

ABSTRACT
Budding of filoviruses, arenaviruses, and rhabdoviruses is facilitated by subversion of host proteins, such as Nedd4 E3 ubiquitin ligase, by viral PPxY late (L) budding domains expressed within the matrix proteins of these RNA viruses. As L domains are important for budding and are highly conserved in a wide array of RNA viruses, they represent potential broad-spectrum targets for the development of antiviral drugs. To identify potential competitive blockers, we used the known Nedd4 WW domain-PPxY interaction interface as the basis of an in silico screen. Using PPxY-dependent budding of Marburg (MARV) VP40 virus-like particles (VLPs) as our model system, we identified small-molecule hit 1 that inhibited Nedd4-PPxY interaction and PPxY-dependent budding. This lead candidate was subsequently improved with additional structure-activity relationship (SAR) analog testing which enhanced antibudding activity into the nanomolar range. Current lead compounds 4 and 5 exhibit on-target effects by specifically blocking the MARV VP40 PPxY-host Nedd4 interaction and subsequent PPxY-dependent egress of MARV VP40 VLPs. In addition, lead compounds 4 and 5 exhibited antibudding activity against Ebola and Lassa fever VLPs, as well as vesicular stomatitis and rabies viruses (VSV and RABV, respectively). These data provide target validation and suggest that inhibition of the PPxY-Nedd4 interaction can serve as the basis for the development of a novel class of broad-spectrum, host-oriented antivirals targeting viruses that depend on a functional PPxY L domain for efficient egress.
IMPORTANCE There is an urgent and unmet need for the development of safe and effective therapeutics against biodefense and high-priority pathogens, including filoviruses (Ebola and Marburg) and arenaviruses (e.g., Lassa and Junin) which cause severe hemorrhagic fever syndromes with high mortality rates. We along with others have established that efficient budding of filoviruses, arenaviruses, and other viruses is critically dependent on the subversion of host proteins. As disruption of virus budding would prevent virus dissemination, identification of small-molecule compounds that block these critical viral-host interactions should effectively block disease progression and transmission. Our findings provide validation for targeting these virus-host interactions as we have identified lead inhibitors with broad-spectrum antiviral activity. In addition, such inhibitors might prove useful for newly emerging RNA viruses for which no therapeutics would be available.
INTRODUCTION
Filoviruses (Ebola [EBOV] and Marburg [MARV]), arenaviruses (e.g., Lassa fever [LFV] and Junin [JUNV]), and rhabdoviruses (e.g., vesicular stomatitis virus [VSV] and rabies virus [RABV]) are enveloped RNA viruses which can cause severe disease in humans and animals. For example, filovirus and arenavirus infections can result in hemorrhagic syndromes with high mortality rates in humans, and, as such, these viruses are classified as NIAID category A priority pathogens (1–4). There are currently no available vaccines or therapeutics to control infection and transmission of EBOV, MARV, LFV, JUNV, and several RABV-related lyssaviruses of phylogroups 2 and 3. In an effort to identify and develop antiviral therapeutics with broad-spectrum activity against these RNA viruses, we focused on the viral matrix proteins and, more specifically, on their interactions with host proteins during the virus life cycle.
The matrix proteins of filoviruses (VP40), arenaviruses (Z), and rhabdoviruses (M) are highly abundant and play key roles in promoting virus assembly and egress (5–7). For example, independent expression of EBOV or MARV VP40 (eVP40 or mVP40, respectively) leads to the production of virus-like particles (VLPs) that accurately mimic the morphology and budding characteristics of infectious virus (5–7). A common feature of these various viral matrix proteins is the presence of one or more motifs referred to as late (L) budding domains. The conservation of L domains within the matrix proteins of filoviruses, arenaviruses, rhabdoviruses, paramyxoviruses, and retroviruses suggests that they are generally important and required for efficient RNA virus budding (8). Viral L domains recruit host ESCRT (endosomal sorting complex required for transport) complexes to mediate efficient virus-cell separation (or “pinching-off”) and consist of core consensus amino acid motifs such as PPxY, P(T/S)AP, YxxL, or FPIV (where x is any amino acid) (for a review, see reference 8). Indeed, a plethora of studies have demonstrated the importance of viral L-domain–host interactions for efficient virus egress and spread (for a review, see references 6 to 13). For example, the PPxY motif mediates interactions with WW domains within mammalian E3 ubiquitin ligase Nedd4 to facilitate virus egress (14–31). Nedd4 is associated with the ESCRT machinery and mono-ubiquitinates ESCRT proteins as well as viral matrix proteins (7, 14–17, 20, 21, 23, 24, 28, 29, 32–40). A functional PPxY motif is present in the matrix proteins of EBOV, MARV, VSV, RABV, LFV, and other viruses (14–31). Thus, recruitment of host proteins such as Nedd4 by viral L domains represents a broad-spectrum target for the identification and advancement of antiviral drugs hypothesized to dampen virus egress from infected cells, thereby reducing virus dissemination and disease progression.
In this report, we employed an in silico screening strategy to identify small molecules that competitively block the interaction between viral PPxY L domains and the WW domain(s) of host Nedd4. Using structure-activity relationship (SAR) analog testing, we dissected initial hit 1 into two fragments (Fig. 1, red dissection line in compound 1) and then searched, acquired, and tested commercially available compounds possessing these two substructures in our VLP and bimolecular complementation (BiMC) assays. This analysis led to the identification of two potential lead series, exemplified by the two current lead compounds 4 and 5, that possess PPxY-dependent antibudding activity in the nanomolar range against VLPs and infectious virus. It is important to note that the PPxY motif is the sole functional L domain within MARV VP40, whereas EBOV VP40 contains functional and overlapping PPxY- and PTAP-type L domains (14, 19, 29, 41–46). Also budding of both VSV and RABV in our cell culture system is driven solely by single PPxY L domains within their M proteins (10, 15, 47–50). Here, we show that lead compounds 4 and 5 can inhibit egress of VLPs formed by MARV VP40, EBOV VP40, and LFV Z and PPxY-dependent budding of VSV and RABV in a dose-dependent manner. In addition, results using a BiMC approach further support our conclusion that the mechanism of action of compounds 4 and 5 involves competitive disruption of the viral PPxY-host Nedd4 interaction, leading to a decrease in VLP and/or virus egress. These findings serve as target validation and suggest that the viral PPxY-host WW domain interaction can be exploited to eventually obtain broad-spectrum antiviral compounds that can be evaluated further in a detailed investigational new drug (IND)-directed manner.
FIG 1.

Rationale and strategy for identifying PPxY budding inhibitors. The diagram shows arenavirus, filovirus, and rhabdovirus virions budding efficiently from the plasma membrane in the absence of inhibitors (A) or remaining tethered to the plasma membrane as a result of PPxY inhibitors blocking the interaction between host Nedd4 and the PPxY L domains present in the Z, VP40, and M viral matrix proteins (B). (C) Flow chart showing an in silico screen and SAR analysis to identify inhibitors of the viral PPxY-host Nedd4 interaction and PPxY-mediated budding. The in silico screen involved computational docking with AutoDock, version 4.0, energy minimization using CHARMM with the MMFF force field, and ranking with Accelrys LigScore2 of 4.8 million drug-like compounds from the ZINC database. The top 20 scoring compounds were tested, as indicated, leading to the identification of our initial lead compound 1. To understand how the structure of compound 1 affected its activity, the molecule was dissected into two fragments (see disconnection point in red), and 20 commercially available compounds having these two fragment substructures (10 compounds from each) were evaluated. Two compounds (2 and 3), one from each substructure search, were found to be more potent than compound 1. Further substructure searches of commercial databases were performed and led to the acquisition and testing of an additional 10 compounds (5 structurally related to compound 2 and 5 related to compound 3). Compound 4 showed improved potency over SAR analog 2, and compound 5 showed improved potency over SAR analog 3. Compound 6 is a structurally related inactive negative control.
MATERIALS AND METHODS
Cell lines, viruses, and antibodies.
HEK293T, BHK-21, and BSR cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), and penicillin (100 U/ml)/streptomycin (100 μg/ml) at 37°C in a humidified 5% CO2 incubator. Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; ab8245) and anti-HSP70 antibodies (BRM-22; ab6535) were purchased from Abcam (Cambridge, MA); mouse anti-Flag monoclonal antibody (F1804-200UG) and mouse anti-β-actin (A1978-200UL) were obtained from Sigma-Aldrich (St. Louis, MO); polyclonal anti-EBOV VP40 antibody was generated by ProSci Incorporated (Poway, CA); anti-VSV-M monoclonal antibody 23H12 was kindly provided by D. Lyles (Winston-Salem, NC); anti-RABV-M is a rabbit polyclonal serum raised against an N-terminal peptide (amino acids [aa] 4 to 19) from SAD B19; anti-LFV-Z is a rabbit polyclonal serum kindly provided by S. Urata (Nagasaki, Japan) (39). VSV wild type (VSV-WT; Indiana strain), VSV recombinant M40, and a VSV mutant in which the PPxY motif was changed to four alanines (VSV-PY>A4) were propagated in BHK-21 cells and have been described previously (22, 41, 51). RABV (SPBN) is derived from the SAD B19 vaccine strain and has been described previously (52).
Test compounds.
All compounds were ≥95% pure, as determined independently at the Fox Chase Chemical Diversity Center (FCCDC) using liquid chromatography-mass spectrometry (LC/MS) (Micromass ZQ mass spectrometer with a Waters 2695 high-performance liquid chromatograph [HPLC] with a 996 diode array detector); compounds were dissolved in dimethyl sulfoxide (DMSO) at concentrations of 10 or 100 mM and stored at −20°C. Compound 1 (Z106187460) was identified from the ZINC database and purchased from ChemDiv (San Diego, CA). Commercial analogs of compound 1, namely, compounds 2 (Amb207302), 3 (Amb21795400), 4 (Amb123203), 5 (Amb21795397), and 6 (Amb21639324), were purchased from Ambinter (Orléans, France).
Plasmids.
pCAGGS-based plasmids expressing EBOV (Zaire) VP40 and Flag-tagged MARV (Musoke) VP40 have been described previously (7, 19, 31, 53–58). Carboxyl-yellow fluorescent protein (CYFP)-mVP40 has been described previously (59). Plasmid NYFP-Nedd4 was generated by amplifying the human Nedd4 open reading frame (ORF) using standard PCR techniques and joining it to the N-terminal YFP (NYFP) fragment. The NYFP-Nedd4 fusion gene was cloned into the SmaI and NheI sites of the pCAGGS vector. The plasmid expressing LFV-Z WT protein (pCLFV-Z) was kindly provided by S. Urata (Nagasaki, Japan) (39). A pCAGGS-based plasmid expressing LFV-Z-ΔPPPY was constructed by standard PCR and cloning techniques.
Bimolecular complementation assay.
A BiMC assay in HEK293T cells was performed as described previously (31, 59, 60). Briefly, HEK293T cells were cotransfected with CYFP-mVP40 and NYFP-Nedd4 for 4 to 5 h, and the cells were then treated with concentrations of compound 4, 5, or 6 as indicated in the figures for an additional 24 h. Cell nuclei were stained with NucBlue Live Cell Stain reagent (Life Technologies) according to the manufacturer's instructions. Cells were examined by fluorescence microscopy and quantified using MetaMorph software (Molecular Devices, CA). Briefly, we used MetaMorph software to mark all regions of YFP fluorescence above a set threshold and quantified this threshold area as a percentage of the entire area of the field. This method indicates the change in overall fluorescence area, regardless of whether it is due to a change in the number of positive cells or to the amount per cell. We also used a similar measurement for area of 4′,6′-diamidino-2-phenylindole (DAPI) stain to ensure that cell number remained roughly the same throughout. In a second set of analyses, we used the cell scoring module of MetaMorph to count the total number of nuclei (taken to also be the total number of cells) and the number of cells positive for YFP fluorescence (i.e., above a set threshold). This is a measure only of percent positive cells and not necessarily of overall amounts of fluorescence per cell.
VLP budding assay and Western blotting.
VLP budding assays and Western blotting were performed as described previously (14, 19, 56, 57).
Virus budding and titration.
To evaluate the effect of compounds on the release of VSV-WT, recombinant VSV-M40, or mutant VSV-PY>A4, HEK293T cells in collagen-coated six-well plates were infected with the appropriate virus at a multiplicity of infection (MOI) of 0.1 for 1 h. The inoculum was removed, and cells were washed three times with phosphate-buffered saline (PBS) and then incubated in serum-free Opti-MEM in the presence of DMSO alone (mock) or the appropriate compounds at the concentrations indicated in the figures. Virions from the culture medium were harvested at 8 h postinfection (p.i.) and centrifuged at 2,500 rpm for 10 min at 4°C to remove cellular debris. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor). VSV-M or M40 protein in cell extracts was analyzed by SDS-PAGE and Western blotting with anti-VSV-M monoclonal antibody 23H12, followed by anti-mouse IgG horseradish peroxidase (HRP)-conjugated secondary antibody. For virus titration, BHK-21 cells in six-well plates were washed one time with PBS and inoculated with 200 μl of 10-fold serial dilutions of virus in serum-free DMEM in triplicate and incubated for 1 h. The inoculum was removed, and the cells were washed three times with PBS and incubated with 2.0 ml of Eagle's minimal essential medium (MEM) containing 5% FBS and 1% methylcellulose at 37°C for 36 to 48 h until plaques were observed. Cells were washed two times with PBS, fixed with methanol, and stained with 1% crystal violet solution.
To evaluate the effect of compounds on the release of RABV, HEK293T cells were infected with SPBN at an MOI of 0.1 in the absence or presence of the probe, and cells were incubated at 34°C. Virus-containing supernatants were harvested at the indicated times postinfection, and titers were determined in duplicate on BSR cells. RABV-M protein was detected by Western blotting of HEK293T cells infected with SPBN at an MOI of 10 for 36 h.
Structure/activity studies.
Compound 1 was identified via an in silico screen involving computational docking of over 4.8 million drug-like compounds from the ZINC database into the published structure between the viral PPxY motif and the host WW domain of Nedd4 (61, 62) with AutoDock, version 4.0, followed by energy minimization using CHARMM with the MMFF force field and ranking with Accelrys LigScore2 (63–66). Experimentally testing the top 20 scoring compounds led to the identification of initial hit 1 as the most active compound. Compound 1 was then dissected conceptually into two fragments, as shown by the red dissection line in Fig. 1, in order to use the two fragments in substructure searches (SSS) of commercial databases to find new SAR analogs to evaluate. More specifically, the 2-piperidin-3-yl-benzothiazole fragment (left side) and 1-acetyl-3-(2,2,2-trifluoroethyl)-urea fragment (right side) were used for substructure searching (SSS) of the Ambinter (Orléans, France) commercial compound database. Ambinter was chosen as the compound vendor because of its large library of compounds containing both substructure fragments of compound 1. Ten commercially available compounds (five from each fragment substructure) were purchased and evaluated. This led to more potent compounds, namely, congener 2 obtained from SSS of the left side fragment and analog 3 from SSS of the right side fragment. Structurally related inactive compound 6, used as a negative control in later studies, also arose from this exercise (SSS analog of the right-hand fragment of compound 1). Substructure/similarity searching for analogs of compound 2 led to the acquisition and testing of an additional two analogs of compound 2. One of these two was compound 4, which proved to be more potent than compound 2. Substructure/similarity searching for analogs of compound 3 led to the acquisition and testing of an additional eight analogs of compound 3. While several active analogs of compound 3 resulted from this exercise, compound 5 stood out as the most potent, exceeding the potency of compound 3.
Statistical analysis.
Statistical analysis was determined using a one-way analysis of variance (ANOVA) test.
RESULTS
Strategy to identify host-oriented inhibitors targeting viral PPxY-host Nedd4 interactions.
As mentioned above, subversion of host Nedd4 E3 ubiquitin ligase by viral PPxY-type L domains is important for efficient budding (virus-cell separation) of numerous RNA viruses including filoviruses, arenaviruses, and rhabdoviruses (Fig. 1A). Small-molecule inhibitors that block this virus-host interaction would be predicted to reduce virus-cell separation and virus spread by concomitantly increasing the number of virions tethered to the plasma membrane (Fig. 1B). We sought to identify such candidate small-molecule compounds by using an in silico screening strategy (63–65) to probe the reported interaction structure between the viral PPxY motif and the host WW domain of Nedd4 (61, 62) (Fig. 1C). This led to the identification of compound 1 as our initial hit. Structural analogs of compound 1 were then found by substructure searching of commercial databases for compounds having the substructure fragments from each side of the dissection line of compound 1 (Fig. 1C), and these compounds were then purchased and evaluated in our assays. This led to the identification of the more potent compounds 2 and 3. Further SAR studies resulted in the discovery of our current most potent lead inhibitors, compounds 4 and 5 (Fig. 1C).
Antibudding activity of initial hit 1.
Compound 1 was tested for its ability to block PPxY-dependent budding of MARV VP40 (mVP40) VLPs in a dose-dependent manner. Briefly, HEK293T cells were transfected with pCAGGS vector alone (Fig. 2A, lane 1) or an mVP40 expression plasmid in the absence (DMSO alone, lane 2) or presence of the compound 1 at the concentrations indicated on the figure (lanes 3 to 6). Compound 1 produced a dose-dependent inhibition of mVP40 VLP formation with an approximate 5-fold decrease in VLP egress at a concentration of 20 μM compared to controls (Fig. 2A, VLPs, lanes 2 and 6, and bar graph). Compound 1 had no effect on expression levels of mVP40 or actin in cells at all drug concentrations tested (Fig. 2A, cells, lanes 1 to 6).
FIG 2.

Effect of 1 on PPxY-dependent budding of MARV VP40 VLPs and infectious VSV. (A) HEK293T cells transfected with pCAGGS vector (lane 1) or mVP40 (lanes 2 to 6) were treated with DMSO alone (lanes 1 and 2) or with the indicated concentrations of compound 1 (lanes 3 to 6). Cells and VLPs were harvested at 24 h posttransfection, and a representative Western blot to detect mVP40 is shown. A Western blot control for β-actin in cells is shown. Data in the bar graph showing mVP40 levels (ImageJ software) in VLPs from samples receiving 0 and 20 μM compound 1 represent the average from three independent experiments. (B) HEK293T cells were infected with VSV-WT or VSV-PY>A4 at an MOI of 0.1 in the absence (DMSO alone) or presence of 10 μM compound 1. Supernatants were harvested at 8 h postinfection, and virions were quantified by standard plaque assay on BHK-21 cells performed in triplicate and graphed as PFU/ml. *, P < 0.005; ns, not significant. Infected cell extracts were harvested at 8 h postinfection, and both VSV M and cellular actin were detected by Western blotting.
We next sought to determine whether compound 1 could inhibit budding of a PPxY-dependent virus from cell culture. We took advantage of the fact that efficient egress of VSV is dependent on a functional PPxY L domain within its M protein (15, 40, 47–50). Briefly, HEK293T cells were infected with VSV-WT or a PPxY mutant virus (VSV-PY>A4) in which the PPxY motif was changed to four alanines, in the absence (DMSO alone) or presence of 10 μM compound 1. Cell extracts and virus-containing supernatants were harvested at 8 h postinfection and analyzed for levels of M protein by Western blotting and viral titers by plaque assay, respectively (Fig. 2B). In the presence of 10 μM compound 1, budding of mutant VSV-PY>A4 was reduced by <2-fold, whereas budding of VSV-WT was reduced significantly by approximately 4-fold (Fig. 2B). Compound 1 had no effect on expression levels of actin or VSV-M at a concentration of 10 μM, as determined by Western blotting of infected cell extracts (Fig. 2B).
Antibudding activity of SAR analogs 2 and 3.
Using the structure of compound 1 as our starting point, we performed SAR analog testing to identify structurally related and progressively more potent small-molecule inhibitors of budding. Toward this end, we identified compounds 2 and 3 and tested their ability to inhibit egress of both MARV and EBOV VP40 VLPs in a dose-dependent manner (Fig. 3). Briefly, HEK293T cells were transfected with plasmids expressing either mVP40 or eVP40 in the absence (DMSO alone) or presence of compound 2 (Fig. 3A and C) or 3 (Fig. 3B and D) at the concentrations indicated on the figure. Both cell extracts and VLPs were harvested at 24 h posttransfection, and levels of mVP40 and eVP40 were assessed by Western blotting. Compound 2 inhibited egress of mVP40 VLPs by 33- and 100-fold at concentrations of 20 μM and 40 μM, respectively (Fig. 3A, VLPs, lanes 4 and 5) with a <2-fold effect on the levels of mVP40 and host GAPDH in cell extracts (Fig. 3A, cells, lanes 1 to 5). Compound 2 inhibited egress of eVP40 VLPs by 2.5- and 50-fold at concentrations of 20 μM and 40 μM, respectively (Fig. 3C, VLPs, lanes 4 and 5) with a <2-fold effect on the levels of eVP40 and host GAPDH in cell extracts (Fig. 3C, cells, lanes 1 to 5). Relative to compound 2, compound 3 was far more potent at inhibiting budding of both mVP40 and eVP40 VLPs. Indeed, compound 3 inhibited egress of mVP40 VLPs by 10-, 100-, and 100-fold at concentrations of 1.0 μM, 10 μM, and 20 μM, respectively (Fig. 3B, VLPs, lanes 2 to 4), and inhibited egress of eVP40 VLPs by <2-, 4-, and 20-fold at concentrations of 1.0 μM, 10 μM, and 20 μM, respectively (Fig. 3D, VLPs, lanes 4 to 6). Compound 3 had no effect on expression levels of mVP40, eVP40, or GAPDH in cell extracts at any concentration tested (Fig. 3B and D, cells). Negative-control compound 6 served to validate the specificity and antibudding activity of compounds 2 and 3 since compound 6 showed no effect on either cell or VLP expression levels of mVP40 at any concentration tested (Fig. 3E, lanes 1 to 6).
FIG 3.

Effect of compounds 2 and 3 on budding of mVP40 and eVP40 VLPs. (A and B) HEK293T cells transfected with mVP40 were treated with DMSO alone (lanes 1) or with the indicated concentrations of compound 2 (A, lanes 2 to 5) or compound 3 (B, lanes 2 to 4). Cells and VLPs were harvested at 24 h posttransfection, and mVP40 was detected by Western blotting. A Western blot control for cellular GAPDH in cells is shown. (C and D) HEK293T cells transfected with eVP40 were treated with DMSO alone (lanes 1) or with the indicated concentrations of compound 2 (C, lanes 2 to 5) or compound 3 (D, lanes 2 to 6). Cells and VLPs were harvested at 24 h posttransfection, and eVP40 was detected by Western blotting. A Western blot control for cellular GAPDH in cells is shown. (E) HEK293T cells transfected with mVP40 were treated with DMSO alone (lane 1) or with the indicated concentrations of compound 6 (lanes 2 to 6) as a negative control. Cells and VLPs were harvested at 24 h posttransfection, and mVP40 was detected by Western blotting. Numbers in parentheses were determined using ImageJ software (NIH), and controls were set at 100%.
Antibudding activity of lead compounds 4 and 5.
Continuing SAR studies led to the identification of our most potent leads, compounds 4 and 5. Compounds 4 and 5 were tested for their ability to inhibit PPxY-dependent budding of both VLPs and infectious virus from cell culture, as well as to competitively block the interaction between mVP40 and host Nedd4 in live mammalian cells using a BiMC approach. Briefly, HEK293T cells were transfected with plasmids expressing either mVP40 or eVP40 in the absence (DMSO alone) or presence of compound 4 (Fig. 4A and B) at the concentrations indicated on the figure. Both cell extracts and VLPs were harvested at 24 h posttransfection, and levels of mVP40 and eVP40 were assessed by Western blotting and quantified. Compound 4 inhibited egress of mVP40 VLPs by 33- and 100-fold at concentrations of 0.5 μM and 1.0 μM, respectively (Fig. 4A, VLPs, lanes 3 and 4, and bar graph). Compound 4 inhibited egress of eVP40 VLPs by 3- and 10-fold at concentrations of 0.5 μM and 1.0 μM, respectively (Fig. 4B, VLPs, lanes 2 and 3, and bar graph). Compound 4 had no effect on expression levels of mVP40, GAPDH, or HSP70 in cell extracts at all concentrations tested (Fig. 4A and B, cells).
FIG 4.

Compound 4 inhibits budding of mVP40 and eVP40 VLPs and blocks mVP40-Nedd4 protein-protein interaction. (A) HEK293T cells transfected with mVP40 were treated with DMSO alone (lane 1) or with the indicated concentrations of compound 4 (lanes 2 to 4). Cells and VLPs were harvested at 24 h posttransfection, and a representative Western blot for mVP40 is shown. Western blot controls for cellular GAPDH and HSP70 in cells are shown. The bar graph represents the average levels of mVP40 VLPs from three independent experiments. *, P < 0.001. Numbers in parentheses were determined using ImageJ software (NIH), and controls were set at 100%. (B) HEK293T cells transfected with eVP40 were treated with DMSO alone (lane 1) or with the indicated concentrations of compound 4 (lanes 2 and 3). Cells and VLPs were harvested at 24 h posttransfection, and a representative Western blot for eVP40 is shown. Western blot controls for cellular GAPDH and HSP70 in cells are shown. The bar graph represents the average levels of eVP40 VLPs from three independent experiments. *, P < 0.005. Numbers in parentheses were determined using ImageJ software (NIH), and controls were set at 100%. (C) BiMC assay and representative images of HEK293T cells coexpressing NYFP-Nedd4 and CYFP-mVP40 fusion proteins in the absence (DMSO alone) or presence of the indicated concentrations of compound 4 or 6. The green signal represents an interaction between mVP40 and Nedd4, and cell nuclei were stained blue with NucBlue. Scale bar, 200 μm. YFP-positive cells were quantified in triplicate using MetaMorph software.
A BiMC assay was used to determine whether compound 4 could specifically block the protein-protein interaction between mVP40 and host Nedd4 in a dose-dependent manner (Fig. 4C). Briefly, the mechanistic basis of the BiMC assay involves the splitting of YFP into N- and C-terminal (NYFP and CYFP, respectively) halves that are then joined to two proteins of interest (mVP40 and Nedd4) (31, 58–60, 67). If mVP40 and Nedd4 interact when coexpressed in mammalian cells, then the N- and C-terminal halves of YFP will come together to reconstitute a functional YFP yielding a fluorescent signal. NYFP-Nedd4 and CYFP-mVP40 were coexpressed in human HEK293T cells for 4 to 5 h, and then cells were treated with DMSO alone or with compound 4 or 6 (as a negative control) at the concentrations of indicated on the figure (Fig. 4C). At 24 h after transfection, cells were examined for YFP fluorescence (Fig. 4C). Total cell counts based on NucBlue staining indicated that equivalent numbers of cells were present in all BiMC assay samples. YFP-positive cells were detected in the presence of vehicle alone, indicating that this assay is capable of detecting mVP40 interactions with host Nedd4 (Fig. 4C). Importantly, YFP-positive cells decreased by approximately 2- and 4-fold in the presence of 0.5 μM and 1.0 μM concentrations of compound 4, respectively, indicating that compound 4 inhibits interactions between mVP40 and host Nedd4 (Fig. 4C). No such inhibition was observed in cells treated with a 10 μM or 20 μM concentration of compound 6, confirming the specificity of the inhibitory activity observed for compound 4 (Fig. 4C). Expression of the NYFP-Nedd4 fusion protein in transfected HEK293T cells was confirmed by Western blotting (Fig. 5), and appropriate controls for BiMC assays were performed, including expression of NYFP-Nedd4 alone, as well as coexpression of NYFP-Nedd4 plus CYFP-eVP40-ΔPT/PY and NYFP-Nedd4 plus CYFP-mVP40-ΔPPPY to confirm the specificity of the observed Nedd4-VP40 interactions (Fig. 5).
FIG 5.

BiMC assay of VP40-Nedd4 interactions. BiMC assay and representative images of HEK293T cells expressing NYFP-Nedd4 alone or with CYFP-eVP40, CYFP-eVP40-ΔPY/PY, CYFP-mVP40, or CYFP-mVP40-ΔPPPY are shown. The green signal represents an interaction between VP40 and Nedd4, and cell nuclei were stained blue with DAPI. A Western blot control is shown for endogenous Nedd4 and exogenous NYFP-Nedd4.
Similar analyses were carried out with compound 5 (Fig. 6). Compound 5 inhibited egress of mVP40 VLPs by 25-, 100-, and 100-fold at concentrations of 0.1 μM, 0.5 μM, and 1.0 μM, respectively (Fig. 6A, VLPs, lanes 2 to 4, and bar graph). Compound 5 inhibited egress of eVP40 VLPs by <2-, 3.3-, and 7-fold at concentrations of 0.1 μM, 0.5 μM, and 1.0 μM, respectively (Fig. 6B, VLPs, lanes 2 to 4, and bar graph). Compound 5 had no effect on expression levels of mVP40, eVP40, GAPDH, or HSP70 in cell extracts at all concentrations tested (Fig. 6A and B, cells). A BiMC assay was used to determine whether compound 5 could specifically block the mVP40-Nedd4 interaction in a dose-dependent manner (Fig. 6C). Equivalent numbers of cells were present in all BiMC assay samples as determined using MetaMorph software (see Materials and Methods). Indeed, the number of YFP-positive cells decreased by approximately 2- and 3-fold in the presence of 0.1 μM and 0.5 μM concentrations of compound 5 compared to those observed in the presence of vehicle alone as determined using MetaMorph software to quantify YFP fluorescence (Fig. 6C).
FIG 6.

Compound 5 inhibits budding of mVP40 and eVP40 VLPs and blocks mVP40-Nedd4 protein-protein interaction. (A) HEK293T cells transfected with mVP40 were treated with DMSO alone (lane 1) or with the indicated concentrations of compound 5 (lanes 2 to 4). Cells and VLPs were harvested at 24 h posttransfection, and a representative Western blot for mVP40 is shown. Western blot controls for cellular GAPDH and HSP70 in cells are shown. The bar graph represents the average levels of mVP40 VLPs from three independent experiments. *, P < 0.001. Numbers in parentheses were determined using ImageJ software (NIH), and controls were set at 100%. (B) HEK293T cells transfected with eVP40 were treated with DMSO alone (lane 1) or with the indicated concentrations of compound 5 (lanes 2 to 4). Cells and VLPs were harvested at 24 h posttransfection, and a representative Western blot for eVP40 is shown. Western blot controls for cellular GAPDH and HSP70 in cells are shown. The bar graph represents the average levels of eVP40 VLPs from three independent experiments. *, P < 0.001. Numbers in parentheses were determined using ImageJ software (NIH), and controls were set at 100%. (C) BiMC assay and representative images of HEK293T cells coexpressing NYFP-Nedd4 and CYFP-mVP40 fusion proteins in the absence (DMSO alone) or presence of the indicated concentrations of compound 5. The green signal represents an interaction between mVP40 and Nedd4, and cell nuclei were stained blue with NucBlue. Scale bar, 200 μm. YFP-positive cells were quantified in triplicate using MetaMorph software.
Compounds 4 and 5 inhibit VLP budding of PPxY-containing LFV-Z protein.
The Z protein of LFV contains both a PPxY and a PTAP motif. We along with others have shown that the PPxY L-domain motif is important for efficient egress of LFV-Z VLPs (Fig. 7A). For example, deletion of the PPxY motif resulted in a 4-fold decrease in VLP production compared to that of WT LFV-Z (Fig. 7A). Thus, we sought to test whether compounds 4 and 5 could inhibit budding of LFV-Z VLPs. Briefly, HEK293T cells were transfected with plasmids expressing LFV-Z in the absence (DMSO alone) or presence of compound 4 (Fig. 7B) or 5 (Fig. 7C) at the concentrations indicated on the figure. Both cell extracts and VLPs were harvested at 24 h posttransfection, and levels of LFV-Z were assessed by Western blotting. Compound 4 inhibited egress of LFV-Z VLPs by >5-fold at a concentration of 0.5 μM and by >10-fold at a concentration of 1.0 μM (Fig. 7B, VLPs). Similarly, compound 5 inhibited egress of LFV-Z VLPs by approximately 2.5-, 5-, and >10-fold at concentrations of 0.1 μM, 0.5 μM, and 1.0 μM, respectively (Fig. 7C, VLPs). Compounds 4 and 5 had no effect on expression levels of LFV-Z or actin in cell extracts at all concentrations tested (Fig. 7B and C, cells).
FIG 7.

Compounds 4 and 5 inhibit budding of LFV-Z VLPs. (A) VLP budding assay and Western blot demonstrating that the PPxY L-domain motif of LFV-Z protein is important for efficient VLP egress from HEK293T cells. Budding of LFV-Z-ΔPPPY VLPs was reduced by 4-fold compared to that of LFV-Z-WT. HEK293T cells transfected with LFV-Z-WT were treated with DMSO alone (0), or with the indicated concentrations of compound 4 (B) or 5 (C). Cells and VLPs were harvested at 24 h posttransfection, and LFV-Z-WT was detected by Western blotting. Western blot loading controls for cellular actin are shown. Numbers in parentheses were determined using ImageJ software (NIH), and controls were set at 100%.
Effect of compounds 4 and 5 on budding of infectious VSV and VSV recombinants.
We next sought to determine whether leads 4 and 5 possessed antibudding activity against infectious viruses that depend on a functional PPxY L domain for efficient egress. Toward this end, HEK293T cells were infected for 8 h at an MOI of 0.1 with either VSV-WT (containing a single PPxY L domain), recombinant VSV-M40 (containing overlapping EBOV PTAP and PPxY L domains), or VSV-PY>A4 (a PPxY mutant and budding defective virus) in the absence (DMSO alone) or presence of compound 4 (Fig. 8A) or 5 (Fig. 8B) at the concentration indicated on the figure. Compound 4 inhibited egress of VSV-WT by >2-fold at 0.1 μM and by 20-fold at 0.5 μM without affecting viral or host protein synthesis (Fig. 8A, left panel). Indeed, titers of VSV-WT averaging 3.3 × 107 PFU/ml at 8 h postinfection were significantly reduced to an average of 1.8 × 106 PFU/ml in the presence of a 0.5 μM concentration of compound 4 (Fig. 8A, left panel). Not surprisingly, compound 4 inhibited egress of VSV-M40 to a lesser degree, i.e., by <2-fold at 0.1 μM and by approximately 3-fold at 0.5 μM without affecting viral or host protein synthesis (Fig. 8A, middle panel). As expected, compound 4 exhibited no significant inhibition of VSV-PY>A4 budding at all concentrations tested (Fig. 8A, right panel). Compound 5 inhibited egress of VSV-WT by >2-fold at 0.1 μM and by 4-fold at 0.5 μM without affecting viral or host protein synthesis (Fig. 8B, left panel). Compound 5 inhibited egress of VSV-M40 by 2-fold at 0.1 μM and by 6-fold at 0.5 μM without affecting viral or host protein synthesis (Fig. 8B, middle panel). Lastly, budding of VSV-PY>A4 was reduced by <2-fold in the presence of 0.1 μM and 0.5 μM concentrations of 5 (Fig. 8B, right panel).
FIG 8.

Compounds 4 and 5 inhibit egress of infectious VSV and VSV recombinants in a PPxY-dependent and dose-dependent manner. (A) HEK293T cells were infected with VSV-WT, VSV-M40, or VSV-PY>A4 at an MOI of 0.1 in the absence (DMSO alone) or presence of 0.1 or 0.5 μM compound 4. Supernatants were harvested at 8 h postinfection, and virions were quantified by standard plaque assay on BHK-21 cells performed in triplicate. **, P < 0.01; ns, not significant (as determined by a one-way ANOVA test). Infected cell extracts were harvested at 8 h postinfection, and VSV-M, GAPDH, and HSP70 were detected by Western blotting. (B) HEK293T cells were infected with VSV-WT, VSV-M40, or VSV-PY>A4 at an MOI of 0.1 in the absence (DMSO alone) or presence of 0.1 or 0.5 μM compound 5. Supernatants were harvested at 8 h postinfection, and virions were quantified by standard plaque assay on BHK-21 cells performed in triplicate. **, P < 0.01; *, P < 0.5. Infected cell extracts were harvested at 8 h postinfection, and VSV-M, GAPDH, and HSP70 were detected by Western blotting. Numbers in parentheses were determined using ImageJ software (NIH), and controls were set at 100%.
Effect of compounds 4 and 5 on budding of PPxY-containing RABV.
Since efficient budding of RABV is also dependent on a single PPxY L domain within its M protein (13, 15, 22, 49, 68), we sought to determine whether lead compounds 4 and 5 possessed antibudding activity against infectious RABV in cell culture. Briefly, HEK293T cells were infected with RABV SPBN at an MOI of 0.1 in the absence (DMSO alone) or presence of compound 4 or 5 at the concentrations indicated on the figure, and virus-containing supernatants were harvested at 24 (data not shown), 36, and 72 h p.i., and titers were determined in duplicate by immunostaining assay on BSR cells (Fig. 9A and B). Compound 4 reduced RABV titers at 36 h postinfection in a dose-dependent manner by approximately 5- and 10-fold at 0.5 μM and 1.0 μM, respectively (Fig. 9A). Compound 4 reduced RABV titers at 72 h postinfection in a dose-dependent manner by approximately 10- and 50-fold at 0.5 μM and 1.0 μM, respectively (Fig. 9A). A similar dose-dependent reduction in RABV titers of approximately 5- and 30-fold was observed in the presence of compound 5 at 36 h postinfection at 0.5 μM and 1.0 μM, respectively (Fig. 9B). Compound 5 reduced RABV titers at 72 h postinfection by approximately 40- and 80-fold at 0.5 and 1.0 μM, respectively (Fig. 9B). Equivalent amounts of RABV-M were detected by Western blotting from cell extracts receiving vehicle alone or compound 4 and 5 at concentrations indicated on the figure, suggesting that these inhibitors did not adversely affect viral protein synthesis at the concentrations tested (Fig. 9C).
FIG 9.

Compounds 4 and 5 inhibit egress of infectious RABV in cell culture. Bar graphs representing multistep growth of RABV in HEK293T cells in the absence (DMSO alone) or presence of the indicated concentrations of compound 4 (A) or compound 5 (B). At the indicated time points, virus-containing supernatant was harvested and titrated in duplicate on BSR cells. **, P < 0.01; *, P < 0.05. (C) Western blot analysis of RABV-infected HEK293T cells in the absence (DMSO alone) or presence of the indicated concentrations of compounds 4 and 5. Cell extracts were harvested at 36 h p.i., and detection of RABV-M protein (24 kDa) by Western blotting is shown.
Compounds 1, 4, and 5 are not cytotoxic.
We wanted to determine whether compounds 1, 4, and 5 were cytotoxic to HEK293T cells under conditions used for transfection experiments. MTT [3-(4,5-dimethylthiazol-2-yl)2 2,5-diphenyl tetrazolium bromide] cell viability assays were performed on HEK293T cells that were treated with DMSO alone or with compounds 1, 4, and 5 at the concentrations indicated on the figure (Fig. 10). We found that compounds 1, 4, and 5 were not cytotoxic to HEK293T cells at the concentrations tested.
FIG 10.
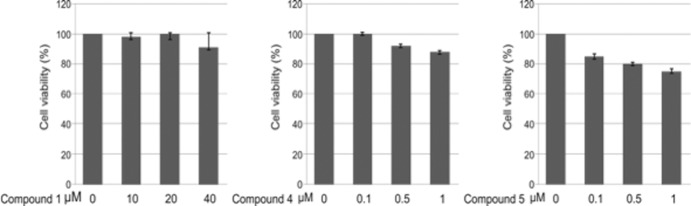
MTT cell viability assays. MTT cell viability assays were performed on HEK293T cells that were treated with DMSO or the indicated concentrations of compounds 1, 4, and 5 under conditions that mimicked those used for VLP transfection. Each concentration was tested in triplicate.
DISCUSSION
In this report, we validate the usefulness of an in silico approach combined with functional VLP and virus budding assays to identify small molecules possessing potent antiviral activity. We targeted the well-established viral PPxY-host Nedd4 interaction. By inhibiting PPxY-dependent recruitment of host Nedd4, a percentage of mature virions will remain tethered to the plasma membrane, unable to bud and spread efficiently to infect new cells (Fig. 1A and B).
Using iterative rounds of analog selection, we progressively identified more potent structural analogs of our initial candidate, leading to identification of our current top candidate inhibitors, compounds 4 and 5. Importantly, compounds 4 and 5 exhibited broad-spectrum antibudding activity against filoviruses, arenavirus, and rhabdovirus VLPs and/or virions with virtually no cytotoxicity by MTT assay (Fig. 10) at the concentrations tested. Our findings suggest that compounds 4 and 5 competitively disrupt the viral PPxY-host Nedd4 interaction. In support, compound 4 significantly inhibited egress of infectious VSV-WT (a PPxY-dependent virus) by more than 1 log at a concentration of 0.5 μM yet had no significant effect on egress of VSV-PY>A4 at the same concentration (Fig. 8A). Our finding that compound 4 inhibited budding of VSV-WT to levels comparable to those of VSV-PY>A4 in the absence of drug strongly suggest that compound 4 specially targets the function of the PPxY motif. It should be noted that we did observe a minimal unanticipated effect of compounds 1 and 5 on budding of VSV-PY>A4 (Fig. 2B and 8B). Since VSV-PY>A4 is still a budding virus, albeit defective, and since we do not yet know whether these compounds bind to the viral and/or the host protein, it may be possible that these compounds hinder other Nedd4 functions and/or interactions (e.g., with Tsg101) in the cell that have a minimal and perhaps indirect impact on egress of VSV-PY>A4. A second possibility is that at higher concentrations, these compounds may simply exhibit off-target effects that we have yet to identify.
The use of VSV recombinant M40 as a surrogate virus allowed us to assess the antiviral activity of compounds 4 and 5 against a biosafety level 2 (BSL-2) virus possessing the PPxY-type L-domain motif originating from a BSL-4 pathogen (EBOV). Importantly, compounds 4 and 5 also inhibited egress of infectious RABV by >1 log at 72 h postinfection without any apparent inhibitory effect on viral protein synthesis (Fig. 9). This finding is of significance since the replicative cycles and pathogenesis of VSV and RABV are quite different, yet both viruses depend on a PPxY L domain for efficient egress. Moreover, the results indicate that these probes might prove useful for RABV-related lyssaviruses such as Mokola virus, for which no vaccines or therapeutics currently exist. In addition to our budding assays, findings from our BiMC analyses indicate that both compounds 4 and 5, but not 6, can inhibit PPxY-dependent binding of MARV VP40 and Nedd4 in live mammalian cells in a dose-dependent manner (Fig. 4 and 6). Indeed, the fraction of positively fluorescing cells, as well as the overall signal intensity, decreased from approximately 20% in the absence of drug to approximately 3% in the presence of a 1.0 μM concentration of compound 4 (Fig. 4).
To our knowledge, small molecules targeting the viral PPxY-host Nedd4 interaction have not been reported; however, a number of previous studies have reported on antivirals targeting virus-host interactions and/or budding of filoviruses, arenaviruses, rhabdoviruses, and others (7, 9, 12, 31, 69–81). Host Tsg101 and its recruitment by viral PTAP type L domains of RNA viruses have been targeted for the development of antivirals in a number of recent studies (7, 9, 31, 39, 71, 82–88). For example, Tavassoli et al. (83) genetically identified cyclic peptides designed to block the interaction between Tsg101 and the PTAP motif within the p6 region of HIV-1 Gag, whereas Liu et al. (31) used the in silico approach, as described here, to identify a small-molecule inhibitor of the interaction between Tsg101 and the PTAP motif within EBOV VP40 to block VLP egress.
Although the concept of host-oriented therapeutics is of great interest, its development still remains in its infancy. However, this report, along with the earlier studies described above, help to illustrate the power and validity of this concept. As these virus L-domain–host interactions are conserved in a range of emerging RNA viruses, we predict that they represent an Achilles heel in the life cycle of these pathogens. Moreover, the chemical tractability of these small-molecule compounds will allow us to rapidly undertake structure-activity analyses to continue to develop and optimize these budding inhibitors. As shown here, iterative rounds of selection successfully led to the identification of structural analogs that exhibited enhanced potency and/or efficacy in blocking PPxY-dependent egress of several RNA viruses. Since many of these RNA viruses contain one or more L domains, it is tempting to speculate that a combination of both PTAP and PPxY inhibitors may have a synergistic effect, resulting in enhanced antiviral potency. Importantly, the antiviral activity of compounds 4 and 5 as well as other L-domain inhibitors against infectious BSL-4 pathogens both in vitro and in vivo remains to be determined. Nevertheless, studies presented here serve to validate our target and position us to transition into a full drug discovery program of study including evaluation of subsequent lead candidate inhibitors in detailed IND-directed pharmacokinetic, pharmacodynamic, and toxicity testing in vivo.
ACKNOWLEDGMENTS
We thank D. Lyles, S. Urata, and S. Becker for generously providing reagents, and J. Pelletier for evaluating the structural integrity and purity of commercial inhibitors 1 to 6 by LC/MS. We also thank D. Argento for assistance in preparation of the manuscript and figures, C. Krummenacher and L. King for helpful suggestions and critical reading of the manuscript, and members of the Harty and Freedman labs for fruitful discussion and suggestions.
This work was supported in part by NIH/NIAID grants AI102104 and U54-AI057168 to R.N.H.
Footnotes
Published ahead of print 16 April 2014
REFERENCES
- 1.Feldmann H, Klenk HD. 1996. Filoviruses, p 877–888 In Baron S. (ed), Medical microbiology, 4th ed. University of Texas Medical Branch at Galveston, Galveston, TX: [PubMed] [Google Scholar]
- 2.Feldmann H, Klenk HD. 1996. Marburg and Ebola viruses. Adv. Virus Res. 47:1–52. 10.1016/S0065-3527(08)60733-2 [DOI] [PubMed] [Google Scholar]
- 3.Grant A, Seregin A, Huang C, Kolokoltsova O, Brasier A, Peters C, Paessler S. 2012. Junin virus pathogenesis and virus replication. Viruses 4:2317–2339. 10.3390/v4102317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters CJ, Liu CT, Anderson GW, Jr, Morrill JC, Jahrling PB. 1989. Pathogenesis of viral hemorrhagic fevers: Rift Valley fever and Lassa fever contrasted. Rev. Infect. Dis. 11(Suppl 4):S743–S749. 10.1093/clinids/11.Supplement_4.S743 [DOI] [PubMed] [Google Scholar]
- 5.Hartlieb B, Weissenhorn W. 2006. Filovirus assembly and budding. Virology 344:64–70. 10.1016/j.virol.2005.09.018 [DOI] [PubMed] [Google Scholar]
- 6.Jasenosky LD, Kawaoka Y. 2004. Filovirus budding. Virus Res. 106:181–188. 10.1016/j.virusres.2004.08.014 [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Harty RN. 2010. Viral and host proteins that modulate filovirus budding. Future Virol. 5:481–491. 10.2217/fvl.10.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen BJ, Lamb RA. 2008. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 372:221–232. 10.1016/j.virol.2007.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harty RN. 2009. No exit: targeting the budding process to inhibit filovirus replication. Antiviral Res. 81:189–197. 10.1016/j.antiviral.2008.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayakar HR, Jeetendra E, Whitt MA. 2004. Rhabdovirus assembly and budding. Virus Res. 106:117–132. 10.1016/j.virusres.2004.08.009 [DOI] [PubMed] [Google Scholar]
- 11.Wolff S, Ebihara H, Groseth A. 2013. Arenavirus budding: a common pathway with mechanistic differences. Viruses 5:528–549. 10.3390/v5020528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urata S, de la Torre JC. 2011. Arenavirus budding. Adv. Virol. 2011:180326. 10.1155/2011/180326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okumura A, Harty RN. 2011. Rabies virus assembly and budding. Adv. Virus Res. 79:23–32. 10.1016/B978-0-12-387040-7.00002-0 [DOI] [PubMed] [Google Scholar]
- 14.Harty RN, Brown ME, Wang G, Huibregtse J, Hayes FP. 2000. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl. Acad. Sci. U. S. A. 97:13871–13876. 10.1073/pnas.250277297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harty RN, Brown ME, McGettigan JP, Wang G, Jayakar HR, Huibregtse JM, Whitt MA, Schnell MJ. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 75:10623–10629. 10.1128/JVI.75.22.10623-10629.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kikonyogo A, Bouamr F, Vana ML, Xiang Y, Aiyar A, Carter C, Leis J. 2001. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. U. S. A. 98:11199–11204. 10.1073/pnas.201268998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasuda J, Hunter E, Nakao M, Shida H. 2002. Functional involvement of a novel Nedd4-like ubiquitin ligase on retrovirus budding. EMBO Rep. 3:636–640. 10.1093/embo-reports/kvf132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gottwein E, Bodem J, Muller B, Schmechel A, Zentgraf H, Krausslich HG. 2003. The Mason-Pfizer monkey virus PPPY and PSAP motifs both contribute to virus release. J. Virol. 77:9474–9485. 10.1128/JVI.77.17.9474-9485.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Licata JM, Simpson-Holley M, Wright NT, Han Z, Paragas J, Harty RN. 2003. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J. Virol. 77:1812–1819. 10.1128/JVI.77.3.1812-1819.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timmins J, Schoehn G, Ricard-Blum S, Scianimanico S, Vernet T, Ruigrok RW, Weissenhorn W. 2003. Ebola virus matrix protein VP40 interaction with human cellular factors Tsg101 and Nedd4. J. Mol. Biol. 326:493–502. 10.1016/S0022-2836(02)01406-7 [DOI] [PubMed] [Google Scholar]
- 21.Yasuda J, Nakao M, Kawaoka Y, Shida H. 2003. Nedd4 regulates egress of Ebola virus-like particles from host cells. J. Virol. 77:9987–9992. 10.1128/JVI.77.18.9987-9992.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irie T, Licata JM, McGettigan JP, Schnell MJ, Harty RN. 2004. Budding of PPxY-containing rhabdoviruses is not dependent on host proteins TGS101 and VPS4A. J. Virol. 78:2657–2665. 10.1128/JVI.78.6.2657-2665.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vana ML, Tang Y, Chen A, Medina G, Carter C, Leis J. 2004. Role of Nedd4 and ubiquitination of Rous sarcoma virus Gag in budding of virus-like particles from cells. J. Virol. 78:13943–13953. 10.1128/JVI.78.24.13943-13953.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segura-Morales C, Pescia C, Chatellard-Causse C, Sadoul R, Bertrand E, Basyuk E. 2005. Tsg101 and Alix interact with murine leukemia virus Gag and cooperate with Nedd4 ubiquitin ligases during budding. J. Biol. Chem. 280:27004–27012. 10.1074/jbc.M413735200 [DOI] [PubMed] [Google Scholar]
- 25.Chung HY, Morita E, von Schwedler U, Muller B, Krausslich HG, Sundquist WI. 2008. NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J. Virol. 82:4884–4897. 10.1128/JVI.02667-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malakhova OA, Zhang DE. 2008. ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J. Biol. Chem. 283:8783–8787. 10.1074/jbc.C800030200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okumura A, Pitha PM, Harty RN. 2008. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. U. S. A. 105:3974–3979. 10.1073/pnas.0710629105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sette P, Jadwin JA, Dussupt V, Bello NF, Bouamr F. 2010. The ESCRT-associated protein Alix recruits the ubiquitin ligase Nedd4-1 to facilitate HIV-1 release through the LYPXnL L domain motif. J. Virol. 84:8181–8192. 10.1128/JVI.00634-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Urata S, Yasuda J. 2010. Regulation of Marburg virus (MARV) budding by Nedd4.1: a different WW domain of Nedd4.1 is critical for binding to MARV and Ebola virus VP40. J. Gen. Virol. 91:228–234. 10.1099/vir.0.015495-0 [DOI] [PubMed] [Google Scholar]
- 30.Zhadina M, Bieniasz PD. 2010. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 6:e1001153. 10.1371/journal.ppat.1001153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Lee MS, Olson MA, Harty RN. 2011. Bimolecular complementation to visualize filovirus VP40-host complexes in live mammalian cells: toward the identification of budding inhibitors. Adv. Virol. 2011:341816. 10.1155/2011/341816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strack B, Calistri A, Accola MA, Palu G, Gottlinger HG. 2000. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. U. S. A. 97:13063–13068. 10.1073/pnas.97.24.13063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sudol M, Hunter T. 2000. NeW wrinkles for an old domain. Cell 103:1001–1004. 10.1016/S0092-8674(00)00203-8 [DOI] [PubMed] [Google Scholar]
- 34.Blot V, Perugi F, Gay B, Prevost MC, Briant L, Tangy F, Abriel H, Staub O, Dokhelar MC, Pique C. 2004. Nedd4.1-mediated ubiquitination and subsequent recruitment of Tsg101 ensure HTLV-1 Gag trafficking towards the multivesicular body pathway prior to virus budding. J. Cell Sci. 117:2357–2367. 10.1242/jcs.01095 [DOI] [PubMed] [Google Scholar]
- 35.Martin-Serrano J, Perez-Caballero D, Bieniasz PD. 2004. Context-dependent effects of L domains and ubiquitination on viral budding. J. Virol. 78:5554–5563. 10.1128/JVI.78.11.5554-5563.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sundquist WI, Schubert HL, Kelly BN, Hill GC, Holton JM, Hill CP. 2004. Ubiquitin recognition by the human TSG101 protein. Mol. Cell 13:783–789. 10.1016/S1097-2765(04)00129-7 [DOI] [PubMed] [Google Scholar]
- 37.Martin-Serrano J, Eastman SW, Chung W, Bieniasz PD. 2005. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J. Cell Biol. 168:89–101. 10.1083/jcb.200408155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bieniasz PD. 2006. Late budding domains and host proteins in enveloped virus release. Virology 344:55–63. 10.1016/j.virol.2005.09.044 [DOI] [PubMed] [Google Scholar]
- 39.Urata S, Noda T, Kawaoka Y, Yokosawa H, Yasuda J. 2006. Cellular factors required for Lassa virus budding. J. Virol. 80:4191–4195. 10.1128/JVI.80.8.4191-4195.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taylor GM, Hanson PI, Kielian M. 2007. Ubiquitin depletion and dominant-negative VPS4 inhibit rhabdovirus budding without affecting alphavirus budding. J. Virol. 81:13631–13639. 10.1128/JVI.01688-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Irie T, Licata JM, Harty RN. 2005. Functional characterization of Ebola virus L-domains using VSV recombinants. Virology 336:291–298. 10.1016/j.virol.2005.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolesnikova L, Bugany H, Klenk HD, Becker S. 2002. VP40, the matrix protein of Marburg virus, is associated with membranes of the late endosomal compartment. J. Virol. 76:1825–1838. 10.1128/JVI.76.4.1825-1838.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolesnikova L, Bohil AB, Cheney RE, Becker S. 2007. Budding of Marburgvirus is associated with filopodia. Cell. Microbiol. 9:939–951. 10.1111/j.1462-5822.2006.00842.x [DOI] [PubMed] [Google Scholar]
- 44.Dolnik O, Kolesnikova L, Stevermann L, Becker S. 2010. Tsg101 is recruited by a late domain of the nucleocapsid protein to support budding of Marburg virus-like particles. J. Virol. 84:7847–7856. 10.1128/JVI.00476-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mittler E, Kolesnikova L, Herwig A, Dolnik O, Becker S. 2013. Assembly of the Marburg virus envelope. Cell. Microbiol. 15:270–284. 10.1111/cmi.12076 [DOI] [PubMed] [Google Scholar]
- 46.Urata S, Noda T, Kawaoka Y, Morikawa S, Yokosawa H, Yasuda J. 2007. Interaction of Tsg101 with Marburg virus VP40 depends on the PPPY motif, but not the PT/SAP motif as in the case of Ebola virus, and Tsg101 plays a critical role in the budding of Marburg virus-like particles induced by VP40, NP, and GP. J. Virol. 81:4895–4899. 10.1128/JVI.02829-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jayakar HR, Murti KG, Whitt MA. 2000. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. J. Virol. 74:9818–9827. 10.1128/JVI.74.21.9818-9827.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Craven RC, Harty RN, Paragas J, Palese P, Wills JW. 1999. Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J. Virol. 73:3359–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harty RN, Paragas J, Sudol M, Palese P. 1999. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J. Virol. 73:2921–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Irie T, Harty RN. 2005. L-domain flanking sequences are important for host interactions and efficient budding of vesicular stomatitis virus recombinants. J. Virol. 79:12617–12622. 10.1128/JVI.79.20.12617-12622.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irie T, Licata JM, Jayakar HR, Whitt MA, Bell P, Harty RN. 2004. Functional analysis of late-budding domain activity associated with the PSAP motif within the vesicular stomatitis virus M protein. J. Virol. 78:7823–7827. 10.1128/JVI.78.14.7823-7827.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gomme EA, Faul EJ, Flomenberg P, McGettigan JP, Schnell MJ. 2010. Characterization of a single-cycle rabies virus-based vaccine vector. J. Virol. 84:2820–2831. 10.1128/JVI.01870-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Licata JM, Johnson RF, Han Z, Harty RN. 2004. Contribution of ebola virus glycoprotein, nucleoprotein, and VP24 to budding of VP40 virus-like particles. J. Virol. 78:7344–7351. 10.1128/JVI.78.14.7344-7351.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han Z, Harty RN. 2005. Packaging of actin into Ebola virus VLPs. Virol. J. 2:92. 10.1186/1743-422X-2-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson RF, Bell P, Harty RN. 2006. Effect of Ebola virus proteins GP, NP and VP35 on VP40 VLP morphology. Virol. J. 3:31. 10.1186/1743-422X-3-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCarthy SE, Johnson RF, Zhang YA, Sunyer JO, Harty RN. 2007. Role for amino acids 212KLR214 of Ebola virus VP40 in assembly and budding. J. Virol. 81:11452–11460. 10.1128/JVI.00853-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, Cocka L, Okumura A, Zhang YA, Sunyer JO, Harty RN. 2010. Conserved motifs within Ebola and Marburg virus VP40 proteins are important for stability, localization, and subsequent budding of virus-like particles. J. Virol. 84:2294–2303. 10.1128/JVI.02034-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu J, Qu Y, Liu Y, Jambusaria R, Han Z, Ruthel G, Freedman BD, Harty RN. 2013. Host IQGAP1 and Ebola virus VP40 interactions facilitate virus-like particle egress. J. Virol. 87:7777–7780. 10.1128/JVI.00470-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Stone S, Harty RN. 2011. Characterization of filovirus protein-protein interactions in mammalian cells using bimolecular complementation. J. Infect. Dis. 204(Suppl 3):S817–S824. 10.1093/infdis/jir293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kerppola TK. 2006. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 1:1278–1286. 10.1038/nprot.2006.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kanelis V, Farrow NA, Kay LE, Rotin D, Forman-Kay JD. 1998. NMR studies of tandem WW domains of Nedd4 in complex with a PY motif-containing region of the epithelial sodium channel. Biochem. Cell Biol. 76:341–350. 10.1139/o98-042 [DOI] [PubMed] [Google Scholar]
- 62.Otte L, Wiedemann U, Schlegel B, Pires JR, Beyermann M, Schmieder P, Krause G, Volkmer-Engert R, Schneider-Mergener J, Oschkinat H. 2003. WW domain sequence activity relationships identified using ligand recognition propensities of 42 WW domains. Protein Sci. 12:491–500. 10.1110/ps.0233203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. 2009. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30:2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang S, Kumar K, Jiang X, Wallqvist A, Reifman J. 2008. DOVIS: an implementation for high-throughput virtual screening using AutoDock. BMC Bioinformatics 9:126. 10.1186/1471-2105-9-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brooks BR, Brooks CL, III, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. 2009. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30:1545–1614. 10.1002/jcc.21287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krammer A, Kirchhoff PD, Jiang X, Venkatachalam CM, Waldman M. 2005. LigScore: a novel scoring function for predicting binding affinities. J. Mol. Graph. Model. 23:395–407. 10.1016/j.jmgm.2004.11.007 [DOI] [PubMed] [Google Scholar]
- 67.Kerppola TK. 2006. Visualization of molecular interactions by fluorescence complementation. Nat. Rev. Mol. Cell Biol. 7:449–456. 10.1038/nrm1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wirblich C, Tan GS, Papaneri A, Godlewski PJ, Orenstein JM, Harty RN, Schnell MJ. 2008. PPEY motif within the rabies virus (RV) matrix protein is essential for efficient virion release and RV pathogenicity. J. Virol. 82:9730–9738. 10.1128/JVI.00889-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harty RN, Schmitt AP, Bouamr F, Lopez CB, Krummenacher C. 2011. Virus budding/host interactions. Adv. Virol. 2011:963192. 10.1155/2011/963192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kolokoltsov AA, Adhikary S, Garver J, Johnson L, Davey RA, Vela EM. 2012. Inhibition of Lassa virus and Ebola virus infection in host cells treated with the kinase inhibitors genistein and tyrphostin. Arch. Virol. 157:121–127. 10.1007/s00705-011-1115-8 [DOI] [PubMed] [Google Scholar]
- 71.Panchal RG, Reid SP, Tran JP, Bergeron AA, Wells J, Kota KP, Aman J, Bavari S. 2012. Identification of an antioxidant small-molecule with broad-spectrum antiviral activity. Antiviral Res. 93:23–29. 10.1016/j.antiviral.2011.10.011 [DOI] [PubMed] [Google Scholar]
- 72.Radoshitzky SR, Dong L, Chi X, Clester JC, Retterer C, Spurgers K, Kuhn JH, Sandwick S, Ruthel G, Kota K, Boltz D, Warren T, Kranzusch PJ, Whelan SP, Bavari S. 2010. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J. Virol. 84:10569–10580. 10.1128/JVI.00103-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Radoshitzky SR, Kuhn JH, de Kok-Mercado F, Jahrling PB, Bavari S. 2012. Drug discovery technologies and strategies for Machupo virus and other New World arenaviruses. Expert Opin. Drug Discov. 7:613–632. 10.1517/17460441.2012.687719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kunz S, de la Torre JC. 2005. Novel antiviral strategies to combat human Arenavirus infections. Curr. Mol. Med. 5:735–751. 10.2174/156652405774962353 [DOI] [PubMed] [Google Scholar]
- 75.Linero FN, Sepulveda CS, Giovannoni F, Castilla V, Garcia CC, Scolaro LA, Damonte EB. 2012. Host cell factors as antiviral targets in arenavirus infection. Viruses 4:1569–1591. 10.3390/v4091569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lingappa UF, Wu X, Macieik A, Yu SF, Atuegbu A, Corpuz M, Francis J, Nichols C, Calayag A, Shi H, Ellison JA, Harrell EK, Asundi V, Lingappa JR, Prasad MD, Lipkin WI, Dey D, Hurt CR, Lingappa VR, Hansen WJ, Rupprecht CE. 2013. Host-rabies virus protein-protein interactions as druggable antiviral targets. Proc. Natl. Acad. Sci. U. S. A. 110:E861–E868. 10.1073/pnas.1210198110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hill-Batorski L, Halfmann P, Neumann G, Kawaoka Y. 2013. The cytoprotective enzyme heme oxygenase-1 suppresses Ebola virus replication. J. Virol. 87:13795–13802. 10.1128/JVI.02422-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pasquato A, Burri DJ, Kunz S. 2012. Current drug discovery strategies against arenavirus infections. Expert Rev. Anti. Infect. Ther. 10:1297–1309. 10.1586/eri.12.117 [DOI] [PubMed] [Google Scholar]
- 79.Garcia CC, Topisirovic I, Djavani M, Borden KL, Damonte EB, Salvato MS. 2010. An antiviral disulfide compound blocks interaction between arenavirus Z protein and cellular promyelocytic leukemia protein. Biochem. Biophys. Res. Commun. 393:625–630. 10.1016/j.bbrc.2010.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Garcia CC, Djavani M, Topisirovic I, Borden KL, Salvato MS, Damonte EB. 2006. Arenavirus Z protein as an antiviral target: virus inactivation and protein oligomerization by zinc finger-reactive compounds. J. Gen. Virol. 87:1217–1228. 10.1099/vir.0.81667-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Garcia M, Cooper A, Shi W, Bornmann W, Carrion R, Kalman D, Nabel GJ. 2012. Productive replication of Ebola virus is regulated by the c-Abl1 tyrosine kinase. Sci. Transl. Med. 4:123ra24. 10.1126/scitranslmed.3003500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perez M, Craven RC, de la Torre JC. 2003. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc. Natl. Acad. Sci. U. S. A. 100:12978–12983. 10.1073/pnas.2133782100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tavassoli A, Lu Q, Gam J, Pan H, Benkovic SJ, Cohen SN. 2008. Inhibition of HIV budding by a genetically selected cyclic peptide targeting the Gag-TSG101 interaction. ACS Chem. Biol. 3:757–764. 10.1021/cb800193n [DOI] [PubMed] [Google Scholar]
- 84.Turpin JA. 2003. The next generation of HIV/AIDS drugs: novel and developmental antiHIV drugs and targets. Expert Rev. Anti. Infect. Ther. 1:97–128. 10.1586/14787210.1.1.97 [DOI] [PubMed] [Google Scholar]
- 85.Martin-Serrano J, Zang T, Bieniasz PD. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313–1319. 10.1038/nm1201-1313 [DOI] [PubMed] [Google Scholar]
- 86.Demirov DG, Ono A, Orenstein JM, Freed EO. 2002. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. U. S. A. 99:955–960. 10.1073/pnas.032511899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen H, Liu X, Li Z, Zhan P, De Clercq E. 2010. TSG101: a novel anti-HIV-1 drug target. Curr. Med. Chem. 17:750–758. 10.2174/092986710790514444 [DOI] [PubMed] [Google Scholar]
- 88.Waheed AA, Freed EO. 2008. Peptide inhibitors of HIV-1 egress. ACS Chem. Biol. 3:745–747. 10.1021/cb800296j [DOI] [PubMed] [Google Scholar]