

ABSTRACT
Most neutralizing antibodies elicited during influenza virus infection or vaccination target immunodominant, variable epitopes on the globular head region of hemagglutinin (HA), which leads to narrow strain protection. In this report, we describe the properties of a unique anti-HA monoclonal antibody (MAb), D1-8, that was derived from human B cells and exhibits potent, broad neutralizing activity across antigenically diverse influenza H3 subtype viruses. Based on selection of escape variants, we show that D1-8 targets a novel epitope on the globular head region of the influenza virus HA protein. The HA residues implicated in D1-8 binding are highly conserved among H3N2 viruses and are located proximal to antigenic site D. We demonstrate that the potent in vitro antiviral activity of D1-8 translates into protective activity in mouse models of influenza virus infection. Furthermore, D1-8 exhibits superior therapeutic survival benefit in influenza virus-infected mice compared to the neuraminidase inhibitor oseltamivir when treatment is started late in infection. The present study suggests the potential application of this monoclonal antibody for the therapeutic treatment of H3N2 influenza virus infection.
IMPORTANCE Recently, a few globular head-targeting MAbs have been discovered that exhibit activity against different subtypes of influenza subtypes, such as H1; however, none of the previously described MAbs showed broadly neutralizing activity against diverse H3 viruses. In this report, we describe a human MAb, D1-8, that exhibits potent, broadly neutralizing activity against antigenically diverse H3 subtype viruses. The genotypic analysis of escape mutants revealed a unique putative epitope region in the globular head of H3 HA that is comprised of highly conserved residues and is distinct from the receptor binding site. Furthermore, we demonstrate that D1-8 exhibits superior therapeutic efficacy in influenza virus-infected mice compared to the neuraminidase inhibitor oseltamivir when treatment is started late in infection. In addition to describing a novel anti-globular head of H3 HA MAb with potent broadly neutralizing activity, our report suggests the potential of D1-8 for therapeutic treatment of seasonal influenza virus H3 infection.
INTRODUCTION
Seasonal influenza virus infection results in ∼200,000 to 500,000 deaths each year, particularly in young children, immunocompromised patients, and the elderly (1, 2). During influenza pandemics, mortality rates can be even higher (3). Currently, vaccination remains the most effective means to prevent morbidity and mortality caused by influenza virus infection. However, vaccination is less effective in the elderly, which is the population at the highest risk for complications from influenza virus infection (4–6). In addition, antigenic drift can reduce the effectiveness of seasonal influenza vaccines, particularly if the vaccine strains are not well matched with circulating strains. Antiviral drugs (e.g., oseltamivir and zanamivir) are effective at reducing the duration of symptoms and complications due to influenza virus infections; however, they are most effective when administered early in infection (i.e., within 48 h after symptom onset) (7, 8). Although providing some benefit, such neuraminidase (NA) inhibitors are less effective at the time of hospitalization, which often occurs >48 h after symptom onset (8). In addition, the effectiveness of current NA inhibitors may be further limited by the emergence of drug-resistant variants of influenza virus (9). Therefore, there remains a significant unmet medical need for new therapies to treat influenza, particularly ones that are effective in high-risk populations (e.g., the elderly) at the time of hospitalization. Recently, a number of preclinical studies with animal models have demonstrated that anti-influenza virus hemagglutinin (HA) monoclonal antibodies (MAbs) have protective activity when administered late in infection, suggesting the potential utility of these agents for treating severe influenza virus infection (10–17).
There are three types of influenza viruses, A, B, and C; however, only influenza A and B viruses cause disease in humans. Influenza A viruses are further classified into subtypes based on the homology of the HA or NA proteins, which are the major glycoproteins expressed on the surface of influenza virus virions. Influenza virus type A contains 18 HA subtypes, which are further divided into two major phylogenetic groups: group 1 (H1, H2, H5, H6, H8, H9, H11, H12, H13, H16, H17, and H18 subtypes) and group 2 (H3, H4, H7, H10, H14, and H15 subtypes). Currently, only H1N1, H3N2, and influenza B viruses are circulating in humans, and the majority of influenza hospitalizations in the United States are typically associated with influenza A virus infections, more specifically with H3N2 (6).
Most neutralizing antibodies elicited by infection or vaccination recognize HA. The mature HA protein is present on virions as a trimer and is comprised of two disulfide-linked subunits, HA1 and HA2. The HA1 subunit forms the globular head region, which contains the sialic acid receptor binding site (RBS). The HA2 subunit and a portion of HA1 form a stalk structure, which drives membrane fusion (18–21). Neutralizing antibodies generated in response to infection or vaccination typically target the globular head. Five distinct antigenic sites have been characterized in the H1 and H3 globular head regions and are designated either as Sa, Sb, Ca1, Ca2, and Cb or as sites A through E (22, 23). Given that these antigenic sites exhibit considerable sequence variability, antibodies generated during vaccination are usually strain specific. In contrast, the RBS on the globular head is relatively well conserved and consists of a shallow pocket located at the apex of HA1 that is surrounded by HA sequences referred to as the 130-loop, the 150-loop, the 190-helix, and the 220-loop (24). Recently, a number of broad-spectrum MAbs targeting the RBS along with adjacent regions in the globular head of HA have been identified (25–31). These MAbs exhibit some cross-reactivity with one or more influenza virus H3 and/or H1 subtypes. However, few of these previously described MAbs targeting the globular head of HA have demonstrated neutralizing activity across large and diverse panels of either group 1 or group 2 influenza A viruses (29).
In this study, we demonstrated that a previously described anti-HA MAb, D1-8 (32), exhibits potent, broadly neutralizing activity across a diverse panel of H3 influenza virus strains. Based on the selection of resistant virus in vitro, we show that D1-8 recognizes a novel, conserved epitope proximal to antigenic site D on the globular head domain of H3 HA. D1-8 protects mice from lethal challenge with a representative H3 influenza virus strain when administered prophylactically or therapeutically. Furthermore, D1-8 exhibits superior protective activity in our therapeutic mouse model of H3 influenza virus infection compared to that of oseltamivir, particularly when administered late in infection.
MATERIALS AND METHODS
Cells, viruses, and antigens.
Madin-Darby canine kidney (MDCK) cells were obtained from the European Collection of Cell Cultures (ECACC) and were used for all cell-based assays. Monolayer cultures of MDCK cells were maintained in minimal essential medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (Invitrogen) plus 2 mM l-glutamine with 1× penicillin-streptomycin. Wild-type influenza virus strains were obtained from the Centers for Disease Control and Prevention (CDC) or purchased from the American Tissue Culture Collection (ATCC) as indicated below. Cold-adapted (ca) live attenuated influenza vaccine viruses were generated either by classical reassortment or by reverse genetics (33). All viruses were propagated in embryonated chicken eggs, and virus titers were determined by a standard tissue culture infectious dose (TCID50) assay on MDCK cells. The H3N2 viruses included A/Perth/16/09, A/Wisconsin/67/05, A/California/7/04, A/Wyoming/03/03, A/Panama/2007/99, A/Sydney/5/97, A/Argentina/3779/94, A/Shangdong/9/93, A/Los Angeles/2/87, A/Victoria/3/75, A/Hong Kong/8/68 (ATCC VR-544), swine origin A/Minnesota/11/2010, and swine origin A/Indiana/10/2011. The live attenuated H7 strains were generated by reverse genetics using the HA and NA genes of A/Netherlands/219/03 (H7N7) and the low-pathogenicity (LP) strain A/chicken/British Columbia/CN-6/2004 (H7N3), respectively, with the six internal protein genes of ca A/Ann Arbor/6/60 (H2N2) virus (34). A reassortant H3 virus (rHK68) containing the H3 HA from A/HK/8/68 (H3N2) with an N165S mutation (glycosylation site) and the remaining 7 gene segments from A/PR8/34 (H1N1) was produced by reverse genetics (33). The reassortant virus containing an antibody resistance mutation in HA was generated by reverse genetics as described previously (33, 35).
Cloning, expression, and purification of recombinant antibody D1-8.
To generate a plasmid that yields a high level of expression of the target gene, VH and Vκ genes were amplified from the original expression plasmid (32) and sequentially cloned into in-house vector pOE. The sequence was confirmed and the plasmid was then transfected into 293F cells using 293fectin (Invitrogen). The antibody was purified from supernatant using a MabSelect protein column (GE Healthcare).
Cloning, expression, and purification of recombinant HA proteins.
The HA expression vector was constructed as described previously (36). The HA cDNAs corresponding to amino acid residues 11 to 329 (HA1) and 1 to 176 (HA2) of the ectodomain of HA0 (A/Perth/16/09 [H3N2], A/California/7/09 [H1N1], or A/Netherlands/219/03 [H7N7]) were cloned into a plasmid vector under the control of the cytomegalovirus (CMV) promoter. The trimerization domain, thrombin cleavage site, and His tag were added to the C-terminal end of the HA protein. The HA protein was purified from transiently transfected 293F cell culture supernatant using a 5-ml nickel-nitrilotriacetic acid (Ni-NTA) Superflow column (Qiagen) equilibrated with phosphate-buffered saline (PBS).
Binding affinity determination.
Affinity measurements were performed using a ForteBio Octet QK 384 kinetic analyzer (Menlo Park, CA) using plates with 384 slanted wells. All reagents were diluted in Octet kinetics buffer. His-tagged HA was immobilized onto anti-His sensors at 10 μg/ml. Anti-HA MAb association and disassociation were then monitored in 2-fold dilutions from 100 nM, and a well without MAb was used as a control. Association and dissociation raw data were corrected for any drift in the zero-MAb controls and then exported to GraphPad Prism (San Diego, CA) for affinity curve fitting. Data were fitted using global association/dissociation fitting with an imposed dissociation limit of >5 × 10−6 s−1.
Western blot analysis of MAb D1-8 under reducing and nonreducing conditions.
Purified trimeric HA protein of A/Perth/09 H3 was evaluated for its reactivity with D1-8 by Western blotting under reducing and nonreducing conditions. The purified trimeric HA protein at 0.5 μg/well was heated at 70°C for 10 min in NuPAGE lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) in the presence or absence of 50 mM dithiothreitol (DTT). Reduced and nonreduced preparations were subjected to electrophoresis in a 4 to 12% bis-tris acrylamide gel in 2-(N-morpholino)ethanesulfonic acid (MES) SDS electrophoresis buffer (Invitrogen). The separated proteins were transferred to nitrocellulose and blocked for 1 h in 5% (vol/vol) powdered milk in PBS containing 0.1% Triton X-100 at room temperature. The blot was probed with D1-8 MAb at 3.0 μg/ml, followed by addition of horseradish peroxidase (HRP)-conjugated anti-human secondary antibody. After incubation followed by several washings, the detection reagent (LumiGLO; KPL) was added to the blot and the blot was exposed to X-ray film.
Microneutralization assay.
The microneutralization assay was modified from a previously described accelerated viral inhibition assay using neuraminidase (NA) activity as a readout (37). Briefly, 100 TCID50s of virus was added to 2-fold dilutions of antibody in a 96-well plate; after 30 min of incubation at room temperature, 4 × 104 cells/well were added to the plate. After incubation in a 33°C, 5% CO2 incubator for approximately 40 h, the NA activity was measured by adding a fluorescently labeled substrate, methylumbelliferyl-N-acetylneuraminic acid (MU-NANA) (Sigma), to each well and incubating the cells at 37°C for 1 h. Virus replication represented by NA activity was quantified by reading fluorescence in an Envision fluorometer (PerkinElmer). The neutralization titer (50% inhibitory concentration [IC50]) is expressed as the final antibody concentration that reduced the fluorescence signal by 50% compared to that of cell control wells.
HAI assay.
The hemagglutination inhibition (HAI) assay was performed as described previously (38). In brief, the antibodies were serially diluted with PBS in 96-well V-bottom plates, and 4 HA units of virus (as determined by incubation with 0.5% chicken red blood cells [RBCs] in the absence of antibody) was added to the well. After 30 to 60 min of incubation at room temperature, 0.5% chicken RBCs (Lampire Biological Laboratories) suspended in PBS was added to each well. After an additional 30 min of incubation at room temperature, the HAI titer was defined as the minimum concentration of antibody that completely inhibited hemagglutination of 0.5% chicken RBCs.
Selection of MARMs.
All of the monoclonal antibody-resistant mutants (MARMs) were selected by incubating the H3N2 influenza viruses A/Wyoming/3/2003 (WY03), A/HongKong/1/1968 (HK68), and A/Perth/16/2009 (Perth09) under increasing concentrations of D1-8 MAb. One hundred PFU of each virus was mixed with 1× IC50 of MAb D1-8 and incubated for 1 h at room temperature. The virus and antibody mixture was added onto confluent MDCK cell monolayers in 12-well tissue culture plates. After 1 h of adsorption, the cell monolayers were washed with PBS and culture medium containing 1× IC50 of MAb D1-8 was added to the wells. The plates were incubated at 33°C for 3 to 7 days and observed daily for signs of cytopathic effects (CPE). Once the infected cells exhibited more than 50% CPE, the culture supernatants were harvested and used to infect fresh MDCK cells as described above. The selection process was repeated four more times with increasing MAb D1-8 concentrations (5×, 10×, 20×, and 100× IC50) in the medium. The viruses from 5 rounds of passage were biologically cloned by plaque assay, and plaques were individually propagated in MDCK cells. The HA gene was amplified by reverse transcription-PCR (RT-PCR) and sequenced to determine the amino acid changes in the HA.
Evaluation of MAb for its prophylactic and therapeutic protective activities in mice.
All animal study protocols were approved and conducted in accordance with MedImmune's Institutional Animal Care and Use Committee and subsequently performed in an Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC)-certified facility. Six- to eight-week-old BALB/c mice (Harlan Laboratories) were used in the studies. The experiments were conducted in accordance with the protocol approved by MedImmune's Institutional Animal Care and Use Committee. Mice were weighed on the day of or 1 day before virus challenge and monitored daily for 14 days for weight loss and survival (mice with body weight loss of ≥25% were euthanized). To determine the prophylactic efficacy of the MAb, mice in groups of 10 were administered either D1-8 intraperitoneally at doses of 0.1, 0.25, 0.5, 1, 5, and 15 mg/kg or a human irrelevant isotype control IgG at 15 mg/kg in 100-μl volumes. Four hours after dosing, mice were inoculated intranasally with 10× the 50% mouse lethal dose (MLD50) of 7:1 A/PR/8-A/HK/8/68 HA reassortant virus (rHK68) in a 0.05-ml volume. All mice were weighed and monitored daily for any signs of morbidity and mortality for 2 weeks. For the study of therapeutic efficacy, mice were infected intranasally with 3× MLD50 of rHK68. At 4, 24, 48, 72, or 96 h postinfection, groups of mice were given intraperitoneal injections of 30 mg/kg of D1-8 or isotype control MAb. To assess virus load in the lungs, five mice from each group were euthanized at 96 h postinfection. Whole lungs were homogenized in 10% (wt/vol) sterile L15 medium (Invitrogen). Virus titers in lung homogenates were determined by TCID50 assay as described above and previously (34). To compare the therapeutic efficacy of D1-8 with that of oseltamivir, antibody-treated groups received a single intraperitoneal injection of 2.5 mg/kg of D1-8 or 30 mg/kg of isotype control MAb on day 4 after infection. Oseltamivir-treated groups of mice were treated with saline as a vehicle control or oseltamivir dosed at 25 mg/kg orally (p.o.) twice daily (BID) starting 96 h after virus inoculation for 5 days. Mice were observed and weighed daily through day 14.
RESULTS
D1-8 exhibits broadly neutralizing activity against H3 viruses.
In this study, we characterized a human MAb, D1-8, which was isolated by molecular cloning of IgG-expressing plasmablasts from a healthy flu vaccine recipient (32). Previously, D1-8 was demonstrated to exhibit high-affinity binding to the H3N2 vaccine strain A/Wisconsin/67/2005, with a Kd value of 286 nM. To determine if D1-8 recognizes other genetically diverse influenza A virus strains, the binding of full-length D1-8 (D1-8 IgG1; bivalent) or the D1-8 antigen binding domain (D1-8 Fab; monovalent) to recombinant trimeric HA proteins (rHA) derived from A/Perth/16/09 (H3N2), A/California/7/09 (H1N1), or A/Netherlands/219/03 (H7N7) was interrogated and Kd values were determined as described in Materials and Methods. D1-8 IgG bound to H3N2 rHA protein with a measured Kd value of <100 pM. In contrast, the monovalent D1-8 Fab exhibited a Kd value that was 5 orders of magnitude greater than that of the bivalent D1-8 IgG (10.9 μM). Binding to H7 and H1 rHA proteins was not detected with either the D1-8 IgG or Fab. These data suggest that D1-8 binds specifically to H3N2 HA and demonstrate that the strong binding exhibited by this antibody is dependent on avidity.
To further characterize the functional activity of D1-8, hemagglutination inhibition (HAI) assays and virus neutralization assays were performed using a diverse group of H3N2 influenza virus strains originally isolated over a span of 40 years. D1-8 demonstrated HAI activity against 11/13 antigenically diverse H3N2 strains tested, with IC50s ranging from 0.2 to 12.5 μg/ml (Table 1). Alternatively, D1-8 exhibited potent neutralizing activity against all 13 H3N2 strains, with a median IC50 of 0.053 μg/ml (mean, 0.18 ± 0.06 μg/ml, and range, 0.01 to 0.79 μg/ml) (Table 1). Thus, D1-8 was found to exhibit 7- to 150-fold-greater potency in neutralization assays than in HAI assays when the same virus strains were compared. Consistent with the binding data, D1-8 did not exhibit antiviral activity against H1 and H7 influenza virus strains (data not shown). Antibodies that exhibit HAI activity typically target the globular head region of the HA. Therefore, these data suggest that D1-8 may recognize a conserved epitope on the globular head of influenza virus H3 HA.
TABLE 1.
D1-8 HAI and neutralization activities against representative influenza H3N2 virus strains
| Strain | HAI activitya (μg/ml) | Neutralization IC50 (μg/ml), mean ± SD |
|---|---|---|
| A/Hong Kong/8/1968 | 1.56 | 0.03 ± 0.02 |
| A/Victoria/3/1975 | 6.25 | 0.04 ± 0.003 |
| A/Los Angeles/2/1987 | 6.25 | 0.10 ± 0.05 |
| A/Shangdong/9/1993 | 12.5 | 0.66 ± 0.15 |
| A/Argentina/3779/1994 | 12.5 | 0.27 ± 0.12 |
| cab A/Sydney/5/1997 | 1.56 | 0.05 ± 0.03 |
| ca A/Panama/2007/1999 | 1.56 | 0.08 ± 0.03 |
| A/Wyoming/03/2003 | 0.4 | 0.03 ± 0.01 |
| ca A/California/7/2004 | 0.78 | 0.04 ± 0.01 |
| ca A/Wisconsin/67/2005 | 0.2 | 0.03 ± 0.003 |
| ca A/Perth/16/2009 | 0.2 | 0.01 ± 0.007 |
| A/Minnesota/11/2010 | >50 | 0.33 ± 0.14 |
| A/Indiana/10/2011 | >50 | 0.63 ± 0.13 |
HAI titers are expressed as the lowest concentrations of purified D1-8 that completely inhibited hemagglutination.
ca, cold adapted.
Epitope identification by selection of monoclonal antibody-resistant mutants (MARMs).
Influenza H3N2 viruses A/Hong Kong/1/68 (HK68), A/Wyoming/03/2003 (WY03), and A/Perth/16/2009 (Perth09) were propagated in the presence of increasing concentrations of D1-8 by serial passage. Potential escape mutants were plaque purified, and their cognate HA genes were subjected to sequence analysis to identify putative amino acid substitutions that conferred resistance to D1-8. When WY03 was propagated in the presence of D1-8 at concentrations up to 20× IC50, single amino acid substitutions at HA residues 240 (G240E) and 207 (K207N) were identified. Similarly, when HK68 was propagated in the presence of D1-8 concentrations up to 20× IC50, a single amino acid substitution was selected at HA residue 207 (R207N) (Table 2). Serial passage of these WY03 or HK68 variants at higher D1-8 concentrations (up to 100× IC50) resulted in the selection of additional amino acid substitutions at HA residue 171 or 173 in combination with the G240E substitution in the case of WY03 (G240E and N171D/E or G240E and K173T) or at HA position 172 in combination with the R207N substitution in the case of HK68 (R207N and D172G). Interestingly, no amino acid substitutions were detected in the HA of Perth09 propagated in the presence of D1-8 at concentrations up to 20× IC50; however, when this virus was subsequently passaged at higher concentrations of D1-8 (100× IC50), the N171K single amino substitution was identified in the HA protein (Table 2).
TABLE 2.
Amino acid changes in the H3 HA of D1-8 MAb-resistant mutants
| H3N2 virus | HA amino acid substitution(s) observed at indicated D1-8 concna |
|
|---|---|---|
| 20× IC50 | 100× IC50 | |
| A/WY/3/2003 | K207N | K207N |
| G240E | G240E | |
| G240E, N171E | ||
| G240E, N171D | ||
| G240E, K173T | ||
| A/HK/1-5/1968 | R207N | R207N, D172G |
| A/Perth/16/2009 | None | N171K |
The concentration of D1-8 MAb corresponding to 20× IC50 was achieved after 3 passages, while 100× IC50 was reached after 2 more passages.
To evaluate the effects of these amino acid substitutions on D1-8 susceptibility, recombinant A/Wyoming/3/2003 (rWY03) viruses encoding either individual mutations or combinations of mutations were generated and evaluated in HAI and neutralization assays. As shown in Table 3, the N171E/K/D and K173T substitutions conferred >2,000-fold and >70,000-fold reductions in susceptibility to D1-8 in HAI and neutralization assays, respectively. Similar changes in susceptibility were observed for the N171K substitution when introduced into either the rHK68 or rPerth09 virus (data not shown). In contrast, the single amino acid changes E172G, K207N, and G240E resulted in more modest (3- to 80-fold) reductions in susceptibility to D1-8 in HAI assays or 59- to 854-fold reductions in susceptibility to D1-8 neutralization. However, when two of these mutations, K207N and E172G, were introduced in combination, the resulting virus was highly resistant to D1-8, with calculated IC50s for HAI and neutralization that were >2,000-fold and >70,000-fold higher, respectively, than those measured with wild-type virus (Table 3). The amino acid changes K207N and K173T resulted in the introduction of a potential N-linked glycosylation site at Asn207 and Asn171 (NXS/T), respectively. This observation suggests that the HA variants may be glycosylated at Asn207 or Asn171 and that this glycosylation may interfere with D1-8 binding. Additional amino acid substitutions (T48K, N165D/S, I406L, and A476T) were identified in the HA proteins of selected MARMs; however, such substitutions did not alter the D1-8 susceptibility of recombinant viruses encoding such substitutions (data not shown).
TABLE 3.
Susceptibilities of rWY03 variants to D1-8 neutralization or HAI
| H3 variant | IC50 (μg/ml) |
Fold change relative to rWY03 |
||
|---|---|---|---|---|
| HAI | Neutralization | HAI | Neutralization | |
| rWY03 (wild type) | 0.55 | 0.017 | 1 | 1 |
| rWY03_N171K | >1,200 | >1,200 | >2,181 | >70,000 |
| rWY03_N171E | >1,200 | >1,200 | >2,181 | >70,000 |
| rWY03_N171D | >1,200 | >1,200 | >2,181 | >70,000 |
| rWY03_E172G | 29 | 4.49 | 53 | 264 |
| rWY03_K173T | >1,200 | >1,200 | >2,181 | >70,000 |
| rWY03_K207N | 1.65 | 1.54 | 3 | 59 |
| rWY03_G240E | 44 | 14.51 | 80 | 854 |
| rWY03_E172G/K207N | >1,200 | >1,200 | >2,181 | >70,000 |
Although not continuous in linear sequence, all resistance-associated substitutions cluster together and are located proximal to antigenic site D on the X-ray crystal structure of H3 HA (18, 21, 22) (Fig. 1A). These structural observations suggest that D1-8 recognizes a conformational epitope on the globular head of H3 HA. Consistent with D1-8 recognizing a structural epitope, the antibody binds HA only under nonreducing, nondenaturing conditions in Western blot analysis (Fig. 1B).
FIG 1.
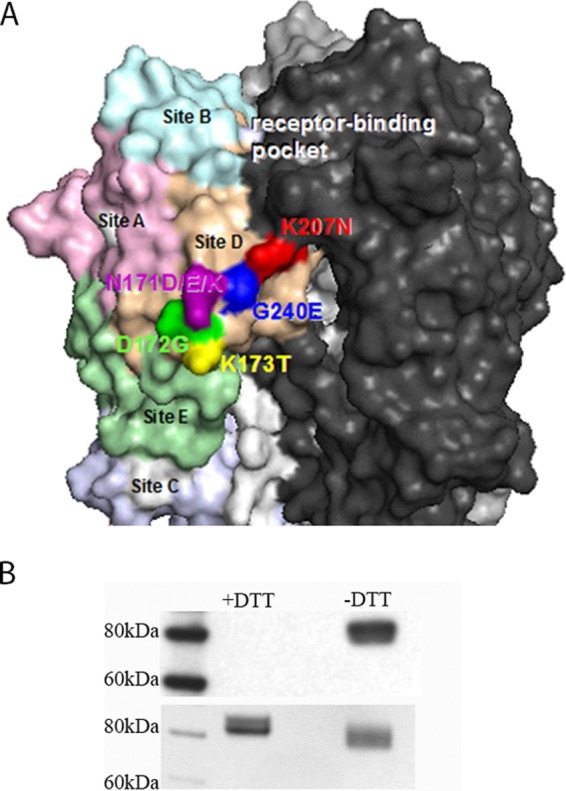
Characteristics of the putative D1-8 antigenic site. (A) D1-8 epitope illustrated on a space-filling model of the trimeric membrane-distal globular head of H3 HA. The H3 HA antigenic sites are colored pink (site A), light blue (site B), light purple (site C), tan (site D), and light green (site E). The D1-8 MAb escape mutations are labeled by amino acid letters and residue numbers. Residues 171 (dark purple), 172 (dark green), 173 (yellow), 207 (red) and 240 (dark blue) are proximal to antigenic site D, which is adjacent to the receptor binding pocket. (B) Western blot analysis of the D1-8/H3 HA interaction. (Top blot) The recombinant trimeric HA of A/Perth/09 proteins were disrupted in 2% SDS without (−) DTT or with (+) 100 mM DTT and then analyzed by Western blotting with MAb D1-8. (Bottom blot) A Coomassie blue-stained duplicate gel to show equal loading of HA proteins.
To investigate the conservation of the proposed D1-8 epitope among recently circulating as well as historical H3N2 isolates, we analyzed 7,432 unique HA amino acid sequences derived from H3N2 strains isolated over the past 44 years (from the Influenza Virus Resource at the National Center for Biotechnology Information [NCBI] database). The amino acid residues Asn at HA position 171, Lys or Arg at position 207, and Gly at position 240 are conserved in >99% of H3N2 HA sequences analyzed (Table 4). Ninety percent of the H3N2 sequences analyzed contained either an Asp or Glu at position 172. The amino acid changes N171E, K207N, and G240E did not affect viral replication kinetics when evaluated in vitro compared to the wild-type recombinant parental virus, rWY03. Alternatively, the amino acid substitution N171K resulted in a >1-log10 reduction in peak viral titer in vitro compared to the parental virus (data not shown). In addition, HK68 recombinant variants encoding either the K207N or G240E substitution were evaluated in vivo. Consistent with in vitro results, the in vivo 50% lethal doses of the K207N and G240E variants were comparable to that of the parental rHK68 virus (data not shown).
TABLE 4.
Conservation of H3N2 HA residues substituted in D1-8 MARMsa
| Residue(s) in D1-8 MARMs | Residue(s) (%) observed in circulating influenza virus H3N2 strains |
|---|---|
| N171K/E/D | N (99.64), T (0.24), S (0.12) |
| E172G | E (77.9), D (12.1), G (9.5), K (0.5) |
| K173T | K (67.7), Q (18.4), N (9.3), E (4.4), R (0.2) |
| K207N | K (96.2), R (3.6), Q (0.2) |
| G240E | G (100.0) |
A total of 7,432 human H3N2 HA protein sequences from the Influenza Research Database from 1968 to 2011 were analyzed.
D1-8 protected mice from lethal challenge with H3 influenza virus when administered prophylactically or therapeutically.
To determine whether potent in vitro neutralizing activity would translate into efficacy in vivo, D1-8 was evaluated in a mouse model of H3N2 influenza virus infection. BALB/c mice were treated with D1-8 either before (prophylactically) or after (therapeutically) challenge with a lethal dose of mouse-adapted reassortant virus (rHK68) as described in Materials and Methods. In the prophylaxis study, D1-8 conferred protection in a dose-dependent manner, providing 100% protection in animals that received D1-8 at a dose of 0.5 mg/kg or greater and 50% or 10% protection in animals that received 0.25 mg/kg or 0.1 mg/kg, respectively (Fig. 2A). As expected, none of the mice that received the isotype control MAb at 15 mg/kg survived the challenge infection. When rHK68-infected mice were treated therapeutically with a single dose of 30 mg/kg, D1-8 provided 90 to 100% protection in all treated animals even when administered as late as 96 h after infection (Fig. 2B). This was remarkable given that mice treated 96 h after infection had already lost >15% of their body weight (data not shown). In addition to surviving the infection, all mice in the D1-8 treatment groups regained body weight and showed a reversal of clinical signs of infection by the end of the observation period (data not shown). In contrast, the mice in the control group succumbed to infection by 9 days postinfection (Fig. 2B).
FIG 2.
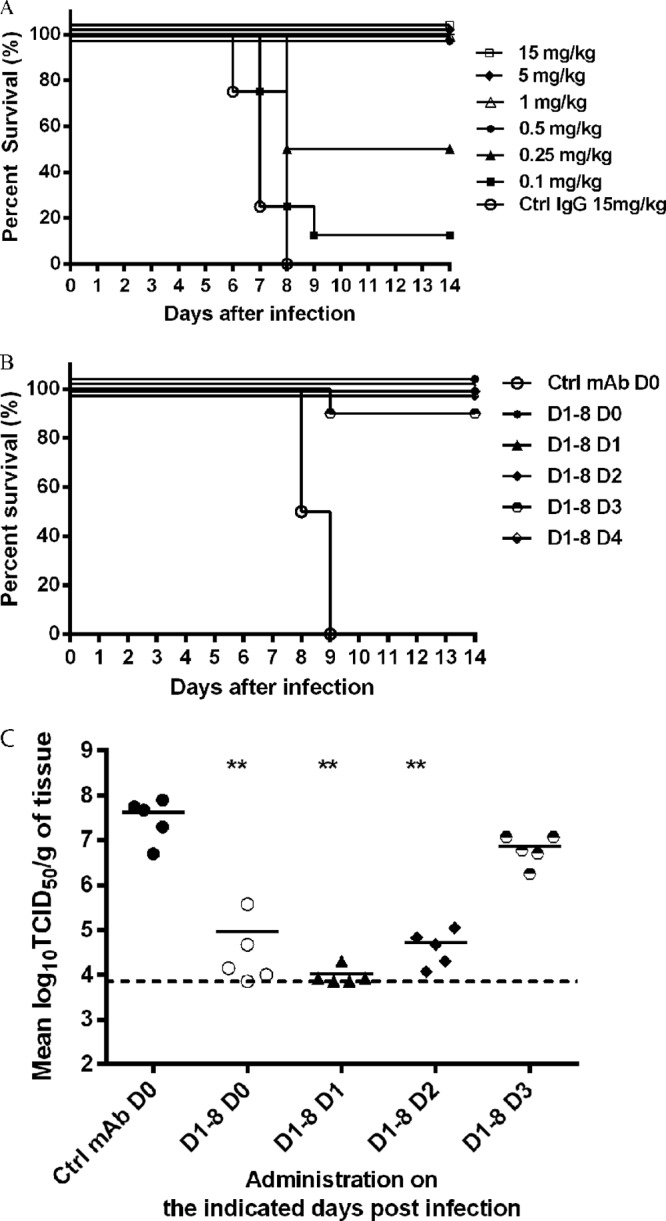
Percent survival of mice in a model of influenza virus infection following prophylactic or therapeutic administration of D1-8. (A) BALB/c mice were treated with D1-8 at the indicated concentrations or with a control MAb and then infected with rHK68 4 h later. Percent survival versus time is presented. (B) BALB/c mice were infected with rHK68 and treated with D1-8 (30 mg/kg) 4 h (D0) or 1 (D1), 2 (D2), 3(D3), or 4 (D4) days after infection. Control mice were treated with an irrelevant control (Ctrl) MAb 4 h prior to infection. Percent survival versus time is presented. (C) Mice were infected as described for panel B and sacrificed 4 days after infection, and virus titers in lungs were measured using the TCID50 method. **, P < 0.01 versus value for control mice (Mann-Whitney test).
To determine whether D1-8 reduced viral load in the lower respiratory tract, mice were infected with rHK68, treated with D1-8 MAb, and sacrificed 4 days after infection. Viral load in the lungs was determined by measuring TCID50. As shown in Fig. 2C, treatment of mice either on the day of infection (day 0) or 1 or 2 days after infection resulted in an ∼100-fold reduction in viral titer in the lungs compared to that in the control group of mice. Mice treated with D1-8 3 days after infection (i.e., treated with MAb for <24 h) exhibited an ∼5-fold reduction in viral titer in the lungs compared to that in the control. Taken together, these results indicate that D1-8 significantly reduces viral replication in the lungs of infected mice, and this correlated with animal survival.
D1-8 is superior to oseltamivir in protecting mice from lethal challenge with H3 influenza virus.
Oseltamivir administered at a dosage of 75 mg twice daily (BID) for 5 days is currently the standard of care for the treatment of influenza virus infections. This clinical dosage is approximately equivalent to a dosage of 12.5 mg/kg BID in mice, based on the body surface area normalization method (39). Our preliminary study showed that therapeutic treatment of influenza virus-infected mice with D1-8 administered 4 days postinfection at concentrations as low as 2.5 mg/kg provided ≥80% protection from lethality (data not shown). To demonstrate the potential utility of antibody therapy late in infection, we compared the therapeutic efficacy of D1-8 when administered as a single dose (2.5 mg/kg) 4 days after infection with oseltamivir dosed at 25 mg/kg BID (total dosage of 50 mg/kg/day) for 5 days, initiated on the 4th day after infection. The oseltamivir dose is equivalent to twice the dose typically administered in the clinic and represents a typical efficacious dose used in mouse models of influenza virus infection (40). As shown in Fig. 3, a single dose of D1-8 resulted in body weight recovery and survival for >80% of treated animals, while limited body weight recovery was observed in the oseltamivir-treated animals and only 20% survived the rHK68 infection. These data show that D1-8 MAb treatment was more effective than oseltamivir in protecting mice late in infection.
FIG 3.

Therapeutic efficacy of D1-8 compared to oseltamivir in an influenza virus mouse model late in infection. Mice received a single intraperitoneal injection of D1-8 (2.5 mg/kg) 4 days after infection or 25 mg/kg of oseltamivir, given orally twice a day (50 mg/kg/day) for 5 days, with dosing initiated 4 days after infection. Mice were monitored for 14 days for body weight loss (A) and survival (B). Mean change in body weight is expressed as a percentage of the baseline body weight. Error bars represent 95% confidence intervals.
DISCUSSION
Several MAbs have been described that neutralize H3 viruses, and some also exhibit cross-reactivity with other subtypes of influenza virus (e.g., H1 strains) (13, 15, 17, 26, 27, 30, 31). Some of these previously described MAbs target the globular head region of HA (26, 27, 30, 31), while others target the more conserved stalk structure (13, 15, 17). However, only a few have demonstrated activity against diverse H3 viruses (>5 viruses), and all such MAbs target the HA stalk structure (13, 15, 17). We show that D1-8 (32) binds to H3N2 HA proteins and potently neutralizes all viruses tested from a diverse panel of H3N2 strains originally isolated over a 40-year period. However, D1-8 did not exhibit binding activity against H1or H7 HA proteins, nor did D1-8 show activity against H1 or H7 influenza virus strains, indicating specificity for H3 influenza virus. In addition, D1-8 exhibited HAI activity, a characteristic typically associated with binding to the globular head region of HA. The antigenic site of D1-8 was further confirmed by the genotypic analysis of escape mutants, which revealed a putative epitope region in the globular head of H3 HA that is unique and comprised of highly conserved residues.
To our knowledge, D1-8 is the first broadly neutralizing MAb described that binds to a conserved epitope on the globular head of HA that appears to be distinct from the RBS. Crystallographic and/or resistant virus studies have shown that previously described broadly neutralizing MAbs that target the HA globular head typically make key interactions with conserved residues overlapping the RBS (29–31). For example, the H1-specific MAb CH65 (29) interacts with the RBS as well as proximal residues in antigenic sites Sa, Sb, and CA2. Alternatively, the mouse MAb S139/1, which neutralizes multiple influenza virus A subtypes (27, 31), binds to highly conserved residues in the RBS and antigenic sites A, B, and D (H3 designation) (31). Another MAb, C05 (30), contains an unusual HCDR3 (24 residues in length) that mediates the majority of its interactions with HA by insertion into the RBS. In contrast, D1-8 appears to interact differently with the globular head of HA than do these other well-characterized MAbs. D1-8 neutralization escape mutations localize proximal to HA antigenic site D, which overlaps with the region of interface between monomers (22), rather than sequences directly surrounding the RBS. The HA residues implicated in D1-8 binding are well conserved across a large number of H3N2 strains, which is consistent with the broad-spectrum H3N2 antiviral activity observed for D1-8. Given that D1-8 was derived directly from the B cells of a vaccinated individual (32), these observations provide proof of concept that antibody responses to conserved epitopes on the influenza H3 HA globular head outside the RBS can be generated in humans.
Consistent with its potent antiviral activity in vitro, D1-8 exhibits significant prophylactic and therapeutic activities in mouse models of influenza virus infection. D1-8 protected mice from lethal H3 influenza virus infection when administered as late as 4 days after infection. More impressively, D1-8 exhibited superior efficacy compared to clinically relevant doses of oseltamivir when treatment was initiated late in infection (4 days) in mice. Such therapeutic efficacy in preclinical models has been demonstrated previously for other MAbs directed against influenza virus HA (10, 12, 13, 17), suggesting that anti-HA MAbs may be effective therapeutics for treating influenza virus infection. These preclinical data also raise the possibility that the anti-HA MAbs such as D1-8 may be more effective than oseltamivir either alone or in combination with oseltamivir, particularly late in infection (17, 41). Anti-HA MAbs have been shown to mediate antiviral activity via multiple mechanisms in preclinical models (in vitro and in vivo), including direct virus neutralization as well as mechanisms dependent on MAb Fc effector function (e.g., antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxicity) (13). It is possible that the additional antiviral mechanisms mediated by anti-HA MAbs could extend the utility of influenza therapies into patient populations with a high unmet medical need, such as hospitalized patients with more advanced infection.
ACKNOWLEDGMENTS
We thank Rafi Ahmed at Emory University and Patrick Wilson at the University of Chicago for providing D1-8 MAb, Sandrina Phipps and Arnita Barnes for generating HA and Fab reagents, Jia Lin for production and purification of D1-8 MAb, and Xing Cheng and James Zengel for providing PR8 reassortant virus.
Footnotes
Published ahead of print 2 April 2014
REFERENCES
- 1.Beigel JH. 2008. Influenza. Crit. Care Med. 36:2660–2666. 10.1097/CCM.0b013e318180b039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen WH, Kozlovsky BF, Effros RB, Grubeck-Loebenstein B, Edelman R, Sztein MB. 2009. Vaccination in the elderly: an immunological perspective. Trends Immunol. 30:351–359. 10.1016/j.it.2009.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, Bandaranayake D, Breiman RF, Brooks WA, Buchy P, Feikin DR, Fowler KB, Gordon A, Hien NT, Horby P, Huang QS, Katz MA, Krishnan A, Lal R, Montgomery JM, Mølbak K, Pebody R, Presanis AM, Razuri H, Steens A, Tinoco YO, Wallinga J, Yu H, Vong S, Bresee J, Widdowson MA. 2012. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modeling study. Lancet Infect. Dis. 12:687–695. 10.1016/S1473-3099(12)70121-4 [DOI] [PubMed] [Google Scholar]
- 4.Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186. 10.1001/jama.289.2.179 [DOI] [PubMed] [Google Scholar]
- 5.Thompson WW, Shay DK, Weintraub E, Brammer L, Bridges CB, Cox NJ, Fukuda K. 2004. Influenza-associated hospitalizations in the United States. JAMA 292:1333–1340. 10.1001/jama.292.11.1333 [DOI] [PubMed] [Google Scholar]
- 6.Zhou H, Thompson WW, Viboud CG, Ringholz CM, Cheng PY, Steiner C, Abedi GR, Anderson LJ, Brammer L, Shay DK. 2012. Hospitalizations associated with influenza and respiratory syncytial virus in the United States, 1993–2008. Clin. Infect. Dis. 54:1427–1436. 10.1093/cid/cis211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aoki FY, Macleod MD, Paggiaro P, Carewicz O, El Sawy A, Wat C, Griffiths M, Waalberg E, Ward P, Study Group IMPACT 2003. Early administration of oral oseltamivir increases the benefits of influenza treatment. J. Antimicrob. Chemother. 51:123–129. 10.1093/jac/dkg007 [DOI] [PubMed] [Google Scholar]
- 8.Louie JK, Yang S, Acosta M, Yen C, Samuel MC, Schechter R, Guevara H, Uyeki TM. 2012. Treatment with neuraminidase inhibitors for critically ill patients with influenza A (H1N1) pdm09. Clin. Infect. Dis. 55:1198–1204. 10.1093/cid/cis636 [DOI] [PubMed] [Google Scholar]
- 9.Poland GA, Jacobson RM, Ovsyannikova IG. 2009. Influenza virus resistance to antiviral agents: a plea for rational use. Clin. Infect. Dis. 48:1254–1256. 10.1086/598989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Throsby M, van den Brink E, Jongeneelen M, Poon LL, Alard P, Cornelissen L, Bakker A, Cox F, van Deventer E, Guan Y, Cinatl J, ter Meulen J, Lasters I, Carsetti R, Peiris M, de Kruif J, Goudsmit J. 2008. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One 3:e3942. 10.1371/journal.pone.0003942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. 2009. Antibody recognition of a highly conserved influenza virus epitope. Science 324:246–251. 10.1126/science.1171491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, Santelli E, Stec B, Cadwell G, Ali M, Wan H, Murakami A, Yammanuru A, Han T, Cox NJ, Bankston LA, Donis RO, Liddington RC, Marasco WA. 2009. Structure and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16:265–273. 10.1038/nsmb.1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, Silacci C, Fernandez-Rodriguez BM, Agatic G, Bianchi S, Giacchetto-Sasselli I, Calder L, Sallusto F, Collins P, Haire LF, Temperton N, Langedijk JP, Skehel JJ, Lanzavecchia A. 2011. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333:850–856. 10.1126/science.1205669 [DOI] [PubMed] [Google Scholar]
- 14.Wrammert J, Koutsonanos D, Li GM, Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig M, Lipkin WI, Mehta A, Razavi B, Del Rio C, Zheng NY, Lee JH, Huang M, Ali Z, Kaur K, Andrews S, Amara RR, Wang Y, Das SR, O'Donnell CD, Yewdell JW, Subbarao K, Marasco WA, Mulligan MJ, Compans R, Ahmed R, Wilson PC. 2011. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 208:181–193. 10.1084/jem.20101352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJ, Brandenburg B, Vogels R, Brakenhoff JP, Kompier R, Koldijk MH, Cornelissen LA, Poon LL, Peiris M, Koudstaal W, Wilson IA, Goudsmit J. 2011. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333:843–850. 10.1126/science.1204839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, van der Vlugt R, Lamrani M, Korse HJ, Geelen E, Sahin Ö, Sieuwerts M, Brakenhoff JP, Vogels R, Li OT, Poon LL, Peiris M, Koudstaal W, Ward AB, Wilson IA, Goudsmit J, Friesen RH. 2012. Highly conserved protective epitopes on influenza B viruses. Science 337:1343–1348. 10.1126/science.1222908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura G, Chai N, Park S, Chiang N, Lin Z, Chiu H, Fong R, Yan D, Kim J, Zhang J, Lee WP, Estevez A, Coons M, Xu M, Lupardus P, Balazs M, Swem LR. 2013. An in vivo human-plasmablast enrichment technique allows rapid identification of therapeutic influenza A antibodies. Cell Host Microbe 14:93–103. 10.1016/j.chom.2013.06.004 [DOI] [PubMed] [Google Scholar]
- 18.Copeland CS, Zimmer KP, Wagner KR, Healey GA, Mellman I, Helenius A. 1988. Folding, trimerization, and transport are sequential events in the biogenesis of influenza virus hemagglutinin. Cell 53:197–209. 10.1016/0092-8674(88)90381-9 [DOI] [PubMed] [Google Scholar]
- 19.Wilson IA, Skehel JJ, Wiley DC. 1981. Structure of the hemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289:366–373 [DOI] [PubMed] [Google Scholar]
- 20.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43. 10.1038/371037a0 [DOI] [PubMed] [Google Scholar]
- 21.Skehel JJ, Bayley PM, Brown EB, Martin SR, Waterfield MD, White JM, Wilson IA, Wiley DC. 1982. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 79:968–972. 10.1073/pnas.79.4.968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiley DC, Wilson IA, Skehel JJ. 1981. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289:373–378. 10.1038/289373a0 [DOI] [PubMed] [Google Scholar]
- 23.Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427. 10.1016/0092-8674(82)90135-0 [DOI] [PubMed] [Google Scholar]
- 24.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569. 10.1146/annurev.biochem.69.1.531 [DOI] [PubMed] [Google Scholar]
- 25.Tsibane T, Ekiert DC, Krause JC, Martinez O, Crowe JE, Jr, Wilson IA, Basler CF. 2012. Influenza human monoclonal antibody 1F1 interacts with three major antigenic sites and residues mediating human receptor specificity in H1N1 viruses. PLoS Pathog. 12:e1003067. 10.1371/journal.ppat.1003067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohshima N, Iba Y, Kubota-Koketsu R, Asano Y, Okuno Y, Kurosawa Y. 2011. Naturally occurring antibodies in humans can neutralize a variety of influenza virus strains, including H3, H1, H2, and H5. J. Virol. 85:11048–11057. 10.1128/JVI.05397-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida R, Igarashi M, Ozaki H, Kishida N, Tomabechi D, Kida H, Ito K, Takada A. 2009. Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog. 5:e1000350. 10.1371/journal.ppat.1000350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krause JC, Tsibane T, Tumpey TM, Huffman CJ, Basler CF, Crowe JE., Jr 2011. A broadly neutralizing human monoclonal antibody that recognizes a conserved, novel epitope on the globular head of the influenza H1N1 virus hemagglutinin. J. Virol. 85:10905–10908. 10.1128/JVI.00700-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whittle JR, Zhang R, Khurana S, King LR, Manischewitz J, Golding H, Dormitzer PR, Haynes BF, Walter EB, Moody MA, Kepler TB, Liao HX, Harrison SC. 2011. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. U. S. A. 108:14216–14221. 10.1073/pnas.1111497108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ekiert DC, Kashyap AK, Steel J, Rubrum A, Bhabha G, Khayat R, Lee JH, Dillon MA, O'Neil RE, Faynboym AM, Horowitz M, Horowitz L, Ward AB, Palese P, Webby R, Lerner RA, Bhatt RR, Wilson IA. 2012. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature 489:526–532. 10.1038/nature11414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee PS, Yoshida R, Ekiert DC, Sakai N, Suzuki Y, Takada A, Wilson IA. 2012. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc. Natl. Acad. Sci. U. S. A. 109:17040–17045. 10.1073/pnas.1212371109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C, James J, Air GM, Capra JD, Ahmed R, Wilson PC. 2008. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453:667–671. 10.1038/nature06890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin H, Lu B, Zhou H, Ma C, Zhao J, Yang CF, Kemble G, Greenberg H. 2003. Multiple amino acid residues confer temperature sensitivity to human influenza virus vaccine strains (FluMist) derived from cold-adapted A/Ann Arbor/6/60. Virology 306:18–24. 10.1016/S0042-6822(02)00035-1 [DOI] [PubMed] [Google Scholar]
- 34.Joseph T, McAuliffe J, Lu B, Vogel L, Swayne D, Jin H, Kemble G, Subbarao K. 2008. A live attenuated cold-adapted influenza A H7N3 virus vaccine provides protection against homologous and heterologous H7 viruses in mice and ferrets. Virology 378:123–132. 10.1016/j.virol.2008.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. U. S. A. 97:6108–6113. 10.1073/pnas.100133697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevens J, Corper AL, Basler CF, Taubenberger JK, Palese P, Wilson IA. 2004. Structure of the uncleaved human H1 hemagglutinin from the extinct 1918 influenza virus. Science 303:1866–1870. 10.1126/science.1093373 [DOI] [PubMed] [Google Scholar]
- 37.Hassantoufighi A, Zhang H, Sandbulte M, Gao J, Manischewitz J, King L, Golding H, Straight TM, Eichelberger MC. 2010. A practical influenza neutralization assay to simultaneously quantify hemagglutinin and neuraminidase-inhibiting antibody responses. Vaccine 28:790–797. 10.1016/j.vaccine.2009.10.066 [DOI] [PubMed] [Google Scholar]
- 38.Chen Z, Wang W, Zhou H, Suguitan AL, Jr, Shambaugh C, Kim L, Zhao J, Kemble G, Jin H. 2010. Generation of live attenuated novel influenza virus A/California/7/09 (H1N1) vaccines with high yield in embryonated chicken eggs. J. Virol. 84:44–51. 10.1128/JVI.02106-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.FDA, Center For Drug Evaluation And Research. 6 July 2005. Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. FDA, Rockville, MD: http://www.fda.gov/downloads/Drugs/Guidance/UCM078932.pdf [Google Scholar]
- 40.Mendel DB, Tai CY, Escarpe PA, Li W, Sidwell RW, Huffman JH, Sweet C, Jakeman KJ, Merson J, Lacy SA, Lew W, Williams MA, Zhang L, Chen MS, Bischofberger N, Kim CU. 1998. Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrob. Agents Chemother. 42:640–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koudstaal W, Koldijk MH, Brakenhoff JP, Cornelissen LA, Weverling GJ, Friesen RH, Goudsmit J. 2009. Pre- and postexposure use of human monoclonal antibody against H5N1 and H1N1 influenza virus in mice: viable alternative to oseltamivir. J. Infect. Dis. 200:1870–1873. 10.1086/648378 [DOI] [PMC free article] [PubMed] [Google Scholar]