

ABSTRACT
Characterizing the cellular factors that play a role in the HIV replication cycle is fundamental to fully understanding mechanisms of viral replication and pathogenesis. Whole-genome small interfering RNA (siRNA) screens have identified positive and negative regulators of HIV replication, providing starting points for investigating new cellular factors. We report here that silencing of the deubiquitinase cylindromatosis protein (CYLD), increases HIV infection by enhancing HIV long terminal repeat (LTR)-driven transcription via the NF-κB pathway. CYLD is highly expressed in CD4+ T lymphocytes, monocyte-derived macrophages, and dendritic cells. We found that CYLD silencing increases HIV replication in T cell lines. We confirmed the positive role of CYLD silencing in HIV infection in primary human CD4+ T cells, in which CYLD protein was partially processed upon activation. Lastly, Jurkat T cells latently infected with HIV (JLat cells) were more responsive to phorbol 12-myristate 13-acetate (PMA) reactivation in the absence of CYLD, indicating that CYLD activity could play a role in HIV reactivation from latency. In summary, we show that CYLD acts as a potent negative regulator of HIV mRNA expression by specifically inhibiting NF-κB-driven transcription. These findings suggest a function for this protein in modulating productive viral replication as well as in viral reactivation.
IMPORTANCE HIV transcription is regulated by a number of host cell factors. Here we report that silencing of the lysine 63 deubiquitinase CYLD increases HIV transcription in an NF-κB-dependent manner. We show that CYLD is expressed in HIV target cells and that its silencing increases HIV infection in transformed T cell lines as well as primary CD4+ T cells. Similarly, reactivation of latent provirus was facilitated in the absence of CYLD. These data suggest that CYLD, which is highly expressed in CD4+ T cells, can control HIV transcription in productive infection as well as during reactivation from latency.
INTRODUCTION
High-throughput RNA interference (RNAi) screening approaches are powerful tools to probe for HIV host interactions. Whole-genome small interfering RNA (siRNA) screens have identified a number of host factors essential for viral replication, but the relative importance of these factors in relevant cells remains to be verified (1–4). Similarly, these screens have the potential to identify cellular molecules which, when silenced, result in increased viral infection. The deubiquitinase cylindromatosis (CYLD) was identified as putative negative regulator of HIV infection in an siRNA screen (L. Pache, J. Marlett, A. Maestre, L. Manganaro, J. Murry, Y. Hwang, K. Olivieri, A. Diaz, P. D. De Jesus, R. König, F. D. Bushman, V. Simon, A. Fernandez-Sesma, J. A. Young, and S. K. Chanda, unpublished data).
CYLD, a multidomain protein, was first studied in association with skin malignancies (5, 6). The amino-terminal region of the 956-amino-acid-long CYLD protein is characterized by three cytoskeleton-associated protein (CAP) domains, two proline-rich (PR) motifs, a phosphorylation region (P), and a TRAF2 binding site (PVQES). The carboxy-terminal region is characterized by the ubiquitin-specific protease (USP) domain, which contains the zinc-binding B box that forms the catalytic domain responsible for CYLD deubiquitinase activity (Fig. 1A) (7). Mutations in this region abolish catalytic activity (8). Naturally occurring mutations are found in the USP and CAP regions (9). Several nonsynonymous mutations in the coding region of this gene have been linked to familial cylindromatosis (also termed “turban syndrome”) as well as to the sporadic Brooke-Spiegler syndrome (5, 10, 11). CYLD controls chronic inflammation during tumor progression, and its expression is downregulated in melanoma, hepatocellular carcinoma, and colon cancer (12, 13). Since CYLD is a lysine 63-linked deubiquitinase, it induces and suppresses different signaling pathways by specifically removing lysine 63-linked ubiquitin moieties (8, 14). Many of these pathways regulate the development (e.g., natural killer cells) and homeostasis (e.g., T cells and dendritic cells [DCs]) of the immune system as well as the quality of the immune responses mounted to different pathogens (15–19). For example, CYLD inhibits type I interferon signaling by deubiquitinating the pattern recognition receptor RIG-I and downregulating Toll-like receptor 2 (TLR2) signaling (20–22).
FIG 1.

CYLD silencing increases HIV infection in single-cycle assays. (A) Domain organization of CYLD (isoform 2), showing the cytoskeleton-associated protein domain (CAP), phosphorylation region (P), proline rich region (PR), and ubiquitin-specific protease domain (USP). (B) HEK 293T cells transfected with CYLD-targeting siRNA (CYLD_2 or CYLD_3) or nontargeting siRNA or mock transfected were infected with VSV-G HIV NL4.3 luciferase reporter virus. Relative light units (RLU) were measured after 2 days. Values represent means plus standard errors of the means (SEM) for five independent experiments. Mock is set at 1, and values represent fold changes over mock. The average RLU for the mock control was 5,732 ± 3,300. *, P < 0.05; **, P < 0.01 (unpaired t tests, Prism software). Knockdown efficiency was quantified by immunoblotting using an antibody against endogenous CYLD and an antibody against GAPDH as a loading control (lower panel). (C) HEK 293T cells stably expressing nontargeting shRNA (NT shRNA) or shRNA targeting CYLD (CYLD shRNA) were transfected with empty plasmid, the short-hairpin-resistant form of Flag-CYLD WT, or the short-hairpin-resistant form of the catalytically inactive mutant of Flag-CYLD (C598A). At 1 day posttransfection, cells were infected with VSV-G HIV NL4.3 luciferase reporter virus (upper panel). Luciferase (relative light units [RLU]) was measured 2 days postinfection. Values represent means plus SEM for three independent experiments. The NT shRNA transfected with an empty vector was set to 1, and values represent fold changes over NT shRNA-expressing cells. The average RLU for the mock control was 1,148 ± 1,093. *, P < 0.05; **, P < 0.01 (unpaired t tests, Prism software). Expression of endogenous or FLAG-tagged CYLD was analyzed by Western blotting (lower panel). GAPDH expression served as loading control.
The best-understood function of CYLD is the repression of the nuclear factor κB (NF-κB) pathway. Several molecules regulating this pathway (e.g., NEMO, TRAF2, TAK1, and TRAF6) are substrates of CYLD (7, 8, 14, 23, 24). Moreover, CYLD itself has been proposed to act as a negative regulator of the NFAT pathway (25, 26).
In the context of HIV infection, NF-κB is essential for efficient HIV transcription in transformed cell lines as well as in primary cells (27–30). NF-κB regulates HIV transcription by binding to two adjacent NF-κB/NFAT sites within the proviral long terminal repeat (LTR) promoter and enhancing the transcription of viral genes from the integrated provirus (31, 32). T lymphocytes are the main target cells for HIV, but only activated T cells support efficient HIV transcription and production of infectious particles (reviewed in reference 33). Resting CD4+ T lymphocytes, in contrast, fail to support productive viral transcription, which has been linked, at least in part, to low levels of active NF-κB (34–39).
Despite the reported importance of CYLD in T cell homeostasis and T cell activation, there are no reports on its effect on HIV replication. Here we describe that silencing of CYLD in a genome-wide siRNA screening resulted in an increase of viral infection. We hypothesized that CYLD modulates HIV infection by limiting viral transcription through negative regulation of the NF-κB pathway. Consistent with that hypothesis, we show here that HIV-1 LTR-driven transcription is increased in the absence of CYLD in an NF-κB-dependent manner. Moreover, we show that activation of T cells leads to cleavage and, presumably, inactivation of CYLD. These findings are important because silencing of CYLD increases HIV infection of T cell lines as well as primary human T lymphocytes. In addition, CYLD was found to negatively regulate phorbol 12-myristate 13-acetate (PMA)-induced reactivation of latent HIV-1 gene expression. In conclusion, the deubiquitinase CYLD is a negative regulator of HIV-1 transcription.
MATERIALS AND METHODS
Cell isolation and cell culture. (i) Transformed cell lines.
Human embryonic kidney 293T (HEK 293T) cells and TZM-bl cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin. The A3R5 and Jurkat JLat (clone 8.4) T cell lines were cultured in Roswell Park Memorial Institute medium (RPMI 1640) supplemented with 10% FBS, 100 IU penicillin, 100 μg/ml streptomycin, 0.1 M HEPES, and 2 mM l-glutamine at 37°C. Jurkat JLat T cell lines were stimulated with 15 nM to 100 nM PMA (Sigma) for 5 h.
(ii) Primary cells.
Human peripheral blood mononuclear cells (PBMCs) were obtained by Ficoll (Ficoll Histopaque; Sigma) density centrifugation from anonymous healthy blood donors (New York Blood Center). CD4+ T cells were negatively selected using magnetic beads (CD4+ T cell isolation kit II; Miltenyi Biotec) as per the manufacturer's instructions. CD4+ T cells were cultured in RPMI 1640 supplemented with 10% FBS, 100 IU penicillin, 100 μg/ml streptomycin, 0.1 M HEPES, 2 mM l-glutamine, and 20 units/ml interleukin-2 (IL-2) (NIH AIDS Reagent Program). Lymphocytes were activated with 1 μg/ml phytohemagglutinin-P (PHA) (Sigma) or with human T-activator CD3/CD28 at a 1:5 bead/cell ratio (Gibco by Life Technologies) for 48 h. CD14+ cells were isolated from PBMCs using the MACS CD14 isolation kit (Miltenyi Biotec) according to the manufacturer's instructions. CD14+ cells were differentiated into dendritic cells (DCs) or macrophages. DCs were obtained by adding 500 U/ml human granulocyte-macrophage colony-stimulating factor (GM-CSF) (PeproTech) and 1,000 U/ml human IL-4 (PeproTech) to the culture medium (RPMI, 10% FBS [HyClone] 100 U/ml l-glutamine, 100 g/ml penicillin-streptomycin, 1 mM sodium pyruvate) and incubating for 5 to 6 days. Macrophages were obtained by incubating CD14+ monocytes with 2,000 U/ml human GM-CSF for 10 days. The GM-CSF containing medium was replenished at days 2, 5, and 8 as previously described (40).
Plasmids.
pEAK Flag-CYLD was obtained by cloning CYLD cDNA into pEAK plasmid. We generated a pEAK Flag-CYLD (transcript variant 2) mutant that is resistant to CYLD short hairpin pGIPZ RHS4430-99158676 by mutating nucleotides 2040, 2043, and 2046 in CYLD (NCBI accession number NM_001042355.1). Mutagenesis was performed using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) and the primers CYLD shmut for (5′-GATTGAGCGCTGCAATCATTAGCATTTGGAG) and CYLD shmut rev (5′-CTCCAAATGCTAATGAATGCAGCGCTCAATC). Mutagenesis to obtain the CYLD C598A mutant was performed using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) with primers CYLD C598A for (5′-GCATCCAGGGTCATTACAATTCTgcTTACTTAGACTCAACCT) and CYLD C598A rev (5′-AGGTTGAGTCTAAGTAAgcAGAATTGTAATGACCCTGGATGC).
The murine leukemia virus (MLV)-LTR-luciferase, HIV-LTR-luciferase, HIV-ΔNF-κB-LTR-luciferase, and MLV-HIV-LTR-luciferase reporter plasmids were previously described (41). The LTR mutant viral clones were previously described (42). The c-fos-luciferase plasmid was previously described (80). The pRL-TK used for the luciferase assay as a control was obtained from Promega.
Silencing of CYLD. (i) Screening.
A set of candidate factors likely to restrict HIV replication were selected based on a previous siRNA-based genome-wide screening approach (2). Multiple siRNAs targeting each of the candidate genes were individually transfected into HEK 293T cells. Cells were subsequently infected with an envelope-deleted, vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped HIV carrying a luciferase reporter gene in place of nef, as previously described (43), and luciferase expression was determined at 24 h postinfection. In the initial round of screening, a gene was considered a hit if at least two independent, nonoverlapping siRNAs increased expression of the luciferase reporter gene by 50% or more compared to that with nontargeting control siRNAs. Among the genes identified in this screen was CYLD. The CYLD-specific siRNAs (Hs_CYLD_2 and Hs_CYLD_3) and nontargeting scrambled control siRNAs were purchased from Qiagen.
(ii) Molecular characterization of CYLD function.
siRNA (Hs_CYLD_2 and Hs_CYLD_3) and nontargeting scrambled control siRNA (Qiagen) were transfected into HEK 293T using Lipofectamine RNAiMax reagent (Invitrogen) by the reverse transfection method. The final siRNA concentration was 12.5 nM.
Primary CD4+ T cells were transfected with the Amaxa Nucleofection Device II (Amaxa), using the Amaxa Human T Cell Nucleofector kit. PHA-stimulated CD4+ T cells (3 × 106 to 5 × 106) were transfected with 300 nM CYLD siRNA or with nontargeting siRNA as a control. The Amaxa program of choice was T-023 for high efficiency.
JLat 8.4 cells were transfected using the Amaxa Nucleofection Device II (Amaxa) in conjunction with the Amaxa Cell Line Nucleofector kit V (program X-001). JLat 8.4 cells (2 × 106) were transfected with 300 nM CYLD siRNA or with nontargeting siRNA as a control.
pGIPZ-CYLD-targeting short hairpin RNA (CYLD shRNA) and pGIPZ-nontargeting (NT) short hairpin RNA lentiviral vectors were obtained from Thermo Scientific. Stably silenced cell lines (HEK 293T, A3R5, and JLat 8.4) were generated by transduction with pGIPZ lentiviral vectors encoding CYLD-targeting short hairpins or nontargeting hairpins followed by puromycin selection (1 μg/ml).
Production of viral stocks.
VSV-G-NL4.3 luciferase viral stocks were generated by transfection of HEK 293T cells with 20 μg of pNL4.3 luciferase and 3 μg VSV-G (pHCMV-G) per 75-cm2 flask using 3 μg/ml polyethylenimine (PEI) (Polysciences) (44). Two days after transfection, supernatants were collected, clarified at 1500 rpm for 5 min, filtered (0.45 μm), aliquoted, and stored at −80°C. The molecular clone pNL4-3 Env-luciferase lacks a functional envelope and encodes luciferase in the position of Nef (pNL-Luc-E−; NIH AIDS Research Program [45]). Once pseudotyped with VSV-G (pHCMV-G), it was used for single-cycle infections (46).
VSV-G-pseudotyped viral stocks of wild-type HIV (HIV WT), HIV ΔUSF, HIV ΔNFIL6 and HIV ΔNF-κB/NFAT viruses were obtained by transfecting HEK 293T cells with the corresponding plasmids (42) together with VSV-G at a ratio of 1:5 using 3 μg/ml PEI.
The replication-competent HIV molecular clones LAI and NL4.3 were obtained from the AIDS Research and Reference Reagent Program (47, 48). LAI and NL4.3 viral stocks were obtained by transfecting HEK 293T cells with the corresponding plasmids using PEI (3 μg/ml).
Infectivity titers of the viral stocks were determined by infecting TZM-bl reporter cells with serial dilutions in triplicate as previously described (44).
Lentiviral stocks used for the generation of stable HEK 293T, JLat 8.4, and A3R5 CYLD shRNAs and nontargeting shRNA were produced by transfecting pGIPZ-CYLD-targeting short hairpin RNA or pGIPZ-nontargeting (NT), psPAX2 (49), and VSV-G in HEK 293T cells using 3 μg/ml PEI at a ratio of 5:5:1.
Single-cycle infection experiments.
HEK 293T cells were plated in 96-well plates and transfected with the indicated siRNA using RNAiMax reagent (Invitrogen). At 48 h posttransfection, cells were infected with VSV-G-pseudotyped NL4.3 Luc virus in the presence of Polybrene (5 μg/ml).
HEK 239T cells stably transduced with short hairpins directed against CYLD or nontargeting short hairpins were plated in a 24-well plates and infected with VSV-G-NL4.3 Luc virus 24 h later. To determine firefly luciferase relative light values, cells were lysed in passive lysis buffer (PLB) at 24 to 48 h postinfection, incubated with luciferase substrate (Promega), and read immediately in a Victor3 multilabel counter (Perkin-Elmer).
Primary CD4+ T cells were PHA activated for 48 h before infection with VSV-G NL4.3 in the presence of Polybrene (2 μg/ml). RNA was collected form these cells at 24 h postinfection.
Spreading-infection experiments.
A3R5 cells (0.5 × 106; transduced with nontargeting control shRNA or CYLD shRNA) were infected (multiplicity of infection [MOI], 0.5) in a 48-well format (0.3 ml) and washed twice at 10 h postinfection. Every 2 days over a 10-day period, 100 μl of culture supernatant was collected, clarified, and stored at −80°C. Cultures were supplemented with 100 μl of fresh medium at each time point. At the end of the infection, the collected culture supernatants were used to infect TZM-bl cells in triplicate (15 μl, 96-well plates), and β-galactosidase activity was quantified 48 h after infection using Tropix (Perkin-Elmer) as previously described (50).
Reactivation experiments.
JLat 8.4 cells (2 × 106) transiently transfected with CYLD or nontargeting siRNAs were activated with PMA (200 ng/ml) at 24 h after AMAXA Nucleofection. Green fluorescent protein (GFP) expression was measured using a BD Biosciences FACSCalibur flow cytometer 48 h after PMA stimulation.
JLat 8.4 cells (2 × 106) stably transduced with nontargeting control shRNA or CYLD shRNA were activated with PMA (200 ng/ml). p24 Gag production was determined at 48 h postactivation by p24 intracellular staining or by immunoblotting.
Immunoblotting.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer supplemented with complete protease inhibitor (Roche). Proteins were separated on 10% SDS-polyacrylamide gels (Invitrogen), transferred onto polyvinylidene difluoride membranes (Pierce), probed with antibodies, and visualized with SuperSignal West Pico or Femto (Pierce) on a Fluorochem E System (Protein Simple) machine. The following antibodies were used: mouse M2 monoclonal antibody to FLAG (Sigma F1804), mouse monoclonal anti-CYLD N-terminal domain (Cell Signaling 4495), mouse monoclonal anti-CYLD N-terminal domain (Invitrogen), monoclonal anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Santa Cruz 32233), monoclonal antiactin (Sigma), monoclonal anti-IκBα (Cell Signaling), and monoclonal anti-p24 (m183 clone; NIH AIDS Research Program). Densitometric analyses of Western blots were performed using Alpha View software (Protein Simple).
Intracellular p24 staining.
JLat cells (3 × 105) were fixed and permeabilized with the Cytofix/Cytoperm kit (BD Biosciences) as per the manufacturer's instructions, followed by staining with anti-p24 antibody KC57 RD1 (1:100 dilution; Coulter Clone). HIV-1 p24 Gag-positive gates were set by comparison with isotype-unrelated antibody control MslgG1-RD1 (1:100 dilution; Coulter Clone). An average of 105 cells were acquired per sample, and data were analyzed using FlowJo software.
RNA isolation and quantitative real-time PCR (qRT-PCR).
Cellular RNA was isolated using TRIzol (Invitrogen) according to the manufacturer's instructions, followed by a treatment with the DNA-free DNase treatment and removal kit (AM1906; Ambion RNA by Life Technologies). cDNA was generated using the iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions.
The expression of host and viral transcripts was measured by quantitative real-time PCR using iQ SYBR green Supermix (Bio-Rad) according to the manufacturer's instructions. The PCR temperature profile was 95°C for 3 min, followed by 40 cycles of 95°C for 10 s and 60°C for 60 s. Expression levels for individual mRNAs were calculated based on their threshold cycle (CT) (ΔΔCT values) using rsp11 as a housekeeping gene to normalize the data. Primers for CYLD (CYLD for, 5′-GGTAATCCGTTGGATCGGTCAG; CYLD rev, 5′-AGTGCCTCTGAAGGTTCCATCC), rsp11 (rsp11 for, 5′-GCCGAGACTATCTGCACTAC; and rsp11 rev, 5′-ATGTCCAGCCTCAGAACTTC), and IκBα (IκBα for, 5′-GCCAGCGTCTGACGTTATGA; IκBα rev, 5′-GAGGGCTGATCCTACCAC) were designed using PrimerDesign software. Primers MH531, MH532, HIV LTP for, and HIV LTP rev were used to measure HIV mRNA expression (51, 52).
Alu PCR.
At 1 day postinfection, cellular DNA was isolated using the DNeasy 96 Blood & Tissue kit (Qiagen) and quantitated using the Quant-iTTM PicoGreen R ds DNA assay kit (Invitrogen) on a Cytofluoro multiwell plate reader series 4000 (Applied Biosystems). In order to measure the amount of total viral cDNA, internal primers annealing between the U5 region and the R region were used. The proviral DNA content was measured using primers specific for the human Alu regions and primers annealing in the gag region together with an LTR-specific molecular beacon. PCRs were carried out using an Applied Biosystems 7500 Fast real-time PCR system. All steps were performed using an Epmotion 5075 robot (Eppendorf) (52).
Luciferase reporter assay.
For MLV and HIV promoter-specific assays, CYLD shRNA and NT shRNA HEK 239T cells were transfected with 100 ng of MLV- and HIV-LTR-luciferase reporter plasmids and 100 ng of p-RL-TK Renilla. Cells were lysed at 24 to 48 h posttransfection with passive lysis buffer (PLB). Firefly luciferase values were measured using the luciferase assay system (Promega), and Renilla luciferase values were measured using the Renilla assay system (Promega). Firefly luciferase data were normalized to Renilla luciferase values, and the data were represented as the fold change over the NT shRNA HEK 293T cell values. All experiments were performed in duplicate or triplicate.
For the NF-κB promoter-specific assays, HEK 239T cells stably transduced with CYLD shRNA and NT shRNA were transfected with 50 ng of c-fos-luciferase reporter plasmids and 25 ng of p-RL-TK Renilla. Cells were lysed at 24 to 48 h posttransfection with PLB (Promega). Firefly luciferase values were measured using the luciferase assay system (Promega), and Renilla luciferase values were measured using the Renilla assay system (Promega). Firefly luciferase data were normalized to Renilla luciferase values, and the data were represented as the fold change over NT shRNA HEK 293T values. All experiments were performed in duplicate or triplicate.
Cell viability was determined using the CellTiter Glo assay system (Promega) as per the manufacturer's instructions.
Immunofluorescence.
HEK 293T cells were plated on coverslips, and 24 h later the cells were stimulated for 2 h with tumor necrosis factor alpha (TNF-α) (10 ng/ml). Cells were washed with PBS, fixed with 3% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 15 min, and blocked in phosphate-buffered saline (PBS), 0.1% Triton X-100, and 1% BSA for 1 h.
Staining for p65 was performed overnight at 4C using anti-p65 (Abcam ab32536). Coverslips were washed three times in PBS–0.1% Triton X-100 and incubated for 1 h with secondary antibody (Alexa Fluor 594; Molecular Probes). Nuclear staining was performed using Hoechst 33342 nucleic acid stain (Invitrogen). Coverslips were washed three times with washing solution and mounted on slides using Vectashield mounting medium (Vector Laboratories).
Fluorescent images of fixed cells were captured on a Leica SP5 DM confocal microscope (Leica Microsystems) at a magnification of ×40. Confocal laser scanning microscopy was performed at the MSSM Microscopy Shared Resource Facility.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism 5 software. P values are two sided, and values of <0.05 were considered to be significant.
RESULTS
CYLD silencing increases HIV infection in single-cycle assays.
Following the identification of CYLD as a putative negative regulator of HIV replication through siRNA screening (Pache et al., unpublished data), we confirmed that transient silencing of CYLD increases HIV infection using two nonoverlapping siRNAs (CYLD_2 and CYLD_3). Figure 1B shows a 12- to 100-fold increase in HIV infection, as measured by luciferase expression, in HEK 293T cells silenced for CYLD (CYLD_3 siRNA or CYLD_2 siRNA) compared to cells transfected with nontargeting siRNAs (NT siRNA) (Fig. 1B, upper panel). Western blot analysis of CYLD_2- and CYLD_3-transfected cells showed efficient knockdown for each siRNA (Fig. 1B, lower panel).
None of the siRNA transfection reagents used induced measurable cell toxicity over the duration of the infection experiments. Viability tests performed by measuring ATP content (CellTiter Glo assay) confirmed that the CYLD siRNAs were not toxic (NT siRNA-transfected cells, 129% of mock-transfected cells; CYLD_3 siRNA-transfected cells, 109%; CYLD_2-siRNA transfected cells, 138%).
We next infected cells stably transduced with shRNA constructs directed against CYLD. A 17-fold increase in HIV infection was observed when infecting HEK 293T cells stably expressing shRNA targeting CYLD (293T CYLD shRNA, CYLD KD) (Fig. 1C, upper panel). Complementation of these CYLD KD cells with an shRNA-resistant CYLD mutant (transcript variant 2) reduced HIV infectivity (Fig. 1C).
To investigate the molecular mechanism by which CYLD knockdown increased HIV infection, we complemented stable 293T CYLD shRNA and 293T NT shRNA (control) cell lines with a catalytically defective CYLD mutant that fails to remove lysine 63-linked ubiquitin moieties from substrates (CYLD C598A [8, 53]) and was rendered resistant to the CYLD shRNA by site-directed mutagenesis. As shown in Fig. 1C, the CYLD C598A mutant failed to reduce HIV infection in 293T cells containing the CYLD shRNA (Fig. 1C), suggesting that the catalytic activity of CYLD is necessary to decrease HIV infection. Of note, expression of the catalytically defective CYLD mutant C598A in 293T NT shRNA cells increases luciferase reporter expression, suggesting that this mutant might function as dominant negative. The efficiency of silencing was on average 80% by quantitative PCR (qPCR) analysis (data not shown) and Western blotting. The stable cell lines were probed with antibodies directed against the endogenous CYLD as well as with anti-FLAG antibodies to confirm expression of the shRNA-resistant FLAG-tagged CYLD variants (Fig. 1C, lower panel).
Taken together, these data suggest that CYLD acts as repressor of HIV infection and that the catalytic activity of CYLD is necessary for the repression.
Identification of the viral step affected by CYLD silencing.
Next we determined the step in the viral replication cycle that is affected by the depletion of CYLD. We measured the production of total viral DNA and integrated provirus at 24 h after infection in HEK 293T cells transiently silenced for CYLD or controls. No differences were detected in cells with and without CYLD (Fig. 2A and B). In contrast, when we analyzed viral transcription upon CYLD silencing, we detected a 9-fold increase in HIV transcription in the CYLD silenced cells compared to the control cells (Fig. 2C). These data indicate that CYLD silencing enhances viral infection by increasing the levels of HIV mRNA transcription without affecting the levels of proviral DNA.
FIG 2.
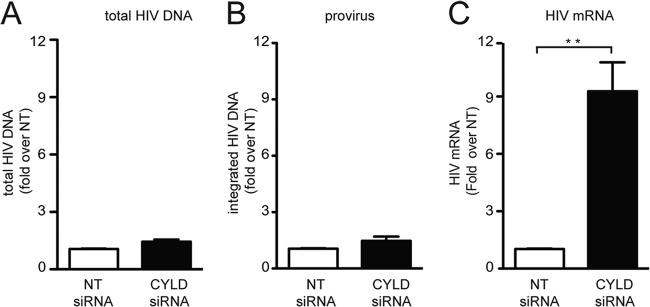
Identification of the viral steps affected by CYLD silencing. (A) HEK 293T cells transiently transfected with CYLD siRNA or nontargeting (NT) siRNA were infected with VSV-G HIV NL4.3 luciferase reporter virus. DNA was extracted at 24 h postinfection, and total HIV DA was quantified by qPCR. The NT siRNA is set at 1, and values represent fold induction over NT siRNA. (B) HEK 293T cells were treated as for panel A, and integrated provirus was quantified by qPCR. The NT siRNA was set to 1, and the plotted values represent the fold induction over NT siRNA. (C) HEK 293T cells transiently transfected with CYLD-targeting siRNA (CYLD siRNA) or nontargeting siRNA (NT siRNA) as a control were infected with VSV-G HIV NL4.3. HIV mRNA expression was analyzed by qPCR using the ΔΔCT method at 24 h postinfection; data were plotted as fold change compared to control cells (NT siRNA). Values represent means plus SEM for three independent experiments. The NT shRNA was set to 1, and values represent fold changes over NT shRNA expressing cells. Expression of HIV mRNA relative to the housekeeping gene rsp11 is 0.09. **, P < 0.01 (unpaired t tests, Prism software).
Increased HIV transcription in CYLD knockdown cells is NF-κB or NFAT dependent.
To dissect the molecular mechanism by which CYLD controls HIV transcription, we took advantage of four HIV molecular clones that bear mutations in transcription factor binding sites in the LTR region: HIV WT, HIV ΔUSF, HIV ΔNFIL6, and HIV ΔNF-κB/NFAT (Fig. 3A) (42). We infected HEK 293T cells silenced for CYLD with VSV-G-pseudotyped viral stocks of these four viruses and analyzed the levels of HIV mRNA by qPCR at 1 day postinfection. All viruses except the one in which the NF-κB/NFAT sites were deleted displayed 6- to 9-fold-increased levels of HIV mRNA expression in the absence of CYLD (Fig. 3B). HIV ΔNF-κB/NFAT showed no increase in infection upon CYLD depletion, indicating that the effects of CYLD on HIV infection are dependent on the NF-κB/NFAT-binding site in the viral LTR.
FIG 3.

The increase in HIV transcription in CYLD knockdown cells is NF-κB/NFAT dependent. (A) Schematic representation of the different viruses used for panel B. (B) HEK 293T cells transiently transfected with CYLD siRNA (CYLD siRNA) or nontargeting siRNA (NT siRNA) were infected with the indicated viruses. HIV mRNA expression was analyzed by qPCR using the ΔΔCT method at 24 h postinfection; data were plotted as fold change compared to nontargeting siRNA-transfected cells. Values are means plus SEM for two independent experiments. The values for the NT siRNA were set to 1, and values represent fold change over NT siRNA-expressing cells. Expression of HIV WT mRNA relative to the housekeeping gene rsp11 is 0.091, that of HIV ΔUSF is 0.046, that of HIV ΔNF-IL-6 is 0.04, and that of HIV ΔNF-κB I,II is 0.09. (C) Schematic representation of the different LTR reporter plasmids used for panel D. (D) HEK 293T cells stably expressing CYLD shRNA or NT shRNA were transfected with luciferase reporter plasmid under the control of HIV LTR, HIV-ΔNF-κB I,II LTR, MLV LTR, or an MLV (HIV U3) LTR chimera (HIV U3 promoter region inserted into MLV) and TK-Renilla as a control. Luciferase and Renilla expression was measured 48 h later. Luciferase values were normalized to the Renilla values and then plotted as fold change over the NT shRNA control. Values are means plus SEM for two independent experiments. The values for the NT shRNA were set to 1, and values represent fold change over NT shRNA-expressing cells. Average raw RLU values for the HIV-LTR WT NT control were 121,155 ± 1,270, those for the HIV-ΔNF-κBI,II LTR NT control were 306,691 ± 36168, those for the MLV-LTR NT control were 83,520 ± 10,650, and those for the MLV (HIV U3) LTR NT control were 131,173 ± 2,352. The average of Renilla control values was 2,180 ± 768.
To further confirm the involvement of the NF-κB pathway in CYLD-dependent HIV transcription, we next used a series of LTR reporter constructs (41). In contrast to that of HIV, MLV transcription is independent of NF-κB (54). Therefore, we compared luciferase reporter expression driven by an MLV LTR to that driven by an HIV LTR after transfection of 293T cells stably silenced for CYLD shRNA as well as control 293T NT shRNA cell lines (Fig. 3C). Additional constructs tested included an MLV with the HIV U3 region (containing the NF-κB/NFAT binding sites) and an HIV LTR lacking the NF-κB/NFAT sites (54). Figure 3D shows a 4-fold increase of luciferase expression driven by the HIV WT LTR and the MLV containing the HIV U3 region in the absence of CYLD. There was no difference in luciferase expression between CYLD knockdown cells and control cells with the MLV LTR and the HIV ΔNF-κB/NFAT LTR construct (Fig. 3D). Thus, CYLD silencing resulted in increased HIV LTR-dependent transcription, while the NF-κB/NFAT-independent MLV LTR-driven transcription was not affected.
CYLD silencing increases NF-κB signaling as a general mechanism.
Several groups reported that CYLD inhibits NF-κB activation by deubiquitinating different factors in this pathway (8, 14, 23). We wanted, therefore, to establish whether CYLD could act as a general inhibitor of NF-κB in our model systems. We first transfected 293T cells stably silenced for CYLD and the corresponding control cells (293T NT shRNA) with a reporter construct carrying the luciferase reporter gene under the control of the c-fos promoter, which contains binding sites for NF-κB. When we measured luciferase activity 24 h later, we found that the activity of the reporter gene was 6-fold increased in CYLD knockdown cells compared to control cells (Fig. 4A). The expression of the short hairpin-resistant form of CYLD decreased c-fos-dependent transcription to the level of the nontargeting control (Fig. 4A).
FIG 4.

CYLD silencing increases NF-κB signaling as a general mechanism. (A) CYLD shRNA HEK 293T cells and NT shRNA HEK 293T cells were transfected with a luciferase reporter plasmid under the control of the c-fos gene promoter and a TK-Renilla vector together with empty plasmid or the short-hairpin-resistant form of Flag-CYLD. Luciferase and Renilla expression was measured 48 h later. Luciferase values were normalized to the Renilla values and then plotted as fold change over the NT shRNA control. Values are means plus SEM for three independent experiments. The value for the NT shRNA cells transfected with an empty vector was set to 1, and values represent fold change over NT shRNA-expressing cells. The average of RLU for the mock control is 10,094 ± 1,748. **, P < 0.01 (unpaired t tests, Prism software). Expression of endogenous, FLAG-tagged CYLD and GAPDH was analyzed by Western blotting (lower panel). (B) CYLD shRNA HEK 293T cells and NT shRNA HEK 293T cells were treated with TNF-α (10 ng/ml) for the indicated times. The rate of degradation of IκBα was analyzed by Western blot using anti-IκBα antibody and anti-GAPDH antibody and quantified by densitometric analysis, where IκBα at time zero is set as 1. Values represent the means plus SEM from three independent experiments. (C) Expression of IκBα mRNA at steady state was measured in CYLD shRNA HEK 293T cells and NT shRNA HEK 293T by qPCR analysis. Expression of IκBα mRNA in the NT shRNA HEK 293T cells was set as 1, and data were plotted as fold change compared to NT shRNA HEK 293T cells. Values represent means plus SEM for three independent experiments. IκBα mRNA expression in the NT shRNA is set at 1, and values represent fold change over NT shRNA. Expression of IκBα mRNA relative to the housekeeping gene rsp11 is 0.02. *, P < 0.05 (unpaired t tests, Prism software). (D) Confocal images of CYLD shRNA HEK 293T cells and NT shRNA HEK 293T cells treated with TNF-α (10 ng/ml) or mock treated for 2 h and stained for NF-κB p65 (red) and with Hoechst stain (blue). (E) The number of cells in which the NF-κB p65 subunit translocated to the nucleus was determined by counting five random fields of view per slide, with an average of 200 cells/field. ***, P < 0.001 (unpaired t tests, Prism software).
We next analyzed the rate of degradation of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IκBα) in response to tumor necrosis factor alpha (TNF-α) stimulation in the stable 293T CYLD shRNA cells compared to the stable 293T NT shRNA control cells. Figure 4B shows that IκBα was degraded more rapidly in the absence of CYLD. To confirm that the NF-κB pathway activation is enhanced in CYLD knockdown cells, we measured the expression level of IκBα, the transcription of which is induced by NF-κB as part of a negative feedback loop (55, 56). We observed that the levels of IκBα mRNA were enhanced (10-fold) in CYLD knockdown cells compared to the control cells (Fig. 4C).
To further investigate the effect of CYLD silencing on the NF-κB pathway, we determined the subcellular localization of the NF-κB p65 subunit upon TNF-α stimulation in the presence or absence of CYLD by confocal microscopy. We observed a significant increase in NF-κB p65 nuclear translocation in CYLD knockdown cells (P < 0.001) (Fig. 4D and E).
Collectively, these data suggest that CYLD silencing generally enhances NF-κB activity in HEK 293T cells.
CYLD is highly expressed in primary CD4+ T lymphocytes and is cleaved upon T cell receptor (TCR)-mediated activation.
In order to determine the CYLD expression profiles in cells of the human immune system relevant to HIV replication, we purified and analyzed CD4+ and CD4− T cell populations from peripheral blood lymphocytes as well as CD14+ cells from peripheral blood lymphocytes obtained from anonymous blood donors. Monocytes were differentiated into immature dendritic cells (DCs) and macrophages. CYLD protein levels in lysates of these three cell populations were analyzed by Western blotting. Figure 5A shows that CD4+ T lymphocytes from both donors tested expressed more CYLD than the CD4− cell population. CYLD was detectable in monocyte-derived macrophages and monocytes derived immature DCs from both donors (Fig. 5B and C).
FIG 5.

CYLD is highly expressed in primary CD4+ T lymphocytes and is cleaved upon TCR-mediated activation. (A) The expression levels of CYLD and GAPDH as a loading control were determined by immunoblot analysis in the CD4+ and CD4− fractions of primary blood lymphocytes (PBLs) purified from two healthy donors. (B) The expression levels of CYLD and GAPDH in monocytic-derived macrophages (MDMs) from two healthy donors were analyzed by immunoblot analysis. (C) Lysates from monocyte-derived dendritic cells (MDDCs) purified from two different healthy donors were analyzed for the expression of CYLD and GAPDH. (D) Primary CD4+ T cells were stimulated for 48 h with IL-2 and anti-CD3 plus anti-CD28 beads, IL-2 and PHA, or IL-2 alone. CYLD and actin (loading control) were analyzed by immunoblotting. The CYLD antibody that was used for detection recognizes an N-terminal epitope and detects an N-terminal fragment of CYLD. *, nonspecific band. (E) JLat 8.4 cells were stimulated for 24 h with PMA/ionomycin or PHA for 24 h. Primary CD4+ T cells were stimulated for 3, 24, or 48 h with IL-2/PHA or IL-2 alone. Cell lysates were analyzed by immunoblotting for CYLD and actin expression. #, nonspecific band.
Previous studies using murine systems or immortalized human T cells suggested that CYLD might undergo proteolytic cleavage upon activation (25, 26, 57). Since productive HIV infection requires T cell activation, we tested whether CYLD was cleaved in primary human CD4+ T cells. Treatment of CD4+ T lymphocytes with either CD3/CD28 or PHA in the presence of IL-2 resulted in CYLD cleavage (Fig. 5D). CYLD cleavage products were detected as early as 24 h after PHA treatment (Fig. 5E).
Taken together, these results indicate that CYLD is expressed in human cells of the hematopoietic lineage and that cellular activation induces its cleavage.
HIV transcription increases upon CYLD knockdown in immortalized and primary CD4+ T lymphocytes.
We next investigated the role of CYLD in spreading viral infection. The A3R5 T cell line was stably transduced with shRNA directed against CYLD or with nontargeting control shRNA. The efficiency of CYLD silencing was approximately 50% at the protein level as determined by Western blotting and densitometric analysis (Fig. 6A). Both cell lines were infected with replication-competent HIV NL4.3 and HIV LAI. Culture supernatants were collected every 2 days over a 10-day period and analyzed on TZM-bl reporter cells at the end of the infection. Both viruses replicated three times more efficiently in the cells expressing less CYLD (Fig. 6B).
FIG 6.

HIV transcription increases upon CYLD knockdown in immortalized and primary CD4+ T lymphocytes. (A) Efficiency of CYLD silencing. A3R5 T cell CYLD shRNA and NT shRNA were determined by Western blot analysis (upper panel) and quantified by densitometric analysis (lower panel). (B) A3R5 T cell CYLD shRNA and NT shRNA were infected with the indicated viruses. Clarified supernatants were collected every 2 days, and infectivity was determined by a TZM-bl infectivity assay. (C) Primary CD4+ T cells from two healthy donors were stimulated for 48 h with IL-2/PHA and transfected with CYLD siRNA and nontargeting siRNA (NT siRNA, control). At 48 h posttransfection, CD4+ T cells were infected with VSV-G-pseudotyped NL4.3/Env− HIV, and expression of HIV mRNA was measured at 24 h postinfection by qPCR. The HIV mRNA expression in NT siRNA is set at 1, and values represent fold induction over NT shRNA. Error bars represent standard deviations for duplicate qPCR analysis. Expression of HIV mRNA relative to the housekeeping gene rsp11 was 0.016 for donor 1 and 0.8 for donor 2. CYLD silencing efficiency was around 60% for both donors as determined by qPCR analysis.
In order to establish whether CYLD also plays a role in human primary CD4+ T cells, we transiently silenced CYLD in purified CD4+ T lymphocytes from two healthy donors. The efficiency of CYLD silencing was approximately 60% for both donors as determined by qPCR. We infected PHA-activated CD4+ T cells silenced for CYLD or transfected with nontargeting siRNAs with a VSV-G-pseudotyped HIV NL4.3 lacking envelope and measured the levels of HIV mRNA at 24 h postinfection. We found that upon CYLD silencing, HIV mRNA expression was increased 2.8- to 5-fold in CYLD-silenced cells compared to control cells (Fig. 6C).
Overall, these data indicate that CYLD limits HIV transcription in transformed T cell lines as well as primary CD4+ T cells.
Reactivation of latent HIV is enhanced in the absence of CYLD.
The NF-κB pathway not only is important during the early stages of the HIV replication cycle to establish a productive infection but also plays a pivotal role in the establishment and maintenance of HIV latency (58). To determine whether CYLD influences reactivation of HIV from latency, we used the well-described Jurkat T cell lines (JLat) that harbor an integrated, latent HIV provirus encoding a GFP reporter (59). Expression of the latent provirus results in GFP expression and can be achieved by stimulation with cytokines (e.g., TNF-α) or mitogens (e.g., PMA) or by silencing of cellular molecules (e.g., promyelocytic leukemia protein) (51, 60, 61).
We first monitored the effect of CYLD silencing on HIV reactivation after PMA treatment of JLat cells (8.4 clone). Flow cytometry analysis showed a 2-fold increase of GFP expression in JLat cells silenced for CYLD compared to cells transfected with a nontargeting (NT) siRNA (Fig. 7A). The efficiency of CYLD silencing was 40% on the protein level (Fig. 7B).
FIG 7.

Reactivation of latent HIV is enhanced in the absence of CYLD. (A) Flow cytometry profiles of JLat 8.4 cells transfected with CYLD siRNA or nontargeting siRNA (NT) in the absence of stimulation or treated with PMA. (B) Efficiency of silencing was determined by Western blot analysis (upper panel) and quantified by densitometric analysis (lower panel). (C) JLat 8.4 cells stably expressing shCYLD or nontargeting (NT) shRNA were stimulated with PMA for the indicated times. Efficiency of CYLD silencing (upper panel), HIV Gag production (middle panel), and actin expression (lower panel) were determined by Western blot analysis. #, nonspecific band. HIV Gag production was quantified by densitometric analysis. The level of Gag p55 expressed in NT shRNA JLat 8.4 cells after 24 h of PMA stimulation was set to 1. Values are means plus SEM from three independent experiments. (D) JLat 8.4 cells stably expressing shCYLD or NT shRNA were stimulated with PMA, and HIV Gag production was determined by flow cytometry using intracellular p24 staining. The plots shown are representative of four independent experiments. (E) Reactivation of HIV upon PMA stimulation of JLat 8.4 cells stably expressing shCYLD or NT shRNA as determined by p24 intracellular staining followed by flow cytometry. Values represent means plus SEM for four independent experiments. *, P < 0.05 (unpaired t tests, Prism software). (F) Quantitative PCR analysis of HIV-1 mRNA levels in JLat 8.4 cells stably expressing shCYLD or NT shRNA with and without PMA stimulation. HIV mRNA expression was analyzed using the ΔΔCT method. Values represent means plus SEM for four independent experiments. HIV mRNA expression in the NT shRNA, PMA-treated group is set to 1, and values represent fold change over NT shRNA. Expression of HIV mRNA relative to the housekeeping gene rsp11 was 0.2. *, P < 0.05 (unpaired t tests, Prism software).
We next stably silenced CYLD in JLat cells (clone 8.4) and analyzed Gag production upon PMA treatment. Both Western blot analysis (Fig. 7C) and intracellular p24 staining (Fig. 7D and E) showed a 4- to 6-fold increase in Gag p55 production. The intracellular Gag staining was required since the shRNA constructs used themselves expressed GFP, making it impossible to monitor HIV reactivation by direct GFP measurements as done in the transient-silencing experiments described above. We verified the levels of CYLD knockdown by immunoblot analysis (Fig. 7C).
We measured HIV mRNA expression by qPCR to ascertain that the enhanced level of viral protein production was caused by an increase in viral transcription. HIV transcription was determined at steady state and after PMA stimulation in JLat cells (clone 8.4) stably silenced for CYLD or transduced with a nontargeting control. We found that CYLD knockdown alone did not induce HIV reactivation, but upon PMA treatment, cells silenced for CYLD expressed 5- to 7-fold more HIV mRNA than the nontargeting control cells (Fig. 7A, upper panels). Thus, silencing of CYLD enhanced reactivation of latent provirus upon PMA reactivation in the JLat model system by increasing HIV transcription.
DISCUSSION
In this study, we dissect the molecular mechanism by which CYLD negatively affects HIV infection. We established that CYLD silencing enhances HIV transcription in different cell types, including primary CD4+ T cells (Fig. 1B and C, 2C, and 6C). The positive effect of CYLD knockdown on HIV infection was specifically linked to the presence of NF-κB/NFAT transcription factor-binding sites within the HIV LTR. Deletion of these sites abolished the effect of CYLD silencing on HIV transcription. Moreover, MLV transcription, which is known to be independent from the NF-κB/NFAT pathways, was not enhanced in cells lacking CYLD (Fig. 3A and B). Silencing of CYLD also improved viral replication in T cell lines (Fig. 6B) and facilitated viral reactivation in the JLat model system (Fig. 7).
In agreement with previous reports, we found that in the absence of CYLD, NF-κB-dependent transcription was enhanced and IκBα degradation upon TNF-α stimulation was accelerated (Fig. 4). We observed, moreover, that the inhibitory effect of CYLD on HIV transcription was dependent on its catalytic activity (Fig. 1B). Overexpression of the short-hairpin-resistant form of CYLD in 293T CYLD shRNA cells decreased NF-κB-dependent transcription of a c-fos reporter to levels comparable to that in control cells (Fig. 4A), while it only partially decreased LTR reporter transcription. This residual activity may be due to the potency of Tat as a transactivator (reviewed in reference 62). In the absence of Tat, the majority of RNA polymerases are paused near the promoter, while in the presence of Tat, the elongation of the transcript is dramatically increased, resulting in more efficient HIV mRNA production (63). Of note, overexpression of the catalytically inactive mutant CYLD C598A in the control NT shRNA cells increased HIV infection (Fig. 1C). This effect could be due to a dominant negative effect of the catalytically inactive version of CYLD. The C598A mutant is still able to bind to its targets but is unable to remove ubiquitin moieties, thus sequestering cellular targets from the endogenous wild-type CYLD. Catalytically inactive proteins that act in a dominant negative manner have been reported, including several deubiquitinases (64–66).
While we clearly show that CYLD acts in an NF-κB-dependent manner, our experimental methods did not distinguish between NF-κB and NFAT protein-specific contributions to HIV transcription. It is well established that CYLD negatively regulates NF-κB signaling (7, 8, 14, 23, 24), while the role of CYLD in the NFAT pathway is less well explored (25, 26). However, recently two groups reported that CYLD also inhibits NFAT signaling (25, 26). CYLD might, thus, act as an inhibitor of HIV transcription through distinct pathways.
Studies using knockout mice revealed a role for CYLD in T cell biology (67–70). In these studies, the absence of CYLD resulted in reduced accumulation of mature thymocytes and peripheral T lymphocytes. In addition, the differentiation of the CD4+ T lymphocytes into T regulatory cell subsets was halted (67–70). Cleavage of CYLD by the cellular paracaspase MALT1 was necessary for proper activation of CD4+ T cells in response to T cell receptor stimulation (57). Conversely, the loss of CYLD caused a hyperresponsive T cell phenotype leading to autoinflammation (24). CYLD can also be cleaved by caspase 8, giving rise to an amino-terminal fragment of 25 kDa (71). Despite the importance of CYLD in CD4+ T cell homeostasis in mouse models, its role in human T cell biology is less well established. We observed that CYLD is highly expressed in the purified CD4+ fraction of human primary lymphocytes. Reminiscent of the findings of Staal et al. (57) and Thuille et al. (25) in mice, we observed CYLD processing in primary human CD4+ T cells upon activation. However, the CYLD fragment that we observed upon T cell activation migrated at a slightly higher size (45 kDa) than what was reported in the murine systems and in Jurkat T cells (40 kDa). Staal and colleagues observed CYLD cleavage within a few hours of stimulation, while in our T cell experiments CYLD cleavage became apparent only after 24 to 48 h of stimulation. Future studies will investigate whether a protease other than MALT1 or caspase 8 cleaves CYLD in primary human lymphocytes or if, alternatively, the described MALT1 fragment is posttranslationally modified during the activation process.
Activated CD4+ T cells support productive HIV infection, while resting T cells are refractory (33). One reason for this phenotype is that quiescent CD4+ T cells have very low levels of cellular deoxynucleoside triphosphates (dNTPs), which are mediated by SAMHD1 (37, 38). Silencing SAMHD1 increases the rate of reverse transcription in resting T cells, but it does not rescue production of infectious viral progeny (37, 38). Thus, additional, SAMHD1-independent blocks limit productive infection in resting T cells. CYLD may contribute to this phenotype, since HIV transcription is still restricted in the absence of SAMHD1. It has recently been reported that regnase-1 (MCPIP1) restricts HIV infection by decreasing viral mRNA through its RNase activity. Similar to CYLD, MCPIP1 is cleaved upon T cell activation by MALT1 (72). In addition, T cell activation has been linked to SAMHD1 phosphorylation and subsequent inactivation of restriction through a still-unclear mechanism (73, 74). Most likely, activation of T cells causes pleiotropic effects (e.g., increased dNTP concentration by inactivation of SAMHD1, MCPIP1 inactivation, increased viral integration, and activation of the NF-κB/NFAT pathways by CYLD cleavage), which collectively render activated T cells suitable for supporting infectious HIV production (73–75).
The global activation status of the NF-κB pathway in the cell at the time of HIV-1 infection may be the key factor determining the outcome of infection (39). Establishment, maintenance, and reactivation of latency correlate with the activation levels of NF-κB (58, 76–78). The stimuli known to activate NF-κB (e.g., PHA and PMA) also induce HIV transcription from latent proviruses (61, 79). CYLD silencing failed to induce spontaneous reactivation of the silent provirus in the JLat model system, but HIV mRNA production was increased when CYLD knockdown was augmented with PMA activation compared to that with PMA activation alone (Fig. 7). These results indicate that for reactivation of the latent provirus, silencing of CYLD was insufficient, hinting at other restrictive cellular factors such as the chromatin environment or transcriptional interference (60). Of note, in a primary T cell model of latency, activation of NFAT instead of NF-κB has been proposed to be the main factor inducing reactivation of the latent provirus (42). Future studies investigating the role of CYLD in a primary model of latency are warranted to validate the observations.
Overall, our study shows that the deubiquitinase CYLD acts as a negative regulator of HIV transcription by inhibiting the NF-κB/NFAT pathway. Future studies will reveal whether modulation of CYLD plays a role in T cell activation and HIV infection in patients.
ACKNOWLEDGMENTS
We thank all the members of the Simon lab, M. A. O'Donnell, and D. Legarda for helpful feedback. We also thank all the participants in the HIV Immune Networks Team for exciting discussions. We thank V. Planelles, A. Bosque (The University of Utah), and W. Mothes (Yale School of Medicine) for reagents.
This work was funded in part by Program Project HIV Immune Networks Team (HINT) P01 AI090935 (V.S., A.F.-S., J.A.T.Y., S.K.C., F.D.B., and A.G.-S.), by NIH grants AI089246 (V.S.), AI064001 (V.S.), AI72645 (J.A.T.Y.), AI052845 (F.D.B.), AI082020 (F.D.B.), 1R01AI073450 (A.F.-S.), AI77780 (S.K.C.), and 052417 (A.T.T.), by DARPA contract HR0011-11-C-0094 (A.F.-S.), and by HIVERA:EURECA, ERANET 01KI1307A (R.K.).
Footnotes
Published ahead of print 23 April 2014
REFERENCES
- 1.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–926. 10.1126/science.1152725 [DOI] [PubMed] [Google Scholar]
- 2.Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK. 2008. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135:49–60. 10.1016/j.cell.2008.07.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeung ML, Houzet L, Yedavalli VS, Jeang KT. 2009. A genome-wide short hairpin RNA screening of Jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 284:19463–19473. 10.1074/jbc.M109.010033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS. 2008. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4:495–504. 10.1016/j.chom.2008.10.004 [DOI] [PubMed] [Google Scholar]
- 5.Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, Jones C, Hansen J, Blair E, Hofmann B, Siebert R, Turner G, Evans DG, Schrander-Stumpel C, Beemer FA, van Den Ouweland A, Halley D, Delpech B, Cleveland MG, Leigh I, Leisti J, Rasmussen S. 2000. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 25:160–165. 10.1038/76006 [DOI] [PubMed] [Google Scholar]
- 6.Poblete Gutierrez P, Eggermann T, Holler D, Jugert FK, Beermann T, Grussendorf-Conen EI, Zerres K, Merk HF, Frank J. 2002. Phenotype diversity in familial cylindromatosis: a frameshift mutation in the tumor suppressor gene CYLD underlies different tumors of skin appendages. J. Invest. Dermatol. 119:527–531. 10.1046/j.1523-1747.2002.01839.x [DOI] [PubMed] [Google Scholar]
- 7.Sun SC. 2010. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 17:25–34. 10.1038/cdd.2009.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. 2003. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 424:793–796. 10.1038/nature01803 [DOI] [PubMed] [Google Scholar]
- 9.Blake PW, Toro JR. 2009. Update of cylindromatosis gene (CYLD) mutations in Brooke-Spiegler syndrome: novel insights into the role of deubiquitination in cell signaling. Hum. Mutat. 30:1025–1036. 10.1002/humu.21024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheinfeld N, Hu G, Gill M, Austin C, Celebi JT. 2003. Identification of a recurrent mutation in the CYLD gene in Brooke-Spiegler syndrome. Clin. Exp. Dermatol. 28:539–541. 10.1046/j.1365-2230.2003.01344.x [DOI] [PubMed] [Google Scholar]
- 11.Zhang G, Huang Y, Yan K, Li W, Fan X, Liang Y, Sun L, Li H, Zhang S, Gao M, Du W, Yang S, Liu J, Zhang X. 2006. Diverse phenotype of Brooke-Spiegler syndrome associated with a nonsense mutation in the CYLD tumor suppressor gene. Exp. Dermatol. 15:966–970. 10.1111/j.1600-0625.2006.00501.x [DOI] [PubMed] [Google Scholar]
- 12.Ahmed N, Zeng M, Sinha I, Polin L, Wei WZ, Rathinam C, Flavell R, Massoumi R, Venuprasad K. 2011. The E3 ligase Itch and deubiquitinase Cyld act together to regulate Tak1 and inflammation. Nat. Immunol. 12:1176–1183. 10.1038/ni.2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hellerbrand C, Bumes E, Bataille F, Dietmaier W, Massoumi R, Bosserhoff AK. 2007. Reduced expression of CYLD in human colon and hepatocellular carcinomas. Carcinogenesis 28:21–27. 10.1093/carcin/bgl081 [DOI] [PubMed] [Google Scholar]
- 14.Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. 2003. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424:801–805. 10.1038/nature01802 [DOI] [PubMed] [Google Scholar]
- 15.Srokowski CC, Masri J, Hovelmeyer N, Krembel AK, Tertilt C, Strand D, Mahnke K, Massoumi R, Waisman A, Schild H. 2009. Naturally occurring short splice variant of CYLD positively regulates dendritic cell function. Blood 113:5891–5895. 10.1182/blood-2008-08-175489 [DOI] [PubMed] [Google Scholar]
- 16.Harhaj EW, Dixit VM. 2012. Regulation of NF-kappaB by deubiquitinases. Immunol. Rev. 246:107–124. 10.1111/j.1600-065X.2012.01100.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moffat JM, Mintern JD, Villadangos JA. 2013. Control of MHC II antigen presentation by ubiquitination. Curr. Opin. Immunol. 25:109–114. 10.1016/j.coi.2012.10.008 [DOI] [PubMed] [Google Scholar]
- 18.Maelfait J, Beyaert R. 2012. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol. Mol. Biol. Rev. 76:33–45. 10.1128/MMBR.05012-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee AJ, Zhou X, Chang M, Hunzeker J, Bonneau RH, Zhou D, Sun SC. 2010. Regulation of natural killer T-cell development by deubiquitinase CYLD. EMBO J. 29:1600–1612. 10.1038/emboj.2010.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, Xavier R, Ting AT. 2008. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9:930–936. 10.1038/embor.2008.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshida H, Jono H, Kai H, Li JD. 2005. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for Toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J. Biol. Chem. 280:41111–41121. 10.1074/jbc.M509526200 [DOI] [PubMed] [Google Scholar]
- 22.Zhang M, Wu X, Lee AJ, Jin W, Chang M, Wright A, Imaizumi T, Sun SC. 2008. Regulation of IkappaB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J. Biol. Chem. 283:18621–18626. 10.1074/jbc.M801451200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. 2003. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 424:797–801. 10.1038/nature01811 [DOI] [PubMed] [Google Scholar]
- 24.Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF, Leonard TO, Norbury CC, Fitzpatrick L, Zhang M, Sun SC. 2007. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J. Exp. Med. 204:1475–1485. 10.1084/jem.20062694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thuille N, Wachowicz K, Hermann-Kleiter N, Kaminski S, Fresser F, Lutz-Nicoladoni C, Leitges M, Thome M, Massoumi R, Baier G. 2013. PKCtheta/beta and CYLD are antagonistic partners in the NFkappaB and NFAT transactivation pathways in primary mouse CD3+ T lymphocytes. PLoS One 8:e53709. 10.1371/journal.pone.0053709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koga T, Lim JH, Jono H, Ha UH, Xu H, Ishinaga H, Morino S, Xu X, Yan C, Kai H, Li JD. 2008. Tumor suppressor cylindromatosis acts as a negative regulator for Streptococcus pneumoniae-induced NFAT signaling. J. Biol. Chem. 283:12546–12554. 10.1074/jbc.M710518200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffin GE, Leung K, Folks TM, Kunkel S, Nabel GJ. 1989. Activation of HIV gene expression during monocyte differentiation by induction of NF-kappa B. Nature 339:70–73. 10.1038/339070a0 [DOI] [PubMed] [Google Scholar]
- 28.Alcami J, Lain de Lera T, Folgueira L, Pedraza MA, Jacque JM, Bachelerie F, Noriega AR, Hay RT, Harrich D, Gaynor RB, et al. 1995. Absolute dependence on kappa B responsive elements for initiation and Tat-mediated amplification of HIV transcription in blood CD4 T lymphocytes. EMBO J. 14:1552–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oeckinghaus A, Ghosh S. 2009. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Persp. Biol. 1:a000034. 10.1101/cshperspect.a000034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nabel G, Baltimore D. 1987. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326:711–713. 10.1038/326711a0 [DOI] [PubMed] [Google Scholar]
- 31.Bielinska A, Krasnow S, Nabel GJ. 1989. NF-kappa B-mediated activation of the human immunodeficiency virus enhancer: site of transcriptional initiation is independent of the TATA box. J. Virol. 63:4097–4100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osborn L, Kunkel S, Nabel GJ. 1989. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. U. S. A. 86:2336–2340. 10.1073/pnas.86.7.2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zack JA, Kim SG, Vatakis DN. 2013. HIV restriction in quiescent CD4(+) T cells. Retrovirology 10:37. 10.1186/1742-4690-10-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korin YD, Zack JA. 1998. Progression to the G1b phase of the cell cycle is required for completion of human immunodeficiency virus type 1 reverse transcription in T cells. J. Virol. 72:3161–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen IS. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213–222. 10.1016/0092-8674(90)90802-L [DOI] [PubMed] [Google Scholar]
- 36.Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. 1990. Hiv-1 replication is controlled at the level of T-cell activation and proviral integration. EMBO J. 9:1551–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, Panitz S, Flory E, Landau NR, Sertel S, Rutsch F, Lasitschka F, Kim B, Konig R, Fackler OT, Keppler OT. 2012. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat. Med. 18:1682–1687. 10.1038/nm.2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, Benkirane M. 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 9:87. 10.1186/1742-4690-9-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan JK, Greene WC. 2012. Dynamic roles for NF-kappaB in HTLV-I and HIV-1 retroviral pathogenesis. Immunol. Rev. 246:286–310. 10.1111/j.1600-065X.2012.01094.x [DOI] [PubMed] [Google Scholar]
- 40.Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, Rodriguez-Madoz JR, Mulder LC, Barber GN, Fernandez-Sesma A. 2012. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 8:e1002934. 10.1371/journal.ppat.1002934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uchil PD, Hinz A, Siegel S, Coenen-Stass A, Pertel T, Luban J, Mothes W. 2013. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 87:257–272. 10.1128/JVI.01804-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bosque A, Planelles V. 2009. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113:58–65. 10.1182/blood-2008-07-168393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konig R, Chiang CY, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y, Chanda SK. 2007. A probability-based approach for the analysis of large-scale RNAi screens. Nat. Methods 4:847–849. 10.1038/nmeth1089 [DOI] [PubMed] [Google Scholar]
- 44.Ooms M, Majdak S, Seibert CW, Harari A, Simon V. 2010. The localization of APOBEC3H variants in HIV-1 virions determines their antiviral activity. J. Virol. 84:7961–7969. 10.1128/JVI.00754-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Connor RI, Chen BK, Choe S, Landau NR. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935–944. 10.1006/viro.1995.1016 [DOI] [PubMed] [Google Scholar]
- 46.Yee JK, Friedmann T, Burns JC. 1994. Generation of high-titer pseudotyped retroviral vectors with very broad host range. Methods Cell Biol. 43:99–112. 10.1016/S0091-679X(08)60600-7 [DOI] [PubMed] [Google Scholar]
- 47.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peden K, Emerman M, Montagnier L. 1991. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology. 185:661–672. 10.1016/0042-6822(91)90537-L [DOI] [PubMed] [Google Scholar]
- 49.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15:871–875. 10.1038/nbt0997-871 [DOI] [PubMed] [Google Scholar]
- 50.Harari A, Ooms M, Mulder LC, Simon V. 2009. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 83:295–303. 10.1128/JVI.01665-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lusic M, Marini B, Ali H, Lucic B, Luzzati R, Giacca M. 2013. Proximity to PML nuclear bodies regulates HIV-1 latency in CD4+ T cells. Cell Host Microbe 13:665–677. 10.1016/j.chom.2013.05.006 [DOI] [PubMed] [Google Scholar]
- 52.Butler SL, Hansen MS, Bushman FD. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7:631–634. 10.1038/87979 [DOI] [PubMed] [Google Scholar]
- 53.Stegmeier F, Sowa ME, Nalepa G, Gygi SP, Harper JW, Elledge SJ. 2007. The tumor suppressor CYLD regulates entry into mitosis. Proc. Natl. Acad. Sci. U. S. A. 104:8869–8874. 10.1073/pnas.0703268104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coffin JM, Hughes SH, Varmus H. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY: [PubMed] [Google Scholar]
- 55.Scott ML, Fujita T, Liou HC, Nolan GP, Baltimore D. 1993. The p65 subunit of NF-kappa B regulates I kappa B by two distinct mechanisms. Genes Dev. 7:1266–1276. 10.1101/gad.7.7a.1266 [DOI] [PubMed] [Google Scholar]
- 56.Sun SC, Ganchi PA, Ballard DW, Greene WC. 1993. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science 259:1912–1915. 10.1126/science.8096091 [DOI] [PubMed] [Google Scholar]
- 57.Staal J, Driege Y, Bekaert T, Demeyer A, Muyllaert D, Van Damme P, Gevaert K, Beyaert R. 2011. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 30:1742–1752. 10.1038/emboj.2011.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Siliciano RF, Greene WC. 2011. HIV latency. Cold Spring Harb. Persp. Med. 1:a007096. 10.1101/cshperspect.a007096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jordan A, Bisgrove D, Verdin E. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 22:1868–1877. 10.1093/emboj/cdg188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Lint C, Bouchat S, Marcello A. 2013. HIV-1 transcription and latency: an update. Retrovirology 10:67. 10.1186/1742-4690-10-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams SA, Greene WC. 2007. Regulation of HIV-1 latency by T-cell activation. Cytokine 39:63–74. 10.1016/j.cyto.2007.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karn J, Stoltzfus CM. 2012. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb. Persp. Med. 2:a006916. 10.1101/cshperspect.a006916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kao SY, Calman AF, Luciw PA, Peterlin BM. 1987. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 330:489–493. 10.1038/330489a0 [DOI] [PubMed] [Google Scholar]
- 64.Sehrawat S, Koenig PA, Kirak O, Schlieker C, Fankhauser M, Ploegh HL. 2013. A catalytically inactive mutant of the deubiquitylase YOD-1 enhances antigen cross-presentation. Blood 121:1145–1156. 10.1182/blood-2012-08-447409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Machida YJ, Machida Y, Vashisht AA, Wohlschlegel JA, Dutta A. 2009. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J. Biol. Chem. 284:34179–34188. 10.1074/jbc.M109.046755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Colleran A, Collins PE, O'Carroll C, Ahmed A, Mao X, McManus B, Kiely PA, Burstein E, Carmody RJ. 2013. Deubiquitination of NF-kappaB by ubiquitin-specific protease-7 promotes transcription. Proc. Natl. Acad. Sci. U. S. A. 110:618–623. 10.1073/pnas.1208446110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reiley WW, Zhang M, Jin W, Losiewicz M, Donohue KB, Norbury CC, Sun SC. 2006. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat. Immunol. 7:411–417. 10.1038/ni1315 [DOI] [PubMed] [Google Scholar]
- 68.Tsagaratou A, Trompouki E, Grammenoudi S, Kontoyiannis DL, Mosialos G. 2010. Thymocyte-specific truncation of the deubiquitinating domain of CYLD impairs positive selection in a NF-kappaB essential modulator-dependent manner. J. Immunol. 185:2032–2043. 10.4049/jimmunol.0903919 [DOI] [PubMed] [Google Scholar]
- 69.Reissig S, Hovelmeyer N, Weigmann B, Nikolaev A, Kalt B, Wunderlich TF, Hahn M, Neurath MF, Waisman A. 2012. The tumor suppressor CYLD controls the function of murine regulatory T cells. J. Immunol. 189:4770–4776. 10.4049/jimmunol.1201993 [DOI] [PubMed] [Google Scholar]
- 70.Zhao Y, Thornton AM, Kinney MC, Ma CA, Spinner JJ, Fuss IJ, Shevach EM, Jain A. 2011. The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor beta (TGF-beta) signaling and the development of regulatory T cells. J. Biol. Chem. 286:40520–40530. 10.1074/jbc.M111.292961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O'Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT. 2011. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol. 13:1437–1442. 10.1038/ncb2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu S, Qiu C, Miao R, Zhou J, Lee A, Liu B, Lester SN, Fu W, Zhu L, Zhang L, Xu J, Fan D, Li K, Fu M, Wang T. 2013. MCPIP1 restricts HIV infection and is rapidly degraded in activated CD4+ T cells. Proc. Natl. Acad. Sci. U. S. A. 110:19083–19088. 10.1073/pnas.1316208110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White TE, Brandariz-Nunez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, Diaz-Griffero F. 2013. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 13:441–451. 10.1016/j.chom.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cribier A, Descours B, Valadao AL, Laguette N, Benkirane M. 2013. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell Rep. 3:1036–1043. 10.1016/j.celrep.2013.03.017 [DOI] [PubMed] [Google Scholar]
- 75.Manganaro L, Lusic M, Gutierrez MI, Cereseto A, Del Sal G, Giacca M. 2010. Concerted action of cellular JNK and Pin1 restricts HIV-1 genome integration to activated CD4+ T lymphocytes. Nat. Med. 16:329–333. 10.1038/nm.2102 [DOI] [PubMed] [Google Scholar]
- 76.Duverger A, Jones J, May J, Bibollet-Ruche F, Wagner FA, Cron RQ, Kutsch O. 2009. Determinants of the establishment of human immunodeficiency virus type 1 latency. J. Virol. 83:3078–3093. 10.1128/JVI.02058-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dahabieh MS, Ooms M, Simon V, Sadowski I. 2013. A doubly fluorescent HIV-1 reporter shows that the majority of integrated HIV-1 is latent shortly after infection. J. Virol. 87:4716–4727. 10.1128/JVI.03478-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan JK, Greene WC. 2011. NF-kappaB/Rel: agonist and antagonist roles in HIV-1 latency. Curr. Opin. HIV AIDS 6:12–18. 10.1097/COH.0b013e32834124fd [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stroud JC, Oltman A, Han A, Bates DL, Chen L. 2009. Structural basis of HIV-1 activation by NF-kappaB—a higher-order complex of p50:RelA bound to the HIV-1 LTR. J. Mol. Biol. 393:98–112. 10.1016/j.jmb.2009.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17:1087–1095. 10.1093/emboj/17.4.1087 [DOI] [PMC free article] [PubMed] [Google Scholar]