

ABSTRACT
Flaviviruses are a major cause of disease in humans and animals worldwide. Tick-borne encephalitis virus (TBEV) is the most important arthropod-borne flavivirus endemic in Europe and is the etiological agent of tick-borne encephalitis, a potentially fatal infection of the central nervous system. However, the contributions of host proteins during TBEV infection are poorly understood. In this work, we investigate the cellular protein TIA-1 and its cognate factor TIAR, which are stress-induced RNA-binding proteins involved in the repression of initiation of translation of cellular mRNAs and in the formation of stress granules. We show that TIA-1 and TIAR interact with viral RNA in TBEV-infected cells. During TBEV infection, cytoplasmic TIA-1 and TIAR are recruited at sites of viral replication with concomitant depletion from stress granules. This effect is specific, since G3BP1, another component of these cytoplasmic structures, remains localized to stress granules. Moreover, heat shock induction of stress granules containing TIA-1, but not G3BP1, is inhibited in TBEV-infected cells. Infection of cells depleted of TIA-1 or TIAR by small interfering RNA (siRNA) or TIA-1−/− mouse fibroblasts, leads to a significant increase in TBEV extracellular infectivity. Interestingly, TIAR−/− fibroblasts show the opposite effect on TBEV infection, and this phenotype appears to be related to an excess of TIA-1 in these cells. Taking advantage of a TBE-luciferase replicon system, we also observed increased luciferase activity in TIA-1−/− mouse fibroblasts at early time points, consistent with TIA-1-mediated inhibition at the level of the first round of viral translation. These results indicate that, in response to TBEV infection, TIA-1 is recruited to sites of virus replication to bind TBEV RNA and modulate viral translation independently of stress granule (SG) formation.
IMPORTANCE This study (i) extends previous work that showed TIA-1/TIAR recruitment at sites of flavivirus replication, (ii) demonstrates that TIAR behaves like TIA-1 as an inhibitor of viral replication using an RNA interference (RNAi) approach in human cells that contradicts the previous hypothesis based on mouse embryonic fibroblast (MEF) knockouts only, (iii) demonstrates that tick-borne encephalitis virus (TBEV) is capable of inducing bona fide G3BP1/eIF3/eIF4B-positive stress granules, (iv) demonstrates a differential phenotype of stress response proteins following viral infection, and (v) implicates TIA-1 in viral translation and as a modulator of TBEV replication.
INTRODUCTION
Flaviviruses include several medically important arboviruses, like dengue virus (DENV), yellow fever virus (YFV), West Nile virus (WNV), Japanese encephalitis virus (JEV), and tick-borne encephalitis virus (TBEV). They have in common an enveloped virion containing a capped, single-stranded, positive-sense RNA genome and comparable genomic organizations and replication strategies (1, 2). TBEV causes around 10,000 cases of severe encephalitis in Europe and Asia annually (3–5). After entry, the incoming capped viral RNA is translated into a polyprotein precursor that is processed by cellular proteases and the viral protease NS2B/3 to obtain three structural and seven nonstructural (NS) proteins. NS5, the RNA-dependent RNA polymerase (RdRp), is required for the synthesis of the negative-strand RNA complementary to genomic RNA, serving as the template for the synthesis of new positive-strand viral RNAs. TBEV infection induces important rearrangements of cytoplasmic membranes, with the formation of vesicles containing double-stranded RNA (dsRNA) and replicative proteins, which are believed to release progeny viral genomes in an extravesicular subcompartment, where newly replicated viral RNA accumulates and RNA translation and virus assembly occur (6).
To detect and respond rapidly to invading pathogens, mammalian cells have evolved a variety of pattern recognition receptors (PRRs) that sense conserved pathogen-associated molecular patterns and induce the interferon response pathway (7, 8). For instance, TBEV is able to trigger the retinoic acid-inducible gene 1 (RIG-I)-dependent antiviral pathway that leads to the activation of the type I interferons (α/β interferon [IFN-α/β]) (9). However, other cellular mechanisms, such as the stress response pathway, are also able to limit viral infection (10). Cells react to various stresses by activating cellular kinases that phosphorylate eukaryotic translation initiation factor 2α (eIF2α), thereby rendering eIF2α inactive and halting cap-dependent translation. The stalled translation preinitiation mRNA complexes, together with the aggregated prion-like T-cell-restricted intracellular antigen 1 (TIA-1), form the cytoplasmic stress granules (SG) that also include the TIA-1-related protein (TIAR), the Ras-GAP SH3 domain binding protein (G3BP), and several other proteins, including initiation of translation factors (11). TIA-1 and TIAR are highly homologous RNA-binding proteins involved in pre-mRNA splicing and mRNA translation inhibition that shuttle between the nucleus and the cytoplasm. TIA-1 has a strong affinity for uridine-rich sequences found in certain cellular transcripts, like β-actin and tumor necrosis factor alpha (TNF-α) (12, 13). G3BP has affinity for RNA, regulates apoptosis, and has a cytoplasmic localization at steady state. The stress response blocks cap-dependent translation, and therefore, DNA viruses and viruses that require cap-dependent translation, such as flaviviruses, may be directly affected by their induction. Recently, the stress response pathway has also been linked to the interferon response by demonstrating localization of PRRs in stress granules (SG) following infection (14, 15). However, although some lines of evidence are in favor of SG formation integrating the interferon response and behaving as a platform for antiviral activity (16), other evidence is challenging this intriguing hypothesis (15).
As often happens in the virus-host relationship, viruses have developed strategies to subvert the antiviral response by targeting SG in various ways. While several viruses target G3BP for SG inhibition, flaviviruses like WNV were proposed to recruit TIA-1 and TIAR through their direct binding to the 3′ stem-loop of the complementary minus-strand RNA [3′(−)SL RNA] (17). TIAR was shown to promote viral replication, as TIAR knockout mouse embryonic fibroblasts (MEF TIAR−/−) displayed reduced viral replication, while MEF TIA-1−/− showed close to normal WNV replication. The opposite phenotype was observed for the positive-strand RNA Sindbis virus and the negative-strand RNA vesicular stomatitis virus (VSV), which both replicate better in MEF TIA−/−. WNV, and also DENV, were initially believed to be unable to induce SG formation, based on TIA-1/TIAR localization (18). However, accumulation of G3BP1 in SG in WNV-infected cells was subsequently demonstrated and closely related to the induction of the interferon response (19).
In this work, we take advantage of our recently developed TBEV replicon, which allows visualization of viral RNA in living cells, to explore selected cellular RNA-binding factors for their association with the viral genomes (20). We found that TIA-1 and TIAR are specifically recruited to perinuclear sites of virus replication, consistent with previous findings for DENV and WNV. SG enriched with G3BP1, eIF3, and eIF4B are formed in infected cells, and their number increases when cells are exposed to heat shock. At variance with this, the number of TIA-1-containing SG is reduced in infected cells upon heat shock compared to uninfected cells. We also show that TIA-1 binds TBEV RNA and that its depletion results in an increase of viral replication and early replicon translation, suggesting a repressive role at the level of viral translation. This evidence suggests that TIA-1 activity on TBEV infection in the cytoplasm is not topologically restricted to SG but is brought to the sites of viral replication.
MATERIALS AND METHODS
Cells, viruses, and replicons.
Cells were grown under standard conditions in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and antibiotics.
TIA-1 knockout MEF (MEF TIA-1−/−) or TIAR knockout MEF (MEF TIAR−/−) and the control wild-type MEF (MEF WT) were obtained from Paul Anderson (12, 21). Human U2OS_EGFP and U2OS_EGFP_TIA-1 cells were cloned after transfection of plasmids encoding enhanced green fluorescent protein (EGFP) (pEGFP-N1; Clontech) or encoding EGFP_TIA-1 (isoform a obtained from Juan Valcarcel) into U2OS cells (22).
Working stocks of TBEV strain Neudoerfl were routinely propagated and titrated on Vero E6 cells. The TBEV replicon pTNd/ΔME_24×MS2; the replication-deficient TNd/ΔME_24×MS2_GAA replicon carrying a GDD-to-GAA mutation in the viral NS5 protein; the TNd/ΔME_C17_fluc replicon expressing the luciferase reporter; and its control, TNd/ΔME_C17_fluc_GAA, have been previously described (C17_fluc_TaV2A backbone) (20, 23).
RNA transcription and transfection.
Subgenomic replicon RNAs were transcribed as described in detail previously (20, 24). Cells (4 × 106) were resuspended in 400 μl ice-cold phosphate-buffered saline (PBS) and mixed in a 0.4-cm gene pulser cuvette with 10 μg of RNA to be electroporated with a Bio-Rad Gene Pulser apparatus at 0.25 kV with a capacitance of 960 μF. After electroporation, the cells were washed in complete growth medium without antibiotics and seeded in the same medium. To assess the efficiency of electroporation, particularly of the TNd/ΔME_24×MS2_GAA replicon that does not express viral proteins, we incorporated fluorescein-12-UTP (Roche) in the transcribed RNA and measured the fluorescence of cells after electroporation with a cytofluorimeter.
IF and immunoblot analyses.
Immunofluorescence (IF) analysis was performed 24 and 48 h after treatment (infection, electroporation, and heat shock). U2OS cells were washed with PBS, fixed with 4% paraformaldehyde (PFA) for 15 min, incubated for 5 min with 100 mM glycine, and permeabilized with 0.1% Triton X-100 for 5 min. Subsequently, the cells were incubated at 37°C for 30 min with PBS, 1% bovine serum albumin (BSA), and 0.1% Tween 20 before incubation with antibodies. The coverslips were rinsed three times with PBS, 0.1% Tween 20 (washing solution) and incubated for 1 h with secondary antibodies. Donkey or goat antibodies specific for rabbit or mouse immunoglobulin G and conjugated to Alexa Fluor 488, 594, or 647 (Invitrogen) were used for this analysis. The coverslips were finally washed three times with washing solution and mounted on slides using Vectashield mounting medium (Vector Laboratories). Fluorescence images of fixed cells were captured on a Zeiss LSM510 Meta confocal microscope with a 63×-numerical-aperture (NA) 1.4 Plan-Apochromat oil objective. The pinhole of the microscope was adjusted to get an optical slice of less than 1.0 μm for any wavelength acquired.
For immunoblotting (IB), whole-cell lysates were resolved by SDS-PAGE and blotted onto nitrocellulose membranes (Reinforced NC; Whatman). The membranes were blocked in 4% nonfat milk in Tris-buffered saline (TBS) plus 0.05% Tween 20 (TBST), followed by incubation with the primary antibody diluted in the same solution at 4°C overnight. After washing three times with TBST, secondary horseradish peroxidase (HRP)-conjugated antibodies (Dako) were incubated for 1 h at room temperature. The blots were developed using a chemiluminescent HRP substrate (Millipore).
The antibodies used in this work were as follows: polyclonal rabbit anti-human eIF2α (Santa Cruz; IB, 1:1,000), polyclonal rabbit anti-human phospho-eIF2α (Ser 51) (Cell Signaling; IB, 1:500), mouse monoclonal antibody anti-NS1 (25) (provided by Connie Schmaljohn; IB, 1:1,000), rabbit polyclonal anti-TBEV serum (26) (provided by Franz X. Heinz; IF, 1:100), mouse monoclonal anti-dsRNA antibody (J2; English and Scientific Consulting; IF, 1:200), goat polyclonal anti-human TIA-1 (Santa Cruz; IF, 1:200; IB, 1:1,000), goat polyclonal anti-human TIAR (Santa Cruz; IF, 1:200; IB, 1:1,000), mouse monoclonal anti-human G3BP1 (BD; IF, 1:100; IB, 1:500), rabbit polyclonal anti-green fluorescent protein (GFP) (Molecular Probes; IB, 1:1,000), goat polyclonal anti-eIF3 (Santa Cruz; IF, 1:200), HRP-conjugated mouse monoclonal β-actin (Sigma; 1:10,000); rabbit polyclonal anti-eIF4B (Abcam; IF, 1:100), and rabbit polyclonal anti-polypyrimidine tract binding protein (PTB) (provided by Francisco Baralle; IF, 1:200).
RNA immunoprecipitation.
Following infection or heat shock, U2OS cells were lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, 1 mM EDTA, RNase/proteinase inhibitors), and the cellular extracts were incubated for 4 h with the TIA-1 rabbit polyclonal antibody (Santa Cruz; Sc1751) bound to A/G Plus agarose beads (Santa Cruz). The immunoprecipitates were spun down and washed six times in ice-cold PBS for 5 min each time at 4°C. The beads were then resuspended in 50 mM Tris, pH 7.5, 5 mM EDTA, 10 mM dithiothreitol (DTT), and 1% SDS for further RNA extraction or IB.
RT-PCR.
Total cellular RNA or RNA coming from TIA-1 immunoprecipitation (IP) was extracted by isol-RNA lysis reagent (5-Prime), treated with DNase (Invitrogen), and quantified; 600 ng of RNA was used for reverse transcription (RT) using Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen). Real-time quantitative PCR (qPCR) using Kapa SYBR-Fast (Kapa Biosystems) was performed from cDNA samples. Signals of inducible cellular mRNAs or viral RNAs were normalized to the β-actin mRNA signal. Amplification and detection were carried out on a CFX96 Real Time system (Bio-Rad).
The primers for quantitative RT-PCR used in this study were as follows: TBEV RNA, 5′NCRA1-fw (5′-GCGTTTGCTTCGGA-3′) and 5′NCRA1-rv (5′-CTCTTTCGACACTCGTCGAGG-3′); β-actin, BA1 (5′-CATGTGCAAGGCCGGCTTCG-3′) and BA4 (5′-GAAGGTGTGGTGCCAGATTT-3′). Those for RT-PCR were as follows: TBEV RNA, 3′NCR-fw (5′-TTGGCAGCTCTCTTCAGGAT-3′) and 3′NCR-rv (5′-AGCGGGTGTTTTTCCGAGTC-3′); U6 RNA, U6-fw (5′-GCTTCGGCAGGACATATACTAAAAT-3′) and U6-rv (5′-CGCTTCACGAATTTGCGTGTCAT-3′); and β-actin primers as described above.
RNA interference.
Pools of small interfering RNAs (siRNAs) were obtained from Dharmacon. Nontargeting siGenome pool 3 was used as a control in all experiments. On-Target plus SmartPool TIA-1 and TIAR were used for the depletion of TIA-1 and TIAR, respectively. U2OS cells were transfected with siRNAs at a concentration of 100 nM, using HiPerFect transfection reagent (Qiagen) according to the manufacturer's instructions.
Luciferase assay.
Beetle-Juice and Renilla-Juice (p.j.k.) were used according to the manufacturer's instructions to simultaneously measure both firefly and Renilla luciferase activity. After electroporation with RNA and/or DNA, cells were washed in culture medium and seeded in 96-well plates. Triplicate wells were lysed at individual time points, followed by measurement with the Envision Multimode Plate Reader (PerkinElmer). For normalization of the firefly luciferase values, replicon RNA containing the firefly luciferase gene was cotransfected with a plasmid encoding Renilla luciferase (phRL-CMV). For only the early time points (1 to 6 h) of the time course, 3 μg of in vitro-transcribed phRL-CMV RNA was cotransfected with the replicon RNA instead. Samples were processed exactly as described previously to obtain normalized values of luciferase activity (23).
Statistics.
A minimum of three independent experiments in triplicate repeats were conducted for each condition examined (in the figure legends, n is the number of independent experiments). Average values are shown with standard deviations and P values, measured with a paired two-tailed t test. Significant P values are indicated by asterisks in the graphs.
RESULTS
Recruitment of TIA-1/TIAR to sites of TBEV replication.
TBEV is a positive-strand RNA virus that replicates in the cytoplasm. Little is known about the host factors that target TBEV RNA. In order to explore cellular proteins that colocalize with viral RNA, we took advantage of a method for the visualization of TBEV replicated RNA previously developed in our laboratory (20, 27). The system consists of a capped viral RNA transcribed in vitro in which the structural proteins have been deleted and an array of 24 binding sites for the MS2 protein have been engineered in the 3′ noncoding region (NCR) (TNd/ΔME_24×MS2). Transfection of the replicon RNA in cells expressing a tagged form of MS2 (enhanced yellow fluorescent protein [EYFP]-MS2nls) allows the visualization of replicated TBEV RNA. A U2OS cell line stably expressing EYFP-MS2nls (U2OS-EYFP-MS2nls) was electroporated with TNd/ΔME_24×MS2 or with a control replicon with a mutation of the catalytic domain of NS5 that is unable to replicate (TNd/ΔME_24×MS2_GAA). The transfection efficiency of replicon RNA was routinely above 70% (data not shown). At 24 h postelectroporation (p.e.), cells were fixed and stained with antibodies for the following host factors. First, we analyzed the localization of the SG component TIA-1. As shown in Fig. 1A, TIA-1 clearly relocalized to perinuclear sites of viral replication defined by the accumulation of EYFP-MS2nls and by costaining with both polyclonal antibody against viral proteins and an antibody against dsRNA replication intermediates (Fig. 1B, top). TIA-1 redistribution was not observed when using the replication-defective TBEV replicon, indicating that viral replication is required for the observed phenotype (Fig. 1B, bottom). To address whether this relocalization was a specific feature of TIA-1, we choose the PTB protein, which has been implicated in the life cycles of DENV and JEV (28–30). PTB shares several features with TIA-1, since both proteins (i) are RNA-binding proteins with several RNA recognition motifs; (ii) are shuttling proteins with a predominant nuclear localization; (iii) are involved in nuclear processes, such as alternative splicing; and (iv) are involved in cytoplasmic processes, such as translation (31). As shown in Fig. 1C, the nucleocytoplasmic distribution of PTB did not change in U2OS cells expressing TNd/ΔME_24×MS2 replication compared to those transfected with TNd/ΔME_24×MS2_GAA, demonstrating that TIA-1 recruitment was not the result of an aspecific accumulation of RNA-binding proteins resulting from the presence of large amounts of viral RNA in the cytoplasm. To confirm the relocalization of TIA-1, we also infected U2OS cells with TBEV at a multiplicity of infection (MOI) of 2 PFU/cell. At 24 h postinfection (p.i.), TIA-1 accumulated in perinuclear sites of viral replication, as has been previously observed for replicon-expressing cells, and this relocalization was maintained at 48 h p.i. (Fig. 1D). Although the efficiency of infection, determined by dsRNA staining, was ∼95% (data not shown), the number of cells showing recruitment of TIA-1 at the perinuclear sites of TBEV replication was approximately 25% of the cells, with a slight increase at 48 h (low-magnification staining at 24 h p.i. in Fig. 1D). The TIA-1-related protein TIAR was also recruited to TBEV replication sites at 24 h p.i. (Fig. 1E) and at 48 h p.i. (data not shown).
FIG 1.

Localization of TIA-1 and TIAR in TBEV-infected cells. (A) U2OS-MS2-YFPnls cells were electroporated with the TNd/ΔME_24×MS2 replicon RNA. At 24 h p.e., the cells were fixed and incubated with anti-TBEV antiserum (Alexa Fluor 624; blue) and with the anti-TIA-1 antibody (TIA-1) (Alexa Fluor 594; red). Colocalization of replicated RNA (MS2-EYFPnls) (yellow) with TIA-1 (top row) or of TBEV proteins with TIA-1 (bottom row) is shown. (B) U2OS-MS2-YFPnls cells were electroporated with the TNd/ΔME_24×MS2 replicon RNA (top row) or with the nonreplicating control TNd/ΔME_24×MS2_GAA (bottom row). At 24 h p.e., the cells were fixed and incubated with the J2 anti-dsRNA antibody (dsRNA) (Alexa Fluor 488; green) and with the anti-TIA-1 antibody (TIA-1) (Alexa Fluor 594; red). Colocalization of dsRNA with TIA-1 in cells electroporated with a replicating TBEV replicon (top row), but not in cells electroporated with a nonreplicating RNA (bottom row), is shown. (C) U2OS-MS2-YFPnls cells were electroporated as for panel B, and 24 h p.e., the cells were fixed and incubated with the monoclonal anti-PTB antibody (PTB) (Alexa Fluor 594; red). Lack of colocalization of replicated RNA (MS2-EYFPnls) (yellow) with PTB is shown (merge). (D) U2OS cells were either infected with TBEV at an MOI of 2 for 24 h (TBEV 24 h p.i.) or 48 h (TBEV 48 h p.i.) or mock infected (mock). The cells were fixed and incubated with the J2 anti-dsRNA antibody (dsRNA) (Alexa Fluor 488; green) and with the anti-TIA-1 antibody (TIA-1) (Alexa Fluor 594; red). Colocalization of dsRNA with TIA-1 in cells infected with TBEV (top and middle), but not in mock-infected cells (bottom), is shown. An additional image at 24 h p.i. was taken at low magnification to show more cells. (E) U2OS cells were infected for 24 h and treated as for panel C except that the anti-TIAR antibody (TIAR) (Alexa Fluor 594; red) was used.
We conclude that TBEV infection induces the specific recruitment of TIA-1 and TIAR to perinuclear sites of viral replication in U2OS cells.
Formation of stress granules in TBEV-infected cells.
SG are defined by the translation initiation factors that form the stalled initiation complexes, which are still bound to mRNA and recruited to SG from disassembling polysomes (32). These factors include the eukaryotic initiation of translation factors eIF3, eIF4F (comprising eIF4E, eIF4A, and eIF4G), and eIF4B; small ribosomal subunits; and PABP-1. These core SG components are universal markers for all SG. Therefore, we analyzed the relocalization of resident SG proteins, such as G3BP1, eIF3, and eIF4B. At 24 h p.i., G3BP1 foci were clearly visible in the cytoplasm of about 30% of infected cells (Fig. 2A). Also, eIF3 and eIF4B colocalized with G3BP1 in SG following viral infection (Fig. 2B and C).
FIG 2.

Formation of stress granules in TBEV-infected cells. (A) U2OS cells were either infected with TBEV at an MOI of 2 (TBEV) (top row) or mock infected (mock) (bottom row). At 24 h p.i., the cells were fixed and incubated with the monoclonal anti-G3BP1 antibody (G3BP1) (Alexa Fluor 488; green) and with the anti-TBEV antiserum (TBEV) (Alexa Fluor 594; red). Formation of G3BP1 SG in TBEV-infected cells (top row), but not in mock-infected cells (bottom row), is shown. (B) U2OS cells were infected as for panel A. At 24 h p.i., the cells were fixed and incubated with the monoclonal anti-G3BP1 antibody (G3BP1) (Alexa Fluor 488; green) and with the polyclonal anti-eIF3 antibody (eIF3) (Alexa Fluor 594; red). Formation of G3BP1 SG in TBEV-infected cells colocalizing with eIF3 (top row), but not in mock-infected cells (bottom row), is shown. (C) U2OS cells were infected as for panel A. At 24 h p.i., the cells were fixed and incubated with the monoclonal anti-G3BP1 antibody (G3BP1) (Alexa Fluor 488; green) and with the polyclonal anti-eIF4B antibody (eIF4B) (Alexa Fluor 594; red). Formation of G3BP1 SG in TBEV-infected cells colocalizing with eIF4B (top row), but not in mock-infected cells (bottom row), is shown. (D) U2OS cells were either infected with TBEV at an MOI of 2 (TBEV) or heat shocked for 40 min at 45°C (heat-shock). The cell lysates were immunoblotted for total eIF2α and phosphorylated eIF2α (top rows). Loading control (β-actin) and infection control (NS1) are also shown.
To confirm that the stress response pathway of translation inhibition and SG formation was activated following infection, we measured eIF2α phosphorylation. Consistent with the induction of G3BP1/eIF3/eIF4B-containing SG, eIF2α appeared to be phosphorylated upon TBEV infection (Fig. 2D). We conclude that TBEV infection of U2OS cells induces eIF2α phosphorylation and the formation of SG containing G3BP1, eIF3, and eIF4B.
TIA-1 recruitment to cytoplasmic sites of viral replication resists heat shock.
SG are formed in response to various kind of stresses. As shown in Fig. 2D, both heat shock and TBEV infection induced eIF2α phosphorylation in U2OS cells, most likely through protein kinase R (PKR) (19, 33). To determine if TBEV prevents SG formation in response to heat shock, U2OS cells were electroporated with TNd/ΔME_24×MS2. At 24 h p.e., the temperature was increased to 45°C for 40 min before fixation and staining for TIA-1. SG were counted in the context of both replication-competent and -incompetent (GAA mutant) replicons. As shown in Fig. 3A, heat shock induced the formation of an average of 6 SG/cell in mock-infected cells (electroporated with TNd/ΔME_24×MS2_GAA), while in cells replicating TNd/ΔME_24×MS2, the number of SG was significantly reduced. Representative images are shown for the two conditions observed following heat shock with TIA-1 either in SG or in viral replication sites. We repeated the experiment, staining with an antibody against G3BP1. G3BP1-SG induced by heat shock showed the opposite behavior, with an increase of granules in the context of TBEV replication compared to the presence of SG induced by heat shock in TNd/ΔME_24×MS2_GAA-electroporated cells (Fig. 3B). The same pattern was observed upon TBEV infection, reinforcing the notion that TBEV-mediated recruitment of TIA-1 to sites of viral replication competes with TIA-1-containing SG formation in response to heat shock (Fig. 3C and D). We have previously shown that TBEV-infected cells already contain SG, and therefore, the number of SG in infected cells following heat shock represents a mixture of those formed prior to the treatment (by infection itself) and those induced afterward. We conclude that TBEV infection induces G3BP1-SG formation but recruits TIA-1 to sites of viral replication that compete with SG for TIA-1 localization, indicating differential responses to viral stress.
FIG 3.

Induction of SG by heat shock following TBEV infection. (A) U2OS cells were electroporated with the TNd/ΔME_24×MS2 replicon RNA (TBEV) or with the nonreplicating control TNd/ΔME_24×MS2_GAA (GAA). At 24 h p.e., the cells were exposed to heat shock and then fixed and stained for TIA-1 and dsRNA. In cells electroporated with the replication-competent RNA, the number of SG per infected cell (stained with dsRNA) was determined, while the number of SG in cells transfected with the control replicon was determined per cell. A total of 100 cells per condition were counted in triplicate. Average values are shown, with standard deviations and P values, measured as described in the text. (B) U2OS cells were treated exactly as described for panel A except that they were stained for G3BP1. (C) U2OS cells were either infected with TBEV (TBEV) (MOI = 2) or mock infected (mock). At 24 h p.i., the cells were exposed to heat shock and then fixed and stained for TIA-1 and dsRNA. In TBEV-infected cells, the number of SG per infected cell (stained with dsRNA) was determined, while the number of SG in mock-infected cells was determined per cell. Counts were performed and represented as for panel A. (D) U2OS cells were infected exactly as described for panel C except that they were stained for G3BP1. *, P < 0.05 (significant); **, P < 0.01 (highly significant).
TIA-1 binds TBEV RNA.
TBEV infection of cells leads to a pronounced accumulation of TIA-1 and TIAR to sites of viral replication that are rich in transcribed viral RNAs. Since both are RNA-binding proteins, we explored their association with viral RNA. To this end, U2OS cells were either heat shocked or infected with TBEV and lysed in RIPA buffer 24 h p.i. Cell extracts were incubated with the specific TIA-1 or TIAR antibody, subjected to IP, and then processed for immunoblotting or RT-PCR. As shown in Fig. 4A, specific enrichment for the TIA-1 protein using a specific antibody, but not using an irrelevant IgG, was obtained. TIAR was found associated with TIA-1 both in heat-shocked and in infected cells while no association could be detected with β-actin control, G3BP1, or TBEV NS1. Both for TIA-1 and TIAR, a doublet of bands was visible in the immunoblot, consistent with the 2 splice variants, which lead to an inclusion in the protein of 11 or 17 amino acids, respectively. Next, we explored the co-IP of RNA. A TBEV 3′ NCR amplification product was detected in TIA-1 beads from infected cells, but not from heat-shocked cells (Fig. 4B). As a negative control, we chose the cellular non-protein-coding small nuclear RNA U6 (U6 snRNA), and as a positive control, β-actin mRNA, which has been shown to coprecipitate with TIA-1 upon heat shock (13). Interestingly, while U6 snRNA was not bound by TIA-1 following heat shock or infection, β-actin mRNA was detected in pulldowns of heat-shocked cells, as previously demonstrated, and also in pulldowns from cells infected by TBEV. This finding is consistent with a described feature of TIA-1, i.e., being able to bind various housekeeping transcripts following different stresses (13). We also conducted a reverse IP with a TIAR-specific antibody. As shown in Fig. 4C, TIA-1 was found associated with TIAR both in heat-shocked and in infected cells, while no association could be detected with the β-actin control. Also for TIAR, infection led to the co-IP of TBEV RNA (Fig. 4D). Intriguingly, in both IPs, the fraction of TIAR/TIA-1 that is coimmunoprecipitated confirms the hypothesis that only a fraction of cellular TIA-1 and TIAR forms a protein-protein interaction. In the specific case of TBEV infection, both TIA-1 and TIAR are recruited at the site of viral replication and interact with viral RNA.
FIG 4.

RNA immunoprecipitation of TIA-1 and TIAR in TBEV-infected cells. (A) U2OS cells were either infected with TBEV or heat shocked. At 24 h p.i. or after 40 min of heat shock, the cells were immunoprecipitated with the TIA-1 antibody. Immunoblotting shows specific TIA-1 IP compared to an irrelevant immunoglobulin (IgG). TIAR was coimmunoprecipitated with TIA-1 following both heat shock and TBEV infection, while TBEV NS1, G3BP1, and control β-actin were not. (B) U2OS cells treated as described for panel A were subjected to total-RNA extraction and RT-PCR with primers specific for the 3′ noncoding region of TBEV (3′-NCR), for β-actin as a positive control for TIA-1 RNA IP, and for the negative-control U6 snRNA. (C) U2OS cells were either infected with TBEV or heat shocked. At 24 h p.i. or after 40 min of heat shock, the cells were immunoprecipitated with the TIAR antibody. Immunoblotting shows specific TIAR IP compared to an irrelevant immunoglobulin (IgG). TIA-1 was coimmunoprecipitated with TIAR following both heat shock and TBEV infection, while control β-actin was not. (D) U2OS cells treated as described for panel C were subjected to total-RNA extraction and RT-PCR with primers specific for the 3′ noncoding region of TBEV (3′-NCR), for β-actin as a positive control for TIAR RNA IP, and for the negative-control U6 snRNA.
Emara et al. mapped the interaction of TIA-1/TIAR within a short AU sequence (UAAUU) located in two internal loops of the WNV3′(−)SL RNA structure, while Lopez de Silanes et al. showed that the TIA-1 motif was a U-rich, 30- to 37-nucleotide-long bipartite element forming loops of variable size and a bent stem, localizing preferentially to the 3′ untranslated region of target mRNAs (13, 34). The common features of these elements are the U-rich stretch and an RNA secondary structure. However, the latter study also showed that this motif applies only to a portion of the TIA-1-bound mRNA, indicating that other, as-yet-unknown signature motifs might exist on TIA-1 target mRNAs. Direct comparison of the 3′(−) untranslated RNAs of WNV and TBEV by M-fold (http://mfold.rit.albany.edu/?q=mfold) analysis did not show obvious similarities, although several U-rich stretches and RNA secondary structures could be found (data not shown).
We conclude that infection of cells with TBEV induces the formation of a subcomplex of viral RNA, TIA-1, and TIAR localized at the sites of viral replication.
TIA-1 is a negative regulator of TBEV replication.
To investigate the functional role of TIA-1 in TBEV replication, we proceeded to siRNA-mediated depletion of TIA-1, TIAR, or both in U2OS cells. As shown in Fig. 5A, transfection of siRNA led to a consistent depletion of TIA-1 and TIAR compared with the nontargeting control siRNA (siNTG). Two days posttransfection, the cells were infected with TBEV and processed to measure extracellular virus release. Viral yields showed a 10-fold increase compared to siNTG at 24 h p.i., which was maintained at 48 h p.i. (Fig. 5B). Knockdown of the single factors showed similar effects, while the combination of siRNAs targeting both TIA-1 and TIAR did not show an improvement of the viral yields, indicating that each protein is equally required for the phenotype.
FIG 5.
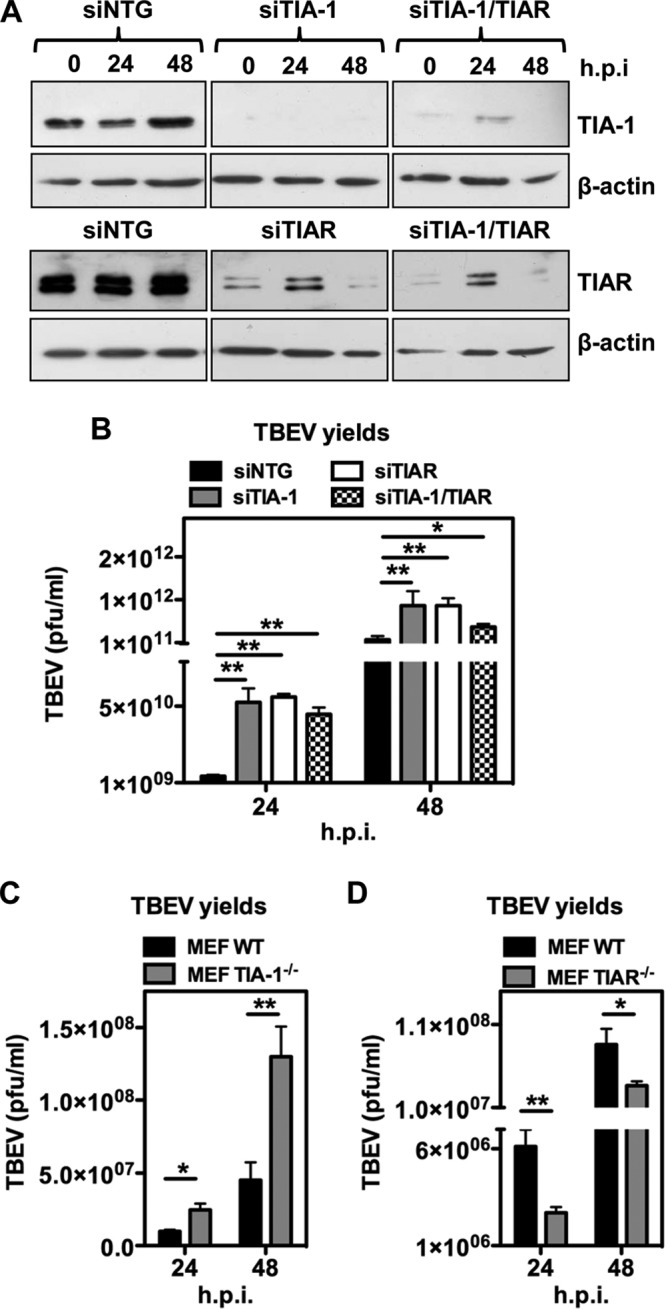
Functional analysis of TIA-1/TIAR depletion in TBEV-infected cells. (A) U2OS cells were transfected with siNTG, with TIA-1/TIAR-specific siRNA (siTIA-1), or with a combination of both. After 48 h, the cells were infected with TBEV (MOI = 2) and harvested for immunoblotting at 24 and 48 h p.i. β-Actin was the loading control. (B) Supernatants from infected cells treated as for panel A were used to infect Vero cells to measure virus yields (PFU/ml) from siTIA-1, siTIAR, siTIA-1/TIAR, or siNTG samples. Average values of triplicate independent experiments (n = 3) are shown, with standard deviations and P values, as described in the text. (C) MEF TIA-1−/− or their control (MEF WT) were infected with TBEV at an MOI of 2. Cell supernatant collected 24 and 48 h p.i. was used to infect Vero cells to measure virus yields (PFU/ml) from infected MEF TIA-1−/− or infected MEF WT samples. Values were plotted as for panel B (n = 6). (D) MEF TIAR−/− or their control (MEF WT) were infected with TBEV at an MOI of 2. Cell supernatant collected 24 and 48 h p.i. was used to infect Vero cells to measure virus yields (PFU/ml) from infected MEF TIAR−/− or infected MEF WT samples. Values were plotted as for panel B (n = 6). *, P < 0.05 (significant); **, P < 0.01 (highly significant).
Next, we investigated TBEV infection in MEF TIA-1−/−. Infection of TIA-1−/− cells with TBEV led to a significant increase in virus yields at 24 to 48 h p.i. (Fig. 5C). On the other hand, infection of TIAR knockout MEF resulted in a decrease in infectious-virus yields (compare Fig. 5D with C). This result could be explained by the upregulation of TIA-1 in TIAR−/− cells, as previously observed (17, 22). Accordingly, we could measure in replica blots a 2.3- ± 0.2-fold increase of TIA-1 expression in MEF TIAR−/−, while TIAR levels were not affected in MEF TIA-1−/− (Fig. 6A). To further confirm that TIA-1 overexpression resulted in inhibition of TBEV replication, we engineered U2OS cells stably expressing EGFP–TIA-1 or EGFP alone (Fig. 6B). These cells showed nuclear localization of EGFP–TIA-1 and responded to heat shock by the formation of SG enriched in endogenous TIA-1 and EGFP–TIA-1 (Fig. 6C). As shown in Fig. 6D, overexpression of TIA-1–GFP resulted in a decrease in viral yields at 24 h p.i. consistent with an inhibitory effect of TIA-1 on TBEV replication. Accordingly, viral-RNA levels were also reduced in U2OS EGFP–TIA-1 compared to U2OS EGFP cells (Fig. 6E). In U2OS cells expressing EGFP–TIA-1, the infection quickly recovers from weak inhibition by EGFP–TIA-1, which could be explained by the presence of the bulky EGFP, which would limit its functionality. However, the effect of EGFP–TIA-1 overexpression was significant at 24 h p.i., in terms of both viral yields and viral RNA (Fig. 6D and E). We conclude that TIA-1, and most likely also TIAR, exerts an inhibitory effect on TBEV replication.
FIG 6.

Functional analysis of TIA-1 overexpression in TBEV-infected cells. (A) Whole-cell extracts of MEF WT, MEF TIA-1−/−, and MEF TIAR−/− were immunoblotted in parallel for TIAR (top) and TIA-1 (middle). β-Actin was the loading control (bottom). (B) Whole-cell extracts from U2OS (mock), U2OS_EGFP_TIA-1, and U2OS_EGFP cells were subjected to immunoblotting with anti-EGFP (left) and anti-TIA-1 (right) antibodies. (C) U2OS_EGFP_TIA-1 cells (top row) and U2OS_EGFP cells (bottom row) show nuclear and nucleocytoplasmic localization of EGFP–TIA-1 and EGFP, respectively (left column). After heat shock, SG are readily formed in the cytoplasm of both cell lines (right columns). EGFP in U2OS_EGFP_TIA-1 (top), but not in U2OS_EGFP (bottom), costained with anti-TIA-1 antibodies. (D) U2OS_EGFP_TIA-1 cells or their control U2OS_EGFP cells were infected with TBEV at an MOI of 2, and 24- and 48-h p.i., the cell supernatants were used to infect Vero cells to measure virus yields (PFU/ml) from infected U2OS_EGFP_TIA-1 or infected MEF WT samples. Average values of triplicate independent experiments (n = 3) are shown, with standard deviations and P values, as described in the text. (E) Total RNA was extracted from cells infected as for panel D and used as a template for real-time qPCR using primers specific for TBEV (5′-NCR). TBEV amplification products were normalized to β-actin RNA. The relative increase in TBEV RNA in U2OS_EGFP_TIA-1 cells with respect to U2OS_EGFP is shown. Values are plotted as in Fig. 5B (n = 3). *, P < 0.05 (significant); **, P < 0.01 (highly significant).
Effect of TIA-1 on TBEV early translation.
The overall effect of TIA-1 depletion/overexpression was consistent with its activity as an inhibitor of TBEV infection. We wished to recapitulate the phenotype using a TBEV replicon expressing the luciferase reporter gene (TNd/ΔME_C17_fluc) (23). U2OS cells expressing EGFP–TIA-1 and control cells expressing EGFP were electroporated with TNd/ΔME_C17_fluc, and luciferase activity was assessed at 24 h p.e. and 48 h p.e. As shown in Fig. 7A, overexpression of EGFP–TIA-1 reduced the luciferase levels at 24 h p.e., but in this case, the reduction was also significant at 48 h p.e. (compare Fig. 7A with 6D). Next, we electroporated MEF TIA-1−/− with TNd/ΔME_C17_fluc and measured luciferase activity at 24 h p.e. and 48 h p.e. To our surprise, we observed a huge increase in luciferase activity in MEF TIA-1−/− that exceeded 100-fold (Fig. 7B). This result gave us a hint as to its possible function. To confirm this observation, we repeated the experiment in U2OS cells treated with TIA-1 siRNA, which also resulted in an increase in luciferase activity compared with cells treated with an irrelevant siRNA (Fig. 7C). The control of siRNA efficiency is shown in Fig. 7D. We reasoned that TIA-1 is a cellular factor with many functions, including translation inhibition. In the course of TBEV infection, there is an early first round of translation of the incoming viral RNA, followed by further rounds after newly synthesized RNA becomes available later in infection. Distinguishing these two phases is not easily accomplished with infectious virus, but it has been previously demonstrated that with a replicon expressing luciferase it is possible to temporally distinguish between the two distinct translation phases (23). Therefore, we proceeded by electroporating TNd/ΔME_C17_fluc in MEF TIA-1−/−, monitoring luciferase activity at various time points. As shown in Fig. 7E, TIA-1 depletion favored early TBEV replicon RNA translation, leading to a sharp increase of the luciferase signal 4 h postelectroporation, which was sustained at later time points. A control replicon carrying a mutation in NS5 (TNd/ΔME_C17_fluc_GAA) was able to translate only the incoming viral RNA, and in this case as well, there was a remarkable increase in luciferase activity at early time points.
FIG 7.

Functional analysis of TIA-1 following TBEV replicon transfection. (A) U2OS_EGFP_TIA-1 cells and U2OS_EGFP cells were electroporated with replicon TNd/ΔME_C17_fluc, encoding the firefly luciferase reporter, and with a plasmid encoding Renilla luciferase for normalization. At 24 and 48 h p.e., the cells were lysed, and dual-luciferase activity was measured (n = 3). Values are shown as normalized firefly/Renilla relative light units (RLU). (B) MEF TIA-1−/− and their control MEF WT were electroporated, and dual-luciferase activity was measured 24 and 48 h p.e,. as described for panel A. (C) U2OS cells were transfected with siNTG or with the TIA-1-specific siRNA (siTIA-1). After 48 h, the cells were electroporated with replicon TNd/ΔME_C17_fluc and with a plasmid encoding Renilla luciferase for normalization. At 24 and 48 h p.e., the cells were lysed, and dual-luciferase activity was measured (n = 3). Values are shown as normalized firefly/Renilla relative light units (RLU). (D) Cells treated as for panel C were harvested for immunoblotting at 24 and 48 h p.e. β-Actin was the loading control. (E) MEF TIA-1−/− and their control MEF WT were electroporated with TNd/ΔME_C17_fluc. A time course of firefly luciferase activity normalized to the internal Renilla luciferase control is shown as relative light units calculated as described previously (n = 3) (23). (F) MEF TIA-1−/− and their control MEF WT were electroporated with TNd/ΔME_C17_fluc_GAA. A time course, as described for panel C, is shown. *, P < 0.05 (significant); **, P < 0.01 (highly significant).
DISCUSSION
In this work, we propose that the host cell stress response protein TIA-1 inhibits TBEV translation and is recruited to sites of viral replication, together with its close homolog, TIAR. We also show that G3BP-enriched SG are formed following TBEV infection lacking TIA-1/TIAR. Our observations stem from the pioneering work of Emara and Brinton, who demonstrated recruitment of TIA-1 and TIAR to sites of WNV and DENV replication (18). They concluded that these viruses inhibit SG formation by targeting TIA-1/TIAR either through the nonstructural protein NS3, which could be immunoprecipitated by both TIA-1 and TIAR, or through the viral 3′(−)SL RNA, which was earlier shown to bind TIAR and, to a lesser extent, TIA-1 (17). However, more recently, they reinterpreted their own data, this time looking at G3BP1 as a marker for SG, showing that natural WNV genotypes, such as Eg101, induced SG less efficiently than the lineage 2/1 chimeric WNV infectious clone W956IC, which produced high levels of early viral RNA (19). They proposed that induction of SG by WNV is closely linked to the availability of dsRNA to PKR signaling and that fast-replicating genotypes, such as W956IC, release more dsRNA from virus-induced vesicles, thus triggering the stress response pathway. In our work, we also show that TBEV infection induces the recruitment of TIA-1/TIAR to sites of TBEV replication, but at the same time, eIF2α becomes phosphorylated, and SG enriched in G3BP1/eIF3/eIF4B are formed in the cytoplasm. Therefore, flaviviruses are indeed capable of inducing genuine SG in infected cells. These viruses may only delay the shutoff of host cell translation and the induction of the interferon response, possibly by shielding viral replication intermediates in the highly organized membranous vesicles that have been described for WNV, DENV, and TBEV (6, 9, 35–37).
In light of these findings, the role of TIA-1/TIAR becomes much less clear, because the simplest explanation, that their recruitment inhibits SG formation, is no longer supported by the data. In order to clarify their roles in flavivirus replication, it would be helpful to review what is known so far. First, TIAR and TIA-1 were identified by affinity purification using synthetic oligonucleotides as host factors that specifically bind the WNV 3′(−)SL RNA (17). In our study, we demonstrate that TIA-1 and TIAR bind viral RNA in infected cell extracts, but we did not address its polarity. By confocal microscopy, we could show that TIA-1 and TIAR colocalized with dsRNA (Fig. 1B, C, and D) and TBEV nonstructural proteins, while newly replicated viral RNA, tagged by MS2-EYFPnls, showed a more diffused distribution (Fig. 1A). Previously, we demonstrated that MS2-tagged viral RNA consisted of replicated RNA not associated with replication vesicles but diffused in the extravesicular space (6). This is consistent with a model in which the replication complex and viral replication intermediates, such as (−)RNA and dsRNA, remain confined in replication vesicles while genomic (+)RNA translocates in the extravesicular space for further rounds of translation or assembly. Therefore, we propose that TIA-1 and TIAR remain associated with viral RNA within replication vesicles. Indeed, heat shock, which potently induces SG, was unable to shift TIA-1 from viral replication sites to SG in infected cells (Fig. 3), despite TIA-1's ability to shuttle continuously in and out of SG (38). However, although this model fits the association of TIA-1 and TIAR with (−)RNA proposed by Brinton's group, it is very difficult to conceive their recruitment to viral RNA in preassembled replication vesicles. Most likely, TIA-1 and TIAR bind viral RNA before the formation of the vesicles at the level of the incoming viral (+)RNA. This interpretation requires that TIA-1/TIAR associate with viral (+)RNA that is then delivered to replication vesicles, where dsRNA intermediates are formed only after complete assembly of the vesicle to protect them from cytoplasmic sensors, such as RIG-I or PKR. TIA-1/TIAR could then be transferred to high-affinity binding sites on (−)SL RNA to exert their putative functions in viral replication. Alternatively, TIA-1 may also act by inhibiting translation of replicated viral RNA to promote assembly. In this case, the differential distribution of TIA-1 and replicated MS2-tagged TBEV RNA could be explained by the low resolution of fluorescence microscopy in detecting a few molecules of TIA-1 bound to RNA compared to 24 doublets of MS2.
Functionally, the effect of the depletion of TIA-1/TIAR on viral replication is controversial. WNV showed a lower virus yield in MEF TIAR−/−, while was slightly affected in MEF TIA-1−/− (17). This observation led to the hypothesis that these proteins provide a necessary function for WNV during its replication cycle. However, when we depleted TIA-1 and TIAR in human cells, we observed an increase in virus production (Fig. 5B). The same phenotype was observed in MEF TIA-1−/− (Fig. 5C), while in MEF TIAR−/−, TBEV growth was inhibited similarly to what has been reported for WNV (Fig. 5D). The latter phenotype could be explained by the well-described upregulation of TIA-1 in MEF TIAR−/− (Fig. 6A) (17, 22). Consistently, infection of U2OS cells overexpressing EGFP–TIA-1 showed impaired virus growth and reduced levels of viral RNA (Fig. 6D and E).
Although the above-mentioned results consistently indicate that TIA-1/TIAR work as inhibitors of TBEV replication, the overall effect of their depletion is rather weak and could be explained by incomplete depletion by siRNA. Indeed, a small quantity of TIA-1/TIAR protein would be sufficient to function on the few viral RNA molecules that become accessible between virion uncoating and genomic-RNA translation/replication. To overcome this limitation, we exploited the TBEV replicon technology, which introduces large amounts of unprotected viral (+)RNA into the cytoplasm. Transfection of the TBEV replicon RNA in MEF TIA-1−/− and in U2OS cells depleted of TIA-1 by siRNA resulted in a significant increase of the luciferase reporter signal 24 h p.i. (Fig. 7B and C). However, at this late time point, we could not distinguish between an effect on translation or on replication. Since the TBEV luciferase replicon can be used as a sensitive reporter of viral translation, we performed a time course of replicon transfection (Fig. 7E and F) (23). By this method, we could distinguish two waves of luciferase signal: the first corresponds to translation of the incoming viral (+)RNA, as demonstrated by the presence of a peak also for the control replicon carrying a mutation in NS5; the second requires viral replication and could be detected only for fully replication-competent replicons. Transfection of MEF TIA-1−/− showed a huge increase in luciferase starting at early time points compared to MEF WT, indicating that TIA-1 acts directly on translation of viral (+)RNA. However, we cannot rule out the possibility that depletion of TIA-1 from the cytoplasmic pools during viral infection also indirectly affects the expression of cellular genes that could positively affect virus growth.
Our data are consistent with a model in which TIA-1 works together with TIAR as a translation inhibitor of TBEV. TIA-1 could function at two steps of the viral life cycle: on incoming genomic viral (+)RNA that is translated into the polyprotein and/or on replicated viral (+)RNA extruded from replication vesicles. Translation competes with replication, with the same RNA also being the template for (−)RNA, and with assembly of new virions. Therefore, TIA-1-dependent modulation of translation could determine the amount of (+)RNA available for replication and/or assembly, suggesting that fine-tuning of this factor may modulate the viral replication cycle.
ACKNOWLEDGMENT
Work on flaviviruses in A.M.'s laboratory is supported by the Beneficientia Stiftung.
Footnotes
Published ahead of print 2 April 2014
REFERENCES
- 1.Lindenbach BD, Thiel HJ, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA. (ed), Fields virology, 5th ed. Lippincott, Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Fernandez-Garcia MD, Mazzon M, Jacobs M, Amara A. 2009. Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe 5:318–328. 10.1016/j.chom.2009.04.001 [DOI] [PubMed] [Google Scholar]
- 3.Gritsun TS, Lashkevich VA, Gould EA. 2003. Tick-borne encephalitis. Antiviral Res. 57:129–146. 10.1016/S0166-3542(02)00206-1 [DOI] [PubMed] [Google Scholar]
- 4.Lindquist L, Vapalahti O. 2008. Tick-borne encephalitis. Lancet 371:1861–1871. 10.1016/S0140-6736(08)60800-4 [DOI] [PubMed] [Google Scholar]
- 5.Mansfield KL, Johnson N, Phipps LP, Stephenson JR, Fooks AR, Solomon T. 2009. Tick-borne encephalitis virus; a review of an emerging zoonosis. J. Gen. Virol. 90:1781–1794. 10.1099/vir.0.011437-0 [DOI] [PubMed] [Google Scholar]
- 6.Miorin L, Romero-Brey I, Maiuri P, Hoppe S, Krijnse-Locker J, Bartenschlager R, Marcello A. 2013. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 87:6469–6481. 10.1128/JVI.03456-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawai T, Akira S. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34:637–650. 10.1016/j.immuni.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 8.Loo YM, Gale M., Jr 2011. Immune signaling by RIG-I-like receptors. Immunity 34:680–692. 10.1016/j.immuni.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miorin L, Albornoz A, Baba MM, D'Agaro P, Marcello A. 2012. Formation of membrane-defined compartments by tick-borne encephalitis virus contributes to the early delay in interferon signaling. Virus Res. 163:660–666. 10.1016/j.virusres.2011.11.020 [DOI] [PubMed] [Google Scholar]
- 10.Beckham CJ, Parker R. 2008. P bodies, stress granules, and viral life cycles. Cell Host Microbe 3:206–212. 10.1016/j.chom.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchan JR, Parker R. 2009. Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36:932–941. 10.1016/j.molcel.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P. 2000. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J. 19:4154–4163. 10.1093/emboj/19.15.4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez de Silanes I, Galban S, Martindale JL, Yang X, Mazan-Mamczarz K, Indig FE, Falco G, Zhan M, Gorospe M. 2005. Identification and functional outcome of mRNAs associated with RNA-binding protein TIA-1. Mol. Cell. Biol. 25:9520–9531. 10.1128/MCB.25.21.9520-9531.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, Takemura A, Sambhara S, Kawaguchi A, Osari S, Nagata K, Matsumiya T, Namiki H, Yoneyama M, Fujita T. 2012. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS One 7:e43031. 10.1371/journal.pone.0043031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langereis MA, Feng Q, van Kuppeveld FJ. 2013. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J. Virol. 87:6314–6325. 10.1128/JVI.03213-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng CS, Jogi M, Yoo JS, Onomoto K, Koike S, Iwasaki T, Yoneyama M, Kato H, Fujita T. 2013. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 87:9511–9522. 10.1128/JVI.03248-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li W, Li Y, Kedersha N, Anderson P, Emara M, Swiderek KM, Moreno GT, Brinton MA. 2002. Cell proteins TIA-1 and TIAR interact with the 3′ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 76:11989–12000. 10.1128/JVI.76.23.11989-12000.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emara MM, Brinton MA. 2007. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. U. S. A. 104:9041–9046. 10.1073/pnas.0703348104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Courtney SC, Scherbik SV, Stockman BM, Brinton MA. 2012. West Nile virus infections suppress early viral RNA synthesis and avoid inducing the cell stress granule response. J. Virol. 86:3647–3657. 10.1128/JVI.06549-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miorin L, Maiuri P, Hoenninger VM, Mandl CW, Marcello A. 2008. Spatial and temporal organization of tick-borne encephalitis flavivirus replicated RNA in living cells. Virology 379:64–77. 10.1016/j.virol.2008.06.025 [DOI] [PubMed] [Google Scholar]
- 21.Beck AR, Miller IJ, Anderson P, Streuli M. 1998. RNA-binding protein TIAR is essential for primordial germ cell development. Proc. Natl. Acad. Sci. U. S. A. 95:2331–2336. 10.1073/pnas.95.5.2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Izquierdo JM, Valcarcel J. 2007. Two isoforms of the T-cell intracellular antigen 1 (TIA-1) splicing factor display distinct splicing regulation activities. Control of TIA-1 isoform ratio by TIA-1-related protein. J. Biol. Chem. 282:19410–19417. 10.1074/jbc.M700688200 [DOI] [PubMed] [Google Scholar]
- 23.Hoenninger VM, Rouha H, Orlinger KK, Miorin L, Marcello A, Kofler RM, Mandl CW. 2008. Analysis of the effects of alterations in the tick-borne encephalitis virus 3′-noncoding region on translation and RNA replication using reporter replicons. Virology 377:419–430. 10.1016/j.virol.2008.04.035 [DOI] [PubMed] [Google Scholar]
- 24.Bartolomei G, Cevik RE, Marcello A. 2011. Modulation of hepatitis C virus replication by iron and hepcidin in Huh7 hepatocytes. J. Gen. Virol. 92:2072–2081. 10.1099/vir.0.032706-0 [DOI] [PubMed] [Google Scholar]
- 25.Iacono-Connors LC, Smith JF, Ksiazek TG, Kelley CL, Schmaljohn CS. 1996. Characterization of Langat virus antigenic determinants defined by monoclonal antibodies to E, NS1 and preM and identification of a protective, non-neutralizing preM-specific monoclonal antibody. Virus Res. 43:125–136. 10.1016/0168-1702(96)01325-1 [DOI] [PubMed] [Google Scholar]
- 26.Orlinger KK, Hoenninger VM, Kofler RM, Mandl CW. 2006. Construction and mutagenesis of an artificial bicistronic tick-borne encephalitis virus genome reveals an essential function of the second transmembrane region of protein E in flavivirus assembly. J. Virol. 80:12197–12208. 10.1128/JVI.01540-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maiuri P, Knezevich A, Bertrand E, Marcello A. 2011. Real-time imaging of the HIV-1 transcription cycle in single living cells. Methods 53:62–67. 10.1016/j.ymeth.2010.06.015 [DOI] [PubMed] [Google Scholar]
- 28.Jiang L, Yao H, Duan X, Lu X, Liu Y. 2009. Polypyrimidine tract-binding protein influences negative strand RNA synthesis of dengue virus. Biochem. Biophys. Res. Commun. 385:187–192. 10.1016/j.bbrc.2009.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anwar A, Leong KM, Ng ML, Chu JJ, Garcia-Blanco MA. 2009. The polypyrimidine tract-binding protein is required for efficient dengue virus propagation and associates with the viral replication machinery. J. Biol. Chem. 284:17021–17029. 10.1074/jbc.M109.006239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agis-Juarez RA, Galvan I, Medina F, Daikoku T, Padmanabhan R, Ludert JE, del Angel RM. 2009. Polypyrimidine tract-binding protein is relocated to the cytoplasm and is required during dengue virus infection in Vero cells. J. Gen. Virol. 90:2893–2901. 10.1099/vir.0.013433-0 [DOI] [PubMed] [Google Scholar]
- 31.Kafasla P, Mickleburgh I, Llorian M, Coelho M, Gooding C, Cherny D, Joshi A, Kotik-Kogan O, Curry S, Eperon IC, Jackson RJ, Smith CW. 2012. Defining the roles and interactions of PTB. Biochem. Soc. Trans. 40:815–820. 10.1042/BST20120044 [DOI] [PubMed] [Google Scholar]
- 32.Anderson P, Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33:141–150. 10.1016/j.tibs.2007.12.003 [DOI] [PubMed] [Google Scholar]
- 33.Anderson P, Kedersha N. 2002. Visibly stressed: the role of eIF2, TIA-1, and stress granules in protein translation. Cell Stress Chaperones 7:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emara MM, Liu H, Davis WG, Brinton MA. 2008. Mutation of mapped TIA-1/TIAR binding sites in the 3′ terminal stem-loop of West Nile virus minus-strand RNA in an infectious clone negatively affects genomic RNA amplification. J. Virol. 82:10657–10670. 10.1128/JVI.00991-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375. 10.1016/j.chom.2009.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gillespie LK, Hoenen A, Morgan G, Mackenzie JM. 2010. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 84:10438–10447. 10.1128/JVI.00986-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Overby AK, Popov VL, Niedrig M, Weber F. 2010. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J. Virol. 84:8470–8483. 10.1128/JVI.00176-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. 2000. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 151:1257–1268. 10.1083/jcb.151.6.1257 [DOI] [PMC free article] [PubMed] [Google Scholar]