

ABSTRACT
The cancer-causing high-risk human papillomavirus (HPV) E6 oncoproteins target a number of cellular proteins that contain PDZ domains. However, the role of many of these interactions in either the HPV life cycle or in HPV-induced malignancy remains to be defined. Previous studies had shown that MAGI-1 was one of the most strongly bound PDZ domain-containing substrates of E6, and one consequence of this interaction appeared to facilitate the perturbation of tight junctions (TJs) by E6. In this study, we describe the generation of a mutation, K499E, within the MAGI-1 PDZ1 domain, which is resistant to E6 targeting. This mutant allows restoration of MAGI-1 expression in HPV-positive cells and defines additional activities of MAGI-1 that are overcome as a consequence of the association with E6. The reexpression of MAGI-1 in HPV-positive cells results in an increased recruitment of ZO-1 and PAR3 to sites of cell-cell contact, repression of cell proliferation, and induction of apoptosis. While the K499E mutation does not significantly affect these intrinsic activities of MAGI-1 in HPV-negative cells, its resistance to E6 targeting in an HPV-positive setting results in more cells expressing the mutant MAGI-1 than the wild-type MAGI-1, with a corresponding increase in TJ assembly, induction of apoptosis, and reduction in cell proliferation. These studies provide compelling evidence of a direct role for the perturbation of MAGI-1 function by E6 in the HPV life cycle and in HPV-induced malignancy.
IMPORTANCE It is clear that the targeting of PDZ-containing substrates by E6 is important for the normal viral life cycle and for the progression to malignancy. Nevertheless, which of these PDZ domain-containing proteins is relevant for HPV pathology is still elusive. In a previous study, we provided evidence that MAGI-1 is a sensitive proteolytic substrate for both the HPV-16 and HPV-18 E6 oncoproteins; however, the biological consequences associated with loss of MAGI-1 expression in HPV-positive cervical cancer cells are still poorly understood. Using a mutant MAGI-1, resistant to E6-mediated degradation, we show that its expression in cervical cancer cells promotes membrane recruitment of the tight junction-associated proteins ZO-1 and PAR3, represses cell proliferation, and promotes apoptosis. These findings suggest that E6-mediated inhibition of MAGI-1 function contributes to HPV pathology by perturbing tight junction assembly with concomitant stimulation of proliferation and inhibition of apoptosis.
INTRODUCTION
Papillomaviruses are a large and heterogeneous group of small nonenveloped DNA viruses able to infect vertebrates, including birds and reptiles (1, 2). The vast majority of human papillomaviruses (HPV) are causative agents of warts and self-remissive papillomas. However, a smaller group of HPV types, known as high-risk types, is associated with cancer onset in humans, where the most commonly caused malignancy is cervical cancer (3). The pathogenesis of cervical cancer is tightly linked to the combined action of E6 and E7, which cooperate efficiently to immortalize human keratinocytes (4) and to promote tumor formation in transgenic mouse models (5, 6). In light of this, it is not surprising that the continuous expression of E6 and E7 is a prerequisite for maintaining the proliferative potential and to prolong the survival of tumor-derived cells (7–10). The first described oncogenic functions of E6 and E7 were their abilities to interact with and promote the inactivation of the tumor suppressors p53 (11, 12) and pRB (13, 14), respectively. However, it is clear that perturbation of other cellular factors is required for the full transforming potential of the two oncoproteins (15, 16).
In this context, a distinctive feature of the E6 oncoproteins of high-risk HPV types is the presence of a PSD95/Dlg/ZO-1 (PDZ) binding motif (PBM) at their C terminus, which is absent from E6 proteins derived from the low-risk virus types. This PBM enables E6 oncoproteins to interact with and, perhaps more importantly, to promote the proteasome-mediated degradation of a pool of cellular PDZ-domain-containing proteins, including known regulators of cell polarity and tumor suppressors, such as hDlg (17, 18), hScrib (19), and members of the MAGI family of proteins (20, 21). Studies in organotypic raft cultures of human foreskin keratinocytes (HFKs) suggested that the presence of a functional E6 PBM in the context of the whole viral genome is important for expanding the population of suprabasal S-phase competent cells, thereby producing an environment suitable for viral genome amplification (22, 23). In addition, the E6-mediated degradation of its PDZ domain-containing substrates has also been associated with the modulation of its transforming properties. Indeed, the loss of a functional PBM was shown to correlate with a weaker propensity of E6 to promote mesenchymal-like morphological changes in immortalized keratinocytes and to induce tumor formation in nude mice (17, 24), and a functional PBM is required to enhance the invasive potential of E6- and E7-expressing tumors in transgenic mouse models (25). In this context, recent studies in HPV-transformed cells suggested that E6 targets a selected pool of PDZ domain-containing proteins for degradation and, among these targets, membrane-associated guanylate kinase (MAGUK) with inverted domain structure 1 (MAGI-1) appears to be a highly sensitive proteolytic substrate for both HPV-16 and -18 E6 oncoproteins (26).
The prototype MAGUK protein is Dlg, the product of the Drosophila lethal(1)discs large-1 tumor suppressor gene, which was the first member of the MAGUK protein family to be characterized (27). The structural features of MAGUK proteins include the presence of a basic core composed of a PDZ domain, an Src homology 3 (SH3) domain, and a guanylate kinase homology (GUK) domain, arranged in a head-to-tail orientation, with additional domains that can be present at the N terminus of the protein (28). Members of the MAGI subfamily, MAGI-1, -2, and -3, are classified as MAGUKs, although their atypical domain composition distinguishes them from other MAGUK proteins: for instance MAGI-1 has 5 or 6 PDZ domains, depending on the isoform, a GUK domain located at the N terminus rather than at the C terminus, and two WW domains substituting the SH3 domain (29). With regard to their interaction with high-risk E6 oncoproteins, the PBMs of HPV-16 and -18 E6 preferentially associate with the PDZ domain 1 of MAGI-1 (30), with HPV-18 E6 displaying the stronger affinity (31, 32). It is interesting to note that recent structural studies suggested that the lysine 499 (K499) is one of the most crucial residues of the MAGI-1 PDZ domain 1 for mediating the interaction with E6, and its mutation to glutamic acid (K499E) strongly reduced the affinity of the isolated PDZ domain for the PBM of HPV-18 E6 (32).
Previous studies suggested that MAGI-1 can colocalize with components of adherens junctions (AJs) and tight junctions (TJs) and that its expression is likely to promote the assembly of macromolecular junctional complexes (26, 33). In addition, MAGI-1 was also shown to interact with important signaling molecules, including the phosphatase PTEN, the RhoA-specific GEF NET1, and β-catenin (33, 34, 35), suggesting that MAGI-1 can function as a modulator of multiple signaling pathways. However, so far the biological function of MAGI-1, as well as the relevance of its E6-mediated degradation in context of HPV pathology, has remained elusive. In an attempt to address this question, we generated a K499E MAGI-1 mutant in order to study its function upon expression in HPV-positive cells. We confirmed that this mutation reduces the affinity of MAGI-1 for HPV-16 and HPV-18 E6 oncoproteins, and, consistent with this, the K499E MAGI-1 mutant is less susceptible to E6-mediated degradation compared with the wild-type (wt) protein. In addition, by expressing mutant MAGI-1 in HeLa cells, we confirmed that MAGI-1 is a strong inducer of junctional assembly and that its expression strongly inhibits proliferation and induces apoptosis of HPV-positive cells. Therefore, this study provides a possible functional explanation for the targeting of MAGI-1 by E6 in the context of HPV-related pathology.
MATERIALS AND METHODS
Plasmids.
pCDNA-3 FLAG-tagged MAGI-1 has been described previously (20). The K499E MAGI-1 mutant was generated using the GeneArt site-directed mutagenesis system (Invitrogen) according to the manufacturer's instruction, using the following primers: forward primer 5′TCCTGCAGATCGAAAGCCTCGTCCTCGATGGTCCT and reverse primer 5′ACGAGGCTTTCGATCTGCAGGAACTCATCAGGCTC.
Untagged HPV-18 E6 and HPV-16 E6 pCDNA-3 expression plasmids have been described previously (18, 36), as have the glutathione transferase (GST) fusion proteins HPV-18 E6 and HPV-16 E6 (37). pCMV MYC-tagged NET1 was described previously (38), and hemagglutinin (HA)-tagged β-catenin was kindly given by Claudio Brancolini.
Cell culture and transfection.
Human embryonic kidney 293 (HEK293), U2OS (human osteosarcoma), HeLa (HPV-18-positive), SiHa (HPV-16 positive), C33I (cervical carcinoma HPV-negative), H1299 (nonsmall cell lung carcinoma), HaCaT (skin keratinocytes), and HT1080 (fibrosarcoma) cells were maintained in Dulbecco's modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin-streptomycin (100 U/ml), and glutamine (300 μg/ml).
DNA transfection in HEK293, HT1080, and H1299 cells was performed using the standard calcium phosphate precipitation protocol as described previously (39). For DNA transfection in U2OS cells followed by immunofluorescent analysis, cells were seeded on glass coverslips at a confluence of 1.5 × 105 and transfected using the calcium phosphate precipitation protocol followed by glycerol shock. Briefly, 5 h after the addition of the DNA precipitate, cells were treated with 15% glycerol in phosphate-buffered saline (PBS) for 1 min. Cells were then washed three times with PBS and left to grow for an additional 24 h in DMEM supplemented with 10% FBS. For DNA transfection in HeLa, SiHa, C33I, and HaCaT cells were seeded on 6-cm dishes at a confluence of 1.5 × 105 and transfected using Fugene HD (Promega). For DNA transfection followed by immunofluorescent analysis, cells were seeded at the same confluence on glass coverslips.
Proteasome inhibitors.
The proteasome inhibitor Z-Leu-Leu-Leu-Al (MG-132; Sigma) was dissolved in dimethyl sulfoxide (DMSO) and used at 50 μM for 3 h.
Antibodies.
The following antibodies were used: mouse monoclonal anti-HA antibody 12CA5 (Roche), mouse monoclonal anti-ZO-1 (ZO-1–1A12) (Invitrogen), and rabbit polyclonal anti-PAR3 (Millipore). The following antibodies were purchased from Santa Cruz Biotechnology: mouse monoclonal anti-p53 (DO-1), mouse monoclonal anti-α-actin (H-2), and mouse monoclonal anti-Myc (9E10). Mouse monoclonal M2 anti-FLAG antibody (F3165) and rabbit polyclonal anti-FLAG (F7425) antibodies were from Sigma.
Western blotting and immunoprecipitation.
For Western blot sample preparation, cells were lysed in 2× SDS sample buffer (100 mM Tris HCl [pH 6.8], 200 mM dithiothreitol [DTT], 4% SDS, 20% glycerol, 0.2% bromophenol blue) and the whole-cell extracts were separated by SDS-PAGE and blotted on 0.22-μm nitrocellulose membranes (Schleicher & Schuell). The membranes were blocked at 37°C for 1 h in 10% milk-PBS and incubated with the appropriate primary antibodies diluted in 10% milk-PBS 0.5%-Tween 20 for 1 to 2 h at room temperature. After several washes, the membranes were incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (Dako) for 1 h at room temperature. After extensive washing, the blots were developed with ECL (GE Healthcare) according to the manufacturer's instructions. Protein band intensities were quantitated where possible using the OptiQuant quantification program.
For coimmunoprecipitation experiments, cells were scraped in ice-cold PBS and extracted in lysis buffer (1% Triton X-100, 50 mM Tris [pH 7.5], 300 mM NaCl, 1 mM EGTA, 1 mM EDTA) supplemented with protease inhibitors (Set1; Calbiochem). The extracts were passed through a 26-gauge needle multiple times and then cleared by centrifugation. Extracts from cells expressing FLAG-tagged MAGI-1 constructs were incubated with anti-FLAG beads (Sigma) for 2 to 3 h on a rotating wheel at 4°C. For the immunoprecipitation of MYC-tagged NET1, cell extracts were incubated with the MYC antibody or the control antibody for approximately 3 h on a rotating wheel at 4°C. Protein A-Sepharose beads (GE Healthcare) were then added for an additional 60 min at 4°C. The beads were then extensively washed, and the immunoprecipitated proteins were analyzed by Western blotting.
Fusion protein purification and in vitro binding assays.
GST-tagged fusion proteins were expressed and purified as described previously (37). Briefly, 40 ml of an overnight culture of Escherichia coli strain DH5α previously transformed with the appropriate expression plasmids was inoculated into Luria broth containing ampicillin (75 μg/ml) and grown at 37°C up to an optical density of 395 nm (OD395) of 0.6. Recombinant protein expression was induced for 3 h with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma). The cells were harvested by centrifugation and disrupted by sonication in lysis buffer (1% Triton X-100–1× PBS), and the lysates were cleared of cell debris by centrifugation. The GST fusion proteins were then incubated for 1 h with glutathione-conjugated agarose beads at 4°C. The purity of all fusion proteins was determined by SDS-PAGE and Coomassie brilliant blue R (Sigma) staining.
For in vitro binding and degradation assays, proteins were transcribed and translated in vitro in rabbit reticulocyte lysate using the Promega TNT system according to the manufacturer's instructions. The HPV-18 and HPV-16 E6 proteins were radiolabeled with [35S]cysteine, while MAGI-1 proteins were radiolabeled with [35S]methionine.
Equal amounts of in vitro-translated proteins were added to GST fusion proteins bound to glutathione resin and incubated for 1 h at 4°C. After extensive washing with PBS containing 0.5% NP-40, the bound proteins were analyzed by SDS-PAGE and autoradiography.
For GST pulldown assays using cell extracts, FLAG-tagged wild-type and K499E mutant MAGI-1 were transfected into HEK293 cells. Twenty-four hours after transfection, cells were scraped in ice-cold PBS and extracted in lysis buffer (1% Triton X-100, 50 mM Tris [pH 7.5], 300 mM NaCl, 1 mM EGTA, 1 mM EDTA) supplemented with protease inhibitors (Set1; Calbiochem). The extracts were then passed through a 26-gauge needle multiple times and cleared by centrifugation. Extracts from cells expressing FLAG-tagged MAGI-1 constructs were incubated with the indicated GST fusion proteins for 1 to 2 h on a rotating wheel at 4°C. The beads were then extensively washed, and the immunoprecipitated proteins were analyzed by Western blotting.
In vivo degradation assays.
HEK293 cells were transfected with 1 μg of either wild-type or K499E MAGI-1 constructs along with 0.3 μg of LacZ. pCDNA3 HPV-18 E6 plasmid was also included at increasing concentrations: 2, 5, 10 μg. Twenty-four hours posttransfection, the cells were harvested and analyzed by Western blotting.
In vitro degradation assays.
Degradation assays were performed as previously described (30). Briefly, radiolabeled proteins were mixed and incubated for the indicated times at 30°C. Volumes were adjusted using water-primed lysate. The residual MAGI-1 proteins were analyzed by SDS-PAGE and autoradiography.
Immunofluorescence microscopy and EdU staining.
Cells were fixed with 3.7% paraformaldehyde in PBS for 20 min and permeabilized with 0.1% Triton X-100 in PBS for 5 min. Slides were incubated with primary antibodies for 2 h at 37°C, extensively washed in PBS, and incubated for 30 min at 37°C with secondary anti-rabbit or anti-mouse antibody conjugated to fluorescein or rhodamine (Molecular Probes). Samples were washed several times with water and mounted with Vectashield mounting medium (Vector Laboratories) on glass slides. Slides were analyzed with either a Leica DMLB fluorescence microscope with a Leica photo camera (A01M871016) or a Zeiss LSM 510 confocal microscope with two lasers giving excitation lines at 480 and 510 nm. The data were collected with a 60× objective oil immersion lens.
For EdU staining, cells were seeded on glass coverslips and transfected with FLAG-tagged wild-type or K499E mutant MAGI-1 constructs. Twenty-four hours after transfection, EdU was added to the culture medium at a final concentration of 20 μM for 2 h. After labeling, cells were fixed with 3.7% paraformaldehyde in PBS for 20 min and permeabilized with 0.1% Triton X-100 in PBS for 5 min. Primary antibodies were incubated for 2 h at 37°C, extensively washed in PBS, and incubated for 30 min at 37°C with secondary anti-rabbit or anti-mouse antibody conjugated to fluorescein or rhodamine (Molecular Probes). After several washes in PBS, incorporated EdU was detected by incubating coverslips with the reaction mix solution [5 mM (+)-Na l-ascorbate (Sigma), 1 mM copper sulfate, 0.05 mM 6-carboxyfluorescein-TEG azide] for 30 min at room temperature. Samples were washed several times with water and mounted with Vectashield mounting medium (Vector Laboratories) on glass slides. Slides were analyzed with a Zeiss LSM 510 confocal microscope with two lasers, giving excitation lines at 480 and 510 nm. The data were collected with a 60× objective oil immersion lens.
Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assays.
Cells were seeded on glass coverslips and transfected with FLAG-tagged wild-type or K499E mutant MAGI-1 constructs. Twenty-four hours after transfection, cells were fixed with 3.7% paraformaldehyde in PBS for 20 min and permeabilized with 1× PBS-0.1% sodium citrate and 0.1% Triton X-100 for 5 min. The coverslips were incubated with the rabbit polyclonal FLAG antibody for 2 h at 37°C, followed by incubation with the rhodamine-conjugated anti-rabbit antibody (Molecular Probes) for 30 min at 37°C. After several washes in PBS, apoptotic cells were detected using the fluorescein-conjugated in situ cell death detection kit (Roche) according to the manufacturer's instructions. Samples were washed several times with water and mounted with Vectashield mounting medium (Vector Laboratories) on glass slides. Slides were analyzed with a Zeiss LSM 510 confocal microscope, with two lasers giving excitation lines at 480 and 510 nm. The data were collected with a 60× objective oil immersion lens.
RESULTS
The K499E mutation reduces MAGI-1 affinity for E6.
The fact that MAGI-1 is a sensitive proteolytic substrate of E6 prompted us to perform a more detailed analysis of its possible function in the context of HPV pathology, and we reasoned that using a MAGI-1 mutant that was resistant to E6 targeting might be one way to do this. We made use of previous studies showing that E6 interacts with MAGI-1 specifically through its PDZ domain 1 (30) and of the more recent structural data defining lysine 499 (K499) as a critical residue within the PDZ1 domain for mediating interaction with E6 (32). However, these studies were not performed in the context of either the full-length E6 or MAGI-1 proteins, nor did they assess the impact of the K499E mutation on the susceptibility of MAGI-1 to E6-directed degradation. In an attempt to address these points, we introduced the K499E mutation into the full-length MAGI-1 cDNA. For this purpose, a FLAG-tagged wild-type MAGI-1 (M1wt) expression construct was used as a template to generate the K499E MAGI-1 mutant (M1K499E) by site-directed mutagenesis. Figure 1a shows a comparison between the protein sequences of the wild-type and the K499E MAGI-1 PDZ1 domain, and the structural elements of the PDZ domain that mediate the interaction of MAGI-1 with its PBM-containing partners are highlighted. Once the FLAG-tagged M1K499E was generated, we first wanted to verify that the wild-type and mutant MAGI-1 proteins were expressed at comparable levels. To assess this, HEK293 cells were transiently transfected with the two constructs. Twenty-four hours after transfection, cells were harvested and the MAGI-1 expression levels were analyzed by SDS-PAGE and Western blotting using anti-FLAG antibody. As can be seen in Fig. 1B, wild-type and mutant MAGI-1 proteins are expressed at comparable levels in HEK293 cells, suggesting that the K499E mutation does not alter the steady-state levels of MAGI-1. In order to test whether the K499E mutation could indeed reduce the affinity of MAGI-1 for HPV-16 and -18 E6 oncoproteins, we first performed GST pulldown assays. To do this, HPV-16 and -18 E6 were expressed as GST fusion proteins and purified using glutathione-coated agarose beads. The beads were incubated for 1 h at 4°C with cell extracts from HEK293 cells that had been transiently transfected with either the M1wt or M1K499E expression plasmids. After extensive washing, the amount of MAGI-1 bound to GST-E6 was assessed by SDS-PAGE and Western blotting using anti-FLAG antibody. As can be seen in Fig. 1C, GST-18 E6 bound strongly to M1wt and retained about 70% of the input protein, whereas the K499E mutation exhibited a dramatically reduced ability to interact with E6. Similar results were also obtained with GST-16 E6 (Fig. 1D), although the wild-type MAGI-1 was bound less efficiently by HPV-16 E6 compared with HPV-18 E6, and this is in agreement with previous studies (30). These results confirm that the K499 residue within the MAGI-1 PDZ1 domain is important for the recognition of MAGI-1 by both the HPV-16 and -18 E6 oncoproteins.
FIG 1.

The K499E mutation perturbs E6 interaction with MAGI-1. (A) Diagram showing the domain composition of MAGI-1 and the location of the K499E mutation. Elements of secondary structure that compose the PDZ1 are also shown (βA to βE, β-strands A to E; αA and αB, α-helix A and B), and those involved in the interaction with E6 and other target proteins are highlighted in red (adapted from Fournane et al. [32]). (B) HEK293 cells were transfected with 1 μg of FLAG-tagged wild-type or mutant MAGI-1 expression plasmids and grown for 24 h prior to harvesting. The levels of MAGI-1 expression were assessed by Western blotting. β-Galactosidase was included to monitor the transfection efficiency. (C and D) Extracts from HEK293 cells transfected with 3 μg of wild-type or mutant MAGI-1 expression plasmids were subjected to GST-pulldown assays with the indicated GST fusion proteins, and bound MAGI-1 was detected by Western blotting using anti-FLAG antibody. Numbers represent the percentage of wild-type and mutant MAGI-1 proteins bound to the indicated GST fusion protein relative to the input control. The lower panel shows the Ponceau staining of the membrane, confirming the equal loading of GST proteins.
The MAGI-1 K499E mutant is resistant to E6-mediated degradation.
To determine whether the reduced ability of E6 to interact with the MAGI-1 K499E mutant also resulted in decreased rates of degradation, we first compared the relative susceptibilities of wild-type and mutant MAGI-1 to E6-mediated degradation in vitro. To do this, the two MAGI-1 proteins were in vitro translated in the presence of 35S-labeled methionine and then incubated at 30°C for different periods of time with in vitro translated and radiolabeled HPV-16 and -18 E6, and the pattern of MAGI-1 expression was assessed by SDS-PAGE and autoradiography. The results in Fig. 2A show that the levels of wild-type and mutant MAGI-1 are stable over a period of 120 min when incubated in the absence of E6. In contrast, wild-type MAGI-1 was degraded upon the addition of HPV-16 or -18 E6, with HPV-18 E6 being the most efficient (30). Consistent with the reduced level of interaction, M1K499E was also significantly more resistant to E6-mediated degradation, although low levels of degradation were observed at the later time points, which is consistent with the residual binding of E6 to the K499E mutant.
FIG 2.

The K499E mutation renders MAGI-1 resistant to E6-mediated degradation. (A) Wild type and the K499E mutant, together with HPV-16 and HPV-18 E6 oncoproteins, were in vitro translated in the presence of 35S-labeled methionine or cysteine. They were then incubated together at 30°C for the times indicated. Residual MAGI-1 protein was detected by SDS-PAGE and autoradiography. Numbers are the band intensities expressing the percentage of residual MAGI-1 protein relative to the control (100%). (B) HEK293 cells were cotransfected with 1 μg of FLAG-tagged wild-type (WT) or K499E mutant MAGI-1 expression plasmids and either 0, 2, 5 or 10 μg of HPV-18 E6 expression plasmid as indicated. After 24 h, cells were harvested and the expression levels of MAGI-1 and β-galactosidase were detected by Western blot analysis. (C) HeLa cells were transfected with 3 μg of FLAG-tagged wild-type or K499E mutant MAGI-1 expression plasmids. After 24 h, the cells were treated with MG-132 for an additional 3 h prior to harvesting as indicated. The expression levels of MAGI-1, p53, and β-galactosidase were assessed by Western blot analysis.
To investigate whether similar results could also be obtained in vivo, we compared the steady-state levels of FLAG-tagged wild-type and mutant MAGI-1 in HEK293 cells when expressed alone or in combination with increasing amounts of HPV-18 E6. The results are shown in Fig. 2B, and, as can be seen, in good agreement with the in vitro assay, M1wt was highly susceptible to E6-induced degradation, whereas the K499E mutant was significantly more resistant. We then wanted to know whether the K499E mutant was resistant to E6 targeting in the more physiologically relevant setting of cervical cancer-derived cells that express endogenous levels of E6 oncoprotein. To address this point, we expressed the FLAG-tagged MAGI-1 constructs in HeLa cells. Twenty-four hours after transfection, cells were left untreated or treated with the proteasome inhibitor MG-132 for an additional 3 h before harvesting, in order to determine whether differences in the MAGI-1 level of expression were due to proteasomal degradation. Total cell extracts were separated by SDS-PAGE, and the patterns of MAGI-1 and, for comparison, of p53 expression were analyzed by Western blotting. As can be seen in Fig. 2C, MG-132 treatment produced a strong recovery in the levels of p53 expression, confirming the efficient inhibition of the proteasome. In agreement with our degradation assays in HEK293 cells, the expression levels of the K499E mutant in HeLa were significantly higher than those of the wild-type protein, again reflecting their differential susceptibility to HPV-18 E6-mediated degradation. Proteasome protection produced an increase in the levels of expression of both M1wt and M1K499E, confirming that M1K499E is nonetheless still susceptible to HPV-18 E6-mediated degradation, although to a much lesser extent than the wild-type MAGI-1.
The K499E mutation perturbs the functionality of MAGI-1 PDZ1 but does not affect its subcellular localization.
As can be seen from Fig. 1A, the K499 residue maps to the MAGI-1 PDZ1 domain in its β-strand C, one of the structural components in close proximity to the substrate-binding groove of the PDZ domain (32, 40), indicating that the alteration of this domain could potentially perturb the functionality of the PDZ domain rather than selectively block the interaction with E6. Several PBM-containing cellular proteins have been identified as potential binding partners for MAGI-1 PDZ1, although only the Rho GEF NET1 has been confirmed in vivo (34, 41). To determine whether the K499E MAGI-1 mutation affected the overall functionality of the PDZ1 domain, we monitored the interaction of M1wt and M1K499E with NET1 by coimmunoprecipitation. To do this, HEK293 cells were transiently transfected with the FLAG-tagged M1wt and M1K499E constructs, either alone or in combination with MYC-tagged NET1. After 24 h, the cells were harvested and cell extracts were immunoprecipitated using anti-MYC antibody, and the coimmunoprecipitated M1wt and M1K499E were detected by Western blotting using anti-FLAG antibody (Fig. 3A). The results of these assays demonstrate that wild-type MAGI-1 specifically coimmunoprecipitates with NET1, confirming previous results (34). In contrast, the interaction between NET1 and the K499E mutant was significantly reduced, suggesting that this mutation does introduce structural changes into the substrate binding groove of the PDZ1 domain so that the ability of MAGI-1 PDZ1 to interact with the PBM of NET1 is perturbed.
FIG 3.

The K499E mutation affects the interaction of MAGI-1 with NET1 but does not alter its subcellular localization. (A) HEK293 cells were transfected with 3 μg of the FLAG-tagged MAGI-1 plasmids together with 5 μg of MYC-tagged mNET1, as indicated. After 24 h, cells were extracted and immunoprecipitated using anti-MYC antibody. NET1-bound MAGI-1 was then detected by Western blotting using anti-FLAG antibody. (B) HEK293 cells were transfected with 3 μg of wild-type or mutant MAGI-1 expression plasmids together with 5 μg of HA-tagged β-catenin. After 24 h, cell extracts were immunoprecipitated using anti-FLAG antibody-conjugated agarose beads, and MAGI-1-bound β-catenin was then detected by Western blotting using anti-HA antibody. The arrow indicates the position of the HA-tagged β-catenin in the top panel. (C) U2OS cells were transfected with the FLAG-tagged MAGI-1 constructs, and after 24 h the cells were fixed and incubated with anti-FLAG antibody to detect the MAGI-1 pattern of expression. Arrows indicate sites of MAGI-1 junctional accumulation.
Since the K499E mutation might affect the functionality of the PDZ1 domain toward some MAGI-1 binding proteins, we proceeded to investigate whether this might also impinge on the pattern of MAGI-1 subcellular localization, as the integrity of the MAGI-1 PDZ domains has been reported to be important for membrane targeting (33, 42). A previously identified membrane-bound interaction partner of MAGI-1 is β-catenin, and although the binding site for β-catenin is spatially segregated from the PDZ1 domain, as it interacts with the PDZ5 domain of MAGI-1 (33), we speculated that this association could be a marker to indirectly test the functionality of the K499E MAGI mutant. We repeated the coimmunoprecipitation experiments in HEK293 cells by overexpressing HA-tagged β-catenin alone or in combination with the FLAG-tagged M1wt or M1K499E constructs. After 24 h, cells were harvested and M1wt and M1K499E were immunoprecipitated by incubating cell extracts with anti-FLAG-conjugated agarose beads. Coimmunoprecipitated β-catenin was detected by Western blotting using anti-HA antibody, and the results of this assay are shown in Fig. 3B. As can be seen, β-catenin coimmunoprecipitated with similar efficiencies with both the wild-type and mutant MAGI-1, suggesting that the interaction between β-catenin and MAGI-1 is not affected by the K499E mutation and further suggesting that MAGI-1 membrane targeting is also unaffected. To confirm this, we also compared the expression pattern of wild-type and mutant MAGI-1 by immunofluorescence analysis. To do this, FLAG-tagged M1wt and M1K499E were transfected into U2OS cells, and, after 24 h, the cells were fixed and stained with anti-FLAG antibody. As can be seen from Fig. 3C, and in agreement with previous studies (33), wild-type MAGI-1 displayed a differential pattern of subcellular localization, with different pools of MAGI-1 detectable in the nucleus as well as in the cytoplasm of transfected cells, whereas a prominent membrane staining was also apparent in cells forming intercellular junctions, where it has also been shown previously to colocalize with β-catenin (33). In good agreement with the β-catenin coimmunoprecipitation experiments, the subcellular distribution of M1K499E was very similar to the wild-type protein, confirming that the K499E mutation does not adversely affect the subcellular distribution of MAGI-1.
Taken together, these data suggest that the K499E mutation, which blocks HPV-16 and -18 E6 recognition, is also likely to affect the ability of MAGI-1 to interact with its cellular partners that also recognize the PDZ1 domain. However, the K499E mutation does not appear to affect the subcellular localization of MAGI-1.
The K499E mutation potentiates the ability of MAGI-1 to establish tight junctions in HeLa cells.
MAGI-1 has been shown to have an important role in the establishment of TJs, promoting the recruitment of TJ-associated proteins such as ZO-1 and occludin (41), and recently the silencing of HPV-18 E6 in HeLa cells was shown to promote the junctional accumulation of ZO-1 through MAGI-1 (26). These observations prompted us to investigate whether the expression of MAGI-1 was sufficient to promote the junctional recruitment of TJ-associated proteins in HeLa cells and to define whether the K499E mutant could potentiate this activity of MAGI-1. To do this, we decided to monitor the expression pattern of the TJ-associated proteins ZO-1 and PAR3 (43, 44). HeLa cells were grown on glass coverslips and transfected with the FLAG-tagged MAGI-1 constructs; 24 h after transfection, cells were fixed and the expression patterns of MAGI-1, ZO-1, and PAR3 were analyzed by confocal microscopy. In order to evaluate whether the expression of M1wt and M1K499E could confer an advantage in terms of junctional assembly, we counted the cells in multiple confocal fields of transfected and untransfected HeLa cells until at least 100 FLAG-positive cells were analyzed. Within each sample, HeLa cells were divided into subpopulations based on their positivity for the FLAG staining (MAGI-1 transfected or untransfected) and on their ability to form ZO-1 and PAR3-positive cellular junctions. As can be seen in Fig. 4A, only a few HeLa cells display junctional ZO-1 staining, and a similar pattern of staining was also observed for PAR3 (Fig. 4B), suggesting that HeLa cells are largely unable to form intact cellular junctions. This was confirmed by cell counts: within the untransfected cell population, about 10 to 20% of HeLa cells displayed ZO-1- and PAR3-positive junctional staining (Fig. 5A). In contrast, the expression of wild-type or mutant MAGI-1 significantly increased the junctional recruitment of both ZO-1 and PAR3 with similar efficiency (Fig. 4 and 5A [P = 0.0009]). Furthermore, compared with the wild-type MAGI-1, the K499E mutant was present in a much higher percentage of HeLa cells (Fig. 4A and B and 5B [P < 0.0001]), which is consistent with its increased resistance to E6-mediated degradation. This increased proportion of K499E MAGI-1-expressing cells was also reflected in the fact that when ZO-1-positive (P < 0.001) and PAR3-positive (P < 0.0001) cells were scored as a percentage of the total cell population, then HeLa cells transfected with the mutant MAGI-1 showed a significant increase over the wild-type MAGI-1-transfected cells (Fig. 4A and B and 5C). However, there did not appear to be any significant correlation between the strength of MAGI-1 staining and the level of ZO-1/PAR3 junctional recruitment. This is most likely a reflection of the fact that junctional recruitment of ZO-1 and PAR3 can be continuous between some cells, indicating fully established TJs, while in other cases it has a more bead-like appearance, indicating TJs that are still in the process of assembly.
FIG 4.
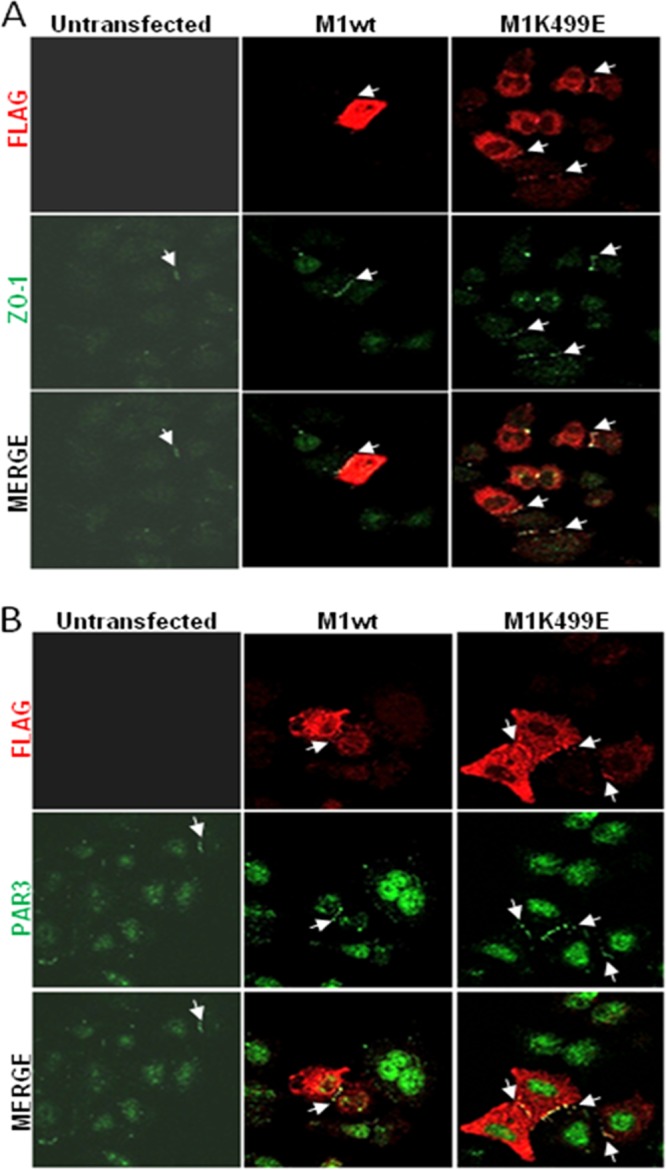
The expression of wild-type and mutant MAGI-1 promotes junctional assembly in HeLa cells. (A) HeLa cells were seeded on glass coverslips and transfected either with wild-type or mutant MAGI-1. After 24 h, the cells were fixed and incubated with anti-FLAG and anti-ZO-1 antibodies and counterstained with rhodamine-conjugated (FLAG) and fluorescein-conjugated (ZO-1) secondary antibodies. Confocal images were taken at 480- and 510-nm wavelengths. (B) Cells were seeded and transfected as described for panel A. After fixation, cells were incubated with anti-FLAG and anti-PAR3 antibodies and counterstained with rhodamine-conjugated (FLAG) and fluorescein-conjugated (PAR3) secondary antibodies. Confocal images were taken at 480- and 510-nm wavelengths. Arrows indicate sites of ZO-1 and PAR3 junctional accumulation.
FIG 5.

The resistance of MAGI-1 K499E to E6-mediated degradation correlates with an increased junctional assembly in HeLa cells. The total numbers of HeLa cells showing M1wt or K499E mutant-positive staining were quantified within the total cell population visualized by immunofluorescence. Represented are the mean values calculated from multiple independent experiments in which at least 100 wild-type and mutant MAGI-1-positive cells were counted. (A) The number of ZO-1 and PAR3-positive junctions formed in untransfected and in wild-type and mutant MAGI-1-transfected cells was calculated as the percentage of cells displaying positive ZO-1 and PAR3 junctional staining in each subpopulation (one-way analysis of variance [ANOVA]). (B) The total population of wild-type and mutant MAGI-1-expressing cells was calculated as the percentage of FLAG-positive cells present in each sample relative to the total population of untransfected cells (P < 0.0001; unpaired two-sample t test). (C) Percentage of MAGI-1-positive cells displaying ZO-1-positive (P = 0.0006) and PAR3-positive (P < 0.0001) junctional staining as a percentage of the total population of cells (unpaired two-sample t test). Diagrams represent mean values together with standard deviation, as calculated from multiple independent experiments. The corresponding P values are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
These data suggest that wild-type and mutant MAGI-1 proteins display comparable efficiencies in recruiting ZO-1 and PAR3 to cell contact sites. However, the resistance of the K499E mutant to E6-mediated degradation increases the number of HeLa cells expressing this mutant, which in turn increases the numbers of cells with intact cell-cell junctions, confirming that MAGI-1 expression enhances the ability of HeLa cells to form junctional complexes.
A well-known effect associated with the establishment of junctional complexes is the inhibition of cell proliferation (45–47). Previous studies indicated that both ZO-1 and PAR3 can inhibit cell proliferation when localized at cell contact sites by modulating the activity of multiple proteins implicated in G1/S cell cycle transition (47, 48). Therefore, having shown that the expression of K499E mutant MAGI-1 can increase the junctional recruitment of ZO-1 and PAR3, we were interested in determining whether this could also affect cell proliferation in HPV-18-positive HeLa cells and HPV-16-positive SiHa cells. To do this, we labeled proliferating cells with the thymidine analogue 5-ethynyl-2′-deoxyuridine (EdU), which allows the visualization of proliferating cells while not affecting the overall structural integrity of the cell (49). HeLa and SiHa cells were grown on glass coverslips and transfected with the FLAG-tagged M1wt and M1K499E constructs. Twenty-four hours posttransfection, and prior to fixation, the cells were pulsed with EdU for an additional 2 h, and the pattern of MAGI-1 and ZO-1 expression and the proportion of EdU-positive cells were visualized by confocal microscopy. As can be seen in Fig. 6A and D, a high proportion of cells display positive nuclear EdU staining. The expression of wild-type MAGI-1 led to a decrease in the EdU staining, with fewer than 5% of HeLa cells staining positive for both MAGI-1 and EdU, while over 70% of the nontransfected cells stained positive for EdU (Fig. 6B). In SiHa cells, around 40% of nontransfected cells were positive for EdU, and this was reduced to around 1% in the wild-type MAGI-1-expressing cells (Fig. 6D). In contrast, the K499E mutant did not have such a dramatic effect upon cell proliferation as the wild-type MAGI-1, although the number of EdU-positive cells was still much lower than the number of nontransfected cells, with approximately 20% of K499E-expressing HeLa cells and 7% of K499E-expressing SiHa cells retaining EdU positivity (Fig. 6B and D). This suggests that the integrity of the MAGI-1 PDZ1 domain contributes toward the ability of MAGI-1 to inhibit cell proliferation in HPV-positive cells. However, when EdU positivity is scored as a percentage of the total cell population, the greater proportion of cells expressing K499E is also reflected in a significant increase in the overall population of EdU-negative cells (P < 0.001), with approximately 15% of the total HeLa cell population and 21% of the SiHa cell population being EdU negative in the wild-type MAGI-1-transfected cells, while this increases to over 30% in the K499E-transfected HeLa and SiHa cells (Fig. 6C and D). We also compared the EdU positivity with the ability of MAGI-1 to induce ZO-1 junctional accumulation, and it can be seen from Fig. 6A that wild-type MAGI-1 both inhibited EdU positivity and stimulated ZO-1 junctional accumulation; however, the K499E mutant, although stimulating ZO-1 junctional recruitment, still showed evidence of EdU positivity in some cells, suggesting that the capacity of MAGI-1 to induce ZO-1 junctional recruitment in HeLa cells is separate from its ability to inhibit EdU incorporation.
FIG 6.

MAGI-1 inhibits proliferation of HeLa and SiHa cells. (A) HeLa cells were seeded on glass coverslips and transfected either with FLAG-tagged wild-type or mutant MAGI-1 K499E expression plasmids. After 24 h, and prior to fixation, cells were incubated with EdU for an additional 2 h in order to allow its incorporation by proliferating cells. After fixation, cells were incubated with anti-FLAG and anti-ZO-1 antibodies and counterstained with rhodamine-conjugated (FLAG) and fluorescein-conjugated (ZO-1) secondary antibodies. After incubation with the indicated antibodies, cells were processed for the detection of EdU-labeled DNA (green nuclei). Confocal images were taken at 480- and 510-nm wavelengths. Arrows indicate sites of MAGI-1 junctional accumulation. (B) The percentage of EdU-positive cells was quantified by direct counting of untransfected and wild-type and K499E mutant MAGI-1-transfected cells displaying positive nuclear EdU staining within each subpopulation. Shown are the mean values with standard deviations calculated from multiple independent experiments (one-way ANOVA). (C) Cells were counted as described for panel B, and shown is the percentage of MAGI-1-positive cells displaying EdU-negative staining relative to the total population of cells in each sample (P = 0.0008; unpaired two-sample t test). (D) SiHa cells were processed as described for panel A, where rhodamine shows MAGI-1-positive cells and EdU-labeled DNA (green nuclei) in the top panels. The left-hand-side histogram represents the total population of wild-type and mutant MAGI-1-expressing cells calculated as the percentage of FLAG-positive cells (∼100 cells per sample) present in each sample relative to the total population of untransfected cells (P = 0.002; unpaired two-sample t test). For the central and right-hand-side histograms, SiHa cells were counted as described for panels B and C, and shown are the percentage of untransfected and wild-type and K499E mutant MAGI-1-transfected cells displaying positive nuclear EdU staining within each subpopulation (one-way ANOVA) and the percentage of MAGI-1-positive cells displaying EdU-negative staining relative to the total population of cells in each sample (P = 0,001; unpaired two-sample t test). The corresponding P values are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Taken together, these data suggest that MAGI-1 is a negative regulator of cell proliferation and that its expression is capable of reducing the proportion of HPV-16- and HPV-18-positive proliferating cells. However, this regulation of proliferative potential in HPV-positive cells appears to be independent of MAGI-1's ability to restore TJs, since it seems to depend, at least in part, upon an as-yet-unidentified function associated with the MAGI-1 PDZ1 domain.
To assess whether MAGI-1 can inhibit cell proliferation in HPV-negative cells, we repeated the EdU staining following transfection of wild-type and K499E MAGI-1 into H1299 cells (p53-negative non-small cell lung carcinoma derived), HaCaT keratinocytes, and HPV-negative C33I cervical carcinoma-derived cells. The results in Fig. 7 demonstrate that, in comparison with their effects in HPV-positive cells, the two MAGI-1 proteins produced much weaker effects on cell proliferation in H1299 (Fig. 7A) and HaCaT and C33I (Fig. 7E), where both the wt and mutant MAGI-1 reduced the percentage of EdU-positive cells from approximately 70% in the nontransfected H1299 cells to approximately 50% in the MAGI-1 expressing cells (Fig. 7B), from over 50% in nontransfected HaCaT cells to around 40% in the MAGI-1-expressing cells (Fig. 7E) and from 70% in the nontransfected C33I cells to approximately 50% in the MAGI-1-expressing cells (Fig. 7E). Consistent with the lack of E6 in H1299 and HaCaT cells, both MAGI-1 proteins were expressed at similar levels, and when the EdU positivity was scored across the total cell population there was no significant difference between the MAGI-1 proteins (Fig. 7C, D, and E). Similar results were also obtained in the HPV-negative U2OS and HT1080 cells (data not shown).
FIG 7.

Analysis of MAGI-1 effects on cell proliferation in HPV-negative cells. (A) H1299 cells were seeded on glass coverslips, and transfection was carried out as described for Fig. 6. After 24 h, and prior to fixation, cells were incubated with EdU for an additional 2 h. After fixation, cells were incubated with anti-FLAG antibody and counterstained with rhodamine-conjugated secondary antibody. After incubation with the indicated antibodies, cells were processed for the detection of EdU-labeled DNA (green nuclei). Confocal images were taken at 480- and 510-nm wavelengths. (B) The percentage of EdU-positive cells was quantified by direct cell count of untransfected and wild-type and K499E mutant MAGI-1-transfected cells displaying positive nuclear EdU staining within each subpopulation. Shown are the mean values with standard deviations calculated from multiple independent experiments (one-way ANOVA). (C) The total population of wild-type and mutant MAGI-1-expressing cells was calculated as the percentage of FLAG-positive cells (∼100 cells per sample) present in each sample relative to the total population of untransfected cells (P = 0.56; unpaired two-sample t test). (D) Cells were counted as described for panel B, and shown is the percentage of MAGI-1-positive cells displaying EdU-negative staining relative to the total population of cells counted in each sample. Shown are the mean values with standard deviations calculated from multiple independent experiments (P = 0.48; unpaired two-sample t test). (E) HaCaT and C33I cells were processed as described for panel A. Immunofluorescent images are of HaCaT cells, with white arrows indicating sites of MAGI-1 and ZO-1 junctional accumulation. The top left and right histograms show the percentage of EdU-positive HaCaT and C33I cells quantified by direct cell count of untransfected and wild-type and K499E mutant MAGI-1-transfected cells displaying positive nuclear EdU staining within each subpopulation (one-way ANOVA). The bottom histograms show the total population of wild-type and mutant MAGI-1-expressing HaCaT cells calculated as described for panel C (left) and the percentage of MAGI-1-positive HaCaT cells displaying EdU-negative staining relative to the total population of untransfected cells counted in each sample (right). Each panel represents the mean values and standard deviations calculated from three independent experiments. The corresponding P values are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; N.S., not significant.
MAGI-1 induces apoptosis in HeLa cells.
The examination of HeLa cells following transfection of the MAGI-1 expression plasmids suggested a possible involvement of this protein in the regulation of apoptosis, since a proportion of the MAGI-1-expressing HeLa cells had morphological features resembling those of cells undergoing apoptosis, including blebbing and rounding up of the cells (50). Therefore, to determine whether the expression of wild-type and K499E mutant MAGI-1 could promote apoptosis of HeLa cells, we monitored their pattern of DNA fragmentation by performing terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assays. We also included HT1080 and H1299 cells in the analysis, to determine whether MAGI-1 could also induce apoptosis in HPV-negative cells. Cells were seeded on glass coverslips and transfected either with wild-type or K499E MAGI-1 constructs. Since the induction of DNA fragmentation is a rather late event during induction of apoptosis (51), TUNEL assays were performed 48 h after transfection. The cells were then fixed, and the pattern of wild-type and K499E mutant MAGI-1 expression as well as the number of cells positive for the TUNEL reaction were assessed by immunofluorescence and confocal microscopy. Results are shown in Fig. 8 and, as can be seen, untransfected HeLa (Fig. 8A) and to a much lesser extent HT1080 (Fig. 8B) cells showed low levels of apoptosis, with approximately 10% of control transfected HeLa cells showing TUNEL staining (Fig. 9A). The expression of wild-type and mutant MAGI-1 led to the appearance of MAGI-1/TUNEL-positive cell subpopulations in HeLa cells (Fig. 8A) in 40 to 50% of the MAGI-1-positive cells (Fig. 9A) but not in HT1080 (Fig. 8B) or H1299 cells (data not shown). While there was little difference in the induction of apoptosis between MAGI-1 wt and the K499E mutant (Fig. 9A), the increased proportion of cells expressing E6-resistant K499E (P < 0.001) (Fig. 9B) again produced a significant effect upon the total cell population (P < 0.001) (Fig. 9C). Therefore, these data indicate that the increased resistance of K499E mutant MAGI-1 to E6-mediated degradation also correlates with an increased induction of apoptosis.
FIG 8.
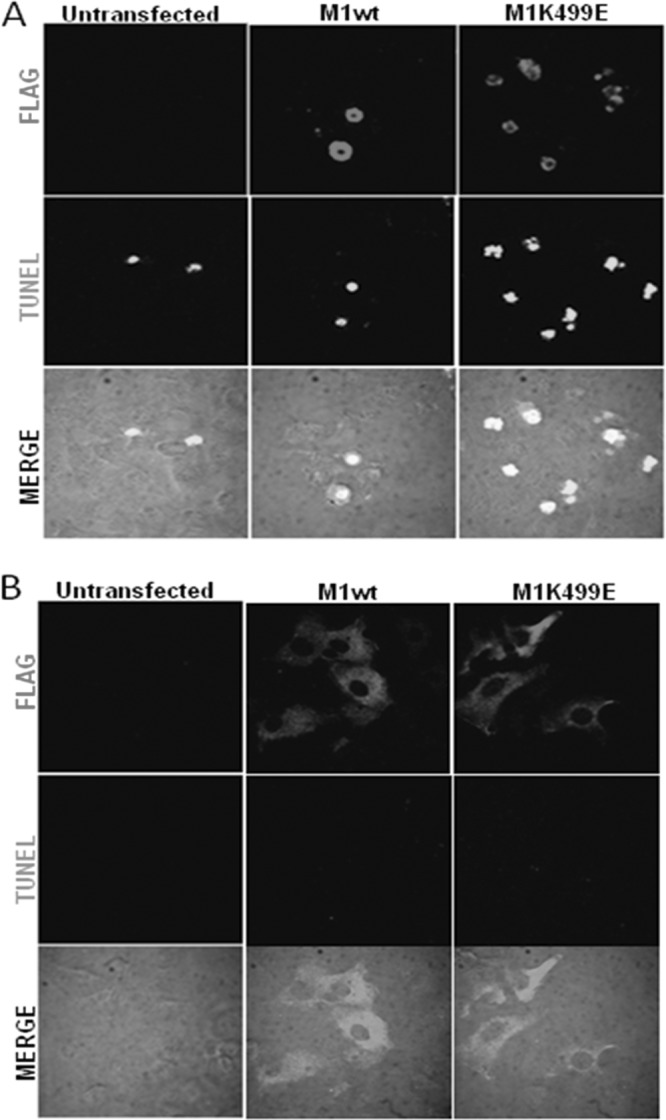
The expression of MAGI-1 induces apoptosis in HeLa cells but not in HT1080 cells. (A) HeLa cells were seeded on glass coverslips and transfected either with wild-type or mutant K499E MAGI-1 expression plasmids. After 48 h, the cells were fixed and incubated with the anti-FLAG antibody and counterstained with rhodamine-conjugated secondary antibodies. After incubation with the indicated antibodies, cells were processed for the identification of TUNEL-positive cells using the in situ cell death detection kit according to the manufacturer's instructions. Confocal images were taken at 480- and 510-nm wavelengths. (B) HT1080 cells were seeded on glass coverslips, and the immunodetection of MAGI-1 and the TUNEL assay were performed as described for panel A.
FIG 9.

Quantitation of cells exhibiting signs of apoptosis following expression of MAGI-1. (A) The percentage of TUNEL-positive HeLa cells was quantified by direct cell count of untransfected, wild-type, or K499E mutant MAGI-1-transfected cells displaying positive TUNEL staining within each subpopulation. Shown are the mean values with standard deviations calculated from multiple independent experiments. (B) The total population of wild-type and mutant MAGI-1-expressing cells was calculated as the percentage of FLAG-positive cells (∼100 cells per sample) present in each sample relative to the total population of untransfected cells. (C) Cells were counted as described for panels A and B, and shown is the percentage of MAGI-1-positive cells displaying TUNEL-positive staining relative to the total population of untransfected cells counted in each sample. Shown are the mean values with standard deviations calculated from multiple independent experiments. The corresponding P values are as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
DISCUSSION
Previous studies had highlighted the potential of the MAGI-1 K499 residue to be critical for the ability of HPV-16 and HPV-18 E6 to recognize the MAGI-1 PDZ1 domain (32). By generating the K499E mutation in the context of the full-length MAGI-1 protein, we were able to significantly perturb the recognition of MAGI-1 by both HPV-16 and HPV-18 E6. This, in turn, resulted in a reduced ability of E6 to induce the proteasome-mediated degradation of MAGI-1, both in vitro and in vivo. Most importantly, this allowed us to reexpress the MAGI-1 protein in HPV-positive cells, under conditions where the wild-type MAGI-1 protein would normally be degraded by E6. These studies now provide compelling evidence for the functional relevance of the E6–MAGI-1 interaction in the regulation of cell-cell contact, proliferation, and apoptosis.
While previous studies had suggested that the K499E mutant could specifically inhibit HPV-16 and HPV-18 E6 recognition of the MAGI-1 PDZ1 domain (32), it was nonetheless important to ascertain its effects upon the ability of MAGI-1 to interact with its known cellular interacting partners. One such binding partner is NET1 (34), which is an RhoA-specific guanine nucleotide exchange factor involved in a number of cancer-associated biological processes, including cell migration, proliferation, and matrix invasion (52, 53). By performing coimmunoprecipitation assays, we confirmed that NET1 is a binding partner for wild-type MAGI-1 in vivo and also that the K499E mutation strongly affects the interaction of MAGI-1 with NET1. This suggests that this mutation has a general effect upon the functional integrity of the MAGI-1 PDZ1 domain. However, this does not seem to affect MAGI-1's ability to interact with other PBM-containing targets, such as β-catenin, nor to adversely affect its membrane localization.
The generation of the K499E MAGI-1 thus provided a valuable tool for exploring its role in the context of HPV-transformed cells, since it allowed us to evaluate the effects of reintroducing MAGI-1 protein into HPV-positive cells without the need to silence E6 and E7 expression and thereby generate a range of potentially unrelated effects. By transiently expressing wild-type and mutant MAGI-1 in HeLa cells, we confirmed that MAGI-1 reexpression alone is sufficient to drive TJ reassembly in these cells, which was assessed by monitoring the expression of ZO-1 and another TJ marker, PAR3. However, both ZO-1 and PAR3 have a dynamic pattern of junctional localization and have been shown to colocalize with AJ components during the induction of primordial AJ structures following initial cell-cell contact (54, 55). Therefore, at this stage, we cannot rule out the possibility that the ZO-1- and PAR3-positive junctional structures observed in MAGI-1-expressing cells are indeed primordial AJs. Nevertheless, these data demonstrate that MAGI-1 directly participates in the establishment of macromolecular complexes that localize at the TJ plaque. PAR3 is part of the PAR complex which localizes and controls the maturation of TJs, and PAR3 is one of the prime regulators of TJ formation. Thus, the fact that MAGI-1 enhances PAR3 junctional localization strongly suggests the involvement of MAGI-1 in the establishment of cell polarity by inducing the formation of apical junctional structures (56). These results demonstrate that the loss of TJs in HPV-18-positive HeLa cells is a direct consequence of the ability of E6 to direct the degradation of MAGI-1 and might provide an explanation as to why this protein is targeted by the virus during the life cycle and malignant progression.
TJs play an important role in differentiation where their correct assembly promotes the exit from the cell cycle and contributes to keratinocyte differentiation (46, 57, 58). Loss of TJs can therefore be expected to delay the differentiation process. In addition, TJs directly participate in the regulation of cell proliferation by modulating signaling cascades such as MAPK, PKB/Akt, and RhoA signaling (35, 47, 59). Interestingly, both ZO-1 and PAR3 are believed to be involved in controlling cell proliferation; while ZO-1 binds and sequesters the transcription factor ZONAB/DbpA at the cell membrane (45), PAR3 is believed to potentially regulate cell proliferation through the modulation of the p53-binding partner ASPP2 (47). In our cell proliferation assays, we found that the expression of wild-type MAGI-1 had dramatic effects upon the proliferative potential of both HeLa and SiHa cells. Unexpectedly, the effects on cell proliferation were somewhat independent of the junctional recruitment of ZO-1 and PAR3, since the K499E mutant was as effective as wild-type MAGI-1 in promoting TJ formation but somewhat weaker at inhibiting cell proliferation in HPV-positive cells. An intriguing possibility is that MAGI-1 might regulate cell proliferation, at least in part, through the modulation of RhoA activity. This is supported by the fact that MAGI-1 interacts with the RhoA-specific activator NET1 through the PDZ1 domain and that MAGI-1 had already been shown to modulate the activity of another small GTPase, Rap1, with consequent stabilization of vascular-endothelial cell adhesion structures (60, 61). In addition, both RhoA and NET1 have already been described as positive regulators of cell cycle progression (53, 62), and the decreased ability of the K499E mutant MAGI-1 to inhibit cell proliferation would then be consistent with its reduced capacity to interact with NET1. It is also interesting to note that recent studies have shown that NET1 is localized mainly in the nucleus of epithelial cells, where its activity is required to maintain RhoA in an active GTP-bound state (38, 63). Although how MAGI-1 can modulate NET1 activity is still unclear, these data suggest one biological function for nuclear MAGI-1 in regulating cell proliferation through NET1/RhoA and provides an explanation as to why such nuclear pools of MAGI-1 might be targeted by E6 (26).
Our data also suggest a new function for MAGI-1 in the control of apoptosis in HPV-positive cells. We hypothesized a possible involvement of MAGI-1 in the regulation of apoptosis, since MAGI-1-positive HeLa cells often display a morphology typically associated with apoptosis. To investigate this, we performed TUNEL assays on HeLa cells transiently transfected either with wild-type or K499E MAGI-1 expression plasmids. Our data suggest that both the wild-type and mutant MAGI-1 can promote apoptosis in HeLa cells. It is also interesting to note that the K499E mutant MAGI-1 displayed an efficiency of apoptosis induction that was comparable to that of the wild-type protein. Therefore, this indicates that the regulation of apoptosis by MAGI-1 is not dependent upon functions associated with the PDZ1 domain. Furthermore, as for the induction of junctional assembly, the increased resistance of the K499E mutant MAGI-1 to E6-mediated degradation also correlated with an increased subpopulation of HeLa cells undergoing apoptosis. In contrast, the effects of MAGI-1 on cell proliferation and apoptosis were much less pronounced in a number of HPV-negative cell lines.
Since MAGI-1 is the most strongly bound PDZ domain-containing target of the high-risk HPV E6 oncoproteins, one would expect it to be of significant biological relevance to the virus. Our studies demonstrate that the E6 interaction with MAGI-1 has diverse biological consequences for the HPV-infected cell. Perturbation of TJ assembly and the consequent alteration of polarity and cell-cell contact is one such chain of consequences. However, independent of this activity, E6 would also appear to perturb MAGI-1 function as a means of increasing cell proliferation and avoiding apoptosis. Understanding how MAGI-1 contributes to the regulation of cell proliferation and apoptosis is now a subject for further analysis.
ACKNOWLEDGMENTS
We are very grateful to Miranda Thomas for providing valuable comments on the manuscript and to Sally Roberts for insightful discussions and advice.
This work was supported in part by research grants from the Associazione Italiana per la Ricerca sul Cancro and the Wellcome Trust.
Footnotes
Published ahead of print 2 April 2014
REFERENCES
- 1.de Villiers E-M, Fauquet C, Broker TR, Bernard H-U, zur Hausen H. 2004. Classification of papillomaviruses. Virology 324:17–27. 10.1016/j.virol.2004.03.033 [DOI] [PubMed] [Google Scholar]
- 2.Bernard H-U, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers E-M. 2010. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401:70–79. 10.1016/j.virol.2010.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.zur Hausen H. 2009. Papillomaviruses in the causation of human cancers—a brief historical account. Virology 384:260–265. 10.1016/j.virol.2008.11.046 [DOI] [PubMed] [Google Scholar]
- 4.Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. 1989. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 8:3905–3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griep AE, Herber R, Jeon S, Lohse JK, Dubielzig RR, Lambert PF. 1993. Tumorigenicity by human papillomavirus type 16 E6 and E7 in transgenic mice correlates with alterations in epithelial cell growth and differentiation. J. Virol. 67:1373–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambert PF, Pan H, Pitot HC, Liem A, Jackson M, Griep AE. 1993. Epidermal cancer associated with expression of human papillomavirus type 16 E6 and E7 oncogenes in the skin of transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 90:5583–5587. 10.1073/pnas.90.12.5583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshinouchi M, Yamada T, Kizaki M, Fen J, Koseki T, Ikeda Y, Nishihara T, Yamato K. 2003. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol. Ther. 8:762–768. 10.1016/j.ymthe.2003.08.004 [DOI] [PubMed] [Google Scholar]
- 8.Jonson AL, Rogers LM, Ramakrishnan S, Downs LS. 2008. Gene silencing with siRNA targeting E6/E7 as a therapeutic intervention in a mouse model of cervical cancer. Gynecol. Oncol. 111:356–364. 10.1016/j.ygyno.2008.06.033 [DOI] [PubMed] [Google Scholar]
- 9.Qi Z, Xu X, Zhang B, Li Y, Liu J, Chen S, Chen G, Huo X. 2010. Effect of simultaneous silencing of HPV-18 E6 and E7 on inducing apoptosis in HeLa cells. Biochem. Cell Biol. 88:697–704. 10.1139/O10-005 [DOI] [PubMed] [Google Scholar]
- 10.Khairuddin N, Gantier MP, Blake SJ, Wu SY, Behlke MA, Williams BR, McMillan NA. 2012. siRNA-induced immunostimulation through TLR7 promotes antitumoral activity against HPV-driven tumors in vivo. Immunol. Cell Biol. 90:187–196. 10.1038/icb.2011.19 [DOI] [PubMed] [Google Scholar]
- 11.Werness BA, Levine AJ, Howley PM. 1990. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 248:76–79. 10.1126/science.2157286 [DOI] [PubMed] [Google Scholar]
- 12.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. 10.1016/0092-8674(90)90409-8 [DOI] [PubMed] [Google Scholar]
- 13.Dyson N, Howley PM, Münger K, Harlow E. 1989. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243:934–937. 10.1126/science.2537532 [DOI] [PubMed] [Google Scholar]
- 14.Boyer SN, Wazer DE, Band V. 1996. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 56:4620–4624 [PubMed] [Google Scholar]
- 15.Helt A-M, Funk JO, Galloway DA. 2002. Inactivation of both the retinoblastoma tumor suppressor and p21 by the human papillomavirus type 16 E7 oncoprotein is necessary to inhibit cell cycle arrest in human epithelial cells. J. Virol. 76:10559–10568. 10.1128/JVI.76.20.10559-10568.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klingelhutz AJ, Foster SA, Mcdougall JK. 1996. Telomerase activation by the E6 gene-product of human papillomavirus type-16. Nature 380:79–82. 10.1038/380079a0 [DOI] [PubMed] [Google Scholar]
- 17.Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. 1997. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. U. S. A. 94:11612–11616. 10.1073/pnas.94.21.11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardiol D, Kühne C, Glaunsinger B, Lee SS, Javier R, Banks L. 1999. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 18:5487–5496. 10.1038/sj.onc.1202920 [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa S, Huibregtse JM. 2000. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 20:8244–8253. 10.1128/MCB.20.21.8244-8253.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R. 2000. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 19:5270–5280. 10.1038/sj.onc.1203906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas M, Laura R, Hepner K, Guccione E, Sawyers C, Lasky L, Banks L. 2002. Oncogenic human papillomavirus E6 proteins target the MAGI-2 and MAGI-3 proteins for degradation. Oncogene 21:5088–5096. 10.1038/sj.onc.1205668 [DOI] [PubMed] [Google Scholar]
- 22.Lee C, Laimins LA. 2004. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 78:12366–12377. 10.1128/JVI.78.22.12366-12377.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delury CP, Marsh EK, James CD, Boon SS, Banks L, Knight GL, Roberts S. 2013. The role of protein kinase A regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. J. Virol. 87:9463–9472. 10.1128/JVI.01234-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watson RA, Thomas M, Banks L, Roberts S. 2003. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J. Cell Sci. 116:4925–4934. 10.1242/jcs.00809 [DOI] [PubMed] [Google Scholar]
- 25.Shai A, Brake T, Somoza C, Lambert PF. 2007. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res. 67:1626–1635. 10.1158/0008-5472.CAN-06-3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kranjec C, Banks L. 2011. A systematic analysis of human papillomavirus (HPV) E6 PDZ substrates identifies MAGI-1 as a major target of HPV type 16 (HPV-16) and HPV-18 whose loss accompanies disruption of tight junctions. J. Virol. 85:1757–1764. 10.1128/JVI.01756-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woods DF, Bryant PJ. 1991. The discs-large tumor suppressor gene of Drosophila encodes a guanylate kinase homolog localized at septate junctions. Cell 66:451–464. 10.1016/0092-8674(81)90009-X [DOI] [PubMed] [Google Scholar]
- 28.Anderson JM. 1996. Cell signalling: MAGUK magic. Curr. Biol. 6:382–384. 10.1016/S0960-9822(02)00501-8 [DOI] [PubMed] [Google Scholar]
- 29.Dobrosotskaya I, Guy RK, James GL. 1997. MAGI-1, a membrane-associated guanylate kinase with a unique arrangement of protein-protein interaction domains. J. Biol. Chem. 272:31589–31597. 10.1074/jbc.272.50.31589 [DOI] [PubMed] [Google Scholar]
- 30.Thomas M, Glaunsinger B, Pim D, Javier R, Banks L. 2001. HPV E6 and MAGUK protein interactions: determination of the molecular basis for specific protein recognition and degradation. Oncogene 20:5431–5439. 10.1038/sj.onc.1204719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas M, Massimi P, Navarro C, Borg J-P, Banks L. 2005. The hScrib/Dlg apico-basal control complex is differentially targeted by HPV-16 and HPV-18 E6 proteins. Oncogene 24:6222–6230. 10.1038/sj.onc.1208757 [DOI] [PubMed] [Google Scholar]
- 32.Fournane S, Charbonnier S, Chapelle A, Kieffer B, Orfanoudakis G, Travé G, Masson M, Nominé Y. 2011. Surface plasmon resonance analysis of the binding of high-risk mucosal HPV E6 oncoproteins to the PDZ1 domain of the tight junction protein MAGI-1. J. Mol. Recognit. 24:511–523. 10.1002/jmr.1056 [DOI] [PubMed] [Google Scholar]
- 33.Dobrosotskaya IY, James GL. 2000. MAGI-1 interacts with beta-catenin and is associated with cell-cell adhesion structures. Biochem. Biophys. Res. Commun. 270:903–909. 10.1006/bbrc.2000.2471 [DOI] [PubMed] [Google Scholar]
- 34.Dobrosotskaya IY. 2001. Identification of mNET1 as a candidate ligand for the first PDZ domain of MAGI-1. Biochem. Biophys. Res. Commun. 283:969–975. 10.1006/bbrc.2001.4880 [DOI] [PubMed] [Google Scholar]
- 35.Kotelevets L, van Hengel J, Bruyneel E, Mareel M, van Roy F, Chastre E. 2005. Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J. 19:115–117. 10.1096/fj.04-1942fje [DOI] [PubMed] [Google Scholar]
- 36.Pim D, Storey A, Thomas M, Massimi P, Banks L. 1994. Mutational analysis of HPV-18 E6 identifies domains required for p53 degradation in vitro, abolition of p53 transactivation in vivo and immortalisation of primary BMK cells. Oncogene 9:1869–1876 [PubMed] [Google Scholar]
- 37.Pim D, Thomas M, Javier R, Gardiol D, Banks L. 2000. HPV E6 targeted degradation of the discs large protein: evidence for the involvement of a novel ubiquitin ligase. Oncogene 19:719–725. 10.1038/sj.onc.1203374 [DOI] [PubMed] [Google Scholar]
- 38.García-Mata R, Dubash AD, Sharek L, Carr HS, Frost JA, Burridge K. 2007. The nuclear RhoA exchange factor Net1 interacts with proteins of the Dlg family, affects their localization, and influences their tumor suppressor activity. Mol. Cell. Biol. 27:8683–8697. 10.1128/MCB.00157-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matlashewski G, Schneider J, Banks L, Jones N, Murray A, Crawford L. 1987. Human papillomavirus type 16 DNA cooperates with activated ras in transforming primary cells. EMBO J. 6:1741–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. 1996. Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell 85:1067–1076. 10.1016/S0092-8674(00)81307-0 [DOI] [PubMed] [Google Scholar]
- 41.Hirabayashi S, Tajima M, Yao I, Nishimura W, Mori H, Hata Y. 2003. JAM4, a junctional cell adhesion molecule interacting with a tight junction protein, MAGI-1. Mol. Cell. Biol. 23:4267–4282. 10.1128/MCB.23.12.4267-4282.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laura RP, Ross S, Koeppen H, Lasky LA. 2002. MAGI-1: a widely expressed, alternatively spliced tight junction protein. Exp. Cell Res. 275:155–170. 10.1006/excr.2002.5475 [DOI] [PubMed] [Google Scholar]
- 43.Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. 1986. Identification of ZO-1: a high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J. Cell Biol. 103:755–766. 10.1083/jcb.103.3.755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Izumi Y, Hirose T, Tamai Y, Hirai S, Nagashima Y, Fujimoto T, Tabuse Y, Kemphues KJ, Ohno S. 1998. An atypical PKC directly associates and colocalizes at the epithelial tight junction with ASIP, a mammalian homologue of Caenorhabditis elegans polarity protein PAR-3. J. Cell Biol. 143:95–106. 10.1083/jcb.143.1.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balda MS, Garrett MD, Matter K. 2003. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 160:423–432. 10.1083/jcb.200210020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aijaz S, D'Atri F, Citi S, Balda MS, Matter K. 2005. Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev. Cell 8:777–786. 10.1016/j.devcel.2005.03.003 [DOI] [PubMed] [Google Scholar]
- 47.Sottocornola R, Royer C, Vives V, Tordella L, Zhong S, Wang Y, Ratnayaka I, Shipman M, Cheung A, Gaston-Massuet C, Ferretti P, Molnár Z, Lu X. 2010. ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors during CNS development. Dev. Cell 19:126–137. 10.1016/j.devcel.2010.06.003 [DOI] [PubMed] [Google Scholar]
- 48.Balda MS, Matter K. 2000. Transmembrane proteins of tight junctions. Sem. Cell Dev. Biol. 11:281–289. 10.1006/scdb.2000.0177 [DOI] [PubMed] [Google Scholar]
- 49.Salic A, Mitchison TJ. 2008. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. U. S. A. 105:2415–2420. 10.1073/pnas.0712168105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor RC, Cullen SP, Martin SJ. 2008. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9:231–241. 10.1038/nrm2312 [DOI] [PubMed] [Google Scholar]
- 51.Collins JA, Schandi CA, Young KK, Vesely J, Willingham MC. 1997. Major DNA fragmentation is a late event in apoptosis. J. Histochem. Cytochem. 45:923–934. 10.1177/002215549704500702 [DOI] [PubMed] [Google Scholar]
- 52.Murray D, Horgan G, Macmathuna P, Doran P. 2008. NET1-mediated RhoA activation facilitates lysophosphatidic acid-induced cell migration and invasion in gastric cancer. Brit. J. Cancer 99:1322–1329. 10.1038/sj.bjc.6604688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han X, Cheng W, Jing H, Zhang J-W, Tang L-L. 2012. Neuroepithelial transforming protein 1 short interfering RNA-mediated gene silencing with microbubble and ultrasound exposure inhibits the proliferation of hepatic carcinoma cells in vitro. J. Ultrasound Med. 31:853–861 http://www.jultrasoundmed.org/content/31/6/853.long [DOI] [PubMed] [Google Scholar]
- 54.Ando-Akatsuka Y, Yonemura S, Itoh M, Furuse M, Tsukita S. 1999. Differential behavior of E-cadherin and occludin in their colocalization with ZO-1 during the establishment of epithelial cell polarity. J. Cell. Physiol. 179:115–125. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki A, Ishiyama C, Hashiba K, Shimizu M, Ebnet K, Ohno S. 2002. aPKC kinase activity is required for the asymmetric differentiation of the premature junctional complex during epithelial cell polarization. J. Cell Sci. 115:3565–3573. 10.1242/jcs.00032 [DOI] [PubMed] [Google Scholar]
- 56.Murata M, Kojima T, Yamamoto T, Go M, Takano K, Chiba H, Tokino T, Sawada N. 2005. Tight junction protein MAGI-1 is up-regulated by transfection with connexin 32 in an immortalized mouse hepatic cell line: cDNA microarray analysis. Cell Tissue Res. 319:341–347. 10.1007/s00441-004-1017-0 [DOI] [PubMed] [Google Scholar]
- 57.Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T, Tsukita S. 2000. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 11:4131–4142. 10.1091/mbc.11.12.4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bordin M, D'Atri F, Guillemot L, Citi S. 2004. Histone deacetylase inhibitors up-regulate the expression of tight junction proteins. Mol. Cancer Res. 2:692–701 http://mcr.aacrjournals.org/content/2/12/692.long [PubMed] [Google Scholar]
- 59.Li D, Mrsny RJ. 2000. Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J. Cell Biol. 148:791–800. 10.1083/jcb.148.4.791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mino A, Ohtsuka T, Inoue E, Takai Y. 2000. Membrane-associated guanylate kinase with inverted orientation (MAGI)-1/brain angiogenesis inhibitor 1-associated protein (BAP1) as a scaffolding molecule for Rap small G protein GDP/GTP exchange protein at tight junctions. Genes Cells 5:1009–1016. 10.1046/j.1365-2443.2000.00385.x [DOI] [PubMed] [Google Scholar]
- 61.Sakurai A, Fukuhara S, Yamagishi A, Sako K, Kamioka Y, Masuda M, Nakaoka Y, Mochizuki N. 2006. MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol. Biol. Cell 17:966–976. 10.1091/mbc.E05-07-0647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leyden J, Murray D, Moss A, Arumuguma M, Doyle E, McEntee G, O'Keane C, Doran P, MacMathuna P. 2006. Net1 and Myeov: computationally identified mediators of gastric cancer. Brit. J. Cancer 94:1204–1212. 10.1038/sj.bjc.6603054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dubash AD, Guilluy C, Srougi MC, Boulter E, Burridge K, García-Mata R. 2011. The small GTPase RhoA localizes to the nucleus and is activated by Net1 and DNA damage signals. PLoS One 6:e17380. 10.1371/journal.pone.0017380 [DOI] [PMC free article] [PubMed] [Google Scholar]