reorganization of the proteinaceous network underlying the inner nuclear membrane and the egression of the DNA-filled capsids (26, 43, 52). By combining molecular-virological analyses with biochemical and pharmacological approaches, we demonstrate that pUL97 binds and phosphorylates IFI16 in vitro, triggering its relocalization from the nuclei into the cytoplasm of HCMV-infected cells. This assertion is based on the finding that the lack of viral pUL97 expression (BAC ΔUL97) and/or the inhibition of its kinase activity substantially reduce IFI16 relocalization. Together with the observation that IFI16 is phosphorylated both in vitro and in HCMV-infected cells by pUL97 kinase, our results demonstrate that one of the viral candidates responsible for IFI16 subcellular relocalization and the inhibition of its restriction activity could be pUL97. In HCMV infection, the transmembrane protein pUL50 anchors the NEC within the inner nuclear membrane and associates with core NEC components, such as pUL53. As a consequence, the NEC is able to recruit regulatory kinases, like pUL97, to disassemble the nuclear lamina and to facilitate nuclear capsid egression. Since IFI16 has been shown to interact with pUL97, we can speculate that IFI16 might interact with further components of the NEC.
Posttranslational modification provides a possible means of regulating IFI16 subcellular localization. The acetylation and phosphorylation of different IFI16 motifs have been demonstrated to regulate its subcellular localization in lymphocytes and macrophages (11, 34). In particular, acetylation of the nuclear localization sequence promotes the cytoplasmic accumulation of IFII6 by inhibiting its nuclear import (34). In HCMV-infected cells, IFI16 interacts with viral pUL97 and undergoes in vitro phosphorylation. Moreover, the nuclear accumulation of IFI16 can be observed upon treatment with Gö6976, an inhibitor of pUL97 phosphorylation (26, 29). Together, these results suggest that phosphorylation by pUL97 may regulate the relocalization of IFI16 from the nucleus to the cytoplasm.
Although the replication of all herpesviruses includes nuclear and cytoplasmic maturation events, the AC is a unique feature of betaherpesvirus-infected cells (53). Virus particles congregate in the AC during the late phases of infection (42), a finding consistent with its important role in controlling final tegumentation, envelopment, and egress from the cell. Immunofluorescence analysis starting at 24 hpi demonstrates that IFI16 colocalizes outside the nucleus of infected cells in a structure that seems to be the AC. The AC was recognized based on the hallmark morphology of CMV-infected cells which consist of an enlargement of the nucleus that transforms into a kidney-shaped form, with the AC pressing against the newly formed depression in the nucleus (54). To confirm definitively the nature of the AC, confocal laser microscopy was performed using antibodies against host proteins of the MVBs included in the AC, such as the ATPase Vps4A involved in the ESCRT pathway, the TGN marker TGN46, and the viral protein gB, known to colocalize to the AC during the late phase of HCMV infection. Moreover, using coimmunoprecipitation experiments, we found that IFI16 interacts with Vps4A upon HCMV infection. Overall, these results demonstrate that HCMV induces IFI16 to mislocalize to MVBs, where the virus undergoes final maturation.
To provide a functional significance of the colocalization experiments, we evaluated the impact of inhibiting the final component of the ESCRT machinery, Vps4A, using a dominant-negative mutant (Vps4AE228Q) known to impair MVB biogenesis and HCMV plaque formation (28). The nuclear retention of IFI16 in cells where MVB biogenesis was inhibited suggests a strict dependence of IFI16 subcellular localization on HCMV replication and designates Vps4A as a key player in the evasion mechanism used by HCMV to escape IFI16.
The presence of IFI16 in the AC and the lack of protein degradation in the late stages of infection could suggest that IFI16 is incorporated into the maturing virion particles. To investigate this possibility, we purified HCMV virions and applied Western blot and electron microscopy analysis to ascertain whether HCMV virions may actually contain the mislocalized IFI16. Intriguingly, Western blot analysis demonstrates the presence of IFI16, but not p53, in the viral protein extract. Moreover, the immunolocalization results confirmed the incorporation of IFI16 into purified virions, and in particular in the outer layer of the tegument in the proximity of pp65, as shown by coprecipitation experiments. This finding leans toward excluding the possibility that IFI16 nonspecifically aggregates to virions during their maturation. Consistent with our results, previous studies based on mass spectrometry approaches have demonstrated the presence of at least 70 host cellular and 71 HCMV proteins in mature virions (55, 56). More recently, it has been demonstrated that HCMV may include both TGN and endosomal markers when undergoing final envelopment (32). Altogether, these findings raise some important questions. The first one asks how specific the inclusion of host proteins in HCMV virions is, since only a percentage of virus particles contain IFI16. The observation that IFI16, but not p53, another protein egressing from the nucleus during HCMV infection, is included in the virion suggests that some mechanisms of selecting host proteins must exist. The second important question that needs addressing is whether HCMV includes IFI16 in the virion in order to evade its restriction activity. At the moment, no evidence exists suggesting a functional consequence of IFI16 embedded within the viral particles, thus only speculations can be put forward. However, some observations may help to explain why the virus hijacks IFI16 and embeds it into the outer layer of the tegument. First, IFI16 triggers the activity of transcription factor NF-κB that is needed in the first steps of HCMV infection (57, 58). Second, it has been demonstrated that pp65 (pUL83) triggers the expression of the viral immediate-early promoter through its interaction with IFI16 protein (11, 18). Finally, overexpression of IFI16 upregulates immediate-early protein expression during the first hours of infection (21). Altogether, these observations suggest that HCMV may hijack IFI16 in order to exploit its capacity to enhance the transcription of IE genes during the early steps of infection, followed by the relocalization of IFI16 into the cytoplasmic AC with the scope of concealing its restriction activity during the late steps of infection.
Supplementary Material
ACKNOWLEDGMENTS
We thank Edward S. Mocarski for his critical review of the manuscript and Thomas Mertens for providing us the anti-UL97 antibodies.
This study was supported by grant MIUR PRIN 2012 to S.L. (2012SNMJRL) and V.D. (20127MFYBR); grant MIUR FIRB 2010 to M.D.A. (RBFR08UUTP); research funding from the University of Turin in 2013 to S.L., M.D.A., and V.D.O.; and ESCMID Research Grant 2013 to V.D.O. M.M. was supported by the Deutsche Forschungsgemeinschaft (SFB 796/C3), Bayerische Forschungsstiftung (grant MM/4SC), and Wilhelm Sander-Stiftung (grant 2011.085.1).
Footnotes
Published ahead of print 2 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00384-14.
REFERENCES
- 1.Bieniasz PD. 2004. Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol. 5:1109–1115. 10.1038/ni1125 [DOI] [PubMed] [Google Scholar]
- 2.Roy CR, Mocarski ES. 2007. Pathogen subversion of cell-intrinsic innate immunity. Nat. Immunol. 8:1179–1187. 10.1038/ni1528 [DOI] [PubMed] [Google Scholar]
- 3.Douglas JL, Gustin JK, Viswanathan K, Mansouri M, Moses AV, Fruh K. 2010. The great escape: viral strategies to counter BST-2/tetherin. PLoS Pathog. 6:e1000913. 10.1371/journal.ppat.1000913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeuchi H, Matano T. 2008. Host factors involved in resistance to retroviral infection. Microbiol. Immunol. 52:318–325. 10.1111/j.1348-0421.2008.00040.x [DOI] [PubMed] [Google Scholar]
- 5.Duggal NK, Emerman M. 2012. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat. Rev. Immunol. 12:687–695. 10.1038/nri3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tavalai N, Stamminger T. 2011. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 157:128–133. 10.1016/j.virusres.2010.10.002 [DOI] [PubMed] [Google Scholar]
- 7.Ahn JH, Hayward GS. 2000. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 274:39–55. 10.1006/viro.2000.0448 [DOI] [PubMed] [Google Scholar]
- 8.Tavalai N, Stamminger T. 2009. Interplay between herpesvirus infection and host defense by PML nuclear bodies. Viruses 1:1240–1264. 10.3390/v1031240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gariglio M, Mondini M, De Andrea M, Landolfo S. 2011. The multifaceted interferon-inducible p200 family proteins: from cell biology to human pathology. J. Interferon Cytokine Res. 31:159–172. 10.1089/jir.2010.0106 [DOI] [PubMed] [Google Scholar]
- 10.Ansari MA, Singh VV, Dutta S, Veettil MV, Dutta D, Chikoti L, Lu J, Everly D, Chandran B. 2013. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J. Virol. 87:8606–8623. 10.1128/JVI.00805-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cristea IM, Moorman NJ, Terhune SS, Cuevas CD, O'Keefe ES, Rout MP, Chait BT, Shenk T. 2010. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 84:7803–7814. 10.1128/JVI.00139-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, Ellermann-Eriksen S, Kurt-Jones E, Landolfo S, Damania B, Melchjorsen J, Bowie AG, Fitzgerald KA, Paludan SR. 2013. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 190:2311–2319. 10.4049/jimmunol.1202749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson KE, Chikoti L, Chandran B. 2013. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 87:5005–5018. 10.1128/JVI.00082-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. 2011. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi's sarcoma-associated herpesvirus infection. Cell Host Microbe 9:363–375. 10.1016/j.chom.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U. S. A. 109:E3008–E3017. 10.1073/pnas.1211302109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh VV, Kerur N, Bottero V, Dutta S, Chakraborty S, Ansari MA, Paudel N, Chikoti L, Chandran B. 2013. Kaposi's sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J. Virol. 87:4417–4431. 10.1128/JVI.03282-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11:997–1004. 10.1038/ni.1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li T, Chen J, Cristea IM. 2013. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14:591–599. 10.1016/j.chom.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg RK, Rahbek SH, Kofod-Olsen E, Holm CK, Melchjorsen J, Jensen DG, Hansen AL, Jorgensen LB, Ostergaard L, Tolstrup M, Larsen CS, Paludan SR, Jakobsen MR, Mogensen TH. 2013. T cells detect intracellular DNA but fail to induce type I IFN responses: implications for restriction of HIV replication. PLoS One 9:e84513. 10.1371/journal.pone.0084513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. 2014. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 24:428-32. 10.1126/science.1243640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gariano GR, Dell'Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, Gribaudo G, Gariglio M, Landolfo S. 2012. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 8:e1002498. 10.1371/journal.ppat.1002498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863–3871. 10.1128/JVI.80.8.3863-3871.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baggetta R, De Andrea M, Gariano GR, Mondini M, Ritta M, Caposio P, Cappello P, Giovarelli M, Gariglio M, Landolfo S. 2010. The interferon-inducible gene IFI16 secretome of endothelial cells drives the early steps of the inflammatory response. Eur. J. Immunol. 40:2182–2189. 10.1002/eji.200939995 [DOI] [PubMed] [Google Scholar]
- 24.Revello MG, Lilleri D, Zavattoni M, Stronati M, Bollani L, Middeldorp JM, Gerna G. 2001. Human cytomegalovirus immediate-early messenger RNA in blood of pregnant women with primary infection and of congenitally infected newborns. J. Infect. Dis. 184:1078–1081. 10.1086/323425 [DOI] [PubMed] [Google Scholar]
- 25.Marschall M, Marzi A, aus dem Siepen P, Jochmann R, Kalmer M, Auerochs S, Lischka P, Leis M, Stamminger T. 2005. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J. Biol. Chem. 280:33357–33367. 10.1074/jbc.M502672200 [DOI] [PubMed] [Google Scholar]
- 26.Milbradt J, Webel R, Auerochs S, Sticht H, Marschall M. 2010. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 285:13979–13989. 10.1074/jbc.M109.063628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689–699. 10.1016/S0092-8674(03)00653-6 [DOI] [PubMed] [Google Scholar]
- 28.Tandon R, AuCoin DP, Mocarski ES. 2009. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J. Virol. 83:10797–10807. 10.1128/JVI.01093-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marschall M, Stein-Gerlach M, Freitag M, Kupfer R, van Den Bogaard M, Stamminger T. 2001. Inhibitors of human cytomegalovirus replication drastically reduce the activity of the viral protein kinase pUL97. J. Gen. Virol. 82:1439–1450 [DOI] [PubMed] [Google Scholar]
- 30.Costa S, Borgogna C, Mondini M, De Andrea M, Meroni PL, Berti E, Gariglio M, Landolfo S. 2011. Redistribution of the nuclear protein IFI16 into the cytoplasm of ultraviolet B-exposed keratinocytes as a mechanism of autoantigen processing. Br. J. Dermatol. 164:282–290. 10.1111/j.1365-2133.2010.10097.x [DOI] [PubMed] [Google Scholar]
- 31.Gugliesi F, Mondini M, Ravera R, Robotti A, de Andrea M, Gribaudo G, Gariglio M, Landolfo S. 2005. Upregulation of the interferon-inducible IFI16 gene by oxidative stress triggers p53 transcriptional activity in endothelial cells. J. Leukoc. Biol. 77:820–829. 10.1189/jlb.0904507 [DOI] [PubMed] [Google Scholar]
- 32.Cepeda V, Esteban M, Fraile-Ramos A. 2010. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell. Microbiol. 12:386–404. 10.1111/j.1462-5822.2009.01405.x [DOI] [PubMed] [Google Scholar]
- 33.Cuchet-Lourenco D, Anderson G, Sloan E, Orr A, Everett RD. 2013. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 87:13422–13432. 10.1128/JVI.02474-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li T, Diner BA, Chen J, Cristea IM. 2012. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. U. S. A. 109:10558–10563. 10.1073/pnas.1203447109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gariglio M, Azzimonti B, Pagano M, Palestro G, De Andrea M, Valente G, Voglino G, Navino L, Landolfo S. 2002. Immunohistochemical expression analysis of the human interferon-inducible gene IFI16, a member of the HIN200 family, not restricted to hematopoietic cells. J. Interferon Cytokine Res. 22:815–821. 10.1089/107999002320271413 [DOI] [PubMed] [Google Scholar]
- 36.Veeranki S, Choubey D. 2012. Interferon-inducible p200-family protein IFI16, an innate immune sensor for cytosolic and nuclear double-stranded DNA: regulation of subcellular localization. Mol. Immunol. 49:567–571. 10.1016/j.molimm.2011.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy EA, Streblow DN, Nelson JA, Stinski MF. 2000. The human cytomegalovirus IE86 protein can block cell cycle progression after inducing transition into the S phase of permissive cells. J. Virol. 74:7108–7118. 10.1128/JVI.74.15.7108-7118.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noris E, Zannetti C, Demurtas A, Sinclair J, De Andrea M, Gariglio M, Landolfo S. 2002. Cell cycle arrest by human cytomegalovirus 86-kDa IE2 protein resembles premature senescence. J. Virol. 76:12135–12148. 10.1128/JVI.76.23.12135-12148.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alwine JC. 2012. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog. 8:e1002878. 10.1371/journal.ppat.1002878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Atalay R, Zimmermann A, Wagner M, Borst E, Benz C, Messerle M, Hengel H. 2002. Identification and expression of human cytomegalovirus transcription units coding for two distinct Fcγ receptor homologs. J. Virol. 76:8596–8608. 10.1128/JVI.76.17.8596-8608.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buchkovich NJ, Maguire TG, Paton AW, Paton JC, Alwine JC. 2009. The endoplasmic reticulum chaperone BiP/GRP78 is important in the structure and function of the human cytomegalovirus assembly compartment. J. Virol. 83:11421–11428. 10.1128/JVI.00762-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol. 20:392–401. 10.1016/j.tim.2012.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marschall M, Feichtinger S, Milbradt J. 2011. Regulatory roles of protein kinases in cytomegalovirus replication. Adv. Virus Res. 80:69–101. 10.1016/B978-0-12-385987-7.00004-X [DOI] [PubMed] [Google Scholar]
- 44.Prichard MN, Gao N, Jairath S, Mulamba G, Krosky P, Coen DM, Parker BO, Pari GS. 1999. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol. 73:5663–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prichard MN, Sztul E, Daily SL, Perry AL, Frederick SL, Gill RB, Hartline CB, Streblow DN, Varnum SM, Smith RD, Kern ER. 2008. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J. Virol. 82:5054–5067. 10.1128/JVI.02174-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Utama B, Shen YH, Mitchell BM, Makagiansar IT, Gan Y, Muthuswamy R, Duraisamy S, Martin D, Wang X, Zhang MX, Wang J, Wang J, Vercellotti GM, Gu W, Wang XL. 2006. Mechanisms for human cytomegalovirus-induced cytoplasmic p53 sequestration in endothelial cells. J. Cell Sci. 119:2457–2467. 10.1242/jcs.02974 [DOI] [PubMed] [Google Scholar]
- 47.Yan N, Chen ZJ. 2012. Intrinsic antiviral immunity. Nat. Immunol. 13:214–222. 10.1038/ni.2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu SY, Sanchez DJ, Cheng G. 2011. New developments in the induction and antiviral effectors of type I interferon. Curr. Opin. Immunol. 23:57–64. 10.1016/j.coi.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. 2011. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 11:143–154. 10.1038/nri2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Jin T, Laustsen A, Hansen K, Ostergaard L, Fitzgerald KA, Xiao TS, Mikkelsen JG, Mogensen TH, Paludan SR. 2013. From the cover: IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. U. S. A. 110:E4571–E4580. 10.1073/pnas.1311669110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orzalli MH, Conwell SE, Berrios C, Decaprio JA, Knipe DM. 2013. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. U. S. A. 110:E4492–E4501. 10.1073/pnas.1316194110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM. 2013. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J. Virol. 88:249–262. 10.1128/JVI.02358-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 81:11861–11869. 10.1128/JVI.01077-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Das S, Pellett PE. 2011. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol. 85:5864–5879. 10.1128/JVI.00155-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baldick CJ, Jr, Shenk T. 1996. Proteins associated with purified human cytomegalovirus particles. J. Virol. 70:6097–6105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, II, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966. 10.1128/JVI.78.20.10960-10966.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caposio P, Gugliesi F, Zannetti C, Sponza S, Mondini M, Medico E, Hiscott J, Young HA, Gribaudo G, Gariglio M, Landolfo S. 2007. A novel role of the interferon-inducible protein IFI16 as inducer of proinflammatory molecules in endothelial cells. J. Biol. Chem. 282:33515–33529. 10.1074/jbc.M701846200 [DOI] [PubMed] [Google Scholar]
- 58.McCormick AL, Mocarski ES., Jr 2007. Viral modulation of the host response to infection, p 324–341 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.