

ABSTRACT
Microfold (M) cells are specialized intestinal epithelial cells that internalize particulate antigens and aid in the establishment of immune responses to enteric pathogens. M cells have also been suggested as a portal for pathogen entry into the host. While virus particles have been observed in M cells, it is not known whether viruses use M cells to initiate a productive infection. Noroviruses (NoVs) are single-stranded RNA viruses that infect host organisms via the fecal-oral route. Murine NoV (MNV) infects intestinal macrophages and dendritic cells and provides a tractable experimental system for understanding how an enteric virus overcomes the intestinal epithelial barrier to infect underlying target cells. We found that replication of two divergent MNV strains was reduced in mice depleted of M cells. Reoviruses are double-stranded RNA viruses that infect hosts via respiratory or enteric routes. In contrast to MNV, reovirus infects enterocytes in the intestine. Despite differences in cell tropism, reovirus infection was also reduced in M cell-depleted mice. These data demonstrate that M cells are required for the pathogenesis of two unrelated enteric viruses that replicate in different cell types within the intestine.
IMPORTANCE To successfully infect their hosts, pathogens that infect via the gastrointestinal tract must overcome the multilayered system of host defenses. Microfold (M) cells are specialized intestinal epithelial cells that internalize particulate antigens and aid in the establishment of immune responses to enteric pathogens. Virus particles have been observed within M cells. However, it is not known whether viruses use M cells to initiate a productive infection. To address this question, we use MNV and reovirus, two enteric viruses that replicate in different cell types in the intestine, intestinal epithelial cells for reovirus and intestinal mononuclear phagocytes for MNV. Interestingly, MNV- and reovirus-infected mice depleted of M cells showed reduced viral loads in the intestine. Thus, our work demonstrates the importance of M cells in the pathogenesis of enteric viruses irrespective of the target cell type in which the virus replicates.
INTRODUCTION
The gastrointestinal (GI) tract, being the largest mucosal surface in the body, forms a barrier between the interior and exterior milieu. Although multiple protective mechanisms are present, enteric viruses have evolved strategies to overcome this barrier and infect the host. Some enteric viruses enter the host by directly infecting enterocytes, e.g., rotavirus (1). Alternatively, microfold (M) cells have been proposed as a route of viral entry after visualization of selective uptake of poliovirus and reovirus particles by Peyer's patch (PP) M cells (2, 3). However, direct evidence demonstrating that M cells are required for the establishment of a productive virus infection is lacking.
M cells are specialized epithelial cells that are mostly located in the follicle-associated epithelium (FAE) of organized lymphoid tissues like PPs. However, M cells also are found in intestinal villi, although villous M cells are less abundant than PP M cells (4). M cells selectively bind and endocytose IgA (5) and selectively express glycosylphosphatidylinositol-anchored glycoprotein 2 (GP2) (6). Mouse M cells also react with the Ulex europaeus agglutinin-I (UEA-I) lectin, which recognizes α1,2 fucose (7). M cells arise from individual stem cells in the crypt (8). Development of M cells depends on the receptor activator of NF-κB ligand (RANKL), which is expressed by subepithelial stromal cells in the PP domes (9, 10). Antibody-mediated neutralization of RANKL in wild-type mice transiently eliminates most PP M cells, while systemic administration of RANKL to RANKL-deficient mice restores PP M cells and induces differentiation of villous M cells (9). M cells function to sample antigens in the intestinal lumen for immune surveillance, including microorganisms and inert particles (e.g., latex beads) (11–13). For example, the bacterial pathogens Listeria monocytogenes, Salmonella enterica serovar Typhimurium, Shigella flexneri, and Yersinia enterocolitica exploit M cells to invade the host and establish infections (14–17). In the case of S. Typhimurium, selective M cell uptake is mediated by a specific ligand-receptor interaction between FimH, a component of type I pili on the bacterial outer membrane, and GP2, a protein specifically expressed on M cells (6). Binding of secretory IgA to its receptor on M cells is important to facilitate the sampling of commensal bacteria (18). Remarkably, transient depletion of M cells in mice by RANKL antibody treatment inhibits prion accumulation and subsequent neuroinvasion (19), suggesting that M cells are also the sites of prion uptake. While collectively these studies provide evidence that M cells contribute to the pathogenesis of bacterial and prion diseases, the role of M cells in the initiation of productive virus infection is less clear.
Noroviruses (NoVs) are nonenveloped, highly stable, positive-sense RNA viruses that infect hosts via the fecal-oral route (20). Little is known about NoV pathogenesis, including early events during infection of the intestine. Murine noroviruses (MNVs) efficiently replicate in macrophages and dendritic cells (DCs) in cell culture (21) and in mice (22–24), providing a tractable experimental system for understanding how an enteric virus overcomes the intestinal epithelial barrier to reach its target cells in the intestinal lamina propria. Despite the high sequence similarity of MNV strains (>75%), they differ in biological phenotypes (25, 26). For example, the MNV strain CR3, which was isolated from the feces of mice, persists in wild-type mice for at least 35 days, while MNV-1 establishes acute infections and virus is not detectable in fecal contents 7 days postinoculation (dpi) (25, 27). Studies using an in vitro model of the FAE demonstrate that MNV is transported across a polarized intestinal epithelial monolayer using M-like cells (28). However, how MNV crosses the intestinal epithelial barrier in vivo to reach the underlying permissive macrophages and dendritic cells is not known.
Mammalian reoviruses are another widely used model for studies of viral pathogenesis (29). Reoviruses are nonenveloped, segmented, double-stranded RNA viruses that cause disease in the very young but do not produce symptoms in adults (30). Reoviruses are classified into three serotypes represented by the prototype strains, type 1 Lang (T1L), type 2 Jones (T2J), and type 3 Dearing (T3D). While T1L and T3D differ in pathways of virus spread (hematogenous versus neural, respectively) (31), the primary site of replication for both strains in perorally inoculated newborn mice are intestinal enterocytes at the villus tips (32). T1L binds to α2,3-linked sialic acid-containing glycans on the apical surface of M cells via the attachment protein σ1 (33, 34). Visualization of virus particles by transmission electron microscopy during the first hours of infection suggests that following binding to the apical surface of M cells, reovirus is internalized into and replicates in M cells prior to infecting enterocytes from the basolateral surface (35). However, it is not apparent whether reovirus can establish productive infection in the host in the absence of M cells, for example, via apical infection of enterocytes.
In this study, we used an M cell depletion protocol to investigate whether M cells are required for infection by MNV and reovirus, which replicate in different cell types (macrophages and dendritic cells versus enterocytes) in the murine intestine. Using a light-sensitive MNV to distinguish between input and replicated virus, we found that two MNV strains have different replication kinetics, and both depend on M cells for efficient intestinal infection. Despite the differences in cell tropism, reovirus infection of the murine intestine also depends on M cells. Thus, intestinal M cells are used as a portal of entry for two unrelated enteric viruses to initiate productive infection in the murine host.
MATERIALS AND METHODS
Mice.
All animal studies were performed in accordance with local and federal guidelines as outlined in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (36). The protocol was approved by the University of Michigan Committee on Use and Care of Animals (UCUCA number 09710).
Wild-type BALB/c (000651) and 129S6/SvEv STAT1−/− (2045) mice were purchased from Jackson Laboratory (Bar Harbor, ME) and Taconic Farms (Hudson, NY), respectively. Six- to 8-week-old mice were used for MNV studies, and 3- to 4-week-old mice were used for reovirus studies. All mice used in the study came from a reovirus-free colony and were further tested for anti-MNV antibodies by enzyme-linked immunosorbent assay (ELISA) as described previously (21) and found to be seronegative.
To transiently deplete M cells in vivo, BALB/c mice were inoculated intraperitoneally with 250 μg of IK22-5 rat anti-mouse RANKL monoclonal antibody every 2 days for a total of four doses prior to infection, as described previously (9). A parallel group of mice were similarly treated with a rat isotype control IgG (Sigma) or left untreated.
Virus stocks and plaque assays.
The plaque-purified MNV-1 clone (GV/MNV1/2002/USA) MNV-1.CW3 and the fecally isolated MNV strain CR3 (GV/CR3/2005/USA) were used at passage 6 for all experiments (25). Viral titers were quantified by plaque assay after visualizing plaques by staining cells with a 0.01% neutral red (NR) solution in phosphate-buffered saline (PBS) for 1 to 3 h as described previously (21, 37). Reovirus T1L stocks were prepared using reverse genetics (38, 39). Viral titers were quantified by plaque assay as described previously (40). The limit of the plaque assay is 10 PFU/ml or 1 log10.
Infection of mice with NR-containing MNV.
NR-containing MNV stocks were generated by infecting RAW 264.7 cells with either MNV strain in the presence of the NR dye as described previously (41 and http://www.bio-protocol.org/wenzhang.aspx?id=415). The NR dye, when exposed to light, cross-links the RNA genome to the capsid by an unknown mechanism, preventing viral uncoating (41, 42). All NR virus preparations displayed 100- to 1,000-fold reductions in viral titers upon light exposure. Mice were inoculated perorally with 105 PFU MNV (NR), and tissues were aseptically removed using a red safety light at 12 (MNV-1) or 18 (CR3) hpi, approximate time points for the first round of viral replication. Regions of the GI tract, including the stomach (ST), jejunum/duodenum (J/D), proximal ileum (PI), distal ileum (DI), cecum (CE), and colon (CO), were harvested, and one fecal pellet (FE) was collected. Tissue samples were flash-frozen in a dry ice/ethanol bath and stored at −80°C. Tissues were homogenized in 1 ml of medium with 1.0-mm-diameter zirconia-silica beads (BioSpec Products) using a MagNALyser (Roche Applied Sciences, Hague Road, IN). Plaque assays were performed in duplicate, one in the dark and the other following a 10-min light exposure. Viral plaques were enumerated 48 h later.
Infection of mice with reovirus.
Three- to 4-week-old BALB/c mice were inoculated with 106 PFU of reovirus T1L by oral gavage. Tissue samples were collected 24 hpi and processed as described for MNV, with the exception that tissues were homogenized in PBS with MgCl2 and CaCl2. Viral titers were determined by plaque assay using L929 cells (32).
Immunostaining of whole mounts, cryosections, and paraffin-embedded tissue.
For whole mounts, PP were harvested from untreated, IgG isotype control-treated, and anti-RANKL-treated mice at various intervals postinoculation and vortexed in 1 ml PBS containing 0.05% Tween 20 for 30 s. After vortexing, PPs were washed once with PBS, fixed in 4% paraformaldehyde in PBS for 15 min, and permeabilized with 0.1% Triton X-100 for 15 min. PPs were blocked with 10% (vol/vol) FBS and 1% (vol/vol) normal goat serum (NGS; Gibco) in PBS for 30 to 60 min. GP2 staining was performed by incubating PPs with a primary rat anti-mouse glycoprotein 2/GP2 (MBL, Woburn, MA) antibody in PBS containing 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) for 1 h, followed by three consecutive PBS washes. PPs were incubated with fluorescein isothiocyanate (FITC)-conjugated secondary rat anti-IgG antibody (eBiosciences, San Diego, CA) for 1 h, along with rhodamine-conjugated Ulex europaeus agglutinin I (UEA-1; Vector Laboratories, Burlingame, CA). PPs were washed three times with PBS and mounted with ProLong Gold antifade reagent containing DAPI (Invitrogen, Grand Island, NY) between two coverslips separated by double-stick tape or clay. Images were captured by laser scanning confocal microscopy (LSM) using LSM software with a Zeiss confocal microscope. Immunofluorescence images were quantified from 5 to 6 individual PPs using the scoring system of intensities by the Metamorph Premier v6.3 image analysis software (Molecular Devices, Downington, PA).
For cryosections, PPs were harvested from BALB/c or STAT1−/− mice orally infected with MNV (6 × 107 PFU/mouse). Tissues were processed as described for whole mounts. M cells were detected with primary rat anti-mouse glycoprotein 2/GP2 antibody (MBL, Woburn, MA), and MNV was detected with a rabbit polyclonal antibody raised against the MNV nonstructural protein N-term (43), generously provided by Vernon Ward (Otago University, Dunedin, New Zealand). Images were captured using an Olympus BX60 upright microscope.
For paraffin-embedded sections from reovirus-infected mice, PPs were harvested from isotype control-treated or anti-RANKL-treated mice, fixed with 10% formalin, and embedded in paraffin. Tissues were then sectioned, deparaffinized, and immunostained as described above for whole mounts using a primary rat anti-mouse IgG2a isotype control or a rat anti-mouse glycoprotein 2/GP2 antibody (MBL, Woburn, MA) and a rabbit polyclonal antibody against the sigma nonstructural protein for reovirus T1L (44, 45). Images were captured using an Olympus BX60 upright microscope.
Statistical analysis.
Data are presented as means ± standard errors of the means (SEM). Statistical analysis was performed using Prism software, version 5.01 (GraphPad Software, CA). The two-tailed Student's t test was used to determine statistical significance.
RESULTS
MNV strains have different replication kinetics.
Analysis of early events in the virus-host encounter benefits from the ability to distinguish between input and replicated virus. In addition, the intestinal transit time in mice is similar to the length of a replicative cycle of MNV (21, 46). Therefore, we adapted the light-sensitive, neutral red (NR)-containing MNV (41) for in vivo studies. The basic premise of this technology is that NR-labeled virions from the inoculum are inactivated upon exposure to light, whereas newly replicated progeny virions are no longer labeled with NR and become light insensitive. To verify that NR-containing viruses remain light sensitive when present in a tissue homogenate, we mixed distal ileum homogenates from wild-type mice with approximately 104 PFU of either NR-containing MNV-1 or CR3 and quantified the total (exposed in the dark) and replicated (after light exposure) viral titers by plaque assay (Fig. 1A). A three-log reduction was observed for both NR-containing virus stocks after light exposure, demonstrating that the NR-containing virus strains are sensitive to light inactivation. As a proof of concept for in vivo use, we next inoculated BALB/c mice perorally with MNV-1 (NR), and the intestine was harvested 3 or 72 hpi as representative time points that do not or do permit replication, respectively. As anticipated, progeny virus at or below the limit of detection was observed in the distal ileum and colon at 3 hpi, while a significant amount of input virus was observed in the colon at this time point (Fig. 1B). In contrast, newly replicated virus was observed at 72 hpi in the distal ileum, the primary site of MNV-1 replication (25), but not in the colon (Fig. 1B). Therefore, NR-containing MNV enables the study of early events during pathogenesis and allows input virus to be distinguished from newly formed virus.
FIG 1.

Neutral red assay distinguishes between input and replicated MNV. (A) Distal ileum (tissue) homogenate from BALB/c mice (7 to 8 weeks old) were mixed with ∼104 PFU of neutral red (NR) containing MNV-1 or CR3 stock. Viral titers were determined by plaque assay in the dark (total) or light (replicated). (B) BALB/c mice (7 to 8 weeks old) were inoculated perorally with 105 PFU of a neutral red-containing MNV-1. Distal ileum and colon were harvested at the times shown. Viral titers were determined by plaque assay in the light (replicated) or dark (total). (C to E) MNV strains differ in replication kinetics. BALB/c mice were inoculated perorally with 105 PFU of either MNV-1 or CR3 containing neutral red. The GI tract was dissected 12, 24, or 72 h later, and viral titers were determined by plaque assay as described for panels A and B. Data are expressed as means ± SEM for three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ND, not detectable; J/D, jejunum/duodenum; PI, proximal ileum; DI, distal ileum; CE, cecum; CO, colon; FE, feces.
To identify an approximate time point at which laboratory-adapted MNV strain MNV-1 and persistent MNV strain CR3 undergo the first round of viral replication, BALB/c mice were inoculated perorally with NR-containing MNV-1 or CR3, and viral titers in the intestine were quantified by plaque assay at 12, 24, or 72 hpi (Fig. 1C to E). Maximum levels of newly replicated virus from mice infected with MNV-1 (NR) were detected in the small intestine (jejunum/duodenum, proximal ileum, and distal ileum) or large intestine (cecum) at 12 hpi, indicating MNV-1 undergoes its first round of replication within 12 h. In contrast, CR3 (NR) replicated mostly in the cecum at 12 hpi and reached its maximum titer in the cecum at 24 hpi, while maximum levels of replicated virus in other regions of the GI tract were reached at 72 hpi. This suggested the first round of CR3 replication occurred between 12 and 24 hpi. No viral titers were detected in the stomach for either virus (data not shown). These data confirmed previous reports from experiments using C57BL/6 mice that CR3 but not MNV-1 replicates in the colon (27) and demonstrated that CR3 has slower replication kinetics than MNV-1 in vivo. More importantly, these data demonstrate that the light-sensitive virus is a powerful tool to investigate early events in viral pathogenesis.
MNV requires M cells for productive infection.
To determine whether M cells are required to initiate a productive MNV infection in vivo, BALB/c mice were selectively depleted of M cells using an antibody against RANKL (anti-RANKL) administered intraperitoneally (i.p.) every 2 days for a total of four doses, as described previously (9) (Fig. 2A). This M cell depletion protocol does not alter the presence and distribution of F4/80+ and CD11c+ cells in the intestine, gastrointestinal lymphoid follicles, and spleen, most likely because macrophages and DCs at these sites express negligible levels of RANK (19). As depletion controls, mice were administered an IgG isotype control antibody or left untreated. At 36 h following the last dose of antibody, mice were inoculated perorally with MNV-1 (NR) or CR3 (NR) (Fig. 2A). The GI tract was excised at 12 hpi for MNV-1 (NR) or 18 hpi for CR3 (NR), time points representing approximately one round of replication, and viral titers were determined by plaque assay. These early time points capturing the first round of viral replication were chosen for each virus, because we reasoned that if MNV enters via M cells, reducing the number of M cells should reduce the number of virus particles capable of reaching the underlying target cells (i.e., macrophages and dendritic cells), resulting in the greatest measurable phenotype. Total MNV-1 titers were significantly decreased in the distal ileum and feces of mice treated with anti-RANKL compared to those of control animals, while replicated MNV-1 titers also were decreased in the cecum (Fig. 2B and C). A significant difference in the total but not replicated MNV-1 titers was observed in the jejunum/duodenum compared to the isotype control (compare Fig. 2B to C). The biological significance of this finding is unclear but suggests differences in attachment to and/or transcytosis by M cells. Anti-RANKL-treated mice perorally inoculated with CR3 (NR) produced significantly decreased total and replicated viral titers in the cecum and feces but not the jejunum/duodenum, proximal ileum, or distal ileum compared to that of untreated and isotype control-treated animals (Fig. 2D and E). Interestingly, the effect of M cell depletion on CR3 infection was greater than that for MNV-1.
FIG 2.

MNV infection is reduced in the GI tract following M cell depletion. (A) Schematic of the experimental design for MNV infection of M cell-depleted mice. BALB/c mice (7 to 8 weeks old) were left untreated or were inoculated i.p. with anti-RANKL or isotype control antibody every other day for a total of four doses and then infected with either MNV-1 (NR) or CR3 (NR) 36 h later, and regions of the GI tract were harvested 12 hpi for MNV-1 and 18 hpi for CR3. (B to E) The number of mice analyzed in each group is indicated in parentheses. Viral titers in indicated tissues were quantified by plaque assay in the dark (total; B and D) or in the light (replicated; C and E). Data are expressed as means ± SEM for two to three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant; ND, not detectable; ST, stomach; J/D, jejunum/duodenum; PI, proximal ileum; DI, distal ileum; CE, cecum; CO, colon; FE, feces.
Thus, we wanted to verify that M cells were successfully depleted. Toward that end, PPs were harvested from each mouse after MNV infection, fixed, and prepared for whole-mount staining using two M cell markers, GP2 and UEA-1 (Fig. 3A to J). Both M cell markers showed significantly reduced staining in the PPs of anti-RANKL-treated mice (Fig. 3G to I and J) compared with isotype control-treated animals (Fig. 3D to F and J) or untreated mice (Fig. 3A to C and J).
FIG 3.

Whole-mount staining of Peyer's patches shows decreased numbers of M cells in anti-RANKL-treated mice. BALB/c mice were left untreated or were inoculated i.p. with anti-RANKL or isotype control antibody every other day for a total of four doses and then infected with either MNV-1 (NR) or CR3 (NR) 36 h later, and Peyer's patches were harvested after infection and immunostained for two M cell markers. (A to I) Representative confocal microscopic images of Peyer's patches stained with M cell markers GP2 (green) and UEA-1 (red) from untreated (A to C), isotype control-treated (D to F), and anti-RANKL-treated (G to I) mice. DAPI was used to stain the nuclei. A 10-μm scale bar is shown in the upper right corner of each image. (J) Quantification of whole-mount staining with M cell markers GP2 and UEA-1 of one Peyer's patch per mouse from a total of 5 to 6 mice per group. Immunofluorescence staining was quantified using the scoring system of intensities by the Metamorph Premier v6.3 image analysis software. Data are expressed as means ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ns, not significant.
MNV-1 does not infect M-like cells in the FAE model in vitro (28). To test whether MNV infects M cells in vivo, PP sections from MNV 1-infected wild-type mice and STAT1−/− mice (mice highly susceptible to MNV infection [22, 23]) were immunostained for the M cell marker GP2 and the viral protein N-term, which is only expressed during active replication (Fig. 4). M cells expressing GP2 at the apical surface were readily observed in wild-type mice (Fig. 4B, also see arrowheads in the inset). Unfortunately, despite using multiple assays (immunofluorescence, immunohistochemistry, and flow cytometry), we were unable to detect viral replication in PPs of wild-type BALB/c mice. This finding most likely reflects the very low number of infected cells in wild-type mice, as has been observed previously (23). In contrast, MNV-1 nonstructural protein expression was observed in STAT1−/− mice in cells located in the subepithelial dome underneath the FAE, but no staining was observed in GP2-positive M cells (Fig. 4D). Taken together, our data suggest that while MNV does not infect M cells, it requires M cells for transport across the intestinal epithelial barrier to permit efficient replication in the host's intestine.
FIG 4.

MNV does not replicate in M cells. BALB/c and STAT1−/− mice were infected with MNV, and Peyer's patches were harvested 24 hpi for cryosectioning and immunostaining. (A and C) The secondary antibody-only control (red) and an isotype control for GP2 (green) were used to identify background staining. (B and D) MNV replication was detected using an antibody against the nonstructural protein N-term (red), and M cells were detected using an anti-GP2 antibody (green). Arrowheads in the inset indicate GP2-positive M cells.
Reovirus requires M cells for a productive infection in mice.
To determine whether an unrelated virus that replicates in a different cell type within the intestine than MNV also traverses the intestinal mucosal barrier using M cells, we tested the effect of M cell depletion on reovirus infection. M cells were depleted from mice prior to peroral inoculation with reovirus strain T1L, and viral titers were determined by plaque assay at 24 hpi. Strikingly, mice treated with anti-RANKL had no detectable reovirus titers in the jejunum/duodenum, proximal ileum, cecum, and colon compared to isotype control-treated or untreated animals (Fig. 5). In addition, significantly lower titers were produced in the distal ileum and feces (Fig. 5). To determine whether reovirus replicates in M cells, PPs from isotype- and anti-RANKL-treated mice were immunostained with the M cell-specific marker GP2 and a polyclonal rabbit antibody against the sigma nonstructural (σNS) protein (Fig. 6). Reovirus σNS protein expression was observed in the isotype-treated but not anti-RANKL-treated mice. Specifically, reovirus antigen was detected in enterocytes adjacent to (arrowhead) and within (star) cells expressing GP2 at their apical surface, indicative of M cells (Fig. 6A). The lack of reovirus staining in anti-RANKL-treated animals (Fig. 6B) is consistent with the absence of reovirus PFU in the small intestine of these mice (Fig. 5). Taken together, these data demonstrate that reovirus, like MNV, requires M cells to establish a productive infection in the murine host, but unlike MNV, it can replicate in M cells.
FIG 5.
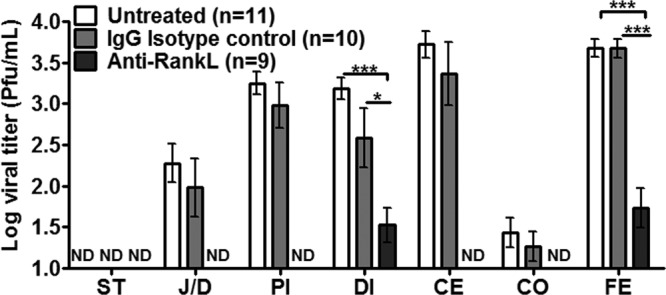
Reovirus infection is reduced in the GI tract following M cell depletion. BALB/c mice (3 to 4 weeks old) were depleted of M cells as described in the legend to Fig. 1 and inoculated perorally with T1L reovirus 24 h later, and regions of the GI tract were harvested 24 hpi. The number of mice analyzed in each group is indicated in parentheses. Viral titers were quantified by plaque assay. Data are expressed as means ± SEM from two independent experiments. *, P < 0.05; ***, P < 0.001; ND, not detectable; ST, stomach; J/D, jejunum/duodenum; PI, proximal ileum; DI, distal ileum; CE, cecum; CO, colon; FE, feces.
FIG 6.
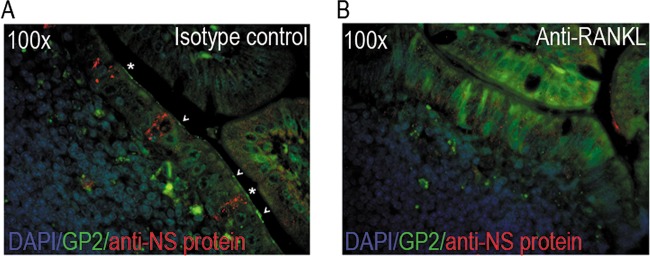
Reovirus replicates in enterocytes adjacent to and in M cells of control but not M cell-depleted mice. Isotype control-treated (A) or anti-RANKL-treated (B) BALB/c mice were infected with reovirus for 24 h. Peyer's patches were harvested for paraffin embedding and immunostaining. Reovirus replication was detected using a polyclonal antibody against the sigma nonstructural (σNS) protein (red), and M cells were detected using an anti-GP2 antibody (green). Arrowheads indicate uninfected GP2-positive M cells, while stars indicate reovirus-infected M cells.
DISCUSSION
The initial steps of virus entry into an infected animal can dictate host range and pathogenesis and offer a point of potential intervention. However, little is known about how enteric viruses cross the intestinal barrier and initiate a productive infection. Virus particles have been observed within M cells, implicating this cell type as a portal of entry into the host, but whether these particles actually initiate a productive infection in addition to contributing to the development of antiviral immune responses is not clear. The M cell depletion studies presented here collectively point to the importance of M cells in the pathogenesis of enteric viruses that replicate within intestinal epithelial cells (reovirus) or intestinal mononuclear phagocytes (MNV) (Fig. 7).
FIG 7.

Proposed mechanisms of MNV and reovirus entry into the intestinal mucosa. MNV (black circles) and reovirus (red circles) in the intestinal lumen initially interact with M cells within the follicle-associated epithelium (FAE) overlying Peyer's patches (PP) to establish a productive infection in untreated animals. In mice depleted of M cells, reovirus infection in the gastrointestinal tract is substantially diminished, while MNV infection is partially reduced, suggesting alternative route(s) of entry for MNV, for example, via transepithelial dendritic cells.
Adapting the use of neutral red-labeled MNV for pathogenesis studies enabled us to determine that the MNV strain CR3 has slower replication kinetics in the GI tract than does strain MNV-1 (Fig. 1). CR3 causes a persistent infection in mice and shares the viral determinant for persistence (Glu94 in the N-terminal protein) identified in CR6, while MNV-1 causes an acute infection (22, 25, 47). A similar correlation between replication kinetics and persistence was observed for lymphocytic choriomeningitis virus (LCMV), leading to the hypothesis that slowly replicating viruses evade immune surveillance to allow persistent infection (48). In addition, MNV-1 and CR3 display different carbohydrate-binding properties in macrophages and differences in colon tropism (27, 49). Thus, it is possible that infection of macrophages, the precise tissue sites of replication, or the effectiveness of immune surveillance contribute to the persistence phenotype of CR3 or the enhanced clearance of MNV-1. Future studies are required to determine the mechanism of MNV persistence.
Our results demonstrate that M cells facilitate the initial steps of productive MNV infection in mice. Viral titers were significantly reduced within the GI tract following depletion of M cells in animals infected with either of two MNV strains (Fig. 2). However, M cells are not susceptible to MNV replication in vivo (Fig. 4), consistent with our previous observation that M-like cells do not become infected in an in vitro model of the FAE (28). Together, these data point to M cells as a conduit for MNV transport across the mucosa but not as a site for replication, as is seen with some viruses (50). However, residual MNV was still detectable within the GI tract of anti-RANKL-treated mice. One reason for this occurrence could be the incomplete depletion of M cells, as weak GP2 expression was detected in PPs from depleted mice (Fig. 3G and J). In addition, we cannot exclude the presence of villous M cells following M cell depletion. We focused on PPs to quantify M cell depletion, because the majority of M cells are located in PPs, while villous M cells are rare (9). The effect of RANKL depletion on villous M cells has not been examined, although villous M cells are induced by RANKL supplementation (9).
In addition, the likelihood of incomplete M cell depletion in our studies suggests that once residual virus has crossed the intestinal epithelium and undergone the first round of replication, progeny virus will be capable of infecting new cells for additional rounds of replication. Thus, differences in viral titers in M cell- and control-depleted mice likely will diminish with increasing rounds of replication. A second possibility is that MNV uses multiple mechanisms to cross the epithelial barrier. Since MNV infects DCs, sentinel DCs that insert dendritic processes between intestinal epithelial cells (51) or M cell-specific transcellular pores (52) may provide an alternate route of MNV entry (Fig. 7). It is unlikely that the DC pathway is upregulated only under these specific experimental conditions. PP DCs and macrophages express negligible levels of Tnfrsf11a, the gene encoding RANK, and mice treated with anti-RANKL antibody have normal numbers and distribution of DCs and macrophages in the intestine and spleen (19, 53). In addition, while RANKL can stimulate the activity of DCs to activate T cells (54), the time frame of our experiments (less than 24 h) is too short for antiviral T cell activity, which is not detectable until 7 to 8 days postinfection (55, 56). However, although unlikely, we cannot rule out that RANKL depletion affects the innate immune response. The incomplete block to MNV replication observed in our study is in contrast to another study in which M cells were depleted using the same anti-RANKL antibody approach. This study shows that prion disease progression in the brain following peroral prion inoculation is abolished following M cell depletion (19). Mice completely lacking M cells will be required to determine whether M cells are the only route used by MNV to infect mice.
Depletion of M cells substantially reduced reovirus titers in the intestine of infected mice (Fig. 5), indicating that M cells also are required for the initiation of a productive infection with reovirus. After proteolytic conversion of reovirus T1L virions to infectious subvirion particles (ISVPs) in the small intestine of mice (57, 58), reovirus binds to α2,3-linked sialic acid on the apical surface of M cells via the attachment protein σ1 (33–35). By electron microscopy, reovirus particles are observed in M cells within vesicles, suggesting that virions are endocytosed into these cells. In suckling mice, viral particles also are observed in M cells within cytoplasmic inclusions, which are a hallmark of reovirus replication (35). Consistent with the ability of reovirus to replicate in M cells, our studies using PPs from isotype-treated adult mice immunostained with the M cell-specific marker GP2 and an anti-reovirus antibody showed cells double positive for both markers (Fig. 6). However, the majority of reovirus antigen-positive cells were enterocytes adjacent to GP2-positive M cells (Fig. 6). Previous studies indicate that reovirus virions preferentially adhere to and are taken up from the basolateral surface of epithelial cells cultivated from the murine small intestine (35). Our data are consistent with those earlier findings and further support the model that reovirus infection is initiated by the uptake of virions into and transport across M cells followed by basolateral infection of enterocytes (32). Conversely, it is highly unlikely that reovirus infects enterocytes from the apical side to initiate a productive infection of the host. Reovirus is also capable of infecting human airway epithelial cells via the basolateral route (59), suggesting common themes of reovirus infection of polarized epithelia.
Reovirus titers in the GI tract following M cell depletion were reduced to a substantially greater extent than titers of MNV in M cell-depleted mice (compare Fig. 2 and 5). While these findings might reflect differences in replication kinetics, we favor an alternate explanation based on the different cell tropisms of these two viruses. Reovirus does not replicate in DCs (44) but is dependent on M cells for transport across the epithelial barrier and as an early target cell for replication (this study). In contrast, MNV replicates in DCs (21), and microbial stimuli are known to increase the sampling of lumenal content by transepithelial DCs (51, 60, 61), upregulating a potential alternative entry route for the virus to access the host (Fig. 7).
Our findings demonstrate that M cells are a gateway for MNV and reovirus infection and serve an important role in the initiation of productive infection in addition to their function in development of immune responses (11). The question of whether M cells are used universally by all enteric viruses during pathogenesis or only by specific enteric viruses remains to be determined. Nonetheless, the induction of mucosal immunity via targeting M cells is an area of intense research (62), and identifying surface molecules involved in MNV and reovirus uptake by M cells may reveal new strategies for the development of mucosal vaccines. Moreover, knowledge that M cells are used as entry portals by at least some enteric virus infections opens up potential therapeutic approaches for the use of noninfectious viral particles as drug delivery vehicles to prevent or treat infectious diseases.
ACKNOWLEDGMENTS
We thank Jason Iskarpatyoti (Vanderbilt University, Nashville, TN) for help with the reovirus experiments and Marta J. Gonzalez-Hernandez (University of Michigan, Ann Arbor, MI), Jeffrey W. Perry (University of Michigan, Ann Arbor, MI), and Nicholas Mantis (Wadsworth Center, Albany, NY) for critical readings of the manuscript. We are also grateful to Nicholas Mantis for providing the template for Fig. 7. Furthermore, we thank the Microscopy & Image Analysis Laboratory (MIL) and the Center for Live-Cell Imaging (CLCI) Cores at the University of Michigan for help with microscopy imaging analysis.
This work was funded by start-up funds from the University of Michigan and NIH grants AI080611 and AI103961 to C.E.W. M.B.G.-H. was funded by a University of Michigan Experimental Immunology Training Grant (NIH T32 A1007413-16), a Molecular Mechanisms of Microbial Pathogenesis Training Grant (NIH T32 A1007528), and the Herman and Dorothy Miller Fund for Innovative Immunology Research. I.R.W. was funded by NIH R01 DK64730. H.Y. was funded by a grant-in-aid (S1201013) from the MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2012 to 2017. Additional funding was provided by Public Health Service award R37 AI38296 and the Elizabeth B. Lamb Center for Pediatric Research to T.S.D. and Public Health Service awards CA68485 for the Vanderbilt-Ingram Cancer Center and DK20593 for the Vanderbilt Diabetes Research and Training Center.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 2 April 2014
REFERENCES
- 1.Starkey WG, Candy DC, Thornber D, Collins J, Spencer AJ, Osborne MP, Stephen J. 1990. An in vitro model to study aspects of the pathophysiology of murine rotavirus-induced diarrhoea. J. Pediatr. Gastroenterol. Nutr. 10:361–370. 10.1097/00005176-199004000-00017 [DOI] [PubMed] [Google Scholar]
- 2.Wolf JL, Rubin DH, Finberg R, Kauffman RS, Sharpe AH, Trier JS, Fields BN. 1981. Intestinal M cells: a pathway for entry of reovirus into the host. Science 212:471–472. 10.1126/science.6259737 [DOI] [PubMed] [Google Scholar]
- 3.Sicinski P, Rowinski J, Warchol JB, Jarzabek Z, Gut W, Szczygiel B, Bielecki K, Koch G. 1990. Poliovirus type 1 enters the human host through intestinal M cells. Gastroenterology 98:56–58 [DOI] [PubMed] [Google Scholar]
- 4.Knoop KA, Miller MJ, Newberry RD. 2013. Transepithelial antigen delivery in the small intestine: different paths, different outcomes. Curr. Opin. Gastroenterol. 29:112–118. 10.1097/MOG.0b013e32835cf1cd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mantis NJ, Cheung MC, Chintalacharuvu KR, Rey J, Corthesy B, Neutra MR. 2002. Selective adherence of IgA to murine Peyer's patch M cells: evidence for a novel IgA receptor. J. Immunol. 169:1844–1851 http://www.jimmunol.org/content/169/4/1844 [DOI] [PubMed] [Google Scholar]
- 6.Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, Kadokura K, Tobe T, Fujimura Y, Kawano S, Yabashi A, Waguri S, Nakato G, Kimura S, Murakami T, Iimura M, Hamura K, Fukuoka S, Lowe AW, Itoh K, Kiyono H, Ohno H. 2009. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature 462:226–230. 10.1038/nature08529 [DOI] [PubMed] [Google Scholar]
- 7.Clark MA, Jepson MA, Simmons NL, Booth TA, Hirst BH. 1993. Differential expression of lectin-binding sites defines mouse intestinal M-cells. J. Histochem. Cytochem. 41:1679–1687. 10.1177/41.11.7691933 [DOI] [PubMed] [Google Scholar]
- 8.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459:262–265. 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- 9.Knoop KA, Kumar N, Butler BR, Sakthivel SK, Taylor RT, Nochi T, Akiba H, Yagita H, Kiyono H, Williams IR. 2009. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J. Immunol. 183:5738–5747. 10.4049/jimmunol.0901563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor RT, Patel SR, Lin E, Butler BR, Lake JG, Newberry RD, Williams IR. 2007. Lymphotoxin-independent expression of TNF-related activation-induced cytokine by stromal cells in cryptopatches, isolated lymphoid follicles, and Peyer's patches. J. Immunol. 178:5659–5667 http://www.jimmunol.org/content/178/9/5659 [DOI] [PubMed] [Google Scholar]
- 11.Kraehenbuhl JP, Neutra MR. 2000. Epithelial M cells: differentiation and function. Annu. Rev. Cell Dev. Biol. 16:301–332. 10.1146/annurev.cellbio.16.1.301 [DOI] [PubMed] [Google Scholar]
- 12.Neutra MR, Mantis NJ, Frey A, Giannasca PJ. 1999. The composition and function of M cell apical membranes: implications for microbial pathogenesis. Semin. Immunol. 11:171–181. 10.1006/smim.1999.0173 [DOI] [PubMed] [Google Scholar]
- 13.Corr SC, Gahan CC, Hill C. 2008. M-cells: origin, morphology and role in mucosal immunity and microbial pathogenesis. FEMS Immunol. Med. Microbiol. 52:2–12. 10.1111/j.1574-695X.2007.00359.x [DOI] [PubMed] [Google Scholar]
- 14.Sansonetti PJ, Arondel J, Cantey JR, Prevost MC, Huerre M. 1996. Infection of rabbit Peyer's patches by Shigella flexneri: effect of adhesive or invasive bacterial phenotypes on follicle-associated epithelium. Infect. Immun. 64:2752–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen VB, Harty JT, Jones BD. 1998. Interactions of the invasive pathogens Salmonella typhimurium, Listeria monocytogenes, and Shigella flexneri with M cells and murine Peyer's patches. Infect. Immun. 66:3758–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones BD, Ghori N, Falkow S. 1994. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer's patches. J. Exp. Med. 180:15–23. 10.1084/jem.180.1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Autenrieth IB, Firsching R. 1996. Penetration of M cells and destruction of Peyer's patches by Yersinia enterocolitica: an ultrastructural and histological study. J. Med. Microbiol. 44:285–294. 10.1099/00222615-44-4-285 [DOI] [PubMed] [Google Scholar]
- 18.Rol N, Favre L, Benyacoub J, Corthesy B. 2012. The role of secretory immunoglobulin A in the natural sensing of commensal bacteria by mouse Peyer's patch dendritic cells. J. Biol. Chem. 287:40074–40082. 10.1074/jbc.M112.405001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donaldson DS, Kobayashi A, Ohno H, Yagita H, Williams IR, Mabbott NA. 2012. M cell-depletion blocks oral prion disease pathogenesis. Mucosal Immunol. 5:216–225. 10.1038/mi.2011.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansman GS, Jiang XJ, Green KY. 2010. Caliciviruses molecular and cellular virology, 1st ed, vol 1 Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 21.Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW. 2004. Replication of norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2:e432. 10.1371/journal.pbio.0020432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW. 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578. 10.1126/science.1077905 [DOI] [PubMed] [Google Scholar]
- 23.Mumphrey SM, Changotra H, Moore TN, Heimann-Nichols ER, Wobus CE, Reilly MJ, Moghadamfalahi M, Shukla D, Karst SM. 2007. Murine norovirus 1 infection is associated with histopathological changes in immunocompetent hosts, but clinical disease is prevented by STAT1-dependent interferon responses. J. Virol. 81:3251–3263. 10.1128/JVI.02096-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ward JM, Wobus CE, Thackray LB, Erexson CR, Faucette LJ, Belliot G, Barron EL, Sosnovtsev SV, Green KY. 2006. Pathology of immunodeficient mice with naturally occurring murine norovirus infection. Toxicol. Pathol. 34:708–715. 10.1080/01926230600918876 [DOI] [PubMed] [Google Scholar]
- 25.Thackray LB, Wobus CE, Chachu KA, Liu B, Alegre ER, Henderson KS, Kelley ST, Virgin HW. 2007. Murine noroviruses comprising a single genogroup exhibit biological diversity despite limited sequence divergence. J. Virol. 81:10460–10473. 10.1128/JVI.00783-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith DB, McFadden N, Blundell RJ, Meredith A, Simmonds P. 2012. Diversity of murine norovirus in wild-rodent populations: species-specific associations suggest an ancient divergence. J. Gen. Virol. 93:259–266. 10.1099/vir.0.036392-0 [DOI] [PubMed] [Google Scholar]
- 27.Taube S, Perry JW, McGreevy E, Yetming K, Perkins C, Henderson K, Wobus CE. 2012. Murine noroviruses bind glycolipid and glycoprotein attachment receptors in a strain-dependent manner. J. Virol. 86:5584–5593. 10.1128/JVI.06854-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez-Hernandez MB, Liu T, Blanco LP, Auble H, Payne HC, Wobus CE. 2013. Murine norovirus transcytosis across an in vitro polarized murine intestinal epithelial monolayer is mediated by M-like cells. J. Virol. 87:12685–12693. 10.1128/JVI.02378-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forrest JC, Dermody TS. 2003. Reovirus receptors and pathogenesis. J. Virol. 77:9109–9115. 10.1128/JVI.77.17.9109-9115.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schiff LA, Nibert ML, Tyler KL. 2007. Orthoreoviruses and their replication, p 1853–1915 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 31.Tyler KL, McPhee DA, Fields BN. 1986. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science 233:770–774. 10.1126/science.3016895 [DOI] [PubMed] [Google Scholar]
- 32.Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71. 10.1016/j.chom.2008.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolf JL, Kauffman RS, Finberg R, Dambrauskas R, Fields BN, Trier JS. 1983. Determinants of reovirus interaction with the intestinal M cells and absorptive cells of murine intestine. Gastroenterology 85:291–300 [PubMed] [Google Scholar]
- 34.Helander A, Silvey KJ, Mantis NJ, Hutchings AB, Chandran K, Lucas WT, Nibert ML, Neutra MR. 2003. The viral sigma1 protein and glycoconjugates containing alpha2-3-linked sialic acid are involved in type 1 reovirus adherence to M cell apical surfaces. J. Virol. 77:7964–7977. 10.1128/JVI.77.14.7964-7977.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bass DM, Trier JS, Dambrauskas R, Wolf JL. 1988. Reovirus type I infection of small intestinal epithelium in suckling mice and its effect on M cells. Lab. Investig. 58:226–235 [PubMed] [Google Scholar]
- 36.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC [Google Scholar]
- 37.Gonzalez-Hernandez MB, Bragazzi Cunha J, Wobus CE. 2012. Plaque assay for murine norovirus. J. Vis. Exp. 2012:e4297. 10.3791/4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200. 10.1016/j.virol.2009.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boehme KW, Ikizler M, Kobayashi T, Dermody TS. 2011. Reverse genetics for mammalian reovirus. Methods 55:109–113. 10.1016/j.ymeth.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 276:2200–2211. 10.1074/jbc.M004680200 [DOI] [PubMed] [Google Scholar]
- 41.Perry JW, Wobus CE. 2010. Endocytosis of murine norovirus 1 into murine macrophages is dependent on dynamin II and cholesterol. J. Virol. 84:6163–6176. 10.1128/JVI.00331-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM. 2007. Imaging poliovirus entry in live cells. PLoS Biol. 5:e183. 10.1371/journal.pbio.0050183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker ES, Luckner SR, Krause KL, Lambden PR, Clarke IN, Ward VK. 2012. Inherent structural disorder and dimerisation of murine norovirus NS1-2 protein. PLoS One 7:e30534. 10.1371/journal.pone.0030534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fleeton MN, Contractor N, Leon F, Wetzel JD, Dermody TS, Kelsall BL. 2004. Peyer's patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J. Exp. Med. 200:235–245. 10.1084/jem.20041132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Becker MM, Goral MI, Hazelton PR, Baer GS, Rodgers SE, Brown EG, Coombs KM, Dermody TS. 2001. Reovirus ςNS protein is required for nucleation of viral assembly complexes and formation of viral inclusions. J. Virol. 75:1459–1475. 10.1128/JVI.75.3.1459-1475.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Padmanabhan P, Grosse J, Asad AB, Radda GK, Golay X. 2013. Gastrointestinal transit measurements in mice with 99mTc-DTPA-labeled activated charcoal using NanoSPECT-CT. EJNMMI Res. 3:60. 10.1186/2191-219X-3-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nice TJ, Strong DW, McCune BT, Pohl CS, Virgin HW. 2013. A single-amino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. J. Virol. 87:327–334. 10.1128/JVI.01864-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bocharov G, Ludewig B, Bertoletti A, Klenerman P, Junt T, Krebs P, Luzyanina T, Fraser C, Anderson RM. 2004. Underwhelming the immune response: effect of slow virus growth on CD8+-T-lymphocyte responses. J. Virol. 78:2247–2254. 10.1128/JVI.78.5.2247-2254.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taube S, Perry JW, Yetming K, Patel SP, Auble H, Shu L, Nawar HF, Lee CH, Connell TD, Shayman JA, Wobus CE. 2009. Ganglioside-linked terminal sialic acid moieties on murine macrophages function as attachment receptors for murine noroviruses. J. Virol. 83:4092–4101. 10.1128/JVI.02245-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buller CR, Moxley RA. 1988. Natural infection of porcine ileal dome M cells with rotavirus and enteric adenovirus. Vet. Pathol. 25:516–517. 10.1177/030098588802500616 [DOI] [PubMed] [Google Scholar]
- 51.Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, Littman DR, Reinecker HC. 2005. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307:254–258. 10.1126/science.1102901 [DOI] [PubMed] [Google Scholar]
- 52.Lelouard H, Fallet M, de Bovis B, Meresse S, Gorvel JP. 2012. Peyer's patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology 142:592–601. 10.1053/j.gastro.2011.11.039 [DOI] [PubMed] [Google Scholar]
- 53.Totsuka T, Kanai T, Nemoto Y, Tomita T, Okamoto R, Tsuchiya K, Nakamura T, Sakamoto N, Akiba H, Okumura K, Yagita H, Watanabe M. 2009. RANK-RANKL signaling pathway is critically involved in the function of CD4+CD25+ regulatory T cells in chronic colitis. J. Immunol. 182:6079–6087. 10.4049/jimmunol.0711823 [DOI] [PubMed] [Google Scholar]
- 54.Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L. 1997. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390:175–179. 10.1038/36593 [DOI] [PubMed] [Google Scholar]
- 55.Tomov VT, Osborne LC, Dolfi DV, Sonnenberg GF, Monticelli LA, Mansfield K, Virgin HW, Artis D, Wherry EJ. 2013. Persistent enteric murine norovirus infection is associated with functionally suboptimal virus-specific CD8 T cell responses. J. Virol. 87:7015–7031. 10.1128/JVI.03389-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bharhani MS, Grewal JS, Pilgrim MJ, Enocksen C, Peppler R, London L, London SD. 2005. Reovirus serotype 1/strain Lang-stimulated activation of antigen-specific T lymphocytes in Peyer's patches and distal gut-mucosal sites: activation status and cytotoxic mechanisms. J. Immunol. 174:3580–3589 http://www.jimmunol.org/content/174/6/3580 [DOI] [PubMed] [Google Scholar]
- 57.Bass DM, Bodkin D, Dambrauskas R, Trier JS, Fields BN, Wolf JL. 1990. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J. Virol. 64:1830–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amerongen HM, Wilson GA, Fields BN, Neutra MR. 1994. Proteolytic processing of reovirus is required for adherence to intestinal M cells. J. Virol. 68:8428–8432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Excoffon KJ, Guglielmi KM, Wetzel JD, Gansemer ND, Campbell JA, Dermody TS, Zabner J. 2008. Reovirus preferentially infects the basolateral surface and is released from the apical surface of polarized human respiratory epithelial cells. J. Infect. Dis. 197:1189–1197. 10.1086/529515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chieppa M, Rescigno M, Huang AY, Germain RN. 2006. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med. 203:2841–2852. 10.1084/jem.20061884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vallon-Eberhard A, Landsman L, Yogev N, Verrier B, Jung S. 2006. Transepithelial pathogen uptake into the small intestinal lamina propria. J. Immunol. 176:2465–2469 http://www.jimmunol.org./content/176/4/2465.long [DOI] [PubMed] [Google Scholar]
- 62.Jepson MA, Clark MA, Hirst BH. 2004. M cell targeting by lectins: a strategy for mucosal vaccination and drug delivery. Adv. Drug Deliv. Rev. 56:511–525. 10.1016/j.addr.2003.10.018 [DOI] [PubMed] [Google Scholar]