

ABSTRACT
Herpes simplex virus 1 (HSV-1) can undergo a productive infection in nonneuronal and neuronal cells such that the genes of the virus are transcribed in an ordered cascade. HSV-1 can also establish a more quiescent or latent infection in peripheral neurons, where gene expression is substantially reduced relative to that in productive infection. HSV mutants defective in multiple immediate early (IE) gene functions are highly defective for later gene expression and model some aspects of latency in vivo. We compared the expression of wild-type (wt) virus and IE gene mutants in nonneuronal cells (MRC5) and adult murine trigeminal ganglion (TG) neurons using the Illumina platform for cDNA sequencing (RNA-seq). RNA-seq analysis of wild-type virus revealed that expression of the genome mostly followed the previously established kinetics, validating the method, while highlighting variations in gene expression within individual kinetic classes. The accumulation of immediate early transcripts differed between MRC5 cells and neurons, with a greater abundance in neurons. Analysis of a mutant defective in all five IE genes (d109) showed dysregulated genome-wide low-level transcription that was more highly attenuated in MRC5 cells than in TG neurons. Furthermore, a subset of genes in d109 was more abundantly expressed over time in neurons. While the majority of the viral genome became relatively quiescent, the latency-associated transcript was specifically upregulated. Unexpectedly, other genes within repeat regions of the genome, as well as the unique genes just adjacent the repeat regions, also remained relatively active in neurons. The relative permissiveness of TG neurons to viral gene expression near the joint region is likely significant during the establishment and reactivation of latency.
IMPORTANCE During productive infection, the genes of HSV-1 are transcribed in an ordered cascade. HSV can also establish a more quiescent or latent infection in peripheral neurons. HSV mutants defective in multiple immediate early (IE) genes establish a quiescent infection that models aspects of latency in vivo. We simultaneously quantified the expression of all the HSV genes in nonneuronal and neuronal cells by RNA-seq analysis. The results for productive infection shed further light on the nature of genes and promoters of different kinetic classes. In quiescent infection, there was greater transcription across the genome in neurons than in nonneuronal cells. In particular, the transcription of the latency-associated transcript (LAT), IE genes, and genes in the unique regions adjacent to the repeats persisted in neurons. The relative activity of this region of the genome in the absence of viral activators suggests a more dynamic state for quiescent genomes persisting in neurons.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) productively replicates in nonneuronal cells at peripheral sites of individuals, where it gains access to nerve endings of the peripheral nervous system. In sensory neurons, the virus can either replicate or establish lifelong latency, where the viral genome is relatively quiescent compared to in productive infection. Therefore, the viral genome can be expressed to produce progeny virions in both nonneuronal and neuronal cells, but it can also establish a reversible silent state in some neuron populations. As some studies suggest (1, 2), there may be differences in how the genome is expressed in neuronal and nonneuronal cells; however, some aspects of the former study may be explained by viral spread in vivo (3).
The transcription of HSV genes in nonneuronal cells by RNA polymerase II (pol II) (4) is sequentially and coordinately regulated (5, 6), generally being described as a cascade where three classes of genes are expressed, the immediate early (IE), early (E), and late (L) genes. IE gene transcription is activated in the absence of prior viral protein synthesis by VP16 present in the infecting virion (7–9). The products of IE genes are required for the transcription of the E genes, which encode the DNA synthetic machinery. IE and E proteins along with viral DNA replication are required for efficient late (L) transcription, the protein products of which mostly comprise the virion structure or are required for its assembly.
Promoters for the genes of the 3 kinetic classes are generally thought to share common features. IE gene promoters contain a TATA box (10, 11), elements that are responsive to VP16 in the incoming virion, and upstream binding sites for cellular transcription factors (7, 8, 12). Early gene promoters that have been characterized contain a TATA box and upstream binding sites for cellular transcription factors, such as found in the thymidine kinase (tk) promoter (13, 14). Late genes that are tightly dependent on viral DNA synthesis contain a TATA box and an initiator element (INR) (15) at the start sites of transcription (16). Despite these similarities, genes of a particular class may be expressed quite differently.
While viral cis- and trans-acting elements are a determinant of how individual viral genes are expressed, the transcriptional environment in different cell types likely influences the expression of the genome as well. When HSV enters sensory neurons, gene expression may be repressed, and the virus can establish a latent infection. The genome of the latent virus persists in a state repressed by heterochromatin (17, 18), where the predominant gene that is expressed is the latency-associated transcript (LAT) (19). The genome can periodically reactivate to produce new virus by a poorly understood process that may involve the initial low-level dysregulated expression of the genome (20, 21).
We and others have utilized a system that employs viral mutants which are defective for the expression of IE genes to model some aspects of latency in vitro (22, 23). d109 is a mutant that does not express the IE proteins and is nontoxic to most cells in culture, and its genome is relatively quiescent in infected Vero cells or diploid human fibroblasts (23). d109 genomes persist in an endless form in cultured cells (24), as does latent virus in vivo (25). Moreover, the expression of select viral genes from d109 genomes persisting in human fibroblasts is several orders of magnitude less than that of genes from wild-type (wt) virus, and the genomes are bound by heterochromatin (26, 27) as they are in latency. While it is reasonable to pursue this model of latency given the similarities with latency in vivo, the expression of mutants such as d109 in cells where latency is naturally established has not been studied in detail.
The intent of this study was to simultaneously quantify the expression of all the viral genes during productive infection with wt virus and during quiescent infection of nonneuronal (diploid fibroblasts) and neuronal (adult trigeminal neurons) cells to provide insight into (i) what determines the kinetics of expression of individual viral genes, (ii) neuron-specific differences in viral gene expression, and (iii) the expression of the viral genome as it persists in trigeminal ganglion (TG) neurons. Previous approaches to simultaneously quantify the expression of the viral genes have used hybridization to microarrays of viral sequences representing the viral genes (28). In the present study, we used RNA sequencing (RNA-seq) of cDNA derived from infected MRC5 cells and neurons. This method proved to be sufficiently sensitive to measure the expression of all the viral genes in the absence of viral activators in MRC5 cells and TG neurons.
MATERIALS AND METHODS
Ethics statement.
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal procedures were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh (protocol number 1201001B, most recently approved on 8 January 2014). Appropriate sedatives, anesthetics, and analgesics were used during handling and surgical manipulations to ensure minimal pain, suffering, and distress to animals.
Virus and cells.
Experiments were performed using human embryonic lung cells (MRC5) and primary mouse trigeminal neurons. MRC5 cells were obtained from and propagated as recommended by the American Type Culture Collection. Neurons were isolated from the trigeminal ganglia of 6-week-old CD1 mice, as described below. The viruses used in this study were d109 (ICP4− ICP0− ICP22− ICP27− ICP47−), d106 (ICP4− ICP22− ICP27− ICP47−), n12 (ICP4−), and the wild type virus KOS. The d109, d106, n12, and KOS viruses were propagated on FO6F1, E11, E5, and Vero cells, respectively (23, 29, 30).
Generation of neuronal cultures.
The trigeminal ganglia (TG) were dissected from 6-week-old CD1 mice, and neurons were isolated following a protocol similar to a previously described procedure (31). Neurons from the TG of 10 mice were used to seed a 24-well plate. Following establishment of cultures, the cells were maintained in Neurobasal-A medium supplemented with 2% B27, 1% penicillin-streptavidin, 0.5 mM l-glutamine, 50 ng/ml nerve growth factor (NGF), glial cell-derived neurotrophic factor (GDNF), and neurturin. The medium was changed once per week.
RNA isolation and reverse transcription (RT).
MRC-5 cells (2 × 106) in 60-mm plates were infected at a multiplicity of infection (MOI) of 10 PFU/cell of d109, d106, n12, or KOS. For the infection of neurons, 106 PFU of d109 or KOS was added to each well. The infection was performed at room temperature for 1 h with rocking every 10 min. Following infection, the inoculum was removed, the cells were washed, and fresh prewarmed medium was added. RNA was isolated with the Ambion RNaqueous-4PCR kit by following the included protocol. RNA was harvested at the indicated time points by removing the medium and adding 500 μl lysis/binding buffer to MRC5 cell cultures and by adding 100 μl lysis/binding buffer per well to neuronal cell culture. For the neuron cultures, 8 wells were pooled per sample. The following steps are the same for both neuron and MRC5 samples. Cells were collected and vortexed. An equal volume of 67% ethanol was added. The solution was applied to a filter spin column and centrifuged at 12,500 rpm for 1 min. The bound RNA was washed with wash buffers 1 and 2/3. The column was centrifuged dry to remove residual wash buffer from the column. RNA was eluted in two steps with 60 μl and 20 μl 75°C elution solution. The RNA was treated with DNase I at 37°C for 30 min to degrade remaining DNA. The DNase was inactivated with the supplied reagent.
To generate cDNA, two micrograms of total RNA was reverse transcribed in a reaction volume of 20 μl containing 0.5 μl Riboguard RNase inhibitor (40 U/μl), 1 μl 10 pM oligo(dT) primer, 0.5 μl Moloney murine leukemia virus (MMLV) high-performance reverse transcriptase (200 U/μl), 2 μl 100 mM dithiothreitol (DTT), 4 μl nucleotide mix (2.5 mM concentration of each deoxynucleoside triphosphate [dNTP]), and 2 μl 10× reaction buffer. The reaction tube was incubated at 65°C for 2 min to remove RNA secondary structure, and the RT reaction was carried out for 1 h at 37°C. Following completion of the RT reaction, the reaction tube was incubated at 85°C for 5 min to inactivate the reverse transcriptase.
qPCR.
RNA was diluted to 1 μg in 60 μl, and cDNA was diluted 1:6 in ultrapure water. A master mix containing 0.3 μl of each primer (stock concentration, 100 μM), 5 μl Applied Biosystems SYBR green mix with 1.0 μM 6-carboxy-X-rhodamine, and 0.4 μl of water for a total of 6 μl for each reaction was made. The primers used in this study were as follows: for gC, GTGACGTTTGCCTGGTTCCTGG and GCACGACTCCTGGGCCGTAACG, and for ICP27, GGGCCCTTTGACGCCGAGACCAGA and ATGGCCTTGGCGGTCGATGCG. The tk primers were the same primers used in previous studies (26). A 96-well plate was prepared with 6 μl master mix and 4 μl of sample or standard. All samples were run in triplicate. Purified viral DNA was used to create a standard curve of 1:10 dilutions from 4,000,000 to 40 copies, which covers the threshold cycle value range for the samples tested. Quantitative PCR (qPCR) was run on a StepOne Plus real-time PCR machine. The conditions for the run were 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. At the end of the run, a dissociation curve was completed to determine the purity of the amplified products. Results were analyzed using the StepOne v2.1 software from Applied Biosystems and compiled in Microsoft Excel.
RNA sequencing.
RNA was harvested using the Ambion kit as described above and prepared for sequencing following the Illumina TruSeq RNA Sample Preparation v2 Guide and accompanying kit (Illumina, San Diego, CA). The libraries were analyzed for length and concentration using the Agilent Bioanalyzer. Samples for an experiment were mixed in equimolar concentration and sent to the Tufts University Core Facility, Boston, MA, for sequencing.
Following sequencing, the Illumina reads were uploaded onto the Galaxy server for analysis (1, 2, 3). The reads were first groomed using the Fastq groomer function and trimmed from the 3′ end to remove low-quality score base reads. The reads were then mapped to the appropriate reference genome using the TopHat read mapper. For wt HSV, the strain KOS sequence was used (GenBank accession number JQ780693). For alignments, the terminal repeats (TRL and TRS) were removed since they are redundant with the internal repeats. For d109, the plasmids used to generate the virus were sequenced (23), and the changes were incorporated into the KOS sequence. The terminal repeats were also removed for the purpose of alignment. Cufflinks was used to quantify the mapped reads, outputting data in FPKM format. Since the number of reads and mapped reads varied between samples, the data were renormalized to the total number of reads.
RESULTS
RNA-seq analysis of productive infection in human fibroblasts and trigeminal neuronal cultures.
In order to determine the kinetics of expression of all the HSV genes in nonneuronal and neuronal cells, monolayers of MRC5 cells and cultures of adult TG neurons were infected with HSV-1 strain KOS. Total RNA was collected at different times postinfection and processed and subjected to Illumina sequencing as described in Materials and Methods. Illumina reads were processed and aligned to a modified KOS sequence. Figure 1A shows the percentage of total reads that map to the viral genome. The percentage of viral reads in the cell rapidly increases from less than 5% at 1 h postinfection (hpi) to over 60% by 6 hpi. This accumulation begins to plateau after 8 hpi, when approximately 80% of the total mRNA in the cell is virally derived. Similar results were obtained in trigeminal neuron cultures, although the percentage of viral reads was somewhat reduced compared to that in MRC5 cells. The results in MRC5 cells are consistent with previous studies using metabolically labeled RNA from infected HeLa cells and hybridization techniques (32), where it was found that 25 to 30% of newly synthesized nuclear poly(A)+ RNA in the nucleus was HSV specific at 2 to 3 hpi, and this increased to 33% between 5 and 6 hpi. The same study found that most of the polysome-associated poly(A)+ RNA was virus specific at both times. While the previous methods differ considerably from ours, the similarity with our results helps validate the approach.
FIG 1.

Evaluation of RNA-seq reads from infected MRC5 cells. Monolayers of MRC5 cells and cultures of adult trigeminal neurons were infected with HSV-1 strain KOS at an MOI of 10 PFU/cell. Total RNA was collected from MRC5 cells at 1, 2, 3, 4, 6, 8, 12, and 16 h postinfection and from TG neurons at 2, 4, and 8 h postinfection. cDNA was prepared and subjected to Illumina sequencing as described in Materials and Methods. Illumina reads were processed and aligned to a modified KOS sequence using the TopHat mapper in the Galaxy Cloud software package. (A) Percentage of sequencing reads mapping to the HSV genome relative to the total number of reads. (B) Comparison of kinetics of ICP27 (UL54) accumulation by RNA-seq and RT-PCR. (C) Comparison of kinetics of tk (UL23) accumulation by RNA-seq and RT-PCR. (D) Comparison of kinetics of gC (UL44) accumulation by RNA-seq and RT-PCR. The units for RT-PCR were cDNA copies per μg RNA as determined by comparison to standards using CsCl-purified viral genomic DNA. The units for RNA-seq are reads mapped to the viral genome per million total reads (viral plus cell) per kilobase pair (MR per MTR per kb).
To further validate our approach, we compared the amounts of signal from representative IE, E, and L genes as determined by RNA-seq and quantitative reverse transcription-PCR (RT-PCR). Figure 1B, C, and D represent the results from ICP27 (IE), tk or UL23 (E), and gC or UL44 (L), respectively. The accumulation of these three genes was similar when measured by RNA-seq and RT-PCR, demonstrating that RNA sequencing is sufficiently sensitive and accurate to detect changes in transcript abundance over time, with the obvious additional ability to quantitatively compare differences in the expression of many genes.
The reads that mapped to the viral genome from the RNA-seq of wt virus (KOS) infection were visualized using the Integrated Genomics Viewer (33). Figure 2 shows the reads as a function of time postinfection mapped to the KOS genome. Changes in the expression of individual genes over time can readily be seen and in many cases appear to reflect the established transcriptional cascade of viral genes. At the 1- and 2-h time points, signals for the immediate early genes, ICP27 (UL54), ICP0 (RL2), ICP22 (US1), and ICP47 (US12), are evident. The accumulation of ICP4 is not readily seen at this level of sensitivity. The three peaks between 111 and 115 kb map to the exons of the ICP0 locus, further validating the precision of RNA sequencing for HSV mRNA profiling.
FIG 2.

Locations of RNA-seq reads across the HSV genome as a function of time postinfection in MRC5 cells. The locations of reads for each of the time points is shown relative to a map of the mRNA for each of the HSV genes. The scales of the graphs were set to normalize to the number of total reads to enable qualitative comparison of transcript abundance between different time points. The reads and the genome are represented without the long and short terminal repeats since their sequences are represented within the internal repeats. The exons of RL2 and UL15 are also shown by darker shading.
At 4 h postinfection, the abundances of ICP27 and ICP22 mRNAs begin to decline. ICP0 abundance continues to increase throughout the infection but constitutes a lower proportion of expressed genes. This is consistent with previous observations (34). Concurrently, early genes begin to be expressed. Thymidine kinase (UL23), located between 37 and 39 kb, peaks between 3 and 4 h postinfection. Expression of DNA replication machinery genes ICP8 (UL29) and polymerase (UL30), between 49 and 52 kb and 53.5 and 57 kb, respectively, also peaks at a time similar to that for UL23. Multiple other early genes show similar expression patterns.
Replication of viral DNA begins around 4 h postinfection, which enables or enhances the expression of late genes. Many late genes, particularly in the region between the genes for gC (UL44) and UL49, predominate at this time. The peak for the mRNA encoding the tegument protein VP16 (UL48) appears between 94.2 and 95.7 kb. It is expressed as early as 2 h postinfection, but its abundance continues to increase following DNA replication. UL46 also shows a similar expression pattern. In contrast, UL44, a true late gene, is not expressed until 4 hpi, after which its expression increases significantly. UL10, another true late gene located between 14 and 15.4 kb, does not have a distinguishable peak until 4 h postinfection and becomes much more abundantly expressed later in infection.
We next aligned the reads from the infections of trigeminal neurons to the virus genome. The aligned reads of cDNAs from mRNAs derived from TG neurons infected for 2, 4 and 8 hpi are shown in Fig. 3. In addition to the reduced abundance of total viral transcripts in TG neurons, there were some notable differences in the accumulation of some individual transcripts over the 8 h. VP16 (UL48) accumulation is more abundant early in infection of TG neurons relative to MRC5 cells. It has been suggested that VP16 possess promoter elements conferring neuron-specific transcription (35). Additionally, there was a larger proportion of the immediate early transcript region expressed, including ICP4, ICP22, and ICP47.
FIG 3.

Locations of RNA-seq reads across the HSV genome as a function of time postinfection in cultured TG neurons, as described in the legend for Fig. 2.
The visual representations of the kinetics of mRNA accumulation (Fig. 2 and 3) provide a picture of the simultaneous transcription of all the HSV genes on the genome, but they lack the sensitivity to accurately reflect the transcription of many of the HSV genes. For example, an abundant transcript, such as UL44, may give rise to 2 ×105 reads, and its peak is clearly evident, while poorly transcribed genes, such as UL28 or UL36, are barely visually evident (Fig. 2). However, there are approximately 2 × 107 total reads in each of the multiplexed samples in Fig. 2, providing sufficient sensitivity to quantify the poorly expressed genes. Therefore, the expression of a subset of immediate early, early, and late genes was quantified in MRC5 cells and neurons using the Cufflinks software package on the Galaxy server. The accumulation of transcripts from the IE genes in MRC5 cells and neurons is shown in Fig. 4A and B, respectively. The expression kinetics for ICP27 and ICP0 were quantitatively similar in both cells types. The kinetics of ICP4 expression were similar in the two cell types; however, ICP4 was 2- to 5-fold more abundant in neurons than in MRC5 cells. There was a considerable difference in the expression profile of ICP22 (US1). In MRC5 cells, ICP22 peaked at 4 hpi; however, transcript abundance continued to increase in neurons.
FIG 4.

Accumulation of HSV transcripts in MRC5 cells and trigeminal neurons. Quantification of IE (α) (A and B), E (β) (C and D), L (γ1) (E and F), and L (γ2) (G and H) transcripts in MRC5 cells (A, C, E, and G) and neurons (B, D, F, and H) is shown. The units for RNA-seq are reads mapped to the viral genome per million total reads (viral plus cell) per kilobase pair (MR per MTR per kb).
Genes specifying the proteins involved in viral DNA synthesis are considered early genes, as is the viral thymidine kinase gene. Figure 4C and D show the kinetics of expression of these genes in MRC5 cells and neurons, respectively. UL23, UL29, and UL30 followed the established expression profile for early genes, with an increase in mRNA abundance early in infection followed by a decrease after 3 to 4 hpi. UL42 was the most highly expressed DNA replication gene, reaching maximal expression by 6 hpi and with the expression subsequently remaining high through the duration of the time course. The number of UL42-specific reads indicates that the abundance of this mRNA comprised over 1% of the total polyadenylated RNA in the cell. The somewhat delayed peak in the accumulation of UL42 mRNA is consistent with earlier studies (36). Unlike those of immediate early genes, the expression patterns of these early genes in MRC5 cells and neurons were similar, although transcript abundance in neurons was less overall. The read abundance of UL5, UL8, UL52, and UL9 was considerably less than the others, and their accumulation continued to increase following replication of DNA, reaching their maximal levels late after infection. Their accumulation appears to be more consistent with late gene expression kinetics.
Late genes (γ) require DNA synthesis for maximum expression and continue to accumulate throughout infection. Some L genes, the γ1 genes, also have characteristics of E genes and are some of the most abundantly expressed genes of the virus. Figure 4E and F show the accumulation of 4 γ1 genes, UL19 (major capsid protein), UL27 (gB), UL48 (VP16), and US6 (gD), in MRC5 cells and trigeminal neurons, respectively. The transcripts for these genes began to accumulate at 2 hpi and continued to accumulate to relatively high levels. VP16 and gB in particular began to accumulate very early in neurons, each eventually comprising 0.4% (4 × 103 reads mapped to the viral genome per million total reads per kb [MR/MTR/kb]) of the total mRNA in the cell. γ2 genes require DNA synthesis for their transcription. The accumulation of transcripts for 9 γ2 genes in MRC5 cells and TG neurons is shown in Fig. 4G and H, respectively. Like the transcripts for early genes (Fig. 4C and D), the expression patterns of these late genes were similar in neurons and MRC5 cells. However, there was a large difference in the levels of accumulation of the different transcripts in this group of genes. The three most highly expressed late genes were UL44, UL45, and UL35. While displaying similar expression kinetics, the accumulation of UL10, UL22, UL34, UL37, UL38, and US8 was considerably less.
Immediate early gene mutants.
Immediate early proteins of HSV are required for the expression of the remainder of the HSV genome (6). Specifically, ICP4 is required for the expression of viral early and late genes (37–39). Mutants deleted in the ICP4 gene are defective for transcription beyond the IE phase (29, 40), such that the expression of a prototypic early gene, UL23 (tk) for example, is approximately two orders of magnitude less than that seen in wt virus infection (41). ICP0 promotes virus growth and reactivation from latency (42–44). The deletion of ICP0 from HSV already deleted for ICP4 results in the further restriction of viral gene expression (23, 26, 30). Abrogating the expression of all IE genes results in a virus that is not toxic to cell and persists for long periods of time (23) in a repressed heterochromatic state (26, 27) resembling that seen with wt virus in vivo (17, 45). To examine the expression across the entire genome of key IE gene mutants, MRC5 cells were infected with KOS, n12, d106, and d109 at an MOI of 10 PFU/cell for 4 h and processed for RNA-seq. n12 contains a nonsense mutation in the coding region of ICP4, rendering it nonfunctional (29). d106 is mutated such that ICP0 is the only intact IE gene, and d109 is defective for the expression of all the IE genes (23). The mapped reads from each virus relative to the KOS genome are shown on the same scale in Fig. 5A to illustrate the disparity in gene expression between the 4 different viruses.
FIG 5.

Transcript accumulation in IE mutant-infected MRC5 cells. MRC5 cells were infected with KOS, n12, d106, and d109 at an MOI of 10 PFU/cell for 4 h and processed for RNA-seq as described in Materials and Methods. (A) Locations of mapped reads relative to the KOS genome. The Illumina reads were aligned to the modified KOS sequence as before. The maximum on the y axis is 30,000 reads. (B) Quantification of the numbers of reads for select HSV genes.
Transcription of numerous genes can be seen across the entire KOS genome, while transcription of the n12 genome appears to be restricted to the UL39 and UL40 loci, ICP27 (UL54), ICP0 (RL2), ICP4 (RS1), ICP22 (US1), and ICP47 (US12). The levels of ICP0, ICP27, and ICP4 transcripts are considerably higher in n12-infected cells than in KOS-infected cells, consistent with the previously defined phenotype of n12 (29). The expression profiles in Fig. 5A of d106 and d109 are also consistent with their published phenotypes (23). However, this representation does not quantitatively describe the expression of the genome in the absence of the IE proteins. Considering that there were ∼20 million reads in each sample and the percent viral reads were 44, 13, 4.9, and 0.07, for KOS, n12, d106, and d109, respectively, then there are a considerable number of reads to accurately quantify the relatively low-abundance transcription across the genome in these mutant backgrounds. For example, while no signal is visible for d109 in Fig. 5A, there are still 1.4 × 104 viral reads in the d109 sample. Figure 5B shows the number of reads mapped to a particular viral gene per million total reads per kilobase pair (MR per MTR per kb) for selected viral genes in the different mutant backgrounds. The genes are for ICP22 (IE), tk (E), ICP8 (E), RR1 (E), dUTPase (E), US3 (L), VP5 (L), UL26 (L), and gB (L). Several observations can be made from the results. (i) IE genes (when present) are highly expressed in the absence of ICP4. (ii) In the absence of ICP4 (n12), early and late gene transcripts accumulate to 0.1% to 5.0% of the levels seen in wt virus infection. An exception is RR1 (UL39), the transcription of which is relatively independent of ICP4. (iii) Early and late gene expression is further reduced 3- to 5-fold in the d106 background at this time postinfection. (iv) In the absence of IE proteins, early and late transcription is 3 to 4 orders of magnitude less than that for wt virus at 4 hpi in MRC5 cells. For example the total number of reads for the tk mRNA is 3 × 105 and 1.5 × 102 for KOS and d109, respectively.
Expression from the d109 genome in MRC5 cells is further reduced with time due to the ongoing formation of heterochromatin (26). To examine how this repression affects expression across the viral genome and to further compare expression in MRC5 cells to that in trigeminal neurons in the absence of IE proteins, MRC5 cells and TG neuronal cultures were infected with d109 at an MOI of approximately 10 PFU/cell. mRNA was isolated at 4, 8, and 24 h and 7 days postinfection and analyzed by RNA-seq as before. The percentage of viral reads from the MRC5 samples decreased from 0.07% at 4 h to just under 0.01% at 7 days, while the percentage of viral reads in trigeminal neurons was 0.5% at 4 h and a little more than 0.1% at 7days (Fig. 6). Therefore, the number of viral reads was about 10-fold higher in d109-infected neurons than in d109-infected MRC5 cells, suggesting a more transcriptionally active genome in trigeminal neurons. This is in contrast to what was seen in KOS-infected cells, where the proportion of viral mRNA in neurons was less than that in MRC5 cells. The locations of the reads are shown in Fig. 7 aligned to a modified d109 genome. Unlike in Fig. 5A, the scale of the graphs was chosen to view the relatively low-level expression across the entire genome. As a consequence, the signals for UL39, UL40, the internal repeats, and GFP are off the scale. They are represented in Fig. 8. Regions where there were no reads are represented by gray areas.
FIG 6.
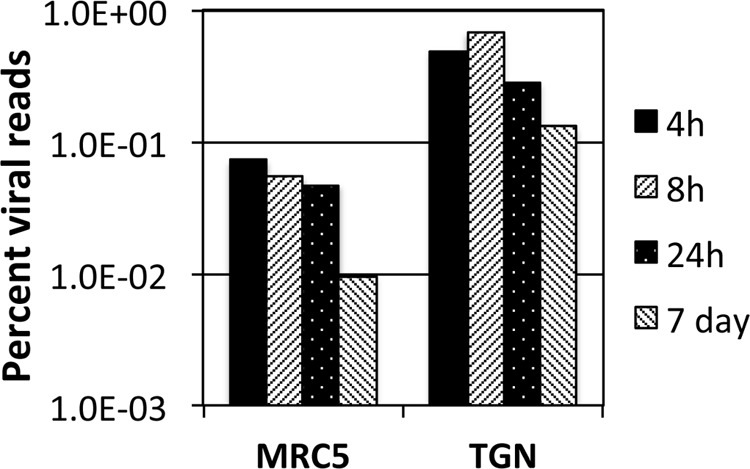
Percent viral reads in d109-infected MRC5 cells and neurons. The numbers of reads that mapped to the d109 genome were divided by the total number of reads and multiplied by 100.
FIG 7.

HSV transcripts synthesized in d109-infected MRC5 cells and TG neurons. MRC cells and cultures of TG neurons were infected with d109 (MOI = 10 PFU/cell) for 4 h, 8 h, 24 h, and 7 days. The RNA synthesized in the cells was analyzed by RNA-seq as before, and the reads were mapped to a modified d109 genome. The reads were aligned to the d109 genome. The GFP gene is shown in red in the deleted ICP27 locus. The maximum on the y axes of the graphs was set to 100 reads. The gray shaded areas indicate regions where there were no reads.
FIG 8.

Transcription of the internal repeat region from persisting genomes. (A) The RNA-seq reads from d109-infected TG neurons mapping to the region encoding the LAT promoter to US2 are shown relative to features in this region of the genome. The XbaI site at 111,151 marks the locus of the ICP0 deletion, and the site 113,549 marks the locus of the deletion that removed the VP16-responsive elements from the region. The numbers shown are in bp from the 5′ end of the modified d109 genome in the parental orientation. The maximum of the y axes for the 4- and 8-h graphs was set at 7,200 reads, while that for the 24-h and 7-day graphs was set at 1,800 reads. (B) The RNA-seq reads for the 1- and 7-day d109-infected TG neuron samples are shown in the region of the LAT TATA box, mRNA start sites, and ICP4 binding site. The maximum on the y axis is 100 reads. The beginning of the signal corresponds to the arrow to the left in the 7-day graph of panel A. (C) Quantification of the reads from the samples in Fig. 5 for select viral genes.
The accumulation of gene-specific reads can readily be seen. Consistent with the higher level of transcription overall, there was a general trend toward higher levels of individual transcripts in neurons than in MRC5 cells. For example, the levels of glycoprotein B (UL27), ICP8 (UL29), UL30, UL41, and UL42 were higher in neurons than in MRC5 cells, even at 7 days postinfection. While transcription across the d109 genome was detected in neurons at 7 days postinfection, no transcription was detected from many loci in MRC5 cells at this time, as indicated by the gray shaded regions. This is not due to a lack of persisting d109 genomes, since it can be demonstrated that the provision of ICP0 can quantitatively “reactivate” the quiescent d109 genomes in MRC5 cells in this system (27).
One interesting observation is the pattern of expression in neurons of genes just outside the long and short repeat regions. In the short unique region of the genome, expression of the US3/4, US5/6/7, and US8/9 clusters is evident, and it decreases over the course of 24 h. However, the abundances of the US3/4 and US8/9 clusters increase from 24 h to 7 days, as that of US5/6/7 decreases, as most of the gene do. Likewise UL1/2 and UL4/5 mRNAs decrease over the course of 24 h but increase at 7 days. This pattern is also in contrast to those of all the other HSV genes in UL.
In neurons, the pattern of expression of the genes adjacent to the long and short repeat regions is also seen within the repeats. Figure 8A shows the region of the d109 genome from the LAT promoter-proximal region through to US2. Figure 8A also shows the RNA-seq reads in this region from d109 in neurons. These signals were off the scale in Fig. 7. Note that the only place in this region where there were no reads was in the US1 intron. The expression in this region overall was considerably greater than that of genes in the unique long and short regions (with the exception of UL39 and GFP). While the numbers of reads in this region generally decreased over 24 h, they were sustained or increased from 24 h to 7 days. The signal just immediately downstream from the previously defined LAT mRNA start site that contains an ICP4 binding site (46, 47) increased considerably from 1 to 7 days postinfection. The LAT start site is indicated by the arrow to the left in Fig. 8A and by the 2 small arrows (previously defined by primer extension analysis [48]) in Fig. 8B. Figure 8B also shows the ICP4 binding site and the TATA box for the LAT transcript along with the RNA-seq reads for d109 in neurons at 1 and 7 days postinfection. The elevated RNA-seq signal begins precisely at the previously described LAT mRNA start site, strongly indicating that it is transcribed from the LAT promoter.
The abundances of transcripts from this region of d109 in MRC5 cells and neurons were quantified and are plotted in Fig. 8C. The XbaI site at 111,151 (Fig. 8A) marks the locus of the ICP0 deletion in d109 (23), and therefore the sequence from the LAT mRNA start site to the XbaI site (111,151) is colinear with the 5′ end of the LAT primary transcript (49). The region quantified and designated LAT in Fig. 8C corresponds to the region from the LAT mRNA start site to 1 kb downstream. The transcription of this region is quite complicated, with the RL1 gene and the L/STs (orf P) transcripts antiparallel to each other. The L/STs TATA box, mRNA start site, and ICP4 binding sites are indicated (Fig. 8A) (50, 51). For the sake of clarity and because there is a distinct increase in reads on the L/STs start site (right arrow), the quantification in Fig. 8C represented as L/STs is the reads from the L/STs start site to 1 kb downstream. Also quantified in Fig. 8C are ΔRS1, the second exons of US1 and US12, GFP, UL39, and UL27. Figure 8C shows several results. (i) The abundance of all the transcripts in this region is higher in TG neurons than in MRC5 cells. (ii) The abundance of all the HSV transcripts in MRC5 cells decreases with time such that the maximum abundance is less than 1 in 106 at 7 days (US1) and LAT is undetectable (<1 in 2 × 107). (iii) In neurons, the abundances of UL27, UL39, and GFP decrease from 1 to 7 days by ∼10-fold, whereas the abundances of ΔRS1, US1 and US12, and LAT all increase from 1 to 7 days postinfection, with LAT increasing the most. (iv) The numbers of reads for LAT, L/STs, ΔRS1, US1, and US12 reflect a transcript abundance for each that is between 1 in 3 × 103 and 1 in 104 of all transcripts in the cell. This suggest that this region of the genome is considerable more transcriptionally active in TG neurons than in MRC5 cells, despite repression of the remainder of the viral genome, even in the absence of viral activators.
DISCUSSION
Next-generation sequencing has emerged as a high-throughput methodology to quantify changes in transcriptome-wide gene expression. RNA sequencing has been shown to be more sensitive for detection of low-abundance transcripts and to have a wider range of detection than traditional microarrays (52). The goal of this study was to determine how the presence and absence of immediate early proteins impacted viral gene expression in neuronal cells (TG neurons) and nonneuronal cells (MRC5). In particular, we were interested in the expression from a virus that establishes a persistent quiescent infection in trigeminal neurons as a model for what might be occurring during latency. From the RNA-seq experiments performed, we have made the following findings: (i) during productive infection, there are deviations from the established cascade paradigm for early genes; (ii) the level of expression of late genes is largely a function of the TATA box and INR; (iii) in the absence of immediate early proteins, transcription from the viral genome occurs at low levels independent of kinetic class; (iv) neurons are less restrictive to viral transcription than MRC5 cells; and (v) genes near the joint/repeat region of the genome, and the LAT locus in particular, are more transcriptionally active during persistence in neurons.
Productive infection, gene regulation, and promoter architecture.
The promoters of HSV early genes are characterized by the presence of upstream elements for cellular transcription factors. Several early genes are quantified in Fig. 4C and D. ICP8 and tk were abundantly expressed. The promoter for the tk gene contains three distinct upstream elements, two Sp1 binding sites and one CCAAT element (13). ICP8 also contains two Sp1 binding sites. The DNA polymerase promoter contains a degenerate TATA box and one Sp1 binding site (53). Expression of this transcript is reduced approximately 4-fold relative to that of tk and 2-fold relative to that of ICP8. UL42 was the most abundantly expressed gene, comprising approximately 1% of the total cellular RNA and peaking at 6 h postinfection, consistent with previous results (36). The abundances of these 4 transcripts decreased late after infection.
The mRNAs for the components of the helicase-primase complex UL5, UL8, and UL52 (54) and the origin binding protein UL9 (55) were expressed with similar kinetics and intensity during early time points prior to DNA replication. Expression of these four transcripts slowly accumulated throughout the time course to low levels. The UL8 and UL9 mRNA start sites have been mapped (56). UL8 and UL9 both have degenerate TATA boxes, and UL8 has an Sp1 site 22 bp upstream of the TATA box. UL9 has been reported to have an upstream Sp1 site; however, none are evident in the KOS sequence (56). The UL5 and UL52 mRNA start sites have not been determined, making it difficult to localize transcription signals; however, the only potential TATA boxes within 400 bp of these genes consist of 4 A and T base pairs.
Therefore, genes with promoters possessing good TATA boxes and multiple sites for upstream factors are abundantly expressed early after infection, and their decline later in infection is pronounced. This represents the early-gene paradigm. However, it is difficult to fit some genes whose products are clearly required for DNA synthesis into this paradigm. Those with poor TATA boxes, lacking upstream elements, are expressed at a low level, their decline later in infection is not as pronounced, and in some cases (UL52 and UL9) accumulation continues at a low level. This is reminiscent of some previous studies with mutant and chimeric promoters (48, 57, 58).
Late genes (γ2) require viral DNA synthesis for their expression, and their promoters consist of a TATA box and an initiator element (59). The promoter elements of the late genes represented in Fig. 4G and H are shown in Table 1. The expression of these genes varies considerably. There are 4 transcripts with abundances above 10,000 mapped fragments per million reads, representing the most highly expressed late genes. The three most highly expressed genes, UL35, UL45, and UL44, have a TATA box that completely matches the consensus sequence and an initiator element with a perfect match to the first 4 bases of the consensus. UL10 is expressed to a lesser extent and has a mismatch in the TATA box. Genes expressed to levels below 10,000 have multiple mismatches the TATA box, the first 4 nucleotides of the initiator element, or a combination of both. There was no apparent correlation to the downstream activation sequence (DAS), though it likely plays an important role on a gene-by-gene basis. Therefore, the expression of late genes varies considerably, and their levels of transcription correlate with the degree of match to both the TATA box and INR element consensus, suggesting that the ability of TFIID to bind to the promoter is the main determinant in late gene transcription.
TABLE 1.
| Gene | Sequencea |
||
|---|---|---|---|
| TATA box | INR | DAS | |
| Consensus | TATAAA | YYANWYY | GAGNNYGAG |
| UL10 | TAAAA | GCACAC | None |
| UL22 | AATAAAA | TCATAAA | GAGTGCCAG |
| UL34 | TATAAA | GCAGAC | CACGGCGAG |
| UL35 | TATAAA | CTAATCG | None |
| UL37 | TATAACA | TCGCAAC | GAGGGCCGC |
| UL38 | TTTAAA | CCAGTCG | GAGGCGGTAG |
| UL44 | TATAAA | CTACCGA | GAGGCCGCA |
| UL45 | TATAAA | TCATCCT | GGCGTGGAG |
| US8 | ATTTAA | CTTCTGG | GAGCCCAAC |
Bold indicates mismatches from the consensus sequence.
γ1 genes are transcribed early after infection, and their synthesis increases after DNA replication. Four γ1 genes are presented in Fig. 4E and F. This group of genes is more uniform in their expression relative to the other groups of genes and is represented by a minimum of 1% of the total reads (virus plus cell). The early and sustained high level of expression is most likely due to the presence of a TATA box INR and binding sites for upstream factors in their promoters (60). The binding sites for upstream factors enable expression early after infection, and the INR allows for the continued expression late after infection.
We hypothesize that late after infection, cellular activator function and the activity of ICP4 on early promoters become less active. We have shown that Sp1 gets phosphorylated after DNA replication and is less active in vitro (61) and that TFIIA, which is required for the activation of tk by ICP4 in vitro, decreases late after infection (62, 63). In addition, upstream activators require a form of the mediator complex for activation, and we have shown that as infection proceeds, ICP4 interacts with a form of the mediator complex that is often associated with repression of transcription (64). Therefore, weaker promoters, such as UL8 or UL9, which are not activated to the same extent by upstream factors such as Sp1, might not show as dramatic a decrease, or any at all, in activity late after infection. The sustained high-level activity of late genes is most likely a function of the ability of ICP4 to activate transcription through the INR sequence present at the start sites of late genes (65, 66).
Gene expression during quiescent infection.
HSV mutants engineered to not express viral activators can establish a quiescent infection in cells in culture such that their genomes are transcriptionally repressed and persist in cells (22, 23, 67). Heterochromatin forms on d109 genomes persisting in MRC5 cells (26, 27), reminiscent of the structure of latent viral genomes (17, 68). We have also shown that d109 genomes may not be as tightly repressed in TG neurons as in nonneuronal cells in culture, based on GFP transgene expression and the ability to reactivate quiescent virus (69). Therefore, we sought to compare the expression of the entire d109 genome in MRC5 cells, as a model for nonneuronal cells, and TG neurons.
Trigeminal neurons were significantly more permissive for the expression of genes across the persisting d109 genome than MRC5 cells (Fig. 7 and 8). The MRC5 cells are presumably more homogeneous than these TG cultures, which are known to contain populations of neurons that differ in their permissiveness for LAT expression (31). Therefore, it is possible that some infected neurons are more permissive for HSV gene expression than others. Moreover, expression bore little relation to kinetic class. This “dysregulated” pattern of HSV gene expression has been seen in two different systems when reactivation stimuli were applied to latent HSV (20, 21). In our experiments, the persisting genomes are devoid of genes that are thought to activate viral early and late genes, and since the infected neuronal cultures are maintained in NGF, known reactivation stimuli were not applied.
The elevated level of transcription of persisting vial genomes in neurons relative to nonneuronal cells most likely is due to the different transcriptional environment in neurons. In mammalian cells, there are three different histone H3 variants, H3.1 H3.2, and H3.3. During differentiation of neurons, the abundances of variants H3.1 and H3.2 decrease, while H3.3 accumulates (70). H3.1 contains a combination of both activating and repressive modifications, H3.2 is enriched in inactive marks, and H3.3 is enriched in activating markers and is localized to transcriptionally active regions (71–73). The association of H3.3 with the viral genome has been shown to facilitate lytic gene expression (74). Perhaps H3.3 facilitates transcription from d109 genomes in TG neurons, while H3.1 and H3.2 restrict gene expression in MRC5 cells. This remains to be studied.
While the d109 genome in general is more transcriptionally active in TG neurons than in MRC5 cells, a large majority of the transcripts in neurons at 7 days postinfection map to loci around the joint regions and in the unique short region of the genome (Fig. 7 and 8). There are several possible explanations for this. (i) Neuron-specific promoters could be directing the expression of all these genes. With the exception of LAT, which is generally thought to possess a neuron-specific promoter, it is unlikely that all of these genes (UL1, UL2, UL4, UL5, US1, US3, US4, US8, US9, US12, RS1, and LAT) have neuron-specific promoters. The underlying basis for their enhanced expression more likely has to do with a general structural property of the region. (ii) Perhaps the high G+C content of the internal repeats relative to rest of the genome results in reduced binding of repressive chromatin in TG neurons. (iii) It is also possible that chromatin insulators, DNA elements that function to separate euchromatic and heterochromatic areas of the genome (75, 76), are involved. Seven distinct regions containing CTCF binding sites have been identified in the HSV genome (77). These are all located in the repeat region of the genome, and 5 remain in d109. Two specific CTCF binding sites surrounding the LAT enhancer have been shown to function as enhancer-blocking elements and maintain the localization of euchromatin to the LAT locus (78). The presence of these elements could explain the specific upregulation of LAT expression during persistence of d109 in neurons. Other insulator elements are located surrounding other immediate early genes near the repeat regions (79). The presence of these insulators could slow the accumulation of heterochromatin in the regions, enabling low-level transcription of IE genes in the absence of VP16.
The physiological consequences of the elevated low-level expression of genes in the joint region are not known. However, an intriguing possibility involves the interplay between HSV gene products and microRNAs (miRNAs) encoded in this region (80, 81). During latent infection, the primary latency-associated transcript serves as a precursor to several miRNAs (81), which may target ICP4 and ICP0 (82). The low-level expression of the IE genes and miRNAs in this region could provide the basis for the ability of the virus to reactivate in a regulated way, independent of viral activators.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01AI030612 and R01AI044821 to N.A.D. J.M.H. was supported by NIH training grant T32 AI049820.
Footnotes
Published ahead of print 9 April 2014
REFERENCES
- 1.Kosz-Vnenchak M, Jacobson J, Coen DM, Knipe DM. 1993. Evidence for a novel regulatory pathway for herpes simplex virus gene expression in trigeminal ganglion neurons. J. Virol. 67:5383–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nichol PF, Chang JY, Johnson EM, Jr, Olivo PD. 1996. Herpes simplex virus gene expression in neurons: viral DNA synthesis is a critical regulatory event in the branch point between the lytic and latent pathways. J. Virol. 70:5476–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pesola JM, Zhu J, Knipe DM, Coen DM. 2005. Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J. Virol. 79:14516–14525. 10.1128/JVI.79.23.14516-14525.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alwine JC, Steinhart WL, Hill CW. 1974. Transcription of herpes simplex type 1 DNA in nuclei isolated from infected HEp-2 and KB cells. Virology 60:302–307. 10.1016/0042-6822(74)90390-0 [DOI] [PubMed] [Google Scholar]
- 5.Honess RW, Roizman B. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honess RW, Roizman B. 1975. Regulation of herpesvirus macromolecular synthesis: sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc. Natl. Acad. Sci. U. S. A. 72:1276–1280. 10.1073/pnas.72.4.1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batterson W, Roizman B. 1983. Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J. Virol. 46:371–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell ME, Palfreyman JW, Preston CM. 1984. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 180:1–19. 10.1016/0022-2836(84)90427-3 [DOI] [PubMed] [Google Scholar]
- 9.Post LE, Mackem S, Roizman B. 1981. Regulation of alpha genes of herpes simplex virus: expression of chimeric genes produced by fusion of thymidine kinase with alpha gene promoters. Cell 24:555–565. 10.1016/0092-8674(81)90346-9 [DOI] [PubMed] [Google Scholar]
- 10.Gannon F, O'Hare K, Perrin F, LePennec JP, Benoist C, Cochet M, Breathnach R, Royal A, Garapin A, Cami B, Chambon P. 1979. Organisation and sequences at the 5′ end of a cloned complete ovalbumin gene. Nature 278:428–434. 10.1038/278428a0 [DOI] [PubMed] [Google Scholar]
- 11.Wasylyk B, Derbyshire R, Guy A, Molko D, Roget A, Téoule R, Chambon P. 1980. Specific in vitro transcription of conalbumin gene is drastically decreased by single-point mutation in T-A-T-A box homology sequence. Proc. Natl. Acad. Sci. U. S. A. 77:7024–7028. 10.1073/pnas.77.12.7024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackem S, Roizman B. 1982. Structural features of the herpes simplex virus alpha gene 4, 0, and 27 promoter-regulatory sequences which confer alpha regulation on chimeric thymidine kinase genes. J. Virol. 44:939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisenberg SP, Coen DM, McKnight SL. 1985. Promoter domains required for expression of plasmid-borne copies of the herpes simplex virus thymidine kinase gene in virus-infected mouse fibroblasts and microinjected frog oocytes. Mol. Cell. Biol. 5:1940–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coen DM, Weinheimer SP, McKnight SL. 1986. A genetic approach to promoter recognition during trans induction of viral gene expression. Science 234:53–59. 10.1126/science.3018926 [DOI] [PubMed] [Google Scholar]
- 15.Smale ST, Baltimore D. 1989. The “initiator” as a transcription control element. Cell 57:103–113. 10.1016/0092-8674(89)90176-1 [DOI] [PubMed] [Google Scholar]
- 16.Guzowski JF, Wagner EK. 1993. Mutational analysis of the herpes simplex virus type 1 strict late UL38 promoter/leader reveals two regions critical in transcriptional regulation. J. Virol. 67:5098–5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kubat NJ, Tran RK, McAnany P, Bloom DC. 2004. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 78:1139–1149. 10.1128/JVI.78.3.1139-1149.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. U. S. A. 102:16055–16059. 10.1073/pnas.0505850102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059. 10.1126/science.2434993 [DOI] [PubMed] [Google Scholar]
- 20.Du T, Zhou G, Roizman B. 2011. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc. Natl. Acad. Sci. U. S. A. 108:18820–18824. 10.1073/pnas.1117203108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 8:e1002540. 10.1371/journal.ppat.1002540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Preston CM, Nicholl MJ. 1997. Repression of gene expression upon infection of cells with herpes simplex virus type 1 mutants impaired for immediate-early protein synthesis. J. Virol. 71:7807–7813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samaniego LA, Neiderhiser L, DeLuca NA. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72:3307–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson SA, DeLuca NA. 2003. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc. Natl. Acad. Sci. U. S. A. 100:7871–7876. 10.1073/pnas.1230643100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rock DL, Fraser NW. 1983. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature 302:523–525. 10.1038/302523a0 [DOI] [PubMed] [Google Scholar]
- 26.Ferenczy MW, DeLuca NA. 2009. Epigenetic modulation of gene expression from quiescent herpes simplex virus genomes. J. Virol. 83:8514–8524. 10.1128/JVI.00785-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferenczy MW, DeLuca NA. 2011. Reversal of heterochromatic silencing of quiescent herpes simplex virus type 1 by ICP0. J. Virol. 85:3424–3435. 10.1128/JVI.02263-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stingley SW, Ramirez JJ, Aguilar SA, Simmen K, Sandri-Goldin RM, Ghazal P, Wagner EK. 2000. Global analysis of herpes simplex virus type 1 transcription using an oligonucleotide-based DNA microarray. J. Virol. 74:9916–9927. 10.1128/JVI.74.21.9916-9927.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeLuca NA, Schaffer PA. 1988. Physical and functional domains of the herpes simplex virus transcriptional regulatory protein ICP4. J. Virol. 62:732–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samaniego LA, Wu N, DeLuca NA. 1997. The herpes simplex virus immediate-early protein ICP0 affects transcription from the viral genome and infected-cell survival in the absence of ICP4 and ICP27. J. Virol. 71:4614–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP. 2011. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J. Virol. 85:6669–6677. 10.1128/JVI.00204-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stringer JR, Holland LE, Swanstrom RI, Pivo K, Wagner EK. 1977. Quantitation of herpes simplex virus type 1 RNA in infected HeLa cells. J. Virol. 21:889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14:178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinheimer SP, McKnight SL. 1987. Transcriptional and post-transcriptional controls establish the cascade of herpes simplex virus protein synthesis. J. Mol. Biol. 195:819–833. 10.1016/0022-2836(87)90487-6 [DOI] [PubMed] [Google Scholar]
- 35.Thompson RL, Preston CM, Sawtell NM. 2009. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog. 5:e1000352. 10.1371/journal.ppat.1000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodrich LD, Rixon FJ, Parris DS. 1989. Kinetics of expression of the gene encoding the 65-kilodalton DNA-binding protein of herpes simplex virus type 1. J. Virol. 63:137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watson RJ, Clements JB. 1980. A herpes simplex virus type 1 function continuously required for early and late virus RNA synthesis. Nature 285:329–330. 10.1038/285329a0 [DOI] [PubMed] [Google Scholar]
- 38.Preston CM. 1979. Control of herpes simplex virus type 1 mRNA synthesis in cells infected with wild-type virus or the temperature-sensitive mutant tsK. J. Virol. 29:275–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dixon RA, Schaffer PA. 1980. Fine-structure mapping and functional analysis of temperature-sensitive mutants in the gene encoding the herpes simplex virus type 1 immediate early protein VP175. J. Virol. 36:189–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLuca NA, McCarthy AM, Schaffer PA. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imbalzano AN, Coen DM, DeLuca NA. 1991. Herpes simplex virus transactivator ICP4 operationally substitutes for the cellular transcription factor Sp1 for efficient expression of the viral thymidine kinase gene. J. Virol. 65:565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leib DA, Coen DM, Bogard CL, Hicks KA, Yager DR, Knipe DM, Tyler KL, Schaffer PA. 1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 63:759–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sacks WR, Schaffer PA. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 61:829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stow ND, Stow EC. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67:2571–2585. 10.1099/0022-1317-67-12-2571 [DOI] [PubMed] [Google Scholar]
- 45.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 83:8182–8190. 10.1128/JVI.00712-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Batchelor AH, O'Hare P. 1990. Regulation and cell-type-specific activity of a promoter located upstream of the latency-associated transcript of herpes simplex virus type 1. J. Virol. 64:3269–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farrell MJ, Margolis TP, Gomes WA, Feldman LT. 1994. Effect of the transcription start region of the herpes simplex virus type 1 latency-associated transcript promoter on expression of productively infected neurons in vivo. J. Virol. 68:5337–5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rivera-Gonzalez R, Imbalzano AN, Gu B, Deluca NA. 1994. The role of ICP4 repressor activity in temporal expression of the IE-3 and latency-associated transcript promoters during HSV-1 infection. Virology 202:550–564. 10.1006/viro.1994.1377 [DOI] [PubMed] [Google Scholar]
- 49.Devi-Rao GB, Goodart SA, Hecht LM, Rochford R, Rice MK, Wagner EK. 1991. Relationship between polyadenylated and nonpolyadenylated herpes simplex virus type 1 latency-associated transcripts. J. Virol. 65:2179–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohenzky RA, Papavassiliou AG, Gelman IH, Silverstein S. 1993. Identification of a promoter mapping within the reiterated sequences that flank the herpes simplex virus type 1 UL region. J. Virol. 67:632–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeh L, Schaffer PA. 1993. A novel class of transcripts expressed with late kinetics in the absence of ICP4 spans the junction between the long and short segments of the herpes simplex virus type 1 genome. J. Virol. 67:7373–7382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. 2014. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One 9:e78644. 10.1371/journal.pone.0078644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yager DR, Coen DM. 1988. Analysis of the transcript of the herpes simplex virus DNA polymerase gene provides evidence that polymerase expression is inefficient at the level of translation. J. Virol. 62:2007–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crute JJ, Tsurumi T, Zhu LA, Weller SK, Olivo PD, Challberg MD, Mocarski ES, Lehman IR. 1989. Herpes simplex virus 1 helicase-primase: a complex of three herpes-encoded gene products. Proc. Natl. Acad. Sci. U. S. A. 86:2186–2189. 10.1073/pnas.86.7.2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olivo PD, Nelson NJ, Challberg MD. 1988. Herpes simplex virus DNA replication: the UL9 gene encodes an origin-binding protein. Proc. Natl. Acad. Sci. U. S. A. 85:5414–5418. 10.1073/pnas.85.15.5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baradaran K, Dabrowski CE, Schaffer PA. 1994. Transcriptional analysis of the region of the herpes simplex virus type 1 genome containing the UL8, UL9, and UL10 genes and identification of a novel delayed-early gene product, OBPC. J. Virol. 68:4251–4261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cook WJ, Gu B, DeLuca NA, Moynihan EB, Coen DM. 1995. Induction of transcription by a viral regulatory protein depends on the relative strengths of functional TATA boxes. Mol. Cell. Biol. 15:4998–5006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Imbalzano AN, DeLuca NA. 1992. Substitution of a TATA box from a herpes simplex virus late gene in the viral thymidine kinase promoter alters ICP4 inducibility but not temporal expression. J. Virol. 66:5453–5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smale ST, Jain A, Kaufmann J, Emami KH, Lo K, Garraway IP. 1998. The initiator element: a paradigm for core promoter heterogeneity within metazoan protein-coding genes. Cold Spring Harbor Symp. Quant. Biol. 63:21–31. 10.1101/sqb.1998.63.21 [DOI] [PubMed] [Google Scholar]
- 60.Lieu PT, Wagner EK. 2000. Two leaky-late HSV-1 promoters differ significantly in structural architecture. Virology 272:191–203. 10.1006/viro.2000.0365 [DOI] [PubMed] [Google Scholar]
- 61.Kim DB, DeLuca NA. 2002. Phosphorylation of transcription factor Sp1 during herpes simplex virus type 1 infection. J. Virol. 76:6473–6479. 10.1128/JVI.76.13.6473-6479.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zabierowski S, DeLuca NA. 2004. Differential cellular requirements for activation of herpes simplex virus type 1 early (tk) and late (gC) promoters by ICP4. J. Virol. 78:6162–6170. 10.1128/JVI.78.12.6162-6170.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zabierowski SE, Deluca NA. 2008. Stabilized binding of TBP to the TATA box of herpes simplex virus type 1 early (tk) and late (gC) promoters by TFIIA and ICP4. J. Virol. 82:3546–3554. 10.1128/JVI.02560-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wagner LM, DeLuca NA. 2013. Temporal association of herpes simplex virus ICP4 with cellular complexes functioning at multiple steps in PolII transcription. PLoS One 8:e78242. 10.1371/journal.pone.0078242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gu B, DeLuca N. 1994. Requirements for activation of the herpes simplex virus glycoprotein C promoter in vitro by the viral regulatory protein ICP4. J. Virol. 68:7953–7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim DB, Zabierowski S, DeLuca NA. 2002. The initiator element in a herpes simplex virus type 1 late-gene promoter enhances activation by ICP4, resulting in abundant late-gene expression. J. Virol. 76:1548–1558. 10.1128/JVI.76.4.1548-1558.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jamieson DR, Robinson LH, Daksis JI, Nicholl MJ, Preston CM. 1995. Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus type 1 Vmw65 mutants. J. Gen. Virol. 76:1417–1431. 10.1099/0022-1317-76-6-1417 [DOI] [PubMed] [Google Scholar]
- 68.Cliffe AR, Knipe DM. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 82:12030–12038. 10.1128/JVI.01575-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Terry-Allison T, Smith CA, DeLuca NA. 2007. Relaxed repression of herpes simplex virus type 1 genomes in Murine trigeminal neurons. J. Virol. 81:12394–12405. 10.1128/JVI.01068-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meshorer E. 2007. Chromatin in embryonic stem cell neuronal differentiation. Histol. Histopathol. 22:311–319 http://www.hh.um.es/pdf/Vol_22/22_3/Meshorer-22-311-319-2007.pdf [DOI] [PubMed] [Google Scholar]
- 71.Ahmad K, Henikoff S. 2002. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 9:1191–1200. 10.1016/S1097-2765(02)00542-7 [DOI] [PubMed] [Google Scholar]
- 72.Chow CM, Georgiou A, Szutorisz H, Maia e Silva A, Pombo A, Barahona I, Dargelos E, Canzonetta C, Dillon N. 2005. Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep. 6:354–360. 10.1038/sj.embor.7400366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hake SB, Garcia BA, Duncan EM, Kauer M, Dellaire G, Shabanowitz J, Bazett-Jones DP, Allis CD, Hunt DF. 2006. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem. 281:559–568. 10.1074/jbc.M509266200 [DOI] [PubMed] [Google Scholar]
- 74.Placek BJ, Huang J, Kent JR, Dorsey J, Rice L, Fraser NW, Berger SL. 2009. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J. Virol. 83:1416–1421. 10.1128/JVI.01276-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Felsenfeld G, Burgess-Beusse B, Farrell C, Gaszner M, Ghirlando R, Huang S, Jin C, Litt M, Magdinier F, Mutskov V, Nakatani Y, Tagami H, West A, Yusufzai T. 2004. Chromatin boundaries and chromatin domains. Cold Spring Harbor Symp. Quant. Biol. 69:245–250. 10.1101/sqb.2004.69.245 [DOI] [PubMed] [Google Scholar]
- 76.Zhao H, Dean A. 2004. An insulator blocks spreading of histone acetylation and interferes with RNA polymerase II transfer between an enhancer and gene. Nucleic Acids Res. 32:4903–4919. 10.1093/nar/gkh832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bloom DC, Giordani NV, Kwiatkowski DL. 2010. Epigenetic regulation of latent HSV-1 gene expression. Biochim. Biophys. Acta 1799:246–256. 10.1016/j.bbagrm.2009.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Q, Lin L, Smith S, Huang J, Berger SL, Zhou J. 2007. CTCF-dependent chromatin boundary element between the latency-associated transcript and ICP0 promoters in the herpes simplex virus type 1 genome. J. Virol. 81:5192–5201. 10.1128/JVI.02447-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Amelio AL, McAnany PK, Bloom DC. 2006. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J. Virol. 80:2358–2368. 10.1128/JVI.80.5.2358-2368.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, Coen DM. 2010. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J. Virol. 84:4659–4672. 10.1128/JVI.02725-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783. 10.1038/nature07103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Flores O, Nakayama S, Whisnant AW, Javanbakht H, Cullen BR, Bloom DC. 2013. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J. Virol. 87:6589–6603. 10.1128/JVI.00504-13 [DOI] [PMC free article] [PubMed] [Google Scholar]