

ABSTRACT
Human cytomegalovirus (HCMV) gene expression during infection is highly regulated, with sequential expression of immediate-early (IE), early (E), and late (L) gene transcripts. To explore the potential role of chromatin regulatory factors that may regulate HCMV gene expression and DNA replication, we investigated the interaction of HCMV with the cellular chromatin-organizing factor CTCF. Here, we show that HCMV-infected cells produce higher levels of CTCF mRNA and protein at early stages of infection. We also show that CTCF depletion by short hairpin RNA results in an increase in major IE (MIE) and E gene expression and an about 50-fold increase in HCMV particle production. We identified a DNA sequence (TTAACGGTGGAGGGCAGTGT) in the first intron (intron A) of the MIE gene that interacts directly with CTCF. Deletion of this CTCF-binding site led to an increase in MIE gene expression in both transient-transfection and infection assays. Deletion of the CTCF-binding site in the HCMV bacterial artificial chromosome plasmid genome resulted in an about 10-fold increase in the rate of viral replication relative to either wild-type or revertant HCMV. The CTCF-binding site deletion had no detectable effect on MIE gene-splicing regulation, nor did CTCF knockdown or overexpression of CTCF alter the ratio of IE1 to IE2. Therefore, CTCF binds to DNA within the MIE gene at the position of the first intron to affect RNA polymerase II function during the early stages of viral transcription. Finally, the CTCF-binding sequence in CMV is evolutionarily conserved, as a similar sequence in murine CMV (MCMV) intron A was found to interact with CTCF and similarly function in the repression of MCMV MIE gene expression mediated by CTCF.
IMPORTANCE Our findings that CTCF binds to intron A of the cytomegalovirus (CMV) major immediate-early (MIE) gene and functions to repress MIE gene expression and viral replication are highly significant. For the first time, a chromatin-organizing factor, CTCF, has been found to facilitate human CMV gene expression, which affects viral replication. We also identified a CTCF-binding motif in the first intron (also called intron A) that directly binds to CTCF and is required for CTCF to repress MIE gene expression. Finally, we show that the CTCF-binding motif is conserved in CMV because a similar DNA sequence was found in murine CMV (MCMV) that is required for CTCF to bind to MCMV MIE gene to repress MCMV MIE gene expression.
INTRODUCTION
Human cytomegalovirus (HCMV) is a human betaherpesvirus that infects a large percentage of the human population and causes serious disease in immunocompromised individuals, especially in the setting of HIV-AIDS (1–3). CMV infection in permissive host cells consists of the following sequence of viral events (2): viral entry, immediate-early (IE) and early (E) gene expression, DNA replication, late gene expression, and finally, viral packaging and release. Major IE (MIE) gene expression is one of the earliest events during CMV infection. The MIE gene is the most abundantly expressed viral gene at the IE stage and gives rise to several nuclear phosphoproteins that are critical for the regulation of viral and cellular gene expression and viral DNA replication (4–14). Hence, the HCMV MIE gene has been the focus of much study.
HCMV MIE gene expression is under the control of the MIE promoter (MIEP). MIEP activity is regulated predominantly by a long upstream DNA sequence referred to as the MIE enhancer. The MIE enhancer contains an array of cis-acting sites that bind cellular transcription factors such as NF-κB, CREB/ATF, NF1, SP-1, AP1, RAR-RXR, ISG, YY1, Gfi-1, ETS, serum response factor, Elk-1, and CAAT/enhancer-binding protein (15). Many of these binding sites (BSs) are repetitive and have complex roles in regulating CMV IE gene transcription. Several transcriptional suppressor proteins can also regulate MIE gene expression, including members of the histone deacetylase family (16, 17), histone chaperone and chromatin remodeling factors (DAXX and ATRX) (18), and antiviral nuclear body/nuclear domain 10 proteins (PML, Sp100, Daxx, and ATRX) (19–22), which block viral transcription initiation. In addition, the CMV MIE gene undergoes complex and essential mRNA splicing. The MIE gene consists of five exons and four introns, and the splicing factors also affect CMV MIE gene expression (23). The first exon and intron A (the first intron) of the MIE gene are noncoding; however, it remains unknown whether any DNA-binding factors that associate with the intron A region regulate MIE gene expression or mRNA splicing.
CCCTC-binding factor (CTCF) is a phosphorylated 11-Zn-finger protein that regulates gene expression through complex architectural functions in DNA and chromatin (24). CTCF is known to provide a barrier (or boundary) function that either prevents repressive heterochromatin from spreading into other domains or provides an enhancer-blocking function when the insulator is positioned between the enhancer and the promoter (25). CTCF is also known to function in the regulation of enhancer-promoter communication and has more recently been implicated in the coordination of RNA polymerase II elongation and mRNA processing (26). Genome-wide chromatin immunoprecipitation and DNA sequencing (ChIP-seq) experiments revealed that CTCF binds to specific DNA sequences, most of which are characterized as chromatin insulator elements (27). The 11 zinc finger domains are required for CTCF to bind to a 20-bp core consensus found throughout the cellular and viral genomes (28, 29). The role of CTCF in gene transcription can be either repressive or activating. Recent studies have discovered that CTCF not only plays important roles in the regulation of latent gene expression programs of Epstein-Barr virus (EBV) (30) and Kaposi's sarcoma-associated herpesvirus (KSHV) (31, 32) but also plays a role in the lytic infection of herpes simplex virus 1 (HSV-1) (33, 34). Whether or not CTCF is involved in HCMV MIE gene expression has yet to be determined.
Here we report a role for CTCF in the negative regulation of MIE gene expression during primary CMV infection. We show that CTCF protein binds directly to a DNA element within intron A of the CMV MIE gene and that this binding is important for its transcriptional attenuation during primary infection. MIE intron A is known to be important for optimal expression of the MIE gene and viral infection (35). So far, there have been only a few investigations that have explored the function of intron A in MIE gene regulation and CMV infection (35–37). An NF-1 binding site (BS) in the DNA that encodes intron A has been shown to be important for proper regulation of the CMV MIE gene (35, 38). Thus, our findings reveal that CTCF is another important component of the transcriptional regulatory factors that regulate CMV MIE gene expression from DNA elements downstream of the transcriptional start site. Our findings also suggest that CTCF binding may have an evolutionarily conserved function in the regulation of herpesvirus infections.
MATERIALS AND METHODS
Tissue culture and viruses.
The human embryonic diploid lung fibroblast cell line MRC-5 (ATCC CCL171), HEK 293T cells, and BJ human foreskin fibroblast cells (ATCC CRL-2522) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (39). HCMV strain AD169 was obtained from ATCC. For immunohistochemical staining, cells were grown on round coverslips (Corning Incorporated, Corning, NY) in 24-well plates (Becton, Dickinson and Company, Franklin Lakes, NJ).
Antibodies.
Monoclonal antibodies (MAbs) against HCMV IE1/2 (MAb 810) and Flag (M2) were purchased from Sigma-Aldrich (Saint Louis, MO). The MAb against tubulin (4G1) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The polyclonal antibody against CTCF (NB500-177) was purchased from Novus Biologicals (Littleton, CO). The MAb against murine CMV (MCMV) IE1 was provided by Stipan Jonjic, Rijeka, Croatia (1:50 for immunofluorescence assay and 1:200 for Western blotting) (40).
Plasmids and transfection.
Plasmid pSVH, carrying the intact HCMV MIE gene, was a gift from R. Stenberg (41) and has been used in our laboratory previously (23). Plasmids expressing Flag-tagged CTCF and Rad21 were described previously (31, 32). Plasmids expressing short hairpin RNAs (shRNAs) against CTCF were purchased from Santa Cruz (catalog no. sc-35124-SH). Plasmid pIE111, carrying the whole MCMV MIE gene, was a generous gift from U. H. Koszinowski (42).
Transfection of plasmids and bacterial artificial chromosome plasmid (BACmids) was performed with Lipofectamine 2000 (Life Technologies, Grand Island, NY), according to the manufacturer's instructions. The AD169-derived HCMV BACmid HB5 (43) was used to generate all of the HCMV mutants described in this report.
Molecular cloning and mutagenesis.
To delete the CTCF-binding sequence from intron A in pSVH or pIE111, we first removed intron A and then inserted a mutated form from which the DNA sequence (for pSVH, TTAACGGTGGAGGGCAGTGT; for pIE111, TTTCCCGTGGAGGACTCCG) had been deleted. The resulting plasmid was named either pSVHdCTCFi or pIE111dCTCFi.
To delete the CTCF-binding sequence from intron A in the HCMV BAC, we employed a seamless BAC technique with galK as the selection marker (44). Briefly, BACmid HB5 was transformed into Escherichia coli SW102. A galK DNA fragment that was made from pgalK by PCR and contains ends that were homologous with intron A of MIE was electroporated into SW102 (harboring HB5) to replace intron A by homologous recombination, which resulted in the BACmid HB5intronAgalK. The galK DNA was then replaced with a PCR fragment made from intron A, from which a fragment containing the CTCF-binding sequence had been deleted, resulting in HB5dCTCFi. The revertant HCMV BAC DNA (HB5dCTCiFRev) was produced from HB5dCTCFi by the same method. The resultant BACmids were transfected into MRC-5 cells to make the viruses HCMVdCTCFi and HCMVdCTCFiRev. The BACmids and viruses were verified by restriction enzyme digestion, DNA sequencing, and PCR. The complete MIE gene was sequenced and confirmed to be correct.
RNA isolation and real-time RT-PCR.
Total RNA was isolated with TRI reagent (Ambion, Inc., Austin, TX) and treated with DNase I (RNase free; Invitrogen catalog no. 18047-019) according to the manufacturer's instructions. About 1 μg of treated RNA was used for reverse transcription (RT), which was carried out with a SuperScript II First-Strand synthesis kit (Invitrogen, Carlsbad, CA) and an oligo(dT) 20mer according to the manufacturer's protocol. To quantitatively examine the CTCF, IE1, and IE2 mRNA levels in HCMV-infected cells, a real-time RT-PCR assay was done with the QuantiTect SYBR green RT-PCR kit (Qiagen, Valencia, CA). Quantitative PCRs (qPCRs) consisted of 50 cycles under optimal conditions of 94°C for 20 s; 50°C for 60 s; 72°C for 30 s; and 80°C for 5 s, an optimized data collection step. Fluorescence captured at 80°C was determined to be absent from the signal generated by primer dimers. All samples were run in triplicate. Data were collected by the iCycler iQ software (Bio-Rad) and expressed as a function of the threshold cycle (CT), which represents the number of cycles at which the fluorescent intensity of the SYBR green dye is significantly greater than the background fluorescence. The CT value is directly correlated to the log10 copy number of the RNA standards. RNA copies were extrapolated from standard curves (CT versus log10 copy number) representing at least a seven-point serial dilution of standard RNA (101 to 107 copies/μl). RNA standards were used as calibrators of the relative quantification of the product generated in the exponential phase of the amplification curve for real-time RT-PCR. The results showed the correlation coefficient for the standard curve to be greater than 0.95. A melting temperature curve analysis was performed by measuring (after the amplification cycles) the fluorescence during a period of warming from 60 to 95°C. The primers used to determine transcriptional levels of CTCF, IE1, and IE2 by real-time RT-PCR are listed in Table 1.
TABLE 1.
Primers used for real-time RT-PCR
| Primer target and direction | Sequence |
|---|---|
| IE1 | |
| Forward | 5′ ATG TCC TGG CAG AAC 3′ |
| Reverse | 5′ CAT CCT CCC ATC ATA TTA 3′ |
| IE2 | |
| Forward | 5′ ATG TCC TGG CAG AAC 3′ |
| Reverse | 5′ GGA TGC CCC GGG GAG AGG −3′ |
| UL112/113 | |
| Forward | 5′ ATG GAT CTC CCT ACT ACC GTC3′ |
| Reverse | 5′ GAA GCC TCG CCG TGC TGC ATA −3′ |
| CTCF | |
| Forward | 5′ ACCAACCAGCCCAAACAGAAC-3′ |
| Reverse | 5′ GTATTCTGGTCTTCAACCTGAATGATAG-3′ |
| MCMVIE1 | |
| Forward | 5′ ATAGCTTCACCATGCCTAGGATCA 3′ |
| Reverse | 5′ GGGCATCTCAGGATCATACT 3′ |
FP assay.
A fluorescence polarization (FP) assay can detect the binding of a small fluorescently labeled oligonucleotide from a protein of interest because of the ability of a fluorescently labeled molecule to emit light with a degree of polarization when excited by polarized light, which is inversely proportional to the rate of molecular rotation (45). FP experiments were used to measure the inhibition constant (Ki) of CTCF BSs and were performed with 384-well black Optiwell plates. All wells were initially filled with 75 μl of assay buffer (100 mM Tris [pH 8.0], 50 mM KCl, 0.6 mg/ml bovine serum albumin, 0.075% Tergitol-type NP-40) for 60 min at room temperature to prevent nonspecific binding. To each well, 2 nM 6-carboxyfluorescein (FAM)-labeled 36-bp double-stranded DNA (dsDNA) probe (TCAGAGTGGCGGCCAGCAGGGGGCGCCCTT GCCAGA) with a known dissociation constant of 17 nM for CTCF's 11-zinc-finger binding domain (CTCF11ZF) recombinant protein (a gift from Ronen Marmorstein, University of Pennsylvania) was added to increasing concentrations of the unlabeled dsDNA probe (0 to 5 μM) (Table 2, CMV CTCF BS). Seventeen nanomolar CTCF11ZF recombinant protein was then added to a final volume of 30 μl per well and incubated for 60 min at 4°C. Polarization values (in millipolarization units) were measured with an Envision 2104 Multilabel Reader (PerkinElmer) with an excitation wavelength of 485 nm and an emission wavelength of 530 nm. Each measurement was done in triplicate. All experimental data were analyzed with Prism 3.0 software, and the inhibition constants were determined by fitting to the following model: log 50% inhibitory concentration (IC50) = log (10l̂[log Ki · (1 + [fluorescent probe]/[fluorescent probe KD]), where Y = [low binding threshold + (high binding threshold − low binding threshold)]/[1 + 10^(X − log IC50)].
TABLE 2.
Oligonucleotides used for EMSA
| Oligonucleotide | Sequence |
|---|---|
| XqYq CTCF BS | GATCCTGCTGTGCCAGGGCGCCCCCTGCTGGCGACTAGGGCAACTA |
| XqYq ΔCTCF | GATCCTGCTGTGCCAGAATACAAAATGCTAATAACTAGGGCAACTA |
| CMV CTCF BS | GTTGCGGTGCTGTTAACGGTGGAGGGCAGTGTAGTCTGAGCAGTA |
| CMV ΔCTCF | GGTAACTCCCGTTGCGGTGCTGAGTCTGAGCAGTACTCGTTGCTG |
Immunoblot analysis.
Proteins were separated by sodium dodecyl sulfate (SDS)–7.5% polyacrylamide gel electrophoresis (PAGE) (10 to 20 μg loaded in each lane), transferred to nitrocellulose membranes (GE Healthcare), and blocked with 5% nonfat milk for 60 min at room temperature. Membranes were incubated overnight at 4°C with a primary antibody and then incubated with a horseradish peroxidase-coupled secondary antibody (Amersham Inc.); this was followed by detection by enhanced chemiluminescence (Pierce, Rockford, IL) by the standard method. Membranes were stripped with stripping buffer (100 mM β-mercaptoethanol, 2% SDS, 62.5 mM Tris-HCl, pH 6.8) for 30 min at 50°C, washed with phosphate-buffered saline–0.1% Tween 20, and used to detect additional proteins.
PFU assay.
To detect the viral growth curve, MRC-5, BJ-kdCTCF, or BJ-kdLuc cells were infected with wild-type HCMV AD169 (HCMVwt), HCMVdCTCFi, or HCMVdCTCFiRev at a multiplicity of infection (MOI) of 0.05 PFU/cell. Medium and cells from infected cultures were collected on different days postinfection, and virus was obtained by three freeze-thaw cycles of the collected culture. Virus titers were determined on MRC-5 or BJ cells after PFU analysis. Student's t test was used to analyze the statistical significance of differences between two groups, and a P value of <0.005 was used as the threshold of significance.
Electrophoretic mobility shift assay (EMSA).
DNA fragments (Table 2) were labeled with T4 polynucleotide kinase (Roche) in the presence of [32P]ATP and purified with G-25 spin columns (GE Healthcare). Purified CTCF protein was a gift from Ronen Marmorstein (University of Pennsylvania). Increasing amounts of purified CTCF protein (0, 50, 150, and 450 ng) were incubated with the same amounts of purified probes for 30 min at room temperature in a total volume of 25 μl of buffer containing 100 μM ZnSO4, 10 mM Tris-Cl (pH 8.0), 60 mM KCl, 1 mM EDTA, 10 mM MgCl2, 0.05 μg/μl poly(dI-dC), 0.5 μg/μl bovine serum albumin, 0.05% NP-40, 35 mM β-mercaptoethanol, and 6% glycerol. The samples were then separated by electrophoresis on a native 5% polyacrylamide gel. Gels were dried and analyzed with a Typhoon PhosphorImager system.
ChIP assay.
HEK 293T cells were transfected with pSVH for 24 h or BJ-kdLuc and BJ-kdCTCF cells were infected with HCMVdCTCFi, HCMVwt, or HCMVdCTCFiRev at an MOI of 0.5 for 12 h and fixed with 1% formaldehyde and then sonicated to shear the DNA into 100- to 300-bp fragments. The anti-CTCF antibody or normal IgG was incubated with the DNA-protein mixture, and ChIP assays were performed with the EZChIP kit (Millipore, Billerica, MA) according to the manufacturer's instructions. The amount of DNA immunoprecipitated by antibodies (normal IgG and anti-CTCF) was examined by real-time PCR in a 25-μl reaction mixture with the primers shown in Table 3. All samples were analyzed in triplicate with SYBR green 2× master mix (Applied Biosystems, Foster City, CA) on an Applied Biosystems 7900HT Fast real-time PCR system. After initial denaturation at 95°C for 5 min, 45 cycles of three-step cycling were performed with an annealing temperature of 60°C. Melting curve analysis was then performed to verify product specificity. The ChIP efficiency with each antibody was calculated as a percentage of the input by the ΔCT method, and the standard deviation was generated from triplicate PCRs.
TABLE 3.
PCR primers used in ChIP assay
| Primer target and direction | Sequence |
|---|---|
| Intron A | |
| Forward | TGGCGGTAGGGTATGTGTCTGAAA |
| Reverse | ACTCAGCTGCCTGCATCTTCTTCT |
| MIE 3′ UTR | |
| Forward | ATTAGTGGTGGCGGTGGTAGGTTT |
| Reverse | ACGATCATCATCCCTGAGGCCAAA |
| XqYq | |
| Forward | ACGGGTGGAACTTCAGTAATCC |
| Reverse | GTGAGCAAGCGGGTCCTGTA |
| 10q | |
| Forward | ACGGTGCTCTGCCATTGC |
| Reverse | GGCGCTGGACACCACTGTA |
| Actin | |
| Forward | GGCTCACCACTGCAGAAATCA |
| Reverse | TTATCTTGGAGGTCCCCT |
| Intron A1 | |
| Forward | ACTGCTCAGATCGTC |
| Reverse | ATATTCTGTATTCTGC |
| Intron A2 | |
| Forward | TGTACTCAGAGGCTG |
| Reverse | AGTACGAGCAACACG |
| Intron A3 | |
| Forward | GCGAAGGATCTCTCTTG |
| Reverse | ACAGACAGAATCCTCT |
| MIE 3′ end | |
| Forward | GAGCGGTGAAGACCTG |
| Reverse | ATCTGCCTCAAACTGG |
kdCTCF.
To make the CTCF knockdown (kdCTCF) cell line, we first cotransfected 293T cells with a lentiviral plasmid expressing an shRNA against CTCF (pLKO.1-puro-CTCF; Sigma) and a packaging plasmid (pHR′8.2deltaR) at an 8:1 ratio, as well as with the envelope plasmid (pCMV-VSV-G) (23). Transfection was performed with METAFECTENE PRO (Biontex Laboratories, Martinsried/Planegg, Germany) according to the manufacturer's instructions. Seventy-two hours after transfection, lentivirus encoding shCTCF was collected from the medium by filtration through a 0.45-μm syringe filter to remove any 293T cells. BJ cells were infected with the lentiviruses and selected with 2 μg/ml puromycin (Sigma) at 24 h postinfection (hpi), and knockdown efficiency was assayed by Western blot and immunofluorescence assays with anti-CTCF antibody. The resulting CTCF-depleted cells were named BJ-kdCTCF. A control cell line named BJ-kdLuc was generated by the same protocol as BJ-kdCTCF, except that pLKO.1-puro-Luc encoding shRNA against luciferase (Sigma) was used to make the lentiviruses. The cell lines were maintained in minimal essential medium with 1 μg/ml puromycin prior to further experiments.
RESULTS
The CTCF level is increased in the early stage of HCMV infection.
As a first step in investigating the role of CTCF in HCMV infection, we tested whether CTCF gene expression is affected by HCMV infection. In our studies of HCMV infection of human fibroblast (MRC-5) cells, we found that CTCF can be upregulated at early times of infection. As shown in Fig. 1A, we infected MRC-5 cells with HCMV at an MOI of 0.5 for different lengths of time (as indicated). Whole-cell lysates were collected and assayed by Western blotting with antibodies against viral proteins (IE1/IE2) and CTCF to examine viral protein production. We found that the CTCF protein level was about 2.5-fold higher at 12 hpi and about 4.5-fold higher after 24 hpi than that in mock-treated cells. To learn whether the increase in CTCF occurs at the gene transcription level, we performed real-time PCR assays to compare CTCF mRNA levels at different time points of after HCMV infection with the level at 0 hpi (Fig. 1B). We found that the CTCF mRNA level was also enhanced by HCMV infection. The increase in CTCF production after HCMV infection might reflect a general response of host cells to viral infection.
FIG 1.

HCMV induces CTCF production at an early time of infection. MRC-5 cells were infected with HCMV at an MOI of 0.5 for different times, as indicated. (A) Whole-cell lysates were collected and subjected to SDS-PAGE to examine cellular and viral protein production by Western blot assay with antibodies against viral proteins (IE1/IE2) and CTCF. Tubulin was used as a sample-loading control. The fold increases in CTCF levels relative to mock-treated and tubulin controls were calculated by Quantity One 4.5.0 software (Bio-Rad Laboratories, Richmond, CA). Quantification of CTCF levels (means ± standard deviations) is shown at the bottom as bar graphs generated from at least three independent viral infections and Western blot assays. (B) Total RNA was isolated from mock-infected (0 hpi) or HCMV-infected cells, and 1 μg of the resulting sample was analyzed by quantitative RT-PCR. CTCF mRNA levels at different time points after HCMV infection relative to the level at 0 hpi are shown. The bar graph represents the mean ± standard deviation from three independent experiments. Statistically significant differences were calculated by Student's t test and are indicated at the top (P < 0.005).
CTCF represses MIE gene expression.
To investigate the potential effect of elevated CTCF levels on HCMV MIE gene expression, we assayed the effects of overexpression of CTCF on the MIE gene locus in transient-transfection assays with plasmid vectors. We cotransfected the HCMV subgenomic fragment containing IE1/IE2 under MIEP control (pSVH) together with different amounts of CTCF expression vector (pFlag-CTCF) or the vector control into 293T cells. pSVH contains the intact MIE gene, including the MIEP, enhancer, introns, and exons. Western blot assays revealed that overexpression of CTCF had significant inhibitory effects on IE1/IE2 production and that the repressive effect of CTCF on IE1/IE2 expression is dose dependent (Fig. 2A). To determine whether the repressive effect was due to the inhibition of MIE gene transcription, we performed a real-time RT-PCR assay and found that the repressive effect of CTCF on MIE gene expression occurred at the mRNA level (Fig. 2B). To further investigate the effect of CTCF on MIE gene expression, we generated stable BJ cells depleted of CTCF by shRNA and transfected pSVH into BJ, BJ-kdLuc, and BJ-kdCTCF cells. CTCF depletion was monitored by Western blotting and demonstrated >80% depletion of CTCF in BJ-kdCTCF cells relative to control cells (BJ-kdLuc or parental BJ cells). We found that the expression of both IE1 and IE2 was greater in kdCTCF cells than that in control cells (Fig. 2C). The inhibition of MIE gene expression by CTCF takes place at the RNA transcription level, as the level of mRNA transcription from BJ or BJ-kdLuc is significantly lower (∼6-fold) than that from BJ-kdCTCF (Fig. 2D). These findings suggest that CTCF has repressive effects on MIE gene expression.
FIG 2.

Effects of CTCF protein levels on MIE gene expression. (A) 293T cells were used for the cotransfection of 1 μg of pSVH (expressing IE1/IE2 under MIEP control) together with different amounts (0.5, 1, 2, and 3 μg) of pFlag-CTCF or the vector control (by using 3 μg of pcDNA3 to supplement the total DNA in the cotransfection system). Whole-cell lysate samples were used for Western blot assays to examine the production of IE1, IE2, and CTCF. Tubulin was used as a sample-loading control. The fold variations in IE1/2 levels relative to pSVH and the vector control were quantified by Quantity One 4.5.0 software and are shown at the bottom as bar graphs (means ± standard deviations) generated from three independent experiments. (B) Same as panel A, except that total RNA samples (1 μg for each assay) were used for real-time RT-PCR to examine IE1 mRNA. The bar graph shows the average value of the IE1 mRNA level of pSVH with pFlag-CTCF relative to that of pSVH alone from three independent experiments (means ± standard deviations). (C) Western blot assays performed to examine CTCF depletion and IE1/IE2 production after the transfection of pSVH into BJ cells or BJ cells stably expressing shRNA against CTCF or control luciferase (Luc). Tubulin was used to control the sample loading. The fold increases in IE1/2 levels relative to BJ cells alone were calculated by Quantity One 4.5.0 software and are shown as bar graphs below the corresponding Western blot assays. (D) Same as panel C, except that real-time RT-PCR was used to examine IE1 mRNA levels in stable knockdown cell lines transfected with pSVH. The levels of IE1 mRNA in different cells were compared to that in control BJ cells. The bar graph represents the means ± standard deviations of three independent experiments.
CTCF binds to a DNA sequence in intron A of the MIE gene.
Since CTCF is a sequence-specific DNA-binding protein, we tested whether CTCF represses MIE gene expression through interaction with a cis element in the MIE gene. As a first approach to search for the DNA sequences interacting with CTCF, we used an in silico CTCF BS prediction tool (46) to analyze the potential CTCF BSs in the HCMV MIE gene locus. One small DNA sequence (TTAACGGTGGAGGGCAGTG), which localizes to the first intron of the MIE gene (referred to as intron A), from site +834 to site +852 (Fig. 3A), received a high score. To validate the prediction experimentally, we tested the ability of purified recombinant CTCF protein to bind to this sequence in an FP assay. In this assay, we measured the ability of a candidate CTCF BS to compete with a known high-affinity BS (47). High-affinity BSs have Kis between 5 and 40 nM, while low-affinity sites have Kis of >100 nM. As shown in Fig. 3B, the CTCF BS derived from the intron A region of CMV can compete efficiently with the high-affinity CTCF BS, with a Ki of 21.4 nM. This result suggests that MIE intron A region contains a high-affinity BS for purified CTCF in vitro.
FIG 3.

CTCF interacts directly with intron A. (A) The DNA sequences of the HCMV MIE gene, from the TATA box to the beginning of exon 2, are shown. The NF-1 and CTCF BSs are underlined. Total exon 1 and partial exon 2 sequences are shown in the enclosing boxes. (B) An FP assay was used to determine whether the DNA fragment was bound by CTCF. Kis were calculated by titrating the HCMV CTCF BS probe against a FAM-labeled probe with a known dissociation constant and measuring changes in CTCF binding via FP assay. The graph represents the mean ± standard deviation of three independent experiments. mP, millipolarization units. (C) An EMSA was used to determine CTCF binding to DNA oligonucleotide probes containing putative BSs from HCMV intron A (HCMV CTCF BS) or XqYq subtelomeres (XqYq CTCF BS), as well as oligonucleotides containing CTCF BS deletion (HCMV ΔCTCF) or point mutations in CTCF recognition sites (XqYq ΔCTCF). The positions of the free probe and the bound probe are indicated on the left. (D) ChIP assay with HEK 293T cells transfected with pSVH at 24 h posttransfection with antibodies specific to CTCF or control IgG. qPCR was used to quantify ChIP efficiency with specific primers in the regions indicated. The bar graph represents the mean percentage of the input for each ChIP from three independent PCRs ± the standard deviation.
To further confirm the direct binding of CTCF to MIE DNA, we performed a DNA EMSA. In the DNA-protein binding reaction system, different amounts of purified CTCF (0, 50, 150, and 450 ng) were added (Fig. 3C) with the same amount, respectively, of the following 32P-labeled DNA probes: CMV CTCF BS with the putative CTCF consensus sites, CMV ΔCTCF with the CTCF BS deleted, XqYq CTCF BS containing the CTCF BS as a positive control, and the CTCF BS substitution mutant XqYq ΔCTCF as a negative control (48). EMSA results showed that purified CTCF protein bound to the CMV CTCF BS probe with an efficiency similar to that of the XqYq CTCF BS probe, and deletion or mutation of CTCF consensus sites diminished the binding by CTCF (Fig. 3C). These finding indicate that CTCF interacts directly with a short DNA sequence (TTAACGGTGGAGGGCAGTG) within intron A of the MIE gene.
To determine whether CTCF interacts with intron A in vivo, we performed a ChIP assay with rabbit anti-CTCF antibody or normal rabbit IgG as a control and 293T cells transfected with pSVH for 24 h (Fig. 3D). As shown, IgG did not pull down any detectable DNA from the samples tested, while anti-CTCF antibody pulled down ∼1% of the input DNA fragments in intron A, similar to that observed for the positive-control CTCF BSs in the cellular subtelomeres for XqYq and 10q (48). Two other fragments, one from the MIE gene exon 5 3′ untranslated region (UTR) and the other from the actin gene, were not immunoprecipitated, indicating the specificity of CTCF ChIP.
The CTCF BS is required for repression of MIE gene expression by CTCF.
To test the functional role of CTCF binding to MIE, we generated an MIE gene-expressing construct (pSVH-dCTCFi) in which the CTCF-bindin sequences were deleted. pSVH-dCTCFi was cotransfected with different amounts of pFlag-CTCF (0, 0.5, 1, 2, and 4 μg) into 293T cells for 24 h. Whole-cell lysates were subsequently used to examine IE1/IE2 protein levels by Western blot assay (Fig. 4A). The expression of Flag-tagged CTCF was examined by using anti-Flag antibody. Total RNAs were used for a real-time RT-PCR assay to determine IE1 mRNA levels (Fig. 4B). Our results indicated that neither IE1/IE2 protein nor mRNA levels were inhibited by CTCF overexpression from pSVH-dCTCFi, whereas CTCF overexpression efficiently repressed IE1/IE2 expression from wild-type pSVH, as was demonstrated previously in Fig. 2. Therefore, the repressive effects of CTCF on IE1/IE2 gene expression were disrupted when the CTCF BS was deleted from pSVH, suggesting that the CTCF-binding motif is necessary for the repression of MIE gene expression by CTCF.
FIG 4.

The CTCF-binding domain in intron A is essential for CTCF-mediated inhibition of MIE gene expression. (A) 293T cells were transfected with pSVH-dCTCFi and different amounts of pFlag-CTCF (0, 0.5, 1, 2, and 4 μg) and assayed for IE1/IE2 expression by Western blotting at 24 h posttransfection. The overexpressed CTCF level was examined by using anti-Flag antibody. Tubulin was used as a sample-loading control. The relative IE1/2 levels calculated by Quantity One 4.5.0 software as described in the legend to Fig. 2A are shown as bar graphs below the corresponding Western blot assays. (B) Same as panel A, except that total RNA samples (1 μg for each assay) were used for real-time RT-PCR in order to detect IE1 mRNA. The bar graph shows the mean IE1 mRNA level of pSVH with pFlag-CTCF relative to that of pSVH alone from three independent experiments ± the standard deviation. (C) 293T cells were cotransfected with pSVH and various amounts (0.5, 1, 2, and 4 μg) of pFlag-Rad21 for 24 h. Whole-cell lysates were assayed by Western blotting with antibodies against IE1/IE2 to detect MIE gene expression, with antibodies against Flag to examine Rad21 overexpression and with antibodies against tubulin as a loading control. The quantification of IE1/2 levels relative to pSVH and the tubulin control is shown as bar graphs below the corresponding Western blot assays. (D) Same as panel C, except that the real-time RT-PCR was performed to detect IE1 mRNA. The bar graph shows the mean IE1 mRNA level of pSVH with pFlag-Rad21 relative to that of pSVH alone from three independent experiments ± the standard deviation. (E) Western blot analysis of 293T cells transfected with pSVH alone, pSVH with pRad21, pSVH with pCTCF, or pSVH with pRad21 and pCTCF with the antibodies indicated. The quantification of IE1/2 levels relative to pSVH alone and the tubulin control is shown as bar graphs below the corresponding Western blot assays. (F) Same as panel E, except that the quantitative RT-PCR was performed to examine MIE gene expression. The bar graph shows the mean IE1 mRNA level relative to that of pSVH alone from three independent experiments ± the standard deviation. Student's t test was used to statistically compare two groups as indicated at the top of the bar graph (*, P < 0.005; **, P > 0.1).
Rad21 alone has no effect on MIE gene expression, but it interacts with CTCF and enhances CTCF's repressive effects on MIE gene expression.
The functional link between CTCF and cohesin has been demonstrated by a series of studies (49–51). The interaction of CTCF with cohesin might be important for CTCF if it is to act as a domain boundary factor or a transcriptional repressor (52). An important component of the cohesin complex is Rad21, which was found to interact directly with CTCF (53). We asked whether Rad21 overexpression alters MIE gene expression by cotransfecting pSVH with various amounts of pFlag-Rad21 into 293T cells (Fig. 4C and D). Whole-cell lysates were assayed by Western blotting with antibodies against IE1/IE2 to detect MIE gene expression and with antibodies against Flag to examine Rad21 overexpression. We also used the total RNA for detection of IE1 mRNA by real-time RT-PCR as shown in Fig. 4D. We found that Rad21 overexpression alone did not affect IE1/IE2 expression and MIE transcription. We next tested whether Rad21 could alter the repressive effect of CTCF on MIE gene expression by cotransfection of Rad21 and CTCF into 293T cells (Fig. 4E and F). As expected, the cotransfection of pSVH with pRad21 had no effect on MIE gene expression and CTCF had a suppressive effect on MIE gene expression. However, the cotransfection of pSVH together with both pCTCF and pRad21 produced an additive reduction of IE1 and IE2 protein and RNA expression. These data indicate that Rad21 can augment CTCF's repressive effects on MIE gene expression when both proteins are expressed ectopically in transient-transfection assays.
HCMV BACmid lacking the CTCF-binding motif presents a higher-replication phenotype.
To determine whether the CTCF-binding motif in intron A of the MIE gene is important for MIE gene expression and HCMV replication in the larger context of the viral genome, we generated CTCF mutations and revertants by using HCMV BACmid technology (44). We have shown that CTCF binds to intron A by using in vitro binding assays and a transient-transfection system (Fig. 3). We next asked whether CTCF is able to interact with intron A in vivo during HCMV infection. MRC-5 cells were infected with HCMVs generated from BACmids at an MOI of 0.5 for 12 h and assayed by CTCF ChIP (Fig. 5A). We found that CTCF bound to intron A with an efficiency similar to that of BSs in the 10q subtelomere when HCMVwt (Fig. 5A, right panel) or HCMVdCTCFiRev (middle panel) was used to infect MRC-5 cells. CTCF did not bind to control DNA elements from either the MIE 3′ UTR or the cellular actin region, further demonstrating that CTCF interacts with intron A in vivo. As expected, the binding of CTCF to intron A was disrupted when the mutant HCMVdCTCFi was used in the ChIP assay (Fig. 5A, left panel). Taken together, these studies indicate that CTCF can bind to intron A in vivo and that the CTCF-binding motif within intron A is required for CTCF to interact with intron A.
FIG 5.
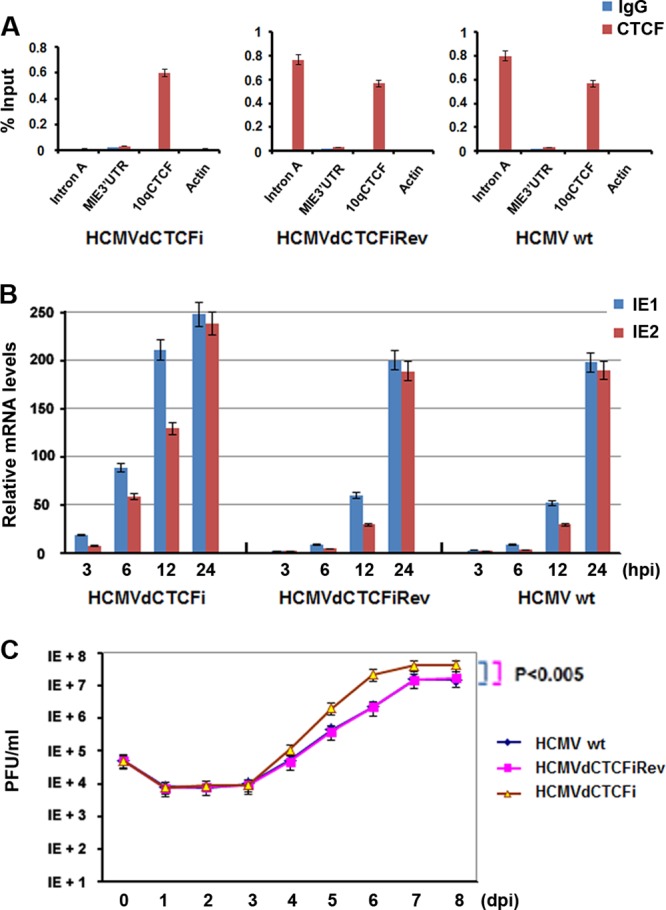
Deletion of the CTCF motif from intron A led to increases in HCMV IE gene expression and viral DNA replication. (A) ChIP-qPCR analysis of CTCF or control IgG with primers specific for the regions indicated in MRC-5 cells infected with HCMVwt, HCMVdCTCFiRev, or HCMVdCTCFi at an MOI of 0.5 for 12 h. The bar graph represents the mean percentage of the input in each ChIP from three independent PCRs ± the standard deviation. (B) Quantitative RT-PCR analysis of IE1 and IE2 mRNAs in MRC-5 cells infected with HCMVs at an MOI of 0.1 at the indicated times after infection. The bar graph represents the relative RT-PCR values from three independent experiments (means ± standard deviations). (C) MRC-5 cells were infected with the HCMVs indicated at an MOI of 0.05, and the viral growth curve was determined by using PFU assays at the indicated times after infection. Student's t test was used to statistically analyze the difference between HCMVdCTCFi infection and HCMVwt (P < 0.005) and HCMVdCTCFiRev (P < 0.005) infections.
We next asked whether deletion of the CTCF-binding motif could affect IE1 and IE2 transcription. The HCMVs were used to infect MRC-5 cells at an MOI of 0.1, and total RNAs taken from the cells at different times after infection were examined for IE1 and IE2 mRNA levels by quantitative RT-PCR (Fig. 5B). We found that the mRNAs of both IE1 and IE2 were present as early as 3 hpi and that their levels were increased by infection with HCMVdCTCFi where the CTCF-binding motif in intron A of the MIE gene was deleted. The IE1 and IE2 mRNA levels of HCMVdCTCFi at 3 hpi (Fig. 5B, left panel) were even higher than those of HCMVwt (right panel) or HCMVdCTCFiRev (middle panel) at 6 hpi; the same accelerated levels of mRNAs were observed at 6 and 12 hpi. However, the differences in the mRNA levels among all of the infected cells were smaller at 24 hpi. Interestingly, the IE1/IE2 ratio remained the same, suggesting that the CTCF-binding motif in intron A did not affect MIE gene splicing.
We then tested the effects of the CTCF-binding motif deletion on viral replication by using PFU assays. As shown in Fig. 5C, the revertant HCMV- and HCMVwt-infected cells show similar viral replication kinetics. In contrast, titers of HCMVdCTCFi were about 10-fold higher than those of the revertant and wild-type viruses from day 5 postinfection and HCMVdCTCFi reached a peak quicker than did the revertant HCMV and HCMVwt. The results suggest that CTCF binding to the MIE gene intron A restricts both HCMV gene expression and viral replication.
Depletion of CTCF enhances HCMV gene expression and viral replication.
We next asked whether the depletion of CTCF could affect HCMV gene expression and viral replication during viral infection. We infected BJ-kdLuc or BJ-kdCTCF cells with HCMVwt, HCMVdCTCFi, or HCMVsCTCFiRev at an MOI of 0.1 for different times, as indicated in Fig. 6A. A real-time PCR assay was performed to determine the relative IE1, IE2, and UL112/113 mRNA levels. We found that the expression of all three genes was largely enhanced by infection with HCMVwt (Fig. 6A, left panel) and HCMVdCTCFiRev (middle panel) at each time point in CTCF-depleted BJ cells (BJ-kdCTCF) relative to that in control cells (BJ-kdLuc). The increase in viral gene expression caused by infection of BJ-kdCTCF cells with HCMVwt and HCMVdCTCFiRev was comparable to that caused by HCMVdCTCFi infection of BJ-KdLuc cells (right panel), further indicating that CTCF mediates a repressive effect on HCMV gene expression during viral infection. In line with these results, no significant differences in IE1 and IE2 expression from BJ-kdCTCF or BJ-kdLuc cells were observed when HCMVdCTCFi was used for infection (Fig. 6A, right panel). Interestingly, viral E protein UL112/113 is expressed at a higher level in BJ-kdCTCF cells than in BJ-kdLuc cells, suggesting that CTCF might play an additional role in regulating viral E gene expression.
FIG 6.

Depletion of CTCF protein enhances HCMV gene expression and viral replication. (A) Quantitative RT-PCR analysis of IE1, IE2, and UL112/113 mRNAs in BJ-kdLuc or BJ-kdCTCF cells infected with HCMVwt (left panel), HSMVdCTCFiRev (middle panel), or HCMVdCTCFi (right panel) at an MOI of 0.1 at the indicated times after infection. The bar graph represents the relative RT-PCR values from three independent experiments (means ± standard deviations). (B) BJ-kdCTCF or BJ-kdLuc cells were infected with the indicated HCMVs at an MOI of 0.05, and the viral growth curve was determined by using PFU assays at the indicated times after infection. Student's t test was used to statistically analyze the difference in viral production between BJ-kdCTCF and BJ-kdLuc cells at the time points indicated (*, P < 0.005; **, P < 0.05).
We then tested whether the depletion of CTCF could affect HCMV replication with plaque formation unit assay. We found that both revertant HCMV and HCMVwt infections of CTCF-depleted cells led to an about 50-fold increase in viral production over that in control cells at 6 and 7 dpi (Fig. 6B, left and middle panels). In contrast, HCMVdCTCFi infection of BJ-kdCTCF cells resulted in an only 3-fold increase in viral replication over that in control cells (Fig. 6B, right panel). These results suggest that CTCF has a repressive effect on HCMV replication and that the CTCF BS in intron A is important for CTCF-mediated inhibition of viral replication. Since HCMVdCTCFi infection in BJ-kdCTCF cells was still able to produce about 3-fold more viral particles than such an infection did in BJ-kdLuc cells, it is likely that CTCF might repress HCMV replication by additional mechanisms besides direct binding to intron A.
Homologous CTCF-binding domain in MCMV intron A.
We next asked whether the CTCF BS was conserved in MCMV MIE gene intron A. Alignment of HCMV intron A with MCMV MIE DNA revealed strong conservation of the core CTCF recognition sequence at a similar position relative to the transcription start site (+810 to +828 in MCMV and +834 to +852 in HCMV) (Fig. 7A). Computational analysis predicted that the MCMV DNA sequence shows a CTCF BS probability score greater than that of HCMV (Fig. 7B). To determine whether CTCF actually interacts with the putative CTCF-binding sequence in MCMV intron A, we performed ChIP assays with 293T cells transfected with pIE111 (expressing the MCMV MIE gene under the control of the MCMV MIEP and containing all four introns and five exons) (Fig. 7C, top panel). We designed three pairs of primers from intron A: A1, for the fragment before the putative CTCF BS; A2, for the fragment containing the putative CTCF BS; and A3, for the fragment after the CTCF BS. We found that anti-CTCF antibody precipitated fragment A2 to a greater extent than fragment A1 or A3 and did not precipitate any detectable DNA from the actin gene region. This ChIP assay result suggests that CTCF interacts with a conserved site in the first intron of the MCMV MIE gene. To determine whether the putative CTCF-binding sequence in the MCMV MIE gene is essential for the interaction of CTCF, we deleted the sequence from plasmid pIE111, resulting in pIE111dCTCFi. The mutant plasmid was transfected into 293T cells and assayed by ChIP for CTCF binding (Fig. 7C, bottom panel). The results showed that CTCF does not bind pIE111dCTCFi, indicating that the CTCF binding consensus site is required for the interaction of CTCF and the MCMV MIE gene.
FIG 7.

The CTCF-binding domain in intron A is conserved in HCMV and MCMV. (A) DNA sequences of the MCMV MIE genes, from the TATA box to the beginning of exon 2. The CTCF BS is underlined. Exon 1 sequences are boxed. (B) Homologous comparison of the CTCF-binding sequence from HCMV and MCMV to the consensus sequence of the canonical CTCF-binding motif. (C) ChIP-qPCR analysis of CTCF or control IgG with primers specific for the indicated regions in 293T cells transfected with either pIE111 (a plasmid expressing the MCMV MIE gene under the control of the MIEP) (top) or pIE111dCTCFi (derived from pIE111 but with the CTCF BS deleted) (bottom) at 24 h posttransfection. (D) Western blotting was used to determine MCMV IE1 expression in 293T cells cotransfected with various amount of pFlag-CTCF (0.5, 1, and 2 μg) and either pIE111 (left) or pIE111dCTCFi (right) at 24 h posttransfection. Flag antibody was used to determine CTCF expression, and tubulin served as a sample-loading control. Quantification of IE1 levels was done by Quantity One 4.5.0 software, and normalized IE1 levels are shown as bar graphs below the corresponding Western blot assays. (E) Same as panel D, except that quantitative RT-PCR was used to determine IE1 mRNA levels. The bar graph represents the relative RT-PCR values (means ± standard deviations) from three independent experiments.
Finally, we tested whether CTCF binding to the MIE gene affects gene expression. We cotransfected different amounts of pFlag-CTCF with pIE111 (Fig. 7D, left panel) or pIE11dCTCFi (Fig. 7D, right panel) into 293T cell for 24 h. Whole-cell lysates were used for Western blot assays to examine IE1 gene production, as shown in the top panels. Total RNA samples were used to detect IE1 mRNA by real-time RT-PCR assay (Fig. 7E). As shown in Fig. 7D, basal levels of IE1 were higher in pIE11dCTCFi than in pIE111. Furthermore, overexpression of CTCF significantly repressed the production of IE1 from pIE111 but not from pIE111dCTCFi. Therefore, CTCF is also able to repress MCMV MIE gene expression via interaction with a BS located within the first intron.
DISCUSSION
A complex combination of regulatory elements has been found to control the expression efficiency of the HCMV MIE gene. The investigative efforts of the past several decades have focused primarily on the upstream enhancer and promoter sites, but the regulatory features of the downstream transcribed regions are less well studied. The transcribed regions of the MIE contain five exons and four introns, with the first exon lacking any protein coding sequence. The earliest experimental investigation of this region found that transcription factor NF-1 has the strongest binding element in intron A among all five of the BSs in the MIE region (38). Further study indicated that the interaction of NF-1 with this site was able to enhance MIE gene expression with a transfection system and a reporter assay (35), suggesting that the noncoding region within the transcribed sequence of the MIE gene contributes to gene regulation at the early stage of HCMV infection.
Intron A is the largest of the introns, measuring about 800 bp in length and residing within the 5′ UTR of the MIE gene. Here, we identified a negative transcriptional control domain within intron A. This domain is positioned in the MIE gene from +834 to +852 (Fig. 3A), approximately 490 bp from a positive regulatory domain, the NF-1-binding domain (+350). This newly identified domain inhibits MIE gene expression through the interaction with the chromatin boundary protein CTCF. Both an EMSA and an FP assay were used to demonstrate the interaction between purified CTCF protein and purified MIE DNA. The affinity of the domain with CTCF is as strong as the positive control used for the EMSA and the FP assay (Fig. 3B and C). The results of the EMSA and the FP assay demonstrated that the interaction of CTCF protein with the intron A domain is direct. Furthermore, a ChIP assay confirmed that the interaction of CTCF with intron A occurs in vivo and that the short motif is required for CTCF-intron A interaction (Fig. 3D and 5A).
Interestingly, we also mapped the CTCF-binding domain in MCMV intron A and discovered a strikingly homologous sequence in MCMV intron A (Fig. 7). With a ChIP assay, we showed that CTCF interacts with MCMV intron A and that the homologous sequence is required for the interaction of CTCF and the MCMV MIE gene (Fig. 7C). Therefore, the CTCF-binding domain in intron A is conserved in CMVs. The conservation of a CTCF BS in approximately the same position within intron A of the CMV MIE gene suggests that these CTCF BSs have an important biological function. Our in vitro cotranscription experiments determined that the 19-bp CTCF-binding motifs of both MCMV and HCMV are required for the downregulation of MIE gene expression by CTCF (Fig. 2, 4, and 7). We also found that CTCF-intron A interaction is important for HCMV infection and affects HCMV replication (Fig. 5 and 6), indicating that the interaction between CTCF and intron A plays a functional role in regulating viral gene expression and DNA replication.
CTCF can function as a chromatin boundary and enhancer-blocking element, as well as contribute to DNA conformation and long-distance interactions between regulatory elements (51). CTCF is essential for embryonic development, as demonstrated in the Drosophila and mouse models (52). The importance of CTCF in the infection of several viruses, including EBV, KSHV, and HSV-1, has been reported, and it is believed that CTCF interaction with the viral genome is important for persistent viral infection and the maintenance of latency (30, 31, 33, 34). Multiple sites for CTCF binding were identified in the EBV and KSHV genomes in genome-wide studies (30). Interestingly, in KSHV, a major CTCF BS was found to colocalize within the first intron of the major latency transcript encoding LANA, vCyclin, and vFLIP (32). In EBV, a major CTCF BS was observed in the first intron of the LMP2A gene, which is expressed at early times during primary infection (54). Here, we demonstrate that CTCF also binds within the first intron of both the HCMV and MCMV MIE genes. This suggests that CTCF plays a universal and conserved role in the regulation of complex transcripts in the herpesvirus family. In agreement with this idea, we found that CTCF downregulates MIE gene expression and inhibits viral replication (Fig. 6 and 7). The function of CTCF binding in EBV and KSHV may be more complex, since these viruses establish long-term latent infections.
CTCF can colocalize with the cohesin complex (55), which has been implicated in long-distance promoter-enhancer interactions. Rad21 is a core component of cohesin and was found to further stabilize CTCF-mediated repression of the MIE gene when overexpressed in transient-transfection assays (Fig. 4E and F). While we have not formally tested the role of cohesin in the regulation of MIE gene expression during viral infection, it is tempting to speculate that CTCF and cohesin may function to coordinate the activity of the MIE enhancer as it acts on RNA polymerase transcribing the MIE gene body. Therefore, CTCF-cohesin may play a major role in regulating the communication between the MIE enhancer and elongating RNA polymerase.
Intron A is a noncoding area of the MIE gene and resides downstream of the gene promoter. Although it is clear that CTCF interacts with the sequence in intron A (Fig. 3), it remains elusive how CTCF represses MIE gene expression. One of the mechanisms CTCF uses to suppress gene expression is associated with chromosomal looping interactions (24). It has been found that CTCF regulates the expression of a variety of genes through CTCF-mediated chromosomal looping interactions, while cohesin is required for the formation of the loops (24, 27). Since repression of MIE expression by CTCF can occur in a reporter plasmid, one might assume that chromatin looping is probably not involved. This assumption is also supported by the fact that overexpression of the cohesin component Rad21 alone has no effects on MIE gene expression (Fig. 4C and D).
Promoter-proximal downstream transcriptional elements, such as those located in intron A of the MIE gene, may function at either the DNA or the RNA level. Elements located within introns may be expected to regulate gene splicing. However, we found no evidence that CTCF protein levels or the CTCF BS within intron A altered the relative IE1 and IE2 mRNA levels. This suggests that CTCF does not affect MIE gene splicing but functions predominantly at the level of transcription initiation or elongation. CTCF has been implicated in the control of mRNA splicing in other systems (26), and we cannot rule out the possibility that CTCF may affect MIE gene splicing in some cell types or under some infection conditions. Nevertheless, we found that the downstream binding of CTCF has a clear role in restricting CMV infection through the downregulation of MIE transcript generation.
In conclusion, we have identified a novel transcription control element located within the first intron of the CMV MIE gene that plays a major role in restricting MIE transcription and viral replication. We have shown that CTCF binds to this control element and is responsible for the attenuation of viral infection. We propose that this CTCF-MIE gene control element constitutes a key mechanism of viral infection regulation. The CTCF BS is conserved in MCMV and has strikingly high homology and similar positions in MIE genes. This downstream control domain may function by blocking RNA polymerase II elongation or MIE enhancer interactions. Intronic CTCF BSs are also observed at complex transcription units of EBV and KSHV, suggesting that CTCF is a universal regulator of herpesvirus gene expression and replication.
ACKNOWLEDGMENTS
We thank Ronen Marmorstein (University of Pennsylvania) for the purified CTCF protein. We are grateful to Bob Ritchie of the Ponce School of Medicine and Health Sciences/RCMI Publications Office (G12 RR003050/8G12MD007579-27) for his help with manuscript preparation.
This study was supported by a pilot grant from the Research Center for Minority Institutes (RCMI) program (2G12RR003050-24/8G12MD007579-27) (Q.T.), an American Cancer Society grant (RSG-090289-01-MPC) (Q.T), and NIH/NIAID SC1AI112785 (Q.T.). Z.D. was supported by an American Heart Association grant (11SDG5330017). P.M.L. was supported by the NIH (CA117830 and DE017336). We acknowledge the instrumental support of the Ponce School of Medicine Molecular Biology Core Laboratory.
Footnotes
Published ahead of print 16 April 2014
REFERENCES
- 1.Landolfo S, Gariglio M, Gribaudo G, Lembo D. 2003. The human cytomegalovirus. Pharmacol. Ther. 98:269–297. 10.1016/S0163-7258(03)00034-2 [DOI] [PubMed] [Google Scholar]
- 2.Mocarski ES, Jr, Shenk T, Pass RF. 2006. Cytomegaloviruses, 5th ed. Lippincott Williams &Wilkins, Philadelphia, PA [Google Scholar]
- 3.Sweet C. 1999. The pathogenicity of cytomegalovirus. FEMS Microbiol. Rev. 23:457–482. 10.1111/j.1574-6976.1999.tb00408.x [DOI] [PubMed] [Google Scholar]
- 4.Ahn JH, Hayward GS. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 71:4599–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Awasthi S, Isler JA, Alwine JC. 2004. Analysis of splice variants of the immediate-early 1 region of human cytomegalovirus. J. Virol. 78:8191–8200. 10.1128/JVI.78.15.8191-8200.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hagemeier C, Walker SM, Sissons PJ, Sinclair JH. 1992. The 72K IE1 and 80K IE2 proteins of human cytomegalovirus independently trans-activate the c-fos, c-myc and hsp70 promoters via basal promoter elements. J. Gen. Virol. 73(Pt 9):2385–2393. 10.1099/0022-1317-73-9-2385 [DOI] [PubMed] [Google Scholar]
- 7.Liu B, Hermiston TW, Stinski MF. 1991. A cis-acting element in the major immediate-early (IE) promoter of human cytomegalovirus is required for negative regulation by IE2. J. Virol. 65:897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meier JL, Stinski MF. 1997. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J. Virol. 71:1246–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadanari H, Yamada R, Yamagoshi T, Ohnishi K, Matsubara K, Fukuda S, Tanaka J. 2000. The major immediate-early genes of human cytomegalovirus induce two novel proteins with molecular weights of 91 and 102 kilodaltons. Arch. Virol. 145:1257–1266. 10.1007/s007050070125 [DOI] [PubMed] [Google Scholar]
- 10.Scully AL, Sommer MH, Schwartz R, Spector DH. 1995. The human cytomegalovirus IE2 86-kilodalton protein interacts with an early gene promoter via site-specific DNA binding and protein-protein associations. J. Virol. 69:6533–6540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stenberg RM. 1996. The human cytomegalovirus major immediate-early gene. Intervirology 39:343–349 [DOI] [PubMed] [Google Scholar]
- 12.Stenberg RM, Thomsen DR, Stinski MF. 1984. Structural analysis of the major immediate early gene of human cytomegalovirus. J. Virol. 49:190–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Q, Li L, Maul GG. 2005. Mouse cytomegalovirus early M112/113 proteins control the repressive effect of IE3 on the major immediate-early promoter. J. Virol. 79:257–263. 10.1128/JVI.79.1.257-263.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Q, Maul GG. 2005. Immediate early interactions and epigenetic defense mechanisms, p 131–150 In Reddehase MJ. (ed), Cytomegaloviruses: molecular biology and immunology. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 15.Meier JL, Stinski MF. 2006. Major immediate-early enhancer and its gene products. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 16.Murphy JC, Fischle W, Verdin E, Sinclair JH. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 21:1112–1120. 10.1093/emboj/21.5.1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nevels M, Paulus C, Shenk T. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. U. S. A. 101:17234–17239. 10.1073/pnas.0407933101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 281:37652–37660. 10.1074/jbc.M604273200 [DOI] [PubMed] [Google Scholar]
- 19.Kim YE, Lee JH, Kim ET, Shin HJ, Gu SY, Seol HS, Ling PD, Lee CH, Ahn JH. 2011. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J. Virol. 85:11928–11937. 10.1128/JVI.00758-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 81:9109–9120. 10.1128/JVI.00827-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tavalai N, Adler M, Scherer M, Riedl Y, Stamminger T. 2011. Evidence for a dual antiviral role of the major nuclear domain 10 component Sp100 during the immediate-early and late phases of the human cytomegalovirus replication cycle. J. Virol. 85:9447–9458. 10.1128/JVI.00870-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tavalai N, Papior P, Rechter S, Leis M, Stamminger T. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 80:8006–8018. 10.1128/JVI.00743-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cosme RS, Yamamura Y, Tang Q. 2009. Roles of polypyrimidine tract binding proteins in major immediate-early gene expression and viral replication of human cytomegalovirus. J. Virol. 83:2839–2850. 10.1128/JVI.02407-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phillips JE, Corces VG. 2009. CTCF: master weaver of the genome. Cell 137:1194–1211. 10.1016/j.cell.2009.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun FL, Elgin SC. 1999. Putting boundaries on silence. Cell 99:459–462. 10.1016/S0092-8674(00)81534-2 [DOI] [PubMed] [Google Scholar]
- 26.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. 2011. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479:74–79. 10.1038/nature10442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barkess G, West AG. 2012. Chromatin insulator elements: establishing barriers to set heterochromatin boundaries. Epigenomics 4:67–80. 10.2217/epi.11.112 [DOI] [PubMed] [Google Scholar]
- 28.Kanduri C, Pant V, Loukinov D, Pugacheva E, Qi CF, Wolffe A, Ohlsson R, Lobanenkov VV. 2000. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol. 10:853–856. 10.1016/S0960-9822(00)00597-2 [DOI] [PubMed] [Google Scholar]
- 29.Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. 2007. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128:1231–1245. 10.1016/j.cell.2006.12.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tempera I, Wiedmer A, Dheekollu J, Lieberman PM. 2010. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog. 6:e1001048. 10.1371/journal.ppat.1001048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang H, Wiedmer A, Yuan Y, Robertson E, Lieberman PM. 2011. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog. 7:e1002140. 10.1371/journal.ppat.1002140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM. 2008. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 27:654–666. 10.1038/emboj.2008.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amelio AL, McAnany PK, Bloom DC. 2006. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J. Virol. 80:2358–2368. 10.1128/JVI.80.5.2358-2368.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Q, Lin L, Smith S, Huang J, Berger SL, Zhou J. 2007. CTCF-dependent chromatin boundary element between the latency-associated transcript and ICP0 promoters in the herpes simplex virus type 1 genome. J. Virol. 81:5192–5201. 10.1128/JVI.02447-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chapman BS, Thayer RM, Vincent KA, Haigwood NL. 1991. Effect of intron A from human cytomegalovirus (Towne) immediate-early gene on heterologous expression in mammalian cells. Nucleic Acids Res. 19:3979–3986. 10.1093/nar/19.14.3979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mariati Ng YK, Chao SH, Yap MG, Yang Y. 2010. Evaluating regulatory elements of human cytomegalovirus major immediate early gene for enhancing transgene expression levels in CHO K1 and HEK293 cells. J. Biotechnol. 147:160–163. 10.1016/j.jbiotec.2010.02.022 [DOI] [PubMed] [Google Scholar]
- 37.Xia W, Bringmann P, McClary J, Jones PP, Manzana W, Zhu Y, Wang S, Liu Y, Harvey S, Madlansacay MR, McLean K, Rosser MP, MacRobbie J, Olsen CL, Cobb RR. 2006. High levels of protein expression using different mammalian CMV promoters in several cell lines. Protein Expr. Purif. 45:115–124. 10.1016/j.pep.2005.07.008 [DOI] [PubMed] [Google Scholar]
- 38.Hennighausen L, Fleckenstein B. 1986. Nuclear factor 1 interacts with five DNA elements in the promoter region of the human cytomegalovirus major immediate early gene. EMBO J. 5:1367–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Bruyn Kops A, Knipe DM. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857–868. 10.1016/0092-8674(88)90141-9 [DOI] [PubMed] [Google Scholar]
- 40.Cosme-Cruz R, Martinez FP, Perez KJ, Tang Q. 2011. H2B homology region of major immediate-early protein 1 is essential for murine cytomegalovirus to disrupt nuclear domain 10, but is not important for viral replication in cell culture. J. Gen. Virol. 92:2006–2019. 10.1099/vir.0.033225-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stenberg RM, Fortney J, Barlow SW, Magrane BP, Nelson JA, Ghazal P. 1990. Promoter-specific trans activation and repression by human cytomegalovirus immediate-early proteins involves common and unique protein domains. J. Virol. 64:1556–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keil GM, Ebeling-Keil A, Koszinowski UH. 1987. Immediate-early genes of murine cytomegalovirus: location, transcripts, and translation products. J. Virol. 61:526–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borst EM, Hahn G, Koszinowski UH, Messerle M. 1999. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J. Virol. 73:8320–8329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warden C., Tang Q, Zhu H. 2011. Herpesvirus BACs: past, present, and future. J. Biomed. Biotechnol. 2011:124595. 10.1155/2011/124595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moerke NJ. 2009. Fluorescence polarization (FP) assays for monitoring peptide-protein or nucleic acid-protein binding. Curr. Protoc. Chem. Biol. 1:1–15. 10.1002/9780470559277.ch090102 [DOI] [PubMed] [Google Scholar]
- 46.Bao L, Zhou M, Cui Y. 2008. CTCFBSDB: a CTCF-binding site database for characterization of vertebrate genomic insulators. Nucleic Acids Res. 36(Database Issue):D83–D87. 10.1093/nar/gkm875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plasschaert RN, Vigneau S, Tempera I, Gupta R, Maksimoska J, Everett L, Davuluri R, Mamorstein R, Lieberman PM, Schultz D, Hannenhalli S, Bartolomei MS. 2014. CTCF binding site sequence differences are associated with unique regulatory and functional trends during embryonic stem cell differentiation. Nucleic Acids Res. 42:774–789. 10.1093/nar/gkt910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng Z, Wang Z, Stong N, Plasschaert R, Moczan A, Chen HS, Hu S, Wikramasinghe P, Davuluri RV, Bartolomei MS, Riethman H, Lieberman PM. 2012. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J. 31:4165–4178. 10.1038/emboj.2012.266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, Cobb BS, Yokomori K, Dillon N, Aragon L, Fisher AG, Merkenschlager M. 2008. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 132:422–433. 10.1016/j.cell.2008.01.011 [DOI] [PubMed] [Google Scholar]
- 50.Rubio ED, Reiss DJ, Welcsh PL, Disteche CM, Filippova GN, Baliga NS, Aebersold R, Ranish JA, Krumm A. 2008. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. U. S. A. 105:8309–8314. 10.1073/pnas.0801273105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, Yahata K, Imamoto F, Aburatani H, Nakao M, Imamoto N, Maeshima K, Shirahige K, Peters JM. 2008. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451:796–801. 10.1038/nature06634 [DOI] [PubMed] [Google Scholar]
- 52.Merkenschlager M, Odom DT. 2013. CTCF and cohesin: linking gene regulatory elements with their targets. Cell 152:1285–1297. 10.1016/j.cell.2013.02.029 [DOI] [PubMed] [Google Scholar]
- 53.Guo Y, Monahan K, Wu H, Gertz J, Varley KE, Li W, Myers RM, Maniatis T, Wu Q. 2012. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proc. Natl. Acad. Sci. U. S. A. 109:21081–21086. 10.1073/pnas.1219280110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. 2012. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 12:233–245. 10.1016/j.chom.2012.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herold M, Bartkuhn M, Renkawitz R. 2012. CTCF: insights into insulator function during development. Development 139:1045–1057. 10.1242/dev.065268 [DOI] [PubMed] [Google Scholar]