

Abstract
FLICE-inhibitory proteins (FLIPs) are a family of viral (poxvirus and herpesvirus) and cellular proteins. The hallmark of this family is the presence of tandem death-effector domains (DEDs). Despite this shared motif, each protein possesses different abilities to modulate apoptosis, NF-κB, and interferon regulatory factor 3 (IRF3). These similarities and differences are discussed and highlighted here. The comparative study of FLIPs provides a unique basis to understand virus-host interactions, viral pathogenesis, and cellular regulation of immune system signal transduction pathways.
INTRODUCTION
My laboratory recently published the discovery of a novel role for FLICE-inhibitory proteins (FLIPs) in the inhibition of interferon regulatory factor 3 (IRF3) activation (1). This represents the third known function for this group of cellular and viral homologs, adding to the previously characterized roles in inhibition of apoptosis and regulation of NF-κB. This Gem highlights new findings as well as some of the foundational work that has led to our current understanding of the functions of FLIPs. Amazingly, each FLIP possesses unique abilities to modulate three intrinsic innate immune responses (IIRs), namely, apoptosis, NF-κB, and IRF3 (Fig. 1). This is surprising because FLIPs are very similar at the levels of domain structure, with each FLIP possessing tandem death effector domains (DEDs) (2).
FIG 1.
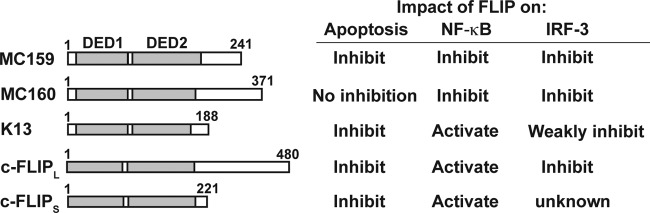
Cartoon of FLIPs and a comparison of their molecular functions. The left portion of the panel shows a cartoon of each FLIP and the position of its tandem death effector domains (DEDs), motifs that are a hallmark of the FLIP family. The right panel shows the functions of each FLIP on three different intrinsic innate immune responses. Publications referring to a FLIP's function are discussed in the text of this Gem.
My article focuses on the most-studied FLIPs, which include the molluscum contagiosum virus (MCV) MC159 and MC160 proteins, the Kaposi's sarcoma-associated herpesvirus (KSHV) K13 protein, and the cellular cFLIPL and cFLIPS proteins (Fig. 1). The comparative study of FLIPs is significant for several reasons. First, it will identify how MCV and herpesviruses manipulate IIRs, which will contribute to our understanding of the molecular mechanisms viruses use to inhibit antiviral responses and cause disease. Second, elevated expression of cFLIPL is observed with many malignant cancers, and increased cFLIPL expression is strongly correlated with poor prognosis in some cancers. Thus, studying this newly identified IRF3-inhibitory function of cFLIPL may give new insights into how apoptosis, NF-κB activation, and IRF3 activation must be balanced in normal versus cancerous cells. Third, I propose that the identification of the molecular mechanisms responsible for each FLIP's distinctive properties is likely to lead to the discovery of novel cellular mechanisms that up- or downregulate each of the IIRs.
DISCOVERY OF THE FLIP FAMILY OF PROTEINS AS INHIBITORS OF APOPTOSIS
FLIPs were discovered because of their homology to cellular death-effector domain (DED)-containing proteins that induce apoptosis, namely, Fas-associated death domain protein (FADD) and procaspase-8/FLICE (2). DED consists of 6 α-helices. A DED does not possess enzymatic activity. Rather, its structure promotes interactions with other proteins (2). In the case of tumor necrosis factor receptor 1 (TNFR1)-induced apoptosis, a large complex is formed in which TNFR1-associated death domain (TRADD) and FADD migrate to the cytoplasmic portion of TNFR1 (3). FADD interacts with procaspase-8 via DED motifs, forming the death-inducing signaling complex (DISC). Next, procaspase-8 undergoes autoproteolysis, in which the C terminus of procaspase-8 is cleaved away from the DISC-associated N-terminal tandem DEDs. The mature, highly active caspase-8 enzyme then initiates the hallmark downstream events of apoptosis, including procaspase-3 activation and poly(ADP-ribose) polymerase (PARP) and DNA cleavage.
The sequencing of several viral genomes revealed the presence of proteins that, like procaspase-8, contain two tandem DEDs (4–6). These viruses include the MCV MC159 and MC160 products and members of the herpesvirus family, including the KSHV K13 and the equine herpesvirus type 2 E8 proteins. The MC159, E8, and K13 proteins inhibit apoptosis (4–6), and these proteins were named FLIPs because they inhibit procaspase-8 (FLICE) activation (Fig. 1). In contrast, MC160 did not inhibit apoptosis in two out of three studies (5, 7, 8), planting the idea that FLIPs may not necessarily have identical biological functions (Fig. 1). The cellular FLIP (cFLIP) was identified soon after the discovery of viral FLIPs (2, 9). There are 3 cFLIP splice variants: cFLIPL, cFLIPS, and cFLIPR. cFLIPS and cFLIPL also inhibit apoptosis when overexpressed, although it should be noted that cFLIPL activates apoptosis when expressed at low levels (2, 9). This same characteristic also is observed under some conditions when MC159 is expressed (10).
Mutational analysis and the solving of the crystal structure of MC159 and other DED-containing proteins identified mechanisms potentially responsible for FLIP antiapoptotic activities (2). In the interest of the present paper, two important findings come from these studies. First, both DEDs of a FLIP must be present for antiapoptotic function (11) (Table 1). Second, there are distinct differences in the mechanisms each of these FLIPs use to inhibit apoptosis. Notably, MC159 prevents FADD-mediated higher-order oligomerization to block apoptosis (11). In contrast, cFLIP and E8 competitively inhibit procaspase-8 recruitment to the DISC. These data suggest that small amino acid differences between FLIPs may have dramatic effects on the surface properties or conformation of a FLIP that can greatly alter the biological properties of a FLIP.
TABLE 1.
Comparison of FLIP DEDs that are required for function
| Protein | Category and DED(s) required |
||
|---|---|---|---|
| Antiapoptosis | NF-κB | Anti-IRF3 | |
| MC159 | DED1 and DED2 (11) | DED1 (inhibit) (10) | DED1 or DED2 (1) |
| MC160 | No effect (8) | DED2 (inhibit) (12) | DED1 (1) |
| cFLIPL | DED1 and DED2 (15) | DED1 and DED2 (activate) (15) | Unknown |
| K13 | DED1 and DED2 (6) | DED1 (activate) (17) | Weak inhibitor (1) |
FLIP REGULATION OF NF-κB
TNFR1 also has the capacity to activate NF-κB (3). In this case, TNFR1 forms a multimeric complex with TRADD, TRAF2, and receptor-interacting protein (RIP), resulting in I kappa B kinase (IKK) activation. IKK phosphorylates IκBα, and phosphorylated IκBα is ubiquitinated. During this process, IκBα is released from NF-κB and directed to the 26S proteosome. Freed NF-κB translocates to the nucleus, becoming a functioning transcription factor to activate expression of genes involved in inflammation and immunity.
In studies querying whether a FLIP could affect TNFR1-induced NF-κB activation, there were several surprising findings. First, the MC159 and MC160 proteins inhibit TNF-induced NF-κB activation (10, 12, 13), which is predicted to aid in the dampening of immune responses during infection (Fig. 1). In quite the opposite manner, K13 instead stimulates NF-κB, and this function is necessary for the transformational properties of KSHV (14) (Fig. 1). Overexpression of cFLIPL also activates NF-κB (14, 15), and this is thought to be a cellular strategy to inhibit death via NF-κB stimulation of genes that are involved in antiapoptosis functions and cellular proliferation. Once again, these findings illustrate the fact that FLIPs have distinct biological functions despite their shared similarities. The second surprise is that the MCV FLIP region that regulates NF-κB is distinct from the antiapoptosis region. For example, while both DEDs are required for MC159 antiapoptosis function (11), only DED1 is required to inhibit NF-κB (10) (Table 1). For MC160, while neither DED of MC160 inhibits apoptosis, the MC160 DED2 or its non-DED-containing C-terminal region is sufficient to inhibit NF-κB (12, 13) (Table 1).
An examination of how FLIPs regulate NF-κB reveals that each protein targets the IKK complex but uses distinct mechanisms to control IKK activation. IKK consists of IKKα, IKKβ, and IKKγ in a 2:2:4 ratio. IKK activation requires IKKγ ubiquitination, a process that allows IKKγ to make extended interactions with IKKβ, which ultimately attracts IκBα to IKKβ and initiates IκBα phosphorylation. Most fascinating are the findings that MC159 and K13 each associate with the IKKγ subunit of IKK (10, 16). Furthermore, the DED1 of MC159 and K13 are each responsible for their NF-κB-modulating function (10, 17) (Table 1). When one considers that MC159 inhibits NF-κB whereas K13 activates NF-κB, one realizes that these differences offer up an incredible system to identify the molecular underpinnings responsible for FLIP regulation of IKKγ and cellular processes that regulate IKKγ. To date, we know that MC159, but not K13, inhibits IKKβ phosphorylation (13). This would suggest that MC159-IKKγ versus K13-IKKγ interactions would result in altered posttranslational modifications of IKKγ. MC160 also inhibits IKK activation, but it has a mechanism different from those of MC159 or K13. MC160 disrupts a functional IKK complex, and its expression induces IKKα degradation (12).
FLIP REGULATION OF IRF3
Beta interferon (IFN-β) is a type I interferon that possesses potent antiviral effects. Major steps in the retinoic acid-inducible gene 1 (RIG-1)/melanoma differentiation-associated protein 5 (MDA5)–interferon regulatory transcription factor 3 (IRF3) activation pathway, which trigger IFN-β production, are known. Viral RNA molecules interact with MDA5, resulting in RIG-I or MDA5 binding to and activating the mitochondrial antiviral signaling protein (MAVS). MAVS clusters on the mitochondrial surface and recruits a complex that includes TRAF3, TANK-binding kinase 1 (TBK1), and IKKε. Activated TBK1-IKKε phosphorylates IRF3, and IRF3 dimerizes either with itself or with IRF7. An IRF3-containing complex migrates to the nucleus and binds to ISRE3 in the IFN-β gene promoter to stimulate IFN-β gene transcription.
Researchers were interested in asking whether FLIPs affected IRF3 activation because two publications provide indirect evidence that FLIPs may inhibit IRF3 or IRF7 activation. One study shows that IRF3 is hyperactivated in cFLIP−/− mouse embryonic fibroblast (MEF) cells (18). In the second study, Balanchandran et al. show that MC159 inhibits IFN-β gene expression at an event upstream of IRF7 activation (19). To follow up these studies, my laboratory probed FLIP-inhibitory functions by using reporter assays in which luciferase genes were controlled by the IFN-β promoter or by a synthetic IRF3 promoter. In both cases, it was observed that the MC159, MC160, and cFLIPL proteins inhibit MAVS-induced IRF3 activation (1). K13 expression also inhibited TBK1-induced IRF3 activation, but not to the extent observed with the other FLIPs (1). Notably, the MC159 and MC160 regions that inhibit IRF3 activation are distinct from the MC159 and MC160 regions required for other inhibitory functions. For example, MC159 DED1 or DED2 possesses IRF3-inhibitory function (1), whereas both MC159 DEDs must be present to inhibit apoptosis (11) (Table 1). For MC160, DED1 or the non-DED-containing C terminus inhibits IRF3 activation (1), but DED1 cannot inhibit NF-κB (12) (Table 1).
Further characterization reveals that MC159 inhibits TBK1 activation (phosphorylation) and also coimmunoprecipitates with TBK1/IKKε (1), suggesting that MC159 interacts with TBK1 in a manner to prevent TBK1 activation. Future studies will assess how MC159 binding to TBK1/IKKε ultimately inhibits IRF3 activation. The mechanism of MC160 remains ill-defined. While MC160 also inhibits TBK1 activation (phosphorylation), MC160 does not immunoprecipitate with TBK1 and IKKε (1). How MC160 inhibits TBK1 activation is a future direction of study.
FLIPs ENCODED BY MCV
Two FLIPs (MC159 and MC160) are encoded by MCV. MCV is a dermatotropic poxvirus that causes one of the most common skin infections (molluscum contagiosum [MC]) among children (20). Childhood MC presents as pearl-like skin neoplasms that persist for weeks or months before spontaneously resolving (20). In immunocompromised patients, MCV spreads unchecked, and larger lesions persist indefinitely (20). Within the context of an MCV infection, perhaps MCV encodes both MC159 and MC160 as a strategy to inhibit the myriad IIR-triggering signals encountered during infection of the skin, thereby aiding in persistence of infection. This possibility currently is difficult to test because all published attempts to cultivate MCV in tissue culture cells or animals are unsuccessful. In fact, little is understood as to how MCV causes disease. One key to filling this gap is the study of MCV immune evasion genes. Of the 182 MCV genes, 77 are predicted to be involved in immune evasion based on (i) their locations at the ends of the linear MCV genome and (ii) the lack of homology to other poxvirus proteins involved in the virus replication cycle. Surprisingly, only 5 of these 77 proteins, including MC159 and MC160, have been studied in any great detail (20). Thus, the remaining MCV genes represent a potential gold mine to discover new strategies that viruses use to counteract innate antiviral immunity to ensure MCV replication and spread.
Footnotes
Published ahead of print 9 April 2014
REFERENCES
- 1.Randall CM, Biswas S, Selen CV, Shisler JL. 2014. Inhibition of interferon gene activation by death-effector domain-containing proteins from the molluscum contagiosum virus. Proc. Natl. Acad. Sci. U. S. A. 111:E265–E272. 10.1073/pnas.1314569111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valmiki MG, Ramos JW. 2009. Death effector domain-containing proteins. Cell. Mol. Life Sci. 66:814–830. 10.1007/s00018-008-8489-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Yin Q, Wu H. 2013. Structural basis of signal transduction in the TNF receptor superfamily. Adv. Immunol. 119:135–153. 10.1016/B978-0-12-407707-2.00005-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertin J, Armstrong RC, Ottilie S, Martin DA, Wang Y, Banks S, Wang GH, Senkevich TG, Alnemri ES, Moss B, Lenardo MJ, Tomaselli KJ, Cohen JI. 1997. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc. Natl. Acad. Sci. U. S. A. 94:1172–1176. 10.1073/pnas.94.4.1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu S, Vincenz C, Buller M, Dixit VM. 1997. A novel family of viral death effector domain-containing molecules that inhibit both CD-95- and tumor necrosis factor receptor-1-induced apoptosis. J. Biol. Chem. 272:9621–9624. 10.1074/jbc.272.15.9621 [DOI] [PubMed] [Google Scholar]
- 6.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer J-L, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. 1997. Viral FLICE inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 386:517–521. 10.1038/386517a0 [DOI] [PubMed] [Google Scholar]
- 7.Shisler JL, Moss B. 2001. Molluscum contagiosum virus inhibitors of apoptosis: The MC159 v-FLIP protein blocks Fas-induced activation of procaspases and degradation of the related MC160 protein. Virology 282:14–25. 10.1006/viro.2001.0834 [DOI] [PubMed] [Google Scholar]
- 8.Shisler JL, Senkevich TG, Berry MJ, Moss B. 1998. Ultraviolet-induced cell death blocked by a selenoprotein from a human dermatotropic poxvirus. Science 279:102–105. 10.1126/science.279.5347.102 [DOI] [PubMed] [Google Scholar]
- 9.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. 1997. Inhibition of death receptor signals by cellular FLIP. Nature 388:190–195. 10.1038/40657 [DOI] [PubMed] [Google Scholar]
- 10.Randall CM, Jokela JA, Shisler JL. 2012. The MC159 Protein from the Molluscum Contagiosum Poxvirus Inhibits NF-kappaB Activation by Interacting with the IkappaB Kinase Complex. J. Immunol. 188:2371–2379. 10.4049/jimmunol.1100136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang JK, Wang L, Zheng L, Wan F, Ahmed M, Lenardo MJ, Wu H. 2005. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol. Cell 20:939–949. 10.1016/j.molcel.2005.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nichols DB, Shisler JL. 2009. Poxvirus MC160 protein utilizes multiple mechanisms to inhibit NF-κB activation mediated via components of the tumor necrosis factor receptor 1 signal transduction pathway. J. Virol. 83:3162–3174. 10.1128/JVI.02009-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichols DB, Shisler JL. 2006. The MC160 protein expressed by the dermatotropic poxvirus molluscum contagiosum virus prevents tumor necrosis factor alpha-induced NF-kappaB activation via inhibition of I kappa kinase complex formation. J. Virol. 80:578–586. 10.1128/JVI.80.2.578-586.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaudhary PM, Jasmin A, Eby MT, Hood L. 1999. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18:5738–5746. 10.1038/sj.onc.1202976 [DOI] [PubMed] [Google Scholar]
- 15.Golks A, Brenner D, Krammer PH, Lavrik IN. 2006. The c-FLIP-NH2 terminus (p22-FLIP) induces NF-kappaB activation. J. Exp. Med. 203:1295–1305. 10.1084/jem.20051556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, Chaudhary PM. 2002. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J. Biol. Chem. 277:13745–13751. 10.1074/jbc.M110480200 [DOI] [PubMed] [Google Scholar]
- 17.Sun Q, Zachariah S, Chaudhary PM. 2003. The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-kappaB activation. J. Biol. Chem. 278:52437–52445. 10.1074/jbc.M304199200 [DOI] [PubMed] [Google Scholar]
- 18.Handa P, Tupper JC, Jordan KC, Harlan JM. 2011. FLIP (Flice-like inhibitory protein) suppresses cytoplasmic double-stranded-RNA-induced apoptosis and NF-kappaB and IRF3-mediated signaling. Cell Commun. Signal. 9:16. 10.1186/1478-811X-9-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balachandran S, Venkataraman T, Fisher PB, Barber GN. 2007. Fas-associated death domain-containing protein-mediated antiviral innate immune signaling involves the regulation of Irf7. J. Immunol. 178:2429–2439 http://www.jimmunol.org/content/178/4/2429.long [DOI] [PubMed] [Google Scholar]
- 20.Randall CMH, Shisler J. 2013. Molluscum contagiosum virus: persistence pays off. Future Virol. 8:561–573. 10.2217/fvl.13.38 [DOI] [Google Scholar]