

ABSTRACT
Human enterovirus 68 (EV68) is a member of the EV-D species, which belongs to the EV genus of the Picornaviridae family. Over the past several years, there have been increasingly documented outbreaks of respiratory disease associated with EV68. As a globally emerging pathogen, EV68 infects both adults and children. However, the molecular basis of EV68 pathogenesis is unknown. Here we report that EV68 inhibits Toll-like receptor 3 (TLR3)-mediated innate immune responses by targeting the TIR domain-containing adaptor inducing beta interferon (TRIF). In infected HeLa cells, EV68 inhibits poly(I·C)-induced interferon regulatory factor 3 (IRF3) activation and beta interferon (IFN-β) expression. Further investigations revealed that TRIF, a critical adaptor downstream of TLR3, is targeted by EV68. When expressed alone, 3Cpro, an EV68-encoded protease, cleaves TRIF. 3Cpro mediates TRIF cleavage at Q312 and Q653, which are sites in the amino- and carboxyl-terminal domains, respectively. This cleavage relies on 3Cpro's cysteine protease activity. Cleavage of TRIF abolishes the capacity of TRIF to activate NF-κB and IFN-β signaling. These results suggest that control of TRIF by 3Cpro may be a mechanism by which EV68 subverts host innate immune responses.
IMPORTANCE EV68 is a globally emerging pathogen, but the molecular basis of EV68 pathogenesis is unclear. Here we report that EV68 inhibits TLR3-mediated innate immune responses by targeting TRIF. Further investigations revealed that TRIF is cleaved by 3Cpro. These results suggest that control of TRIF by 3Cpro may be a mechanism by which EV68 impairs type I IFN production in response to TLR3 activation.
INTRODUCTION
Human enteroviruses (EVs) are small, nonenveloped, positive-sense, single-stranded RNA viruses belonging to the family Picornaviridae. Based on molecular and antigenic properties, EVs can be classified into four species (EV-A to -D) (1). EVs are associated with diverse clinical syndromes, ranging from mild upper respiratory illnesses to severe and potentially fatal pathologies, and are among the most common human pathogens (1). Human enterovirus 68 (EV68), a serotype of EV-D, is a rarely reported virus historically linked to respiratory disease. However, over the past 3 years, outbreaks have occurred in France, Netherlands, the United States, Philippines, Japan, Thailand, the United Kingdom, South Africa, and the Gambia (2–15). Notably, children represent the majority of cases of symptomatic infections.
Although evidence implicates EV68 as an emerging respiratory pathogen, its pathogenic mechanism is largely unknown. With respect to clinical presentation, EV68 is more similar to human rhinoviruses (16), which trigger Toll-like receptor 3 (TLR3)-mediated cytokine expression (17). It is well established that upon recognition of double-stranded RNA (dsRNA), TLR3 recruits the TIR domain-containing adaptor protein inducing beta interferon (TRIF), which then associates with tumor necrosis factor receptor-associated factor 3 (TRAF3) to activate two Iκ-B kinase (IKK)-related kinases, TANK-binding kinase 1 (TBK1) and IKKi. These two kinases phosphorylate interferon regulatory factor 3/7 (IRF3/7), resulting in the induction of type I interferons (IFNs) and expression of IFN-inducible genes. In addition, TRIF stimulates NF-κB activation through receptor-interacting protein 1 (RIP1) and TRAF6, leading to the induction of proinflammatory cytokine genes (18, 19). Therefore, the interplay of EV68 and the TLR3-mediated pathway may represent an interface which determines the outcome of EV68 infection.
This study was undertaken to investigate the mechanism by which EV68 interacts with the TLR3 signaling pathway. We report that EV68 suppresses TLR3-mediated expression of type I IFNs and inflammatory cytokines through cleavage of TRIF in infected cells. We provide evidence that 3Cpro of EV68 mediates TRIF cleavage, resulting in inactive TRIF fragments. This activity requires a functional viral protease. Furthermore, we show that TRIF cleavage occurs at two sites, located in the amino- and carboxyl-terminal domains, thereby inactivating TRIF. Together, these results suggest that modulation of the TLR3 pathway may be a viral mechanism that contributes to EV68 infection.
MATERIALS AND METHODS
Cell lines and viruses.
293T (CRL-11268; ATCC) cells and HeLa (CCL-2; ATCC) cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (HyClone, Logan, UT), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a 5% CO2 humidified atmosphere. The enterovirus 68 strain used in this study is a Beijing strain (GenBank accession number KF726085). Virus infection was carried out as follows. Briefly, cells were infected with EV68. Unbound virus was washed away 2 h after infection, and cells were then cultured in fresh medium supplemented with 10% FBS.
Plasmids and reagents.
The plasmids pEGFPC1, pCDNA3.1-Flag-TRIF, pGL3-IFN-β-Luc, pNF-κB-Luc, pISRE-Luc, and pRL-SV40 have been described elsewhere (20, 21). 3Cpro was amplified from nucleic acids of EV68 and was cloned into the XhoI and BamHI sites of the pEGFPC1 vector, resulting in green fluorescent protein (GFP) fusion proteins. The GFP-3C variants and TRIF mutants were constructed by using Pfu DNA polymerase (Stratagene, La Jolla, CA). All variants were confirmed by subsequent DNA sequencing. Antibodies against Flag, GFP, and β-actin were purchased from Sigma (St. Louis, MO). Rabbit antibody against TBK1 was purchased from Cell Signaling Technology (Danvers, MA). Goat anti-TRIF and anti-MyD88 antibodies were purchased from R&D Systems (Minneapolis, MN). Rabbit antibodies against IRF3 and phospho-IRF3 (pS386) were purchased from Epitomics (Burlingame, CA). IRDye 800-labeled IgG or IRDye 680-labeled IgG secondary antibodies were purchased from Li-Cor Biosciences (Lincoln, NE). Rupintrivir was purchased from Santa Cruz (Santa Cruz, CA). Poly(I·C) was purchased from Sigma (St. Louis, MO).
Preparation of 3Cpro and VP1 antibodies.
3Cpro and VP1 were amplified from nucleic acids of EV68 and cloned into the BamHI and XhoI sites of pET-30a, resulting in His fusion constructs. BALB/c mice were injected subcutaneously with the purified proteins, obtained from Escherichia coli, as described elsewhere (22). Animal experiments were performed in accordance with the animal experiment regulations of the Chinese government in the Institute of Laboratory Animal Sciences (ILAS), Chinese Academy of Medical Sciences (CAMS). All experimental procedures were approved (license number SCXKJ2009-0017) and supervised by the Animal Protection and Usage Committee of ILAS, CAMS. Sera collected from mice were purified using protein G Sepharose columns (GE Healthcare, Waukesha, WI), and purified IgG antibody was quantified using a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific).
Reporter assays.
Luciferase reporter assays were performed as described previously (23). 293T cells were seeded in 24-well plates at a cell density of 3 × 105 cells per well. Sixteen hours after plating, cells were transfected with a control plasmid or plasmids expressing TRIF, TRIF mutants, and 3Cpro or its variants, along with pGL3-IFN-β-luc, NF-κB-luc, ISRE-luc, and pRL-SV40, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Twenty-four hours after transfection, cells were harvested and cell lysates were used to determine luciferase activities, using a dual-luciferase reporter system (Promega, Madison, WI) according to the manufacturer's instructions. The firefly luciferase activities were normalized to the renilla luciferase activities.
Quantitative real-time reverse transcription-PCR (RT-PCR).
Total RNA was extracted using TRIzol reagent (Invitrogen) and treated with DNase I (Pierce, Rockford, IL). Aliquots of RNA were reverse transcribed to cDNA by using a Superscript cDNA synthesis kit (Invitrogen) according to the manufacturer's instructions. Samples were then subjected to quantitative real-time PCR analysis using primers specific for detection of IFN-β, interferon-stimulated gene 56 (ISG56), ISG15, interleukin-6 (IL-6), IL-8, RANTES (regulated on activation, normal T cell expressed and secreted), ISG54, and IFN-γ-inducible protein 10 (IP10), using SYBR green kits (TaKaRa Bio, Otsu, Japan) according to the manufacturer's instructions. Expression of IFN-β, ISG56, ISG15, IL-6, IL-8, RANTES, ISG54, and IP10 mRNAs was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression. The sequences of primers have been described elsewhere (20, 24).
Western blot analysis.
Cells were pelleted by centrifugation and lysed in buffer containing 150 mM NaCl, 25 mM Tris (pH 7.4), 1% NP-40, 0.25% sodium deoxycholate, and 1 mM EDTA with a protease inhibitor cocktail (Roche, Indianapolis, IN). Aliquots of cell lysates were electrophoresed in 12% SDS-PAGE gels and transferred to a nitrocellulose membrane (Pall, Port Washington, NY). The membranes were blocked with 5% nonfat dry milk, and then proteins on the membrane were incubated with the indicated primary antibodies at 4°C overnight. This was followed by incubation with the corresponding IRD Fluor 800-labeled IgG or IRD Fluor 680-labeled IgG secondary antibody (Li-Cor Inc., Lincoln, NE). After washing, the membranes were scanned with an Odyssey infrared imaging system (Li-Cor, Lincoln, NE) at a wavelength of 700 or 800 nm, and the molecular sizes of the developed proteins were determined by comparison with prestained protein markers (Fermentas, CA).
RNA interference.
To generate control or TRIF-knockdown cell lines, HeLa cells were seeded onto 24-well plates. The next day, cells were infected with lentiviruses expressing scrambled or TRIF-specific short hairpin RNA (shRNA) (GenePharma) at a multiplicity of infection (MOI) of 100. After 72 h, cells were selected by use of 1 μg/ml puromycin for 3 weeks. The sequence of the shRNA targeting TRIF was 5′-AAGACCAGACGCCACTCCAAC-3′.
RESULTS
EV68 inhibits the induction of innate antiviral immunity by poly(I·C) in infected cells.
To study the effect of EV68 infection on the TLR3 pathway, we evaluated the induction of antiviral immune responses by poly(I·C), a prototype TLR3 agonist. HeLa cells, with a functional TLR3 pathway, were initially mock infected or infected with EV68 for 4 h. At various time points after incubation with poly(I·C), the cells were examined for the expression of IFN-β, ISG56, ISG15, ISG54, IL-6, IL-8, RANTES (regulated on activation, normal T cell expressed and secreted), IP10 (IFN-γ-inducible protein 10), and GAPDH by quantitative real-time RT-PCR analysis. Poly(I·C) induced IFN-β (Fig. 1A, control), ISG56 (Fig. 1B, control), ISG15 (Fig. 1C, control), ISG54 (Fig. 1D, control), IP10 (Fig. 1G, control), and RANTES (Fig. 1H) expression in mock-infected cells, but the kinetics of expression were different among different genes. The expression of IFN-β peaked around 4 h after poly(I·C) treatment and then decreased rapidly, while the expression of ISG56, ISG15, ISG54, RANTES, and IP10 had apparent elevations 8 h after poly(I·C) treatment and remained at high levels thereafter. EV68 infection reduced the expression of IFN-β, ISG56, ISG15, ISG54, RANTES, and IP10. In parallel, EV68 inhibited IRF3 phosphorylation (a hallmark of IRF3 activation) induced by poly(I·C) compared to that in mock-infected cells (Fig. 1I). Interestingly, like EV71 infection (20), EV68 infection also enhanced IL-6 and IL-8 expression after poly(I·C) stimulation (Fig. 1E and F). These results show that EV68 inhibits the induction of innate antiviral immune responses by poly(I·C) in infected cells.
FIG 1.

EV68 reduces the transcription of IFN-β (A), ISG56 (B), ISG15 (C), ISG54 (D), IP10 (G), and RANTES (H) induced by poly(I·C), and it stimulates the transcription of IL-6 (E) and IL-8 (F). HeLa cells were mock infected or infected with EV68 at an MOI of 4. Four hours after infection, cells were incubated with or without poly(I·C). At the indicated time points, total RNAs extracted from cells and the expression of the IFN-β, ISG56, ISG15, ISG54, IL-6, IL-8, IP10, RANTES, and GAPDH genes were evaluated by quantitative real-time PCR using SYBR green. Results are expressed as increases in mRNA levels relative to those in cells in the absence of poly(I·C) and were normalized by using the GAPDH housekeeping gene. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (I) EV68 inhibits poly(I·C)-induced phosphorylation of IRF3. HeLa cells were mock infected or infected with EV68 (MOI of 4) for 4 h. Cells were then incubated with or without poly(I·C) for an additional 20 h. Cell lysates were then subjected to Western blot analysis with antibodies against IRF3 and phosphorylated IRF3.
EV68 downregulates the expression of TRIF in infected cells.
TLR3 mediates TRIF-dependent signaling that activates NF-κB and IRF3, leading to type I IFN production (19). To determine where EV68 exerts its effect, we analyzed protein levels in EV68-infected cells. HeLa cells were mock infected or infected with increasing doses of EV68. Twenty-four hours after infection, cell lysates were subjected to Western blot analysis. As illustrated in Fig. 2A, expression of MyD88, IRF3, TBK1, and β-actin exhibited no or little reduction in EV68-infected cells. Under these experimental conditions, the level of TRIF expression decreased as the infection dose of EV68 increased (Fig. 2B). This coincided with the appearance of VP1 and 3C expression. In contrast, UV-treated EV68 barely reduced the TRIF protein level (Fig. 2A, lane 2). Therefore, replication of EV68 specifically downregulated TRIF in infected mammalian cells. Notably, small interfering RNA (siRNA) knockdown of TRIF increased viral replication (Fig. 2C). Moreover, ectopic expression of TRIF decreased viral replication (Fig. 2D). These results suggest that interaction between TRIF and EV68 determines the outcome of viral infection.
FIG 2.

Effects of EV68 infection on protein levels. (A) HeLa cells were mock infected or infected with UV-EV68 or EV68. Twenty-four hours after infection, cell lysates were subjected to Western blot analysis with antibodies against TRIF, MyD88, TBK1, IRF3, VP1, 3C, and β-actin. (B) Densitometry analysis. TRIF protein bands from three independent experiments as shown in panel A were quantified and normalized to β-actin by using Odyssey image software. (C) Control or TRIF-knockdown HeLa cells were infected with EV68 (MOI = 3) for 24 h. Cell lysates were subjected to Western blot analysis with antibodies against TRIF, VP1, 3C, and β-actin. (D) HeLa cells were transfected with a control plasmid or with increasing amounts of plasmids expressing TRIF (5 ng, 20 ng, or 50 ng). Twenty-four hours after transfection, cells were infected with EV68 (MOI = 1) for 24 h. Total RNA was extracted, and the viral RNA levels of EV68 were evaluated by quantitative real-time PCR using SYBR green. Results are expressed as viral RNA levels relative to the GAPDH RNA level. ***, P < 0.001.
3Cpro of EV68 suppresses the expression of antiviral or proinflammatory genes induced by poly(I·C).
Previous studies suggest that the 3Cpro proteins of EV71, coxsackievirus B3 (CVB3), and poliovirus (PV) inhibit type I IFN responses (20, 25, 26). Since 3Cpro is a conserved protein (Fig. 3), we assessed whether 3Cpro of EV68 contributed to the inhibition of innate antiviral immune responses. Specifically, HeLa cells were transfected with a GFP or GFP-3C plasmid. Twenty-four hours after transfection, cells were incubated with poly(I·C), and the expression of IFN-β, ISG54, ISG56, IL-6, RANTES, IP10, and GAPDH was determined by quantitative real-time RT-PCR analysis. Overexpression of GFP-3C reduced the expression of IFN-β (Fig. 4A), ISG54 (Fig. 4B), IL-6 (Fig. 4C), ISG56 (Fig. 4D), IP10 (Fig. 4E), and RANTES (Fig. 4F) induced by poly(I·C). Consistently, overexpression of GFP-3C inhibited poly(I·C)-induced IRF3 phosphorylation (Fig. 4G and H). These results suggest that 3Cpro of EV68 impairs the dsRNA-induced innate antiviral immune response.
FIG 3.
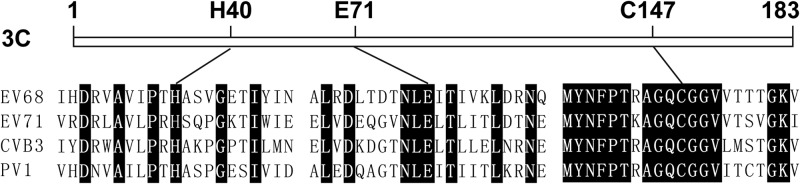
Schematic diagrams of 3Cpro. 3Cpro of EV68 has the same catalytic triad as other members of the genus Enterovirus, such as EV71, CVB3, and PV1. Amino acid residues adjacent to the catalytic site are shown. Conserved residues are shown in black.
FIG 4.

3Cpro of EV68 reduces the transcription of IFN-β (A), ISG54 (B), IL-6 (C), ISG56 (D), IP10 (E), and RANTES (F) by poly(I·C). HeLa cells were transfected with GFP or GFP-3C. Twenty-four hours after transfection, cells were treated with or without poly(I·C). Two, 4, 8, and 12 h after poly(I·C) treatment, total RNAs extracted from cells and the expression of the IFN-β, ISG54, ISG56, IL-6, RANTES, IP10, and GAPDH genes were evaluated by quantitative real-time PCR using SYBR green. Results are expressed as increases in mRNA levels relative to those in cells in the absence of poly(I·C) and were normalized by using the GAPDH housekeeping gene. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (G) 3Cpro of EV68 inhibits poly(I·C)-induced phosphorylation of IRF3. HeLa cells were transfected with GFP or GFP-3C. Twenty-four hours after transfection, cells were treated with or without poly(I·C) for 4 h. Cell lysates were subjected to Western blot analysis with antibodies against IRF3 or phosphorylated IRF3. (H) Densitometry analysis. Phosphorylated IRF3 protein bands from three independent experiments as shown in panel G were quantified and normalized to IRF3 by using Odyssey image software. *, P < 0.05.
3Cpro of EV68 inhibits IFN-β and NF-κB activation by downregulation of TRIF.
Based on the above results, we assessed whether 3Cpro inhibited innate antiviral immune responses by downregulating TRIF. As indicated in Fig. 5A to C, in 293T cells, 3Cpro of EV68 inhibited TRIF-stimulated IFN-β, NF-κB, and interferon-stimulated response element (ISRE) promoter activation in a dose-dependent manner. Western blot analysis showed that 3Cpro of EV68 cleaved TRIF (Fig. 5D), producing a 100-kDa fragment and a 45-kDa fragment. Note that the extent of TRIF cleavage was correlated with the expression level of 3Cpro (Fig. 5D and E).
FIG 5.

(A) Effects of 3Cpro of EV68 on IFN-β promoter activation. 293T cells were transfected with TRIF and GFP-3C along with pGL3-IFN-β-Luc; pRL-SV40 was used as an internal control. Twenty-four hours after transfection, cell lysates were assayed for luciferase activities. The fold activation was calculated by dividing the luciferase activity in cells transfected with pCDNA3.1-TRIF by that in cells transfected with the pCDNA3.1 vector. ***, P < 0.001. (B) Effects of 3Cpro of EV68 on NF-κB promoter activation. 293T cells were transfected with TRIF and GFP-3C along with pNF-κB-Luc. Assays were carried out as described for panel A. **, P < 0.01; ***, P < 0.001. (C) Effects of 3Cpro of EV68 on ISRE promoter activation. 293T cells were transfected with TRIF and GFP-3C along with pISRE-Luc. Assays were carried out as described for panel A. ***, P < 0.001. (D) 3Cpro of EV68 induces TRIF cleavage in a dose-dependent manner. 293T cells were transfected with Flag-TRIF and a control plasmid or increasing amounts of plasmids expressing GFP-3C (5 ng, 50 ng, or 200 ng; indicated with a wedge). Twenty-four hours after transfection, cells were analyzed by Western blotting. (E) Densitometry analysis. TRIF or cleaved TRIF protein bands from three independent experiments as shown in panel D were quantified and normalized to β-actin by using Odyssey image software.
3C protease activity is crucial for suppressing IFN-β and NF-κB activation mediated by TRIF.
The 3Cpro protein of EV68 bears a catalytic triad consisting of Cys147, His40, and Glu71. To investigate its role, we carried out mutational analysis. The H40D variant of 3Cpro has a histidine-to-aspartic acid substitution at amino acid 40, and the C147A variant possesses a mutation of cysteine to alanine at amino acid 147 (Fig. 6A). As illustrated in Fig. 6B, wild-type 3Cpro inhibited IFN-β promoter activation mediated by TRIF. However, the H40D and C147A variants were unable to do so. A similar result was seen with NF-κB promoter activation (Fig. 6C).
FIG 6.

(A) Schematic diagrams of 3Cpro variants. (B and C) Effects of 3Cpro variants of EV68 on TRIF-mediated IFN-β and NF-κB promoter activation. 293T cells were transfected with plasmids encoding TRIF and IFN-β–Luc (B) or NF-κB–Luc (C), along with GFP or GFP-3C variants. pRL-SV40 was included as an internal control. Twenty-four hours after transfection, cells were harvested to determine luciferase activities. The fold activation was calculated by dividing the luciferase activity in cells transfected with pCDNA3.1-TRIF by that in cells transfected with the pCDNA3.1 vector. ***, P < 0.001. (D) Interaction of 3Cpro variants of EV68 with TRIF. 293T cells were transfected with Flag-TRIF and GFP or GFP-3C variants as indicated. Cell lysates were immunoprecipitated (IP) with anti-Flag antibody. Immunoprecipitates and aliquots of cell lysates were subjected to Western blot analysis (WB) with antibodies against TRIF, GFP, and β-actin. (E) Effect of the protease inhibitor rupintrivir on TRIF cleavage. 293T cells were transfected with Flag-TRIF along with GFP or GFP-3C. Four hours after transfection, cells were incubated with the protease inhibitor rupintrivir (1 μM) for 24 h. Cell lysates were then processed for Western blot analysis. In panels D and E, arrows denote EV68-induced cleavage fragments.
Next, we determined TRIF cleavage in mammalian cells. As shown in Fig. 6D, unlike wild-type 3C of EV68, neither the H40D nor C147A variant cleaved TRIF as measured by Western blotting (top panel). Immunoprecipitation assays revealed that both the H40D and C147A variants associated with TRIF (lower panel). These data indicate that 3Cpro forms a complex with TRIF. Notably, wild-type 3Cpro and TRIF were not coimmunoprecipitated, presumably due to cleavage of full-length TRIF (Fig. 6D). Thus, 3Cpro-mediated cleavage is essential for inhibiting promoter activation. To corroborate this, we assessed rupintrivir, which is an inhibitor of 3Cpro with a broad spectrum of activity against picornaviruses (27). Indeed, when cells were treated with rupintrivir, 3Cpro of EV68 failed to mediate cleavage of TRIF (Fig. 6E). Hence, the protease activity of 3Cpro is essential for its function.
3Cpro cleaves TRIF at Gln-312 and Gln-653.
Since TRIF cleavage produced a 45-kDa product and a fragment of about 100 kDa, we inferred that the TRIF cleavage sites might fall in the central and C-terminal regions. Because 3Cpro of EV68 has a preference for glutamine in the P1 position and alanine in the P4 position (28), we focused on the sites with the signature sequence AXXQ ↓ G/S and constructed a series of mutants in which Gln was replaced with Ala. As illustrated in TRIF cleavage assays, the Q312A mutation blocked appearance of the 45-kDa fragment (Fig. 7B, lane 4), and the Q312A/Q653A combined mutation blocked the appearance of both the 45-kDa and 100-kDa fragments (Fig. 7B, lane 6). The Q312A/Q575A/Q653A or Q312A/Q575/Q653A/Q672A combination yielded an effect similar to that seen with the Q312A/Q653A combination. In reporter assays, the Q312A/Q653A mutant was able to activate the IFN-β and NF-κB promoters in the presence or absence of GFP-3C (Fig. 7C and D). Collectively, these data suggest Q312 and Q653 as 3Cpro cleavage sites within TRIF.
FIG 7.

3Cpro of EV68 cleaves the N- and C-terminal domains of TRIF at specific sites. (A) Schematic diagrams of TRIF showing the locations of possible cleavage sites for 3Cpro of EV68. (B) Western blotting for wild-type TRIF or 3Cpro-resistant mutants of TRIF. 293T cells were transfected with wild-type TRIF or TRIF mutants with alanine substitutions for glutamine, along with GFP (lanes 1, 3, 5, 7, and 9) or GFP-3C (lanes 2, 4, 6, 8, and 10), as indicated. Twenty-four hours after transfection, cell lysates were subjected to Western blot analysis with antibodies against TRIF, GFP, and β-actin. (C and D) Effects of 3Cpro of EV68 on IFN-β and NF-κB promoter activation mediated by wild-type TRIF or 3Cpro-resistant mutants of TRIF. 293T cells were transfected with wild-type TRIF or the Q312A/Q653A mutant of TRIF along with IFN-β–Luc (C) or NF-κB–Luc (D). pRL-SV40 was used as a control. Twenty-four hours after transfection, cell lysates were assayed for luciferase activities. The fold activation was calculated by dividing the luciferase activity in cells transfected with pCDNA3.1-TRIF by that in cells transfected with pCDNA3.1 vector. *, P < 0.05. (E and F) Effects of possible 3Cpro-cleaved fragments of TRIF on IFN-β and NF-κB promoter activation. 293T cells were transfected with the full, 1–312, 313–712, 1–653, and 313–653 fragments of TRIF along with IFN-β–Luc (E) or NF-κB–Luc (F). pRL-SV40 was used as a control. Twenty-four hours after transfection, cell lysates were assayed for luciferase activities. (G) TRIF-knockdown HeLa cells were transfected with pCDNA3.1 vector (control) and the full, 1–312, and 313–653 fragments of TRIF (10 ng). Twenty-four hours after transfection, cells were infected with EV68 (MOI = 1) for 24 h. Total RNA was extracted, and the viral RNA levels of EV68 were evaluated by quantitative real-time PCR using SYBR green. Results are expressed as viral RNA levels relative to the GAPDH RNA level. ***, P < 0.001.
3Cpro-mediated cleavage inactivates TRIF.
To test whether TRIF cleavage mediated by 3Cpro has a functional consequence, we generated TRIF variants carrying residues 1 to 653, 1 to 312, 313 to 712, and 313 to 653 (Fig. 7A). In reporter assays, full-length TRIF activated the IFN-β (Fig. 7E) and NF-κB (Fig. 7F) promoters. The 1–653 fragment retained the capacity to activate IFN-β and NF-κB signaling to various degrees. In contrast, the 1–312 and 313–653 fragments lost the ability to activate the IFN-β and NF-κB promoters. Intriguingly, the 313–712 fragment was able to activate the NF-κB promoter. However, it failed to activate the IFN-β promoter. These data suggest that TRIF functions through different domains. Consistently, in TRIF-knockdown cells, ectopic expression of full-length TRIF significantly inhibit EV68 replication, whereas the 1–312 and 313–653 TRIF fragments, which were unable to stimulate the IFN-β and NF-κB promoters, failed to inhibit viral replication (Fig. 7G). Thus, multiple cleavages by 3Cpro are necessary to disrupt TRIF functions.
DISCUSSION
Human enterovirus 68, first isolated in the United States in 1962 (29), is an emerging respiratory pathogen. Over the past 3 years, the occurrence of respiratory diseases due to EV68 infection has increased steadily. Outbreaks occurred in different countries or regions, such as France (14), Netherlands (2, 11, 13), the United States (2), Philippines (5), Japan (3, 7), Thailand (10), the United Kingdom (9), South Africa (15), and the Gambia (15). Recently, we reported that EV68 is the major pathogen of enterovirus-associated acute respiratory tract infections in Beijing, China (30). Nevertheless, the pathogenesis of EV68 infection is poorly understood, and its link to innate immunity has not been reported previously. In this study, we show that EV68 inhibits type I IFN responses. This inhibition depends on 3Cpro, which cleaves TRIF, a critical adaptor of the TLR3 pathway. Collectively, these observations suggest a model whereby EV68 may modulate TLR3-mediated immunity in viral pathogenesis.
Several lines of evidence demonstrate that TLR3 plays a role in the antiviral response against enterovirus infections (31–37). Rhinovirus infection is recognized initially by TLR3 (38). As EV68 shares biological features with the enteroviruses and the rhinoviruses, inhibition of TLR3-mediated responses is probably beneficial for EV68 replication and/or spread. This is consistent with the fact that EV68 reduced the induction of type I IFN, chemokine, and interferon-stimulated genes upon treatment with dsRNA in virus-infected cells. Furthermore, EV68 infection downregulated the expression of TRIF. This coincided with the inhibition of IRF3 phosphorylation. Replication of EV68 increased when TRIF was knocked down. Conversely, viral replication decreased when TRIF was overexpressed. These observations highlight a functional link of EV68 to the TLR3 signaling pathway. Intriguingly, EV68 infection stimulated IL-8 expression, similar to the case in rhinovirus infection (39). This phenomenon may be involved in the pathogenesis of virus-induced acute exacerbations of asthma (3). We hypothesize that inhibition of type I IFN responses may favor EV68 replication, which in turn induces inflammatory cytokines through a distinct pathway. Further work is required to address this issue.
TRIF is a 712-amino-acid protein that transmits signals to IRF3 and NF-κB. This process relies on a dynamic assembly of the TRIF complex. Notably, the amino-terminal domain recruits TRAF6 and TRAF3, whereas the carboxyl-terminal domain binds to TLR3 and RIP1. Our work suggests that EV68 3CPro mediates TRIF cleavage at Q312 and Q653. This perhaps reflects a stringent regulation of TRIF by 3CPro, because multiple cleavages may be required to neutralize host defenses in EV68 infection. TRIF cleavage at Q312 yielded the amino-terminal and carboxy-terminal halves of the molecule, respectively. Further cleavage at Q653 truncated the carboxyl terminus. Interestingly, the amino-terminal domain of TRIF (1–312 fragment) failed to induce type I IFN responses. In addition, the carboxyl-terminal domain (313–712 fragment) partially retained this activity. However, additional cleavage (313–653 fragment) abrogated its activity. Consistently, ectopic expression of the 1–312 or 313–653 fragment was unable to inhibit viral replication in TRIF-knockdown cell lines. This may serve as a viral mechanism to control TRIF upon EV68 infection. Thus, EV68 3CPro-mediated cleavage at multiple sites likely modulates the TRIF complex assembly, resulting in the inhibition of IRF3 activation and IFN responses.
The data presented in the present study suggest that the EV68 3CPro function relies on its enzymatic activity. When treated with a 3C inhibitor, rupintrivir, EV68 3CPro was unable to mediate TRIF cleavage. Similarly, the H40D or C147A substitution in the catalytic triad (C-H-D) eliminated its activity. These results suggest TRIF as a cellular substrate for EV68 3CPro. In this regard, it is notable that the 3C proteases from EV71 and CVB3 also target TRIF (20, 26). EV71 3CPro cleaves TRIF at Q312 only, whereas CVB3 3CPro mediates TRIF cleavage at Q190 and sites in the carboxy-terminal domain (Q653, Q659, Q671, or Q702). This is consistent with the fact that the 3C proteases from EV68, EV71, and CVB3 exhibit considerable amino acid sequence homology. The differences in cleavage sites may be related to elements unique to each 3CPro.
Accumulating evidence indicates that picornavirus 3C proteases have evolved a variety of mechanisms to evade antiviral innate immunity. For example, 3Cpro of poliovirus cleaves retinoic acid-induced gene I (RIG-I) (25). 3Cpro of CVB3 cleaves both TRIF and IFN promoter-stimulating factor 1 (IPS-1) directly to impede host antiviral signaling (26). 3Cpro of EV71 inhibits innate antiviral immune responses not only through disrupting the RIG-I–IPS-1 complex (24) but also through cleaving TRIF (20) and IRF7 (21). 3Cpro of foot-and-mouth disease virus cleaves NF-κB essential modulator (NEMO) to impair innate immune signaling (40). The 3Cpro precursors, 3ABC and 3CD, of hepatitis A virus cleave IPS-1 (41) and TRIF (42) to block antiviral signaling. The TRIF cleavage by EV68 3Cpro observed in this work suggests that it may be a mechanism to impair TLR3-mediated immunity, which contributes to EV68 pathogenesis.
ACKNOWLEDGMENTS
We thank Chao Wu for providing technical assistance.
This work was supported by grants from the 973 Project (grant 2011CB504903), the National Science Foundation for Outstanding Young Scientists (grant 81225014), the National Natural Science Foundation of China (grant 31270200), Program for Changjiang Scholars and Innovative Research Team in University (IRT13007), and the National Institute of Allergy and Infectious Diseases of the United States (grant AI092230).
Footnotes
Published ahead of print 26 March 2014
REFERENCES
- 1.Pallansch M, Roos R. 2007. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, p 840–842 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Centers for Disease Control and Prevention. 2011. Clusters of acute respiratory illness associated with human enterovirus 68—Asia, Europe, and United States, 2008–2010. MMWR Morb. Mortal. Wkly. Rep. 60:1301–1304 [PubMed] [Google Scholar]
- 3.Hasegawa S, Hirano R, Okamoto-Nakagawa R, Ichiyama T, Shirabe K. 2011. Enterovirus 68 infection in children with asthma attacks: virus-induced asthma in Japanese children. Allergy 66:1618–1620. 10.1111/j.1398-9995.2011.02725.x [DOI] [PubMed] [Google Scholar]
- 4.Ikeda T, Mizuta K, Abiko C, Aoki Y, Itagaki T, Katsushima F, Katsushima Y, Matsuzaki Y, Fuji N, Imamura T, Oshitani H, Noda M, Kimura H, Ahiko T. 2012. Acute respiratory infections due to enterovirus 68 in Yamagata, Japan between 2005 and 2010. Microbiol. Immunol. 56:139–143. 10.1111/j.1348-0421.2012.00411.x [DOI] [PubMed] [Google Scholar]
- 5.Imamura T, Fuji N, Suzuki A, Tamaki R, Saito M, Aniceto R, Galang H, Sombrero L, Lupisan S, Oshitani H. 2011. Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg. Infect. Dis. 17:1430–1435. 10.3201/eid1708.101328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobson LM, Redd JT, Schneider E, Lu X, Chern SW, Oberste MS, Erdman DD, Fischer GE, Armstrong GL, Kodani M, Montoya J, Magri JM, Cheek JE. 2012. Outbreak of lower respiratory tract illness associated with human enterovirus 68 among American Indian children. Pediatr. Infect. Dis. J. 31:309–312. 10.1097/INF.0b013e3182443eaf [DOI] [PubMed] [Google Scholar]
- 7.Kaida A, Kubo H, Sekiguchi J, Kohdera U, Togawa M, Shiomi M, Nishigaki T, Iritani N. 2011. Enterovirus 68 in children with acute respiratory tract infections, Osaka, Japan. Emerg. Infect. Dis. 17:1494–1497. 10.3201/eid1708.110028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreuter JD, Barnes A, McCarthy JE, Schwartzman JD, Oberste MS, Rhodes CH, Modlin JF, Wright PF. 2011. A fatal central nervous system enterovirus 68 infection. Arch. Pathol. Lab. Med. 135:793–796 [DOI] [PubMed] [Google Scholar]
- 9.Lauinger IL, Bible JM, Halligan EP, Aarons EJ, MacMahon E, Tong CY. 2012. Lineages, sub-lineages and variants of enterovirus 68 in recent outbreaks. PLoS One 7:e36005. 10.1371/journal.pone.0036005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linsuwanon P, Puenpa J, Suwannakarn K, Auksornkitti V, Vichiwattana P, Korkong S, Theamboonlers A, Poovorawan Y. 2012. Molecular epidemiology and evolution of human enterovirus serotype 68 in Thailand, 2006–2011. PLoS One 7:e35190. 10.1371/journal.pone.0035190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meijer A, van der Sanden S, Snijders BE, Jaramillo-Gutierrez G, Bont L, van der Ent CK, Overduin P, Jenny SL, Jusic E, van der Avoort HG, Smith GJ, Donker GA, Koopmans MP. 2012. Emergence and epidemic occurrence of enterovirus 68 respiratory infections in The Netherlands in 2010. Virology 423:49–57. 10.1016/j.virol.2011.11.021 [DOI] [PubMed] [Google Scholar]
- 12.Piralla A, Lilleri D, Sarasini A, Marchi A, Zecca M, Stronati M, Baldanti F, Gerna G. 2012. Human rhinovirus and human respiratory enterovirus (EV68 and EV104) infections in hospitalized patients in Italy, 2008–2009. Diagn. Microbiol. Infect. Dis. 73:162–167. 10.1016/j.diagmicrobio.2012.02.019 [DOI] [PubMed] [Google Scholar]
- 13.Rahamat-Langendoen J, Riezebos-Brilman A, Borger R, van der Heide R, Brandenburg A, Scholvinck E, Niesters HG. 2011. Upsurge of human enterovirus 68 infections in patients with severe respiratory tract infections. J. Clin. Virol. 52:103–106. 10.1016/j.jcv.2011.06.019 [DOI] [PubMed] [Google Scholar]
- 14.Renois F, Bouin A, Andreoletti L. 2013. Enterovirus 68 in pediatric patients hospitalized for acute airway diseases. J. Clin. Microbiol. 51:640–643. 10.1128/JCM.02640-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tokarz R, Firth C, Madhi SA, Howie SR, Wu W, Sall AA, Haq S, Briese T, Lipkin WI. 2012. Worldwide emergence of multiple clades of enterovirus 68. J. Gen. Virol. 93:1952–1958. 10.1099/vir.0.043935-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oberste MS, Maher K, Schnurr D, Flemister MR, Lovchik JC, Peters H, Sessions W, Kirk C, Chatterjee N, Fuller S, Hanauer JM, Pallansch MA. 2004. Enterovirus 68 is associated with respiratory illness and shares biological features with both the enteroviruses and the rhinoviruses. J. Gen. Virol. 85:2577–2584. 10.1099/vir.0.79925-0 [DOI] [PubMed] [Google Scholar]
- 17.Hewson CA, Jardine A, Edwards MR, Laza-Stanca V, Johnston SL. 2005. Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J. Virol. 79:12273–12279. 10.1128/JVI.79.19.12273-12279.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowie AG, Unterholzner L. 2008. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8:911–922. 10.1038/nri2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. 10.1016/j.cell.2010.01.022 [DOI] [PubMed] [Google Scholar]
- 20.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 85:8811–8818. 10.1128/JVI.00447-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J. 2013. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 87:1690–1698. 10.1128/JVI.01855-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo L, Wang Y, Zhou H, Wu C, Song J, Li J, Paranhos-Baccala G, Vernet G, Wang J, Hung T. 2012. Differential seroprevalence of human bocavirus species 1–4 in Beijing, China. PLoS One 7:e39644. 10.1371/journal.pone.0039644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng Z, Cerveny M, Yan Z, He B. 2007. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA dependent protein kinase PKR. J. Virol. 81:182–192. 10.1128/JVI.01006-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B, Wang J. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation. J. Virol. 84:8051–8061. 10.1128/JVI.02491-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barral PM, Sarkar D, Fisher PB, Racaniello VR. 2009. RIG-I is cleaved during picornavirus infection. Virology 391:171–176. 10.1016/j.virol.2009.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB. 2011. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 7:e1001311. 10.1371/journal.ppat.1001311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Palma AM, Vliegen I, De Clercq E, Neyts J. 2008. Selective inhibitors of picornavirus replication. Med. Res. Rev. 28:823–884. 10.1002/med.20125 [DOI] [PubMed] [Google Scholar]
- 28.Tan J, George S, Kusov Y, Perbandt M, Anemuller S, Mesters JR, Norder H, Coutard B, Lacroix C, Leyssen P, Neyts J, Hilgenfeld R. 2013. 3C protease of enterovirus 68: structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J. Virol. 87:4339–4351. 10.1128/JVI.01123-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schieble JH, Fox VL, Lennette EH. 1967. A probable new human picornavirus associated with respiratory diseases. Am. J. Epidemiol. 85:297–310 [DOI] [PubMed] [Google Scholar]
- 30.Xiang Z, Gonzalez R, Wang Z, Ren L, Xiao Y, Li J, Li Y, Vernet G, Paranhos-Baccala G, Jin Q, Wang J. 2012. Coxsackievirus A21, enterovirus 68, and acute respiratory tract infection, China. Emerg. Infect. Dis. 18:821–824. 10.3201/eid1805.111376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abe Y, Fujii K, Nagata N, Takeuchi O, Akira S, Oshiumi H, Matsumoto M, Seya T, Koike S. 2012. The Toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J. Virol. 86:185–194. 10.1128/JVI.05245-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abston ED, Coronado MJ, Bucek A, Onyimba JA, Brandt JE, Frisancho JA, Kim E, Bedja D, Sung YK, Radtke AJ, Gabrielson KL, Mitzner W, Fairweather D. 2013. TLR3 deficiency induces chronic inflammatory cardiomyopathy in resistant mice following coxsackievirus B3 infection: role for IL-4. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304:R267–R277. 10.1152/ajpregu.00516.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris KG, Coyne CB. 2013. Enter at your own risk: how enteroviruses navigate the dangerous world of pattern recognition receptor signaling. Cytokine 63:230–236. 10.1016/j.cyto.2013.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R, Yanai H, Seko Y, Shitara H, Bishop K, Yonekawa H, Tamura T, Kaisho T, Taya C, Taniguchi T, Honda K. 2008. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. U. S. A. 105:20446–20451. 10.1073/pnas.0810372105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oshiumi H, Okamoto M, Fujii K, Kawanishi T, Matsumoto M, Koike S, Seya T. 2011. The TLR3/TICAM-1 pathway is mandatory for innate immune responses to poliovirus infection. J. Immunol. 187:5320–5327. 10.4049/jimmunol.1101503 [DOI] [PubMed] [Google Scholar]
- 36.Riad A, Westermann D, Zietsch C, Savvatis K, Becher PM, Bereswill S, Heimesaat MM, Lettau O, Lassner D, Dorner A, Poller W, Busch M, Felix SB, Schultheiss HP, Tschope C. 2011. TRIF is a critical survival factor in viral cardiomyopathy. J. Immunol. 186:2561–2570. 10.4049/jimmunol.1002029 [DOI] [PubMed] [Google Scholar]
- 37.Richer MJ, Lavallee DJ, Shanina I, Horwitz MS. 2009. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS One 4:e4127. 10.1371/journal.pone.0004127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slater L, Bartlett NW, Haas JJ, Zhu J, Message SD, Walton RP, Sykes A, Dahdaleh S, Clarke DL, Belvisi MG, Kon OM, Fujita T, Jeffery PK, Johnston SL, Edwards MR. 2010. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 6:e1001178. 10.1371/journal.ppat.1001178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chun YH, Park JY, Lee H, Kim HS, Won S, Joe HJ, Chung WJ, Yoon JS, Kim HH, Kim JT, Lee JS. 2013. Rhinovirus-infected epithelial cells produce more IL-8 and RANTES compared with other respiratory viruses. Allergy Asthma Immunol. Res. 5:216–223. 10.4168/aair.2013.5.4.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang D, Fang L, Li K, Zhong H, Fan J, Ouyang C, Zhang H, Duan E, Luo R, Zhang Z, Liu X, Chen H, Xiao S. 2012. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 86:9311–9322. 10.1128/JVI.00722-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. 2007. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. U. S. A. 104:7253–7258. 10.1073/pnas.0611506104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qu L, Feng Z, Yamane D, Liang Y, Lanford RE, Li K, Lemon SM. 2011. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 7:e1002169. 10.1371/journal.ppat.1002169 [DOI] [PMC free article] [PubMed] [Google Scholar]