

ABSTRACT
Nonstructural protein 5A (NS5A) of hepatitis C virus (HCV) possesses multiple functions in the viral life cycle. NS5A is a phosphoprotein that exists in hyperphosphorylated and basally phosphorylated forms. Although the phosphorylation status of NS5A is considered to have a significant impact on its function, the mechanistic details regulating NS5A phosphorylation, as well as its exact roles in the HCV life cycle, are still poorly understood. In this study, we screened 404 human protein kinases via in vitro binding and phosphorylation assays, followed by RNA interference-mediated gene silencing in an HCV cell culture system. Casein kinase I-α (CKI-α) was identified as an NS5A-associated kinase involved in NS5A hyperphosphorylation and infectious virus production. Subcellular fractionation and immunofluorescence confocal microscopy analyses showed that CKI-α-mediated hyperphosphorylation of NS5A contributes to the recruitment of NS5A to low-density membrane structures around lipid droplets (LDs) and facilitates its interaction with core protein and the viral assembly. Phospho-proteomic analysis of NS5A with or without CKI-α depletion identified peptide fragments that corresponded to the region located within the low-complexity sequence I, which is important for CKI-α-mediated NS5A hyperphosphorylation. This region contains eight serine residues that are highly conserved among HCV isolates, and subsequent mutagenesis analysis demonstrated that serine residues at amino acids 225 and 232 in NS5A (genotype 2a) may be involved in NS5A hyperphosphorylation and hyperphosphorylation-dependent regulation of virion production. These findings provide insight concerning the functional role of NS5A phosphorylation as a regulatory switch that modulates its multiple functions in the HCV life cycle.
IMPORTANCE Mechanisms regulating NS5A phosphorylation and its exact function in the HCV life cycle have not been clearly defined. By using a high-throughput screening system targeting host protein kinases, we identified CKI-α as an NS5A-associated kinase involved in NS5A hyperphosphorylation and the production of infectious virus. Our results suggest that the impact of CKI-α in the HCV life cycle is more profound on virion assembly than viral replication via mediation of NS5A hyperphosphorylation. CKI-α-dependent hyperphosphorylation of NS5A plays a role in recruiting NS5A to low-density membrane structures around LDs and facilitating its interaction with the core for new virus particle formation. By using proteomic approach, we identified the region within the low-complexity sequence I of NS5A that is involved in NS5A hyperphosphorylation and hyperphosphorylation-dependent regulation of infectious virus production. These findings will provide novel mechanistic insights into the roles of NS5A-associated kinases and NS5A phosphorylation in the HCV life cycle.
INTRODUCTION
Hepatitis C virus (HCV) is a major causative agent of liver-related morbidity and mortality worldwide and represents a global public health problem (1). An estimated 130 million individuals are chronically infected with HCV worldwide, and the treatment of HCV infection imposes a large economic and societal burden (2). HCV is an enveloped virus with a positive-sense, single-stranded RNA genome in the Hepacivirus genus within the Flaviviridae family (3). The approximately 9.6-kb genome is translated into a single polypeptide of approximately 3,000 amino acids (aa), which is cleaved by cellular and viral proteases to produce the structural proteins (core, E1, E2, and p7) and nonstructural (NS) proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (4). NS3 to NS5B are sufficient for RNA replication in cell culture (5). NS5B is an RNA-dependent RNA polymerase (RdRp), and NS3 functions as both an RNA helicase and a serine protease (4). NS4A is the cofactor of the NS3 protease, and the NS3-NS4A complex is required for viral precursor processing (4). NS4B induces the formation of a specialized membrane compartment, a sort of membranous web where viral RNA replication may take place (6). NS5A is essential for both viral RNA replication and virion assembly (7–9).
NS5A is an RNA binding protein and exists as a component of the replicase complex (10–13). NS5A is phosphorylated on multiple serine and threonine residues and can be found in hyperphosphorylated (p58) and basally phosphorylated (p56) forms (14–16). Although the distinct mechanisms for generating p56 and p58 forms are still unclear, it has been reported that two regions located around the center and near the C-terminal regions of NS5A are required for basal phosphorylation, while hyperphosphorylation primarily targets serine residues located within low-complexity sequence I (LCS I), which is the linker between domains I and II (15, 17–19). Several phosphorylation sites have been mapped in NS5A by using recombinantly expressed protein and NS5A extracted from cells harboring subgenomic replicons (20–23).
NS5A phosphorylation plays roles in the regulation of viral RNA replication and virion assembly. Some of the cell culture-adaptive mutations in NS4B and NS5A, which reduce NS5A hyperphosphorylation, have been found to confer efficient replication of genotype 1 replicons in Huh-7 cells (17, 18). Similarly, suppression of NS5A hyperphosphorylation through either the use of kinase inhibitors or mutagenesis allows higher RNA replication in non-culture-adapted replicons (18, 24). In contrast, HCV RNA replication is inhibited after treatment of cells carrying adapted replicons with the same kinase inhibitor (24). The C-terminal domain III of NS5A is not essential for viral RNA replication, but it is important for the production of infectious virus. Alanine replacements of the serine cluster in this domain impair NS5A phosphorylation, leading to a decrease in NS5A-core protein interaction, perturbation of the subcellular distribution of NS5A, and disruption of virion production (7–9).
A number of protein kinases have been identified as having the ability to phosphorylate NS5A based on comprehensive screening by using an RNA interference (RNAi) library, recombinantly expressed kinases, and kinase inhibitors (25–28). Among them, casein kinase I-α (CKI-α) and Polo-like kinase 1 (Plk1) have been shown to play roles in viral RNA replication (25, 27). Although silencing of CKI-α inhibits the replication of the genotype 1b subgenomic replicon containing an adaptive mutation (27), its effect on infectious virus production has not been studied to date. Casein kinase II (CKII) has been identified as a positive regulator of virus production via studies with chemical inhibitors and small interfering RNA (siRNA) (9). However, the functional roles of NS5A phosphorylation by its associated kinases in regulation of the viral life cycle are not yet fully understood.
To identify NS5A-associated kinases involved in the HCV life cycle, we developed an in vitro, high-throughput screening system for analyzing protein-protein interactions. Using this system followed by in vitro phosphorylation assays, we screened human protein kinases on a kinome-wide scale and identified several NS5A-associated kinases. siRNA experiments showed that silencing of CKI-α leads to the most marked inhibition of infectious virus production among the candidate kinases. Here, we report a novel function of CKI-α in the viral life cycle. It is more likely that CKI-α has a more profound impact on virion assembly than on viral replication through hyperphosphorylation of NS5A. Hyperphosphorylated NS5A was predominantly localized in low-density membrane structures around lipid droplets (LDs), in which NS5A interacts with the core for virion assembly, while reduction of NS5A hyperphosphorylation by siRNA targeting CKI-α led to a decrease in NS5A abundance in the low-density membrane structures. The present study provides important insights into the regulatory roles of NS5A-associated kinases and NS5A phosphorylation in the viral life cycle, especially as a molecular switch governing the transition between viral replication and virion assembly.
MATERIALS AND METHODS
Plasmids.
Plasmids pJFH1 and pSGR-JFH1/Luc were generated as previously described (29, 30). The JFH-1-based Gaussia princeps luciferase (GLuc) reporter construct, which encodes GLuc followed by the foot-and-mouth disease virus (FMDV) 2A protein between p7 and NS2, was generated in a manner similar to the description in a previous report (31). A 1,042-bp double-stranded DNA fragment containing GLuc (32) and FMDV 2A (33) sequences flanked by BsaI and NotI sites at its ends was synthesized and then inserted into the corresponding sites of pJFH1. Related constructs containing serine-to-alanine or serine-to-aspartic acid mutations in NS5A were generated using oligonucleotide-directed mutagenesis techniques. To construct pCAG-CKI-α, the full-length CKI-α-coding sequence was amplified by PCR using cDNAs prepared from Huh-7 cells. The resulting PCR product was then inserted into the multiple-cloning site of pCAGGS (34). pCAG-CKI-α/m6, which contains six silent point mutations that ablate the binding of CKI-α siRNA but maintain the wild-type amino acid sequence of CKI-α, was generated by oligonucleotide-directed mutagenesis of pCAG-CKI-α. All PCR products were confirmed by automated nucleotide sequencing with an ABI Prism 7000 sequence detection system (Life Technologies, Carlsbad, CA).
Cells.
The human hepatoma cell line Huh-7, its derivative cell lines Huh7.5.1 (35) (a gift from Francis V. Chisari, The Scripps Research Institute) and Huh7–25 (36), and the human embryonic kidney cell line 293T used to generate HCV pseudoparticles (HCVpp), were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with nonessential amino acids, 100 U of penicillin/ml, 100 μg of streptomycin/ml, and 10% fetal bovine serum (FBS) at 37°C in a 5% CO2 incubator. SGR-JFH1/LucNeo cells, which harbor a genotype 2a subgenomic replicon carrying a firefly luciferase reporter gene fused to the neomycin phosphotransferase gene of pSGR-JFH1 (37), and LucNeo#2 cells, which harbor a genotype 1b subgenomic replicon carrying a firefly luciferase/neomycin phosphotransferase fusion reporter gene (38, 39) (a gift from Koichi Watashi, National Institute of Infectious Diseases, and Kunitada Shimotohno, National Center for Global Health and Medicine), were cultured in the above medium supplemented with 300 μg/ml G418.
Antibodies.
Mouse monoclonal antibody against core protein (2H9) was generated as described previously (30). Anti-NS5A mouse monoclonal antibody (9E10) was a kind gift from Charles M. Rice (The Rockefeller University), and anti-NS5A rabbit polyclonal antibody (TB0705#1) was developed by immunization with the recombinant NS5A protein (8, 40). For detection of cellular proteins, the following antibodies were used: mouse monoclonal antibodies directed against Plk1 (Life Technologies) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Millipore, Temecula, CA); rabbit polyclonal antibodies detecting CKI-α (Santa Cruz Biotechnology, Dallas, TX), CKI-ε (Santa Cruz Biotechnology), cyclinc AMP (cAMP)-dependent protein kinase catalytic subunit β (PKACβ; Santa Cruz Biotechnology), phosphatidylinositol 4-kinase IIIα (PI4K-IIIα; Cell Signaling Technology, Danvers, MA), claudin-1 (CLDN1; Life Technologies), calnexin (Enzo Life Sciences, Farmingdale, NY), and GM130 (Sigma-Aldrich, St. Louis, MO); and goat polyclonal antibodies specific for CKII-α′ (Santa Cruz Biotechnology) and apolipoprotein E (ApoE; Millipore). Fluorescence-conjugated secondary antibodies, including Alexa Fluor 488 goat anti-mouse IgG1 and Alexa Fluor 568 goat anti-mouse IgG2a, were purchased from Life Technologies. Horseradish peroxidase (HRP)-conjugated secondary antibodies were from Cell Signaling Technology.
Protein kinase library and cell-free protein synthesis.
The construction and identity of the 404 cDNAs encoding human protein kinases used in this study were previously described (41). In vitro transcription and cell-free protein synthesis were performed as previously reported (42, 43). Briefly, DNA templates containing a biotin-ligating sequence were amplified by split-primer PCR with kinase cDNAs and corresponding primers and then used for protein synthesis with a fully automated protein synthesizer, a GenDecoder (CellFree Sciences, Ehima, Japan). For synthesis of FLAG-tagged full-length NS5A and domain III proteins derived from the JFH-1 isolate, DNA templates containing a FLAG sequence were generated from the NS5A expression plasmid (8) by split-primer PCR and used with a wheat germ expression kit (CellFree Sciences) according to the manufacturer's instructions.
Amplified luminescent proximity homogeneous assay (AlphaScreen).
FLAG-tagged NS5A proteins were mixed with biotinylated kinases in 15 μl of reaction buffer (20 mM Tris-HCl [pH 7.6], 5 mM MgCl2, 1 mM dithiothreitol, and 1 mg/ml bovine serum albumin) in the wells of 384-well OptiPlates (PerkinElmer, Waltham, MA) and incubated at 26°C for 1 h. The mixture was then added to the detection mixture containing 0.1 μl protein A-conjugated acceptor beads (PerkinElmer), 0.1 μl streptavidin-coated donor beads (PerkinElmer), and 5 μg/ml of the anti-FLAG M2 antibody, followed by incubation at 26°C for 1 h. AlphaScreen signals from the mixture were detected using an EnVision device (PerkinElmer) with the AlphaScreen signal detection program.
In vitro phosphorylation assay.
To obtain purified kinases used for in vitro phosphorylation assays, DNA templates containing a glutathione S-transferase (GST)–tobacco etch virus (TEV) sequence were generated by split-primer PCR with kinase cDNAs and corresponding primers and used in a cell-free production system with the wheat germ expression kit as described above. The GST-fusion recombinant proteins were purified on glutathione-Sepharose 4B (GE Healthcare, Buckinghamshire, United Kingdom) and then eluted in 40 μl of phosphate-buffered saline (PBS) containing 5 U of AcTEV protease (Life Technologies) in order to cleave the GST tag from the protein. Biotinylated NS5A proteins were synthesized from DNA templates by using the cell-free BirA system (44). Biotinylated NS5A proteins (40 μl) were coupled on 15 μl of streptavidin Magnesphere Paramagnetics particles (Promega, Madison, WI) and were dephosphorylated by using 10 U of lambda protein phosphatase (New England BioLabs, Ipswich, MA). After washing three times, protein-coupled beads were incubated with 1 μl of purified recombinant kinases at 37°C for 30 min in 15 μl kinase buffer (50 mM Tris-HCl [pH 7.6], 500 mM potassium acetate, 50 mM MgCl2, and 0.5 mM dithiothreitol) containing 1 μCi of [γ-32P]ATP. After the reaction, the beads were washed twice with PBS and then boiled in sample buffer and separated by SDS-PAGE. Phosphorylated NS5A proteins were visualized via autoradiography, and the relative kinase activity of each kinase was determined by normalizing the band intensity of NS5A to that of NS5A incubated with dihydrofolate reductase (DHFR). The band intensities were quantified using Image J software.
Preparation of viral stocks and virus infections.
Cell culture-derived infectious HCV particles (HCVcc) were prepared as described previously (30, 36). HCVpp consisting of HCV envelope glycoproteins, the murine leukemia virus Gag-Pol core proteins, and the luciferase transfer vector and pseudoparticles with the vesiclar stomatitis virus G glycoprotein (VSV-Gpp) were generated in accordance with methods described previously (36, 45). Cells seeded onto 24-well plates were transfected with siRNA and/or plasmid DNA as described below and infected with HCVcc for 4 h at a multiplicity of infection (MOI) of 0.5 to 5 or with diluted supernatant containing HCVpp or VSV-Gpp for 3 h. After infection, the cells were washed with PBS and incubated in fresh complete growth medium for 72 h at 37°C until harvest.
siRNA and plasmid DNA transfections.
siRNAs were purchased from Sigma-Aldrich. The sequences were as follows: CKI-α, 5′-GGCUAAAGGCUGCAACAAAdTdT-3′ and 5′-UUUGUUGCAGCCUUUAGCCdTdT-3′; CKI-γ1, 5′-GAGAUGAUUUGGAAGCCCUdTdT-3′ and 5′-AGGGCUUCCAAAUCAUCUCdTdT-3′; CKI-γ2, 5′-GCGAGAACUUCCCAGAGGAdTdT-3′ and 5′-UCCUCUGGGAAGUUCUCGCdTdT-3′; CKI-γ3, 5′-CUUACAGGAACAGCUAGAUdTdT-3′ and 5′-AUCUAGCUGUUCCUGUAAGdTdT-3′; CKI-ε, 5′-GCGACUACAACGUGAUGGUdTdT-3′ and 5′-ACCAUCACGUUGUAGUCGCdTdT-3′; CKII-α′, 5′-CCUAGAUCUUCUGGACAAAdTdT-3′ and 5′-UUUGUCCAGAAGAUCUAGGdTdT-3′; PKACβ, 5′-CAAAUAGAGCAUACUUUGAdTdT-3′ and 5′-UCAAAGUAUGCUCUAUUUGdTdT-3′; Plk1, 5′-GUCUCAAGGCCUCCUAAUAdTdT-3′ and 5′-UAUUAGGAGGCCUUGAGACdTdT-3′; TSSK2, 5′-CACCUACUGACUUUGUGGAdTdT-3′ and 5′-UCCACAAAGUCAGUAGGUGdTdT-3′; ApoE, 5′-GGAGUUGAAGGCCUACAAAdTdT-3′ and 5′-UUUGUAGGCCUUCAACUCCdTdT-3′; CLDN1, 5′-CAGUCAAUGCCAGGUACGAdTdT-3′ and 5′-UCGUACCUGGCAUUGACUGdTdT-3′; PI4K-IIIα, 5′-CCCUAAAGGCGACGAGAGAdTdT-3′ and 5′-UCUCUCGUCGCCUUUAGGGdTdT-3′. The Mission siRNA universal negative control (Sigma-Aldrich), which is designed to have no homology to known gene sequences, was used as a negative control. Silencer Cy3-labeled GAPDH siRNA (Life Technologies) was used to confirm siRNA delivery efficiency. Basically, 10 nM siRNAs were transfected into cells by using Lipofectamine RNAiMax (Life Technologies) according to the manufacturer's recommended procedures. Plk1 siRNA was transfected at 5 nM because of its cytotoxic effect. Plasmid DNA transfection was carried out by using TransIT-LT1 transfection reagent (Mirus, Madison, WI) according to the manufacturer's protocol. For cotransfection of siRNA and plasmid DNA, 6 pmol of siRNA and 200 ng of plasmid DNA were transfected into Huh7.5.1 cells seeded onto a 24-well cell culture plate by using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions.
RNA synthesis and electroporation.
HCV RNA synthesis and electroporation were basically performed as described previously (8). In the context of coelectroporation of siRNA and an in vitro-synthesized subgenomic reporter replicon, or full-length HCV RNA, a total of 3 × 106 to 5 × 106 Huh-7 cells were electroporated with 120 pmol siRNA and 3 μg SGR-JFH1/Luc RNA or 5 μg JFH-1 RNA at 260 V and 950 μF. After electroporation, the cells were immediately transferred onto 24-well or 6-well culture plates or 10-cm cell culture dishes.
Luciferase assay.
Cells harboring a subgenomic reporter replicon and HCVpp-infected cells were lysed in passive lysis buffer (Promega). The luciferase activity was determined using a luciferase assay system (Promega) as previously described (46). Secreted GLuc activity was measured in 25-μl aliquots of cell culture supernatants by using the BioLux Gaussia luciferase assay kit (New England BioLabs) according to the manufacturer's recommended protocol. The luminescence signal was measured on an Infinite M200 microplate reader (Tecan, Männedorf, Switzerland).
Quantification of HCV core.
HCV core protein in cell lysates and culture supernatants was quantified by using a highly sensitive enzyme immunoassay as described previously (8).
RNA extraction and RT-qPCR.
Total cellular RNA was extracted with TRIzol reagent (Life Technologies) according to the manufacturer's instructions. Quantification of cellular gene expression was performed by reverse transcription-quantitative PCR (RT-qPCR) using an Applied Biosystems 7500 fast real-time PCR system (Life Technologies) as described previously (47, 48). Primer/probe sets for qPCR targeting CKI-γ1, CKI-γ2, CKI-γ3, and TSSK2 genes were selected from validated Assays-on-Demand products (Life Technologies).
Intra- and extracellular infectivity assays.
Intra- and extracellular infectivities of HCVcc were determined as described previously (8). The infectious titers were expressed as focus-forming units (FFU)/ml.
Cell viability assay.
Cell viability was determined using the CellTiter-Glo luminescent cell viability assay (Promega) according to the manufacturer's instructions.
Expression of HCV proteins based on vaccinia virus, immunoprecipitation, immunoblotting, and silver staining.
HCV protein expression based on vaccinia virus, immunoprecipitation, and immunoblotting were performed as previously described (8). pJFH1 was transfected into cells before infection with vaccinia virus expressing the T7 RNA polymerase. NS5A p58 and p56 protein levels were quantified by densitometry using Image J software. Silver staining of proteins in polyacrylamide gels was carried out using a Silver Stain MS kit (Wako Pure Chemical Industries, Osaka, Japan) in accordance with the manufacturer's protocol.
Subcellular fractionation analysis.
Cells were suspended in homogenization buffer (10 mM HEPES-NaOH [pH 7.4], 0.25 M sucrose, and 1 mM EDTA) and disrupted by repeated passages through a 25-gauge needle. After low-speed centrifugation, postnuclear supernatants were layered on linear 11-ml iodixanol gradients from 2.5% to 30% and centrifuged at 40,000 rpm for 3 h in an SW41 rotor (Beckman, Fullerton, CA). Thirteen fractions (0.8 ml in each fraction) were collected from the top of the gradient. Each fraction was concentrated by ultrafiltration units with a 10-kDa molecular mass cutoff (Millipore, Bedford, MA), separated by SDS-PAGE, and immunoblotted with antibodies specific for NS5A, calnexin, and GM130.
Indirect immunofluorescence and microscopy analyses.
Cells incubated for 3 days after infection with HCVcc of JFH-1 were fixed with 4% paraformaldehyde for 15 min at room temperature. After washing with PBS, the cells were permeabilized with 0.05% Triton X-100 in PBS for 15 min at room temperature and subsequently incubated in PBS containing 10% goat serum for 1 h. The cells were then costained with antibodies against core and NS5A, followed by incubation with fluorescent secondary antibodies. Cells were counterstained with Hoechst 33342 (Sigma-Aldrich) to label nuclei and BODIPY 493/503 (Life Technologies) to label lipid droplets and then mounted in Vectashield (Vector Laboratories, Burlingame, CA). Subcellular localization of HCV proteins was observed on a Leica SP2 AOBS laser scanning confocal microscope (Leica, Wetzlar, Germany). Colocalization of NS5A and LDs or core was evaluated quantitatively by using the intensity correlation analysis of the Image J software. To statistically compare degrees of colocalization, we determined the intensity correlation quotient (ICQ) (49). ICQ values are distributed between −0.5 and +0.5, with a value of ∼0 reflecting random staining and values between 0 and +0.5 versus values between 0 and −0.5 indicative of dependent versus segregated immunolabeling, respectively.
Mass spectrometry analysis.
Immunoprecipitated NS5A bands were excised from the gels after silver staining and destained, followed by in gel digestion with trypsin in 50 mM ammonium bicarbonate overnight at 30°C. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed on an LTQ Orbitrap Velos hybrid mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) using Xcalibur (version 2.0.7), coupled to an UltiMate 3000 LC system (Dionex LC Packings, Sunnyvale, CA). The Proteome Discoverer software (version 1.3; Thermo Fisher Scientific) was used to generate peak lists from the raw MS data files. The resulting peak lists were subsequently submitted to a Mascot search engine (version 2.4.1; Matrix Science, London, United Kingdom) and compared against the HCV protein sequences in the NCBI nonredundant protein database (version 20 January 2013; 74,0475 sequences) to identify peptides. The Mascot search parameters were as follows: two missed cleavages permitted in the trypsin digestion; variable modifications including oxidation of methionine, propionamidation of cysteine, and phosphorylation of serine, threonine, and tyrosine; peptide mass tolerance of ±5 ppm; fragment mass tolerance of ±0.5 Da. A minimum Mascot peptide score of 25 was set for peptide selection.
Statistical analyses.
Statistical analyses were performed using the Student t test unless otherwise noted. A P level of <0.05 was considered significant.
RESULTS
A kinome-wide screening of human protein kinases for identification of NS5A-associated kinases.
It has been reported that some protein kinases directly or stably associate with HCV NS5A and phosphorylate it in vitro (25, 50, 51). To search comprehensively to identify novel NS5A-associated kinases, a kinome-wide screening for interactions of full-length NS5A with human kinases was initially performed. We synthesized 404 human kinases with a wheat germ cell-free protein production system and screened them in terms of their association with NS5A by using a high-throughput assay system based on AlphaScreen technology (Fig. 1A; see also Table S1 in the supplemental material). Ninety-five proteins were selected as those that possibly bind to NS5A under the cutoff condition of luminescence signals at ≥1,300. Among them, 84 were serine/threonine kinases, and Plk1 and CKII-α′, the catalytic subunit α′ of CKII whose associations with NS5A have been reported (25, 50), were found in the group as signals at 3,230 (Plk1) and 5,068 (CKII-α′). This suggested that our assay system is highly reliable for screening the NS5A-kinase interaction.
FIG 1.

Identification of NS5A-associated kinases. (A) AlphaScreen-based protein-protein interaction assay. FLAG-tagged full-length NS5A, and each of 404 biotinylated human protein kinases, which were synthesized in a cell-free protein production system, were mixed with the detection mixture containing anti-FLAG antibody, protein A-conjugated acceptor beads, and streptavidin-coated donor beads in 384-well plates. Luminescence signals from the mixture were detected. Ninety-five protein kinases were identified with luminescence signals of ≥1,300, of which 84 were serine/threonine kinases. The assay was performed in duplicate for each sample, and data shown are mean values of duplicate experiments. (B) Exemplary autoradiographic images of NS5A phosphorylated in vitro. Purified kinases were mixed with biotinylated NS5A proteins coupled on streptavidin beads in kinase buffer containing [γ-32P]ATP. After the reaction, samples were subjected to SDS-PAGE and autoradiography. The arrows, black arrowheads, and white arrowheads indicate phosphorylated full-length NS5A, phosphorylated NS5A domain III, and autophosphorylated kinases, respectively.
In vitro phosphorylation of NS5A by the identified NS5A binding serine/threonine was determined. Each kinase that was synthesized in vitro and purified was incubated with either full-length NS5A or domain III of NS5A in the presence of [γ-32P]ATP and separated by SDS-PAGE. Phosphorylated NS5A proteins were then visualized by autoradiography (Fig. 1B). The relative kinase activity was determined by normalizing the band intensity of phosphorylated NS5A with that of NS5A incubated with DHFR, which had no kinase activity and was used as a negative control. Twenty-nine out of 84 serine/threonine kinases were not accurately assessed due to their low levels of expression. As shown in Table 1, among a total of 55 kinases tested (see Table S2 in the supplemental material), nine (CKI-α, CKI-γ1, CKI-γ2, CKI-γ3, CKI-ε, CKII-α′, PKACβ, Plk1, and TSSK2) exhibited efficient phosphorylation of NS5A, defined as a more-than-4-fold or 8-fold increase in activity against the full-length NS5A or domain III of NS5A, respectively, compared to the negative control. Consistent with previous reports (9, 25, 50), Plk1 and CKII-α′ showed apparent kinase activities against NS5A in vitro.
TABLE 1.
Serine/threonine kinases that exhibited efficient phosphorylation of NS5A
| Kinase | LU | Relative kinase activitya |
|
|---|---|---|---|
| FL | D3 | ||
| TSSK2 | 8,310 | 4.24 | 14.94 |
| CKII-α′ | 5,068 | 2.30 | 8.20 |
| Plk1 | 3,230 | 2.70 | 44.65 |
| CKI-γ2 | 2,602 | 4.31 | 3.15 |
| CKI-γ1 | 2,560 | NA | 138.69 |
| CKI-γ3 | 2,218 | 0.23 | 52.51 |
| CKI-ε | 2,012 | 7.95 | 9.48 |
| PKACβ | 1,854 | 0.15 | 69.54 |
| CKI-α | 1,354 | 4.66 | 1.00 |
LU, light units from the AlphaScreen; FL, full-length NS5A; D3, domain III of NS5A. NA, not assessed due to overlap between purified kinases and NS5A on the gel. The relative kinase activity is the fold increase of the in vitro activity of each kinase relative to that of DHFR.
Identification of an NS5A-associated kinase, CKI-α, that is important for the HCV life cycle.
On the basis of the in vitro screenings, nine candidate kinases were further tested as to whether they play roles in the HCV life cycle. We conducted siRNA-based gene silencing of each kinase and assessed its effect on virion production. Huh7.5.1 cells were transfected with siRNAs targeting the kinases and infected with JFH-1 virus at an MOI of 1, 48 h after siRNA transfection. After an additional 72-h incubation, the viral core levels and infectious virus yields in cell culture supernatants were determined. Knockdown efficiencies of the targeted genes at 72 h after JFH-1 infection are shown in Fig. 2A. Efficient knockdown was confirmed either by immunoblotting or RT-qPCR. Figure 2B indicates the effects of gene silencing on virus production (upper panel) and on cell viability by ATP-based luminescence assays (lower panel). An approximately 30-fold reduction in infectious virus yields was observed following knockdown of ApoE, which has been shown to have important roles in HCV assembly and release (52). Among the kinases tested, silencing of CKI-α showed the most profound inhibition of infectious HCV production (∼40-fold) without cytotoxicity. Knock down of CKII-α′ and PKACβ led to a moderate reduction in infectious virus production (∼10-fold), which was consistent with previous reports showing that CKII-α′ and PKA are involved in virion assembly and viral entry, respectively (9, 53). Knockdown of CKI-ε and Plk1 also resulted in a moderate decrease in virion production (∼20-fold), but they induced moderate to severe cell toxicity as well.
FIG 2.
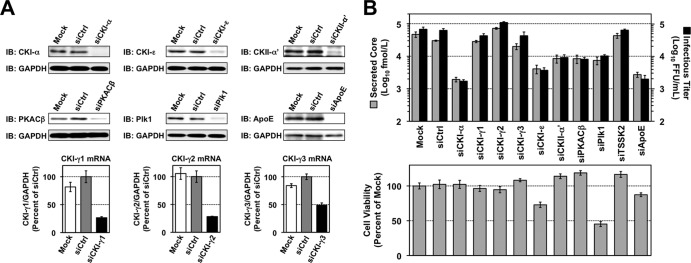
Identification of NS5A-associated kinases involved in the HCV life cycle. (A) siRNA-based gene silencing of NS5A-associated kinases. Huh7.5.1 cells were transfected with siRNAs targeting the indicated genes and were harvested 5 days later for immunoblotting (IB) and RT-qPCR to confirm knockdown efficiencies. mRNA levels of target genes relative to GAPDH mRNA were normalized with values for transfection of control siRNAs (siCtrl), which were set at 100%. Results represent the means ± standard deviations from three independent transfections of siRNA. Mock represents transfection without siRNA. (B) Infectious HCV production and cell viability following knockdown of NS5A-associated kinases. Huh7.5.1 cells were infected with JFH-1 virus at an MOI of 1, 2 days after siRNA transfection. Culture supernatants and cells were harvested 3 days later to determine infectious virus yields (upper panel) and cell viability (lower panel), respectively. Cell viability for each transfection was normalized to that for mock transfection (mock), which was set at 100%. Results shown represent the means ± standard deviations from three independent transfections of siRNA. Mock, transfection without siRNA.
Thus, CKI-α, which phosphorylates NS5A, had the highest impact on HCV production based on in vitro comprehensive screenings for protein kinases and a subsequent siRNA-based assay.
To further demonstrate that impaired virus production results specifically from CKI-α silencing and is not an off-target effect of the siRNA, cells were cotransfected with CKI-α siRNA and a mutated CKI-α expression vector (pCAG-CKI-α/m6) that contained 6 base mismatches within the site targeted by the CKI-α siRNA without a change in amino acids, followed by JFH-1 infection at an MOI of 0.5 on the next day. The cells and culture supernatants were harvested 3 days later for immunoblotting and titrations of virus yields, respectively (Fig. 3A). Transfection of the CKI-α siRNA led to a significant reduction in infectious virus yields and in the p58/p56 ratio of NS5A. Ectopic expression of the siRNA-resistant CKI-α/m6 apparently restored virus yields (P < 0.05), as well as the p58/p56 ratio of NS5A (P < 0.05). Similar results regarding the p58/p56 ratio were obtained from immunoblot analysis following vaccinia virus-T7 polymerase-mediated expression of HCV proteins (Fig. 3B). These results indicated that impaired virion production and reduced NS5A hyperphosphorylation are specifically caused by CKI-α silencing. Infectious virus yields showed a closer correlation with the p58/p56 ratio of NS5A than did the expression level of CKI-α, suggesting that the involvement of CKI-α in HCV production is through hyperphosphorylation of NS5A.
FIG 3.

Restoration of NS5A hyperphosphorylation and infectious virus yields by ectopic expression of siRNA-resistant CKI-α. (A) Huh7.5.1 cells were cotransfected with the indicated siRNAs and plasmid DNAs. The next day, cells were infected with JFH-1 virus at an MOI of 0.5. Culture supernatants and cells were harvested an additional 3 days later for measurement of virus yields and immunoblotting (IB). The p58/p56 ratios were calculated after quantifying the band intensities of NS5A. Values shown represent the means ± standard deviations from three replicate experiments. *, P < 0.05; **, P < 0.01. (B) Huh-7 cells were transfected either with CKI-α siRNA (siCKI-α) or with an irrelevant control siRNA (siCtrl). The next day, cells were retransfected with pJFH1 and pCAG-CKI-α/m6 or empty vector (pCAGGS), followed by infection with vaccinia virus expressing the T7 RNA polymerase at an MOI of 10. NS5A bands were quantified by densitometric analysis, and the p58/p56 ratios were calculated. Immunoblotting images and values shown are representative of two independent experiments.
CKI-α is mainly involved in virion assembly in the HCV life cycle.
Although it has been reported that CKI-α plays roles in the regulation of HCV RNA replication through NS5A phosphorylation, experiments addressing its involvement in the viral life cycle have been performed using the subgenomic replicon system (27). To determine the basic role of CKI-α in the production of infectious HCV, the effect of CKI-α silencing on individual steps in the HCV life cycle was assessed.
First, we used an HCVpp system to analyze viral entry. Two days after the siRNA transfection, the cells were infected with HCVpp derived from JFH-1 or VSV-Gpp and were cultured for a further 3 days (Fig. 4A). Consistent with a previous report (54), CLDN1 knockdown inhibited HCVpp entry by approximately 70%, but not VSV-Gpp entry, compared to transfection with negative control siRNA. CKI-α silencing did not affect HCVpp or VSV-Gpp entry, suggesting that CKI-α is not required for HCV entry.
FIG 4.

Virion assembly is a primary target of CKI-α. (A) Effect of CKI-α knockdown on viral entry. Huh7.5.1 cells were transfected with the indicated siRNAs. Two days later, cells were infected with HCVpp (gray bars) or VSV-Gpp (black bars) and harvested an additional 3 days later for immunoblotting (IB) and luciferase assays. Values were normalized to the value for transfection with control siRNAs (siCtrl), set at 100%. Values shown represent the means ± standard deviations from three independent transfections of siRNA. (B) Transient replication assay for the JFH-1 subgenomic replicon following CKI-α knockdown. Huh-7 cells transfected with the indicated siRNAs were coelectroporated with the identical siRNAs, and JFH-1 subgenomic luciferase reporter replicon RNA, and harvested at the indicated time points for immunoblotting (IB) and luciferase assays. The luciferase activity at each time point was corrected by the luciferase value at 4 h posttransfection to normalize transfection efficiencies. Values shown represent the means ± standard deviations from three replicate experiments. (C) Effects of CKI-α knockdown on replication in replicon cell lines derived from genotypes 1b and 2a. Two cell lines harboring an HCV subgenomic luciferase reporter replicon, LucNeo#2 (genotype 1b, GT1b) and SGR-JFH1/LucNeo (genotype 2a, GT2a), were transfected with the indicated siRNAs and harvested 3 days later for immunoblotting (IB) and luciferase assays. Luciferase activities were normalized to the luciferase values for transfection with control siRNA (siCtrl), set at 100%. Values shown represent the means ± standard deviations from three independent transfections of siRNA. (D) Effects of CKI-α knockdown on viral assembly and release. Huh7-25 cells transfected with the indicated siRNAs were coelectroporated with the identical siRNAs and JFH-1 RNA. Cells and supernatants were harvested 3 days later for immunoblotting (IB) and titrations of extracellular and intracellular infectious virus by focus-forming unit (FFU) assays. Values represent the means ± standard deviations from three replicate experiments. (E) Effect of CKI-α knockdown on the abundance of intracellular core protein. Amounts of core in cells for which results are shown in panel D were measured. Results represent the means ± standard deviations from three replicate experiments. (F) Effect of CKI-α knockdown on intracellular infectivity relative to core protein expression. Intracellular infectivity relative to core expression was determined by normalizing the yield of intracellular infectious virus (shown in panel D) with the amount of intracellular core protein shown in panel E. Results represent the means ± standard deviations from three replicate experiments.
Second, the effect of CKI-α knockdown was tested using an HCV subgenomic replicon system. Three days after the siRNA transfection, the cells were coelectroporated with the identical siRNA and JFH-1 subgenomic luciferase reporter replicon RNA, followed by harvesting at different time points (Fig. 4B). The reporter luciferase activity at each time point was corrected with the luciferase value at 4 h posttransfection to normalize transfection efficiencies. Although efficient knockdown was achieved with siRNAs (Fig. 4B, left panel), CKI-α knockdown led to a marginal but nonnegligible decrease in the luciferase activity over the indicated time period. In contrast, PI4K-IIIα knockdown, as a positive control, resulted in a marked decrease (>200-fold) in activity (Fig. 4B, right panel). The effect of CKI-α silencing on the replication of the subgenomic replicon was further analyzed by using two cell lines derived from genotype 1b (LucNeo#2) (38, 39) and genotype 2a (SGR-JFH1/LucNeo) (Fig. 4C). Both Huh-7-based subgenomic replicon cell lines carry a firefly luciferase reporter gene fused to the neomycin phosphotransferase gene. As shown in the right panel of Fig. 4C, knockdown of CKI-α resulted in a marked (∼65%) decrease in replication of the genotype 1b replicon but only a slight (∼10%) decrease in the genotype 2a replication, although knockdown efficiencies of CKI-α were sufficient and comparable in both cell lines (Fig. 4C, left panels). Our result with the genotype 1b replicon was consistent with a previous report (27). In contrast, the limited impact of CKI-α silencing on the replication of the JFH-1 subgenomic replicon suggests that the RNA replication step may not be a key role for CKI-α in the regulation of HCV JFH-1 production.
Finally, we focused on the late stages of the HCV life cycle and analyzed the involvement of CKI-α in virion assembly and release via a single-cycle virus production assay (55), in which Huh7–25 cells lacking CD81 expression were used. Three days posttransfection with siRNAs, the cells were cotransfected with the identical siRNAs and JFH-1 RNA by electroporation. The cells and culture supernatants were harvested after a further 3 days, and titrations of intra- and extracellular infectious virus were assessed (Fig. 4D to F). Reduced NS5A hyperphosphorylation was observed following CKI-α knockdown, but not following ApoE knockdown or transfection with irrelevant siRNA (Fig. 4D, left panel). Both CKI-α and ApoE knockdown led to an ∼10-fold reduction in the yield of extracellular infectious virus compared to the negative control. Approximately a 9-fold reduction was found in the yield of intracellular infectious virus following CKI-α knockdown (Fig. 4D, right panel), indicating that CKI-α is not required for virus release from cells. Despite the marked decrease in intracellular virion yield, CKI-α knockdown resulted in only a 1.3-fold reduction in the abundance of intracellular core protein (Fig. 4E), supporting a limited impact for CKI-α knockdown on viral replication (Fig. 4B). Furthermore, CKI-α silencing led to approximately an 8-fold reduction in the intracellular infectivity relative to core protein expression, which represents the efficiency of viral assembly expressed as the yield of intracellular infectious virus normalized to the amount of intracellular core protein (Fig. 4F). Collectively, these observations suggest that in the HCV life cycle, CKI-α plays a key role most likely in the assembly of infectious viral particles.
NS5A hyperphosphorylation mediated by CKI-α possibly contributes to recruitment of NS5A to low-density membrane structures around LDs in infected cells.
It has been demonstrated that recruitment of NS5A to cytoplasmic low-density membrane structures surrounding LDs, and the interaction of NS5A with the core protein at the site, are essential to HCV assembly (7, 8, 56). To gain mechanistic insight into the function of CKI-α in virion assembly, we performed a subcellular fractionation assay and examined whether NS5A phosphorylation by CKI-α contributed to the subcellular localization of NS5A. Lysates of cells transfected with JFH-1 RNA in the presence or absence of CKI-α silencing (Fig. 5A, left panel) were fractionated with 2.5 to 30% iodixanol gradients followed by immunoblotting of the fractions (Fig. 5A, right panels). In control cells (siCtrl), hyperphosphorylated p58 NS5A predominantly resided in low-density fractions, such as fractions 1 to 3, while hypophosphorylated p56 NS5A localized not only in the low-density fractions but also in high-density fractions, such as fractions 11 and 12. In contrast, knockdown of CKI-α (siCKI-α) decreased the abundance of hyperphosphorylated NS5A and NS5A in the low-density fractions. NS5A levels in the high-density fractions were not reduced by CKI-α knockdown. These results indicate that CKI-α is involved in the distribution of NS5A in cells as well as in its hyperphosphorylation.
FIG 5.

Effects of CKI-α knockdown on the subcellular localization of NS5A and its interaction with LDs or core protein. (A) Iodixanol density gradient analysis (right). Huh7-25 cells transfected with the indicated siRNAs were coelectroporated with the identical siRNAs and JFH-1 RNA. Cell lysates were prepared 3 days after electroporation and fractionated by iodixanol gradients of 2.5% to 30%. The gradient was collected in 0.8-ml fractions for immunoblotting. Total cell lysates before fractionation were loaded as input controls. Detected bands in fractions 1 to 3 and in fractions 11 and 12 are enclosed by squares. (Left) Immunoblotting (IB) results for CKI-α 3 days after electroporation. GAPDH was included as a loading control. (B) Immunoblot (IB) of NS5A and CKI-α 3 days after HCVcc infection. GAPDH was included as a loading control. (C) siRNA delivery efficiency. Cy3 fluorescence (red) was observed 3 days after transfection of Silencer Cy3-labeled GAPDH siRNA. Nuclei were counterstained with Hoechst 33342 (blue). (D) Colocalization of NS5A and LDs. Confocal microscopy images show cells transfected with either CKI-α siRNA (siCKI-α) or an irrelevant control siRNA (siCtrl), followed by infection with JFH-1 virus (left). Cells were fixed with paraformaldehyde 3 days after infection and labeled with an antibody specific for NS5A (red). Cells were counterstained with BODIPY 493/503 (green) to label lipid droplets and with Hoechst 33342 (blue) to label nuclei. Insets represent enlarged views of portions surrounded by squares. Colocalization of NS5A and LDs pixels were assessed quantitatively by intensity correlation analysis using ImageJ software (right). Plots shown represent the ICQ obtained from each of >60 NS5A/LD double-positive cells. Bars indicate the median ± interquartile range of the plots. **, P < 0.01 by two-sided Mann-Whitney test. (E) Colocalization of NS5A and core protein. Confocal microscopy images show cells transfected either with CKI-α siRNA (siCKI-α) or with a control siRNA (siCtrl), followed by infection with JFH-1 virus (left). Fixed cells were labeled with antibodies specific for NS5A (red) and core (green). Nuclei were counterstained with Hoechst 33342 (blue) in the merged images. Mag images represent enlarged views of portions surrounded by squares in the merged images. Colocalization of NS5A and core pixels was assessed quantitatively by intensity correlation analysis using ImageJ software (right). Plots shown represent ICQs obtained from each of >60 NS5A/core double-positive cells. Bars indicate the median ± interquartile range. *, P < 0.05 by two-sided Mann-Whitney test.
We next assessed whether the intracellular localization of NS5A and its interaction with LDs or the core protein are affected by CKI-α knockdown by using laser-scanning confocal immunofluorescence microscopy. Cells were transfected either with CKI-α siRNA (siCKI-α) or with an irrelevant control siRNA (siCtrl), followed by infection with HCVcc. Efficient knockdown of CKI-α was confirmed by immunoblotting and was associated with decreased p58 expression (Fig. 5B). The delivery of siRNA into nearly 100% of the cells was observed with Cy3-labeled siRNA (Silencer Cy3-labeled GAPDH siRNA) (Fig. 5C). In siCtrl-transfected cells, NS5A was colocalized or closely associated with LDs. In contrast, its association with LDs was decreased following CKI-α depletion (Fig. 5D) (P < 0.0001 by two-sided Mann-Whitney test). Similarly, NS5A and the core protein were clearly colocalized in control cells, while their colocalization was reduced in CKI-α knockdown cells (Fig. 5E) (P = 0.0110 by two-sided Mann-Whitney test). These microscopy findings suggest that CKI-α and/or CKI-α-mediated hyperphosphorylation of NS5A is involved in the NS5A-core colocalization at or around LDs in HCV-infected cells. Taken together with the results of our subcellular fractionation assay (Fig. 5A), it is likely that CKI-α plays a role in recruiting NS5A to low-density membrane structures around LDs through hyperphosphorylation of NS5A, and it may facilitate the NS5A-core interaction at these sites.
Identification of potential phospho-acceptor regions for CKI-α.
The above results prompted us to identify the phospho-acceptor sites for CKI-α by using a proteomics approach. Lysates of cells expressing the HCV JFH-1 genome, transfected with either CKI-α siRNA or an irrelevant control siRNA, were immunoprecipitated with an anti-NS5A antibody followed by SDS-PAGE (Fig. 6A). Immunoblotting showed a marked reduction of NS5A p58 following CKI-α knockdown (Fig. 6A, right panel). Silver-stained gel bands of p58 and p56 (Fig. 6A, left panel) were excised and subjected to in-gel digestion, followed by mass spectrometry analysis (Fig. 6B). A total of 629 peptides were identified from both control NS5A (siCtrl) and NS5A with CKI-α knockdown (siCKI-α) after peptide selection with a Mascot peptide score of ≥25 (see Table S3 in the supplemental material) and yielded 53% proteome coverage in total (49.4% for control NS5A and 41.8% for NS5A with CKI-α knockdown), as indicated in the upper panel of Fig. 6B (red letters). Peptides corresponding to domain III in NS5A were not obtained in this analysis. We identified three kinds of phosphopeptides (1, GSPPSEASSSVSQLSAPSLR; 2, APTPPPR; 3, TVGLSESTISEALQQLAIK [Fig. 6B, upper panel, highlighted in green, blue, and yellow, respectively]) (see also Table S3). However, fine mapping of phosphorylation sites was not completely successful in this assay, probably due to the low abundance of immunoprecipitated NS5A. We next assessed which peptide contained the potential phospho-acceptor sites for CKI-α by comparing the frequencies of phosphopeptides identified with and without CKI-α knockdown. As shown in the lower panel of Fig. 6B, the frequency of phosphopeptide 1 relative to the total number of peptide 1 identified was decreased after CKI-α knockdown (from 26.2% to 19.8%). In contrast, the relative frequencies of phosphopeptides 2 and 3 were unaffected or increased by CKI-α knockdown. We noted that the threonine residue in peptide 2 is unlikely to be a consensus phosphorylation site of CKI (57). Thus, the results suggest that peptide 1 (GSPPSEASSSVSQLSAPSLR) is the peptide most likely to contain the amino acids phosphorylated by CKI-α.
FIG 6.

Identification of NS5A phosphopeptides by a phospho-proteome approach. (A) Silver staining and immunoblotting of immunoprecipitated NS5A. Huh-7 cells transfected with the indicated siRNAs were coelectroporated with the identical siRNAs and JFH-1 RNA. Cell lysates were prepared 3 days after electroporation and immunoprecipitated with an anti-NS5A antibody. Immunoprecipitates were subjected to SDS-PAGE, followed by silver staining and immunoblotting (IB). (B) Phosphopeptide mapping of NS5A by LC-MS/MS analysis. p58 and p56 bands of NS5A were excised from the gel and subjected to in-gel digestion, followed by mass spectrometry analysis. Red letters represent the amino acids identified. Three kinds of phosphopeptides identified are highlighted in green (phosphopeptide 1), blue (phosphopeptide 2), and yellow (phosphopeptide 3). The numbers represent amino acid positions within NS5A.
S225 and S232 are key residues involved in NS5A hyperphosphorylation and hyperphosphorylation-dependent regulation of infectious virus production.
Peptide 1 identified above contains eight serine residues that are highly conserved among HCV isolates and are clustered within LCS I (Fig. 7A). To identify amino acids responsible for CKI-α-mediated hyperphosphorylation, we assessed the impacts of alanine or aspartic acid substitutions for these 8 serine residues on NS5A hyperphosphorylation and virus production. An HCV JFH-1 genome with the reporter luciferase, which enabled us to evaluate viral replication by measuring GLuc activity, and a series of its NS5A mutated constructs (Fig. 7A) were generated. Supernatants of cell cultures transfected with the RNA transcripts were harvested at 4, 24, 48, and 72 h posttransfection and subjected to the GLuc assay. As shown in the left panel of Fig. 7B, serine-to-alanine substitution at either aa 229 (S229A) or at aa 235 (S235A) resulted in severe reduction in the viral replication. In contrast, the replication capacities of S222A, S228A, S230A, and S238A mutant reporter viruses were comparable to that of the wild type (WT). S225A and S232A mutations led to a slight but nonnegligible reduction in replication compared to WT. The phospho-mimetic aspartic acid substitution for aa 235 (S235D) exhibited a much higher replication capacity (∼100-fold) than S235A, indicating that phosphorylation of S235 is required for efficient viral replication. In contrast, the replication capacity of S229D was still more-than-10-fold lower than that of WT, suggesting that introduction of negative charge at this position is not sufficient to enhance viral replication (Fig. 7B, right panel). S225D and S232D mutations restored viral replication capacities and exhibited the same replication phenotype as WT. Interestingly, S222D and S230D resulted in a slight reduction in viral replication compared to S222A and S230A, consistent with previous reports (23, 58) (Fig. 7B, right panel).
FIG 7.

Effects of mutations at potential CKI-α phosphorylation sites on NS5A hyperphosphorylation, viral replication, and infectious virion production. (A) Highly conserved serine residues located within the LCS I region of NS5A and the sequences of NS5A mutants used. Arrowheads represent putative phosphorylation sites replaced with alanine or aspartic acid. The numbers represent amino acid positions within NS5A. (B) Viral replication. Huh-7 cells were transfected with the indicated JFH-1-based GLuc reporter constructs, including the WT and its replication-defective mutant (GND). Culture supernatants were harvested at the indicated time points for luciferase assays. The GLuc activity at each time point was normalized with the activity at 4 h posttransfection, and the fold chnages are shown. Results represent the means ± standard deviations from three independent experiments, each performed in triplicate. (C) Infectious virion production. Huh-7 cells were transfected with the indicated JFH-1 viral RNAs, including the WT and its replication-defective mutant (GND). Culture supernatants were harvested 3 days (72 h) later for titrations of infectious virus in a focus-forming unit (FFU) assay. The infectious virus yield of each NS5A mutant was normalized with that of JFH-1 WT, which was set at 100%. Results represent the means ± standard deviations from multiple independent experiments, each performed at least in triplicate. (D) Immunoblot of NS5A in lysates of cells transfected with JFH-1 viral RNAs carrying the indicated serine-to-alanine mutations (left). GAPDH is a loading control. The p58/p56 ratio of each virus was determined by carrying out densitometric analysis of NS5A bands (right). Results shown represent the means ± standard deviations from multiple independent experiments. *, P < 0.05, compared to WT. N.D., not determined, due to low levels of expression. (E) Vaccinia virus-T7 polymerase-mediated expression of NS5A in cells transfected with pJFH1 carrying the indicated serine-to-alanine mutations (left). NS5A bands were quantified by densitometric analysis, and p58/p56 ratios were calculated (right). Data shown represent the means ± standard deviations from multiple independent experiments. *, P < 0.05; **, P < 0.01 (versus WT). (F) Immunoblot of NS5A in lysates of cells transfected with JFH-1 viral RNA carrying the indicated serine-to-aspartic acid mutation. GAPDH was included as a loading control. The p58/p56 ratio of each virus was determined by carrying out densitometric analysis of NS5A bands. Results shown represent the means ± standard deviations from multiple independent experiments. N.D., not determined, due to the poor separation of p58 and p56 bands. (G) Vaccinia virus-T7 polymerase-mediated expression of NS5A in cells transfected with pJFH1 carrying the indicated serine-to-aspartic acid mutation. NS5A bands were quantified by densitometric analysis, and p58/p56 ratios were calculated. Data shown are representative of two independent experiments. N.D., not determined, due to the poor separation of p58 and p56 bands. S and L in panels D and F represent short exposure and long exposure, respectively.
We next evaluated the effects of the NS5A mutations on infectious virus production by titrations of infectious virus in culture supernatants of cells transfected with RNA transcripts of JFH-1 viruses at 72 h posttransfection (Fig. 7C). S225A and S232A mutations resulted in 4- and 5-fold reductions in the virus infectious titer, respectively, compared to WT, while the abilities of S225D and S232D mutants to produce infectious virus were comparable to that of WT. Little or no virus production was observed with S229A, S229D, and S235A mutations, presumably because of their strong negative impacts on viral replication. A slight reduction in virus production observed with S230D and S235D mutations was most likely due to their replication capacities. S222A, S222D, S228A, S228D, S230A, S238A, and S238D substitutions had no significant effect on virus production (>75% of the WT level).
To determine the effect of the NS5A mutations on NS5A hyperphosphorylation, cells expressing JFH-1 viruses were subjected to immunoblotting, and the p58/p56 ratio of NS5A was estimated (Fig. 7D). The hyperphosphorylated p58 band of NS5A was clearly detected in cells transfected with WT and S222A, S228A, S230A, and S238A mutants, which had mean p58/p56 ratios of 0.42, 0.38, 0.35, 0.50, and 0.60, respectively. These p58/p56 ratios were reproducible in multiple repeated experiments but much lower than the p58/p56 ratios (e.g., 0.93 in siCtrl-transfected cells) in Fig. 3A. This difference may be attributed to the difference in the way by which HCV was introduced into cells (virus infection in Fig. 3A and transfection of viral genome in Fig. 7D and F). The p58 levels were significantly reduced in cells transfected with S225A or S232A mutants, which had mean p58/p56 ratios of 0.11 and 0.15, respectively (Fig. 7D). Since the p58/p56 ratios of the S229A and S235A mutants were not determined due to low levels of NS5A expression, we reevaluated the p58/p56 ratio of each viral muatant by using a vaccinia virus-T7 polymerase-mediated protein expression system (Fig. 7E, left panel). Cells transfected with pJFH1 or a series of its NS5A mutants were infected with vaccinia virus expressing the T7 RNA polymerase and harvested for immunoblotting. Similar to the results shown in Fig. 7D, the p58/p56 ratios of S225A and S232A mutants were significantly reduced, while S229A and S235A mutations had no effects (Fig. 7E, right panel). The hyperphosphorylated band of NS5A was observed in cells transfected with the S222D, S225D, S228D, S229D, or S230D mutant in both experimental settings. Interestingly, the S232D, S235D, and S238D mutations resulted in a slight retardation of p56 mobility (Fig. 7F and G), consistent with previous reports (58, 59).
Collectively, S225 and S232 are key residues involved in NS5A hyperphosphorylation and hyperphosphorylation-dependent regulation of infectious virus production. In addition, S225A and S232A reproduced the viral phenotype following CKI-α knockdown more precisely than the other serine mutants within the peptide 1 region. It is most likely that S225 and S232 of NS5A are important for CKI-α-mediated hyperphosphorylation, which is involved in the robust production of infectious HCV.
DISCUSSION
Phosphorylation at serine and threonine residues in HCV NS5A is critical for regulation of the viral life cycle, including genome replication and infectious virus assembly (8, 9, 14, 16–18, 59–61). Several serine/threonine protein kinases have been identified as enzymes that potentially phosphorylate NS5A (9, 25, 26, 28, 50, 51). To our knowledge, however, this study is the first to identify through a kinome-wide screening protein kinases that interact with and phosphorylate NS5A. The in vitro AlphaScreen and phosphorylation assays, followed by RNAi screening on the HCVcc system, identified CKI-α as a major NS5A-associated kinase involved in NS5A hyperphosphorylation and the production of infectious virus.
In a previous study, CKI-α was reported to be involved in the replication of the subgenomic replicon derived from genotype 1b, with evidence that attenuation of CKI-α expression inhibited viral RNA replication up to 60% 5 days after the CKI-α knockdown (27). However, our study with the HCVcc system, as well as detailed analyses dissecting individual steps in the HCV life cycle, revealed that virion assembly is more affected by CKI-α silencing than is viral genome replication. It is highly likely that the CKI-α-mediated hyperphosphorylation of NS5A plays a role in recruiting NS5A to low-density membrane structures around LDs, leading to the acceleration of the early step(s) of virus particle formation. Mutagenesis analyses of putative CKI-α phosphorylation sites identified by a phospho-proteomic approach demonstrated that serine-to-alanine substitution at aa 225 or aa 232 in NS5A did to some extent reproduce the viral phenotype following CKI-α knockdown, indicating that S225 and S232 may be key residues for CKI-α-mediated NS5A hyperphosphorylation and regulation of virion assembly.
It is commonly held that HCV replication is regulated through the tight and delicate control of the ratio between p58 and p56 levels. Adaptive mutations or kinase inhibitors, which reduce NS5A hyperphosphorylation, enhance the HCV RNA replication of genotype 1 isolates, possibly by modulating its interaction with the host vesicle-associated membrane protein-associated protein subtype A (VAP-A), which is an essential factor for HCV replication (17–19, 24). In contrast, reduction of NS5A hyperphosphorylation by RNAi targeting protein kinases results in inhibition of the replication of genotype 1 adaptive replicons, indicating a role for p58 in efficient viral replication (25, 27). Impaired RNA replication resulting from reduced NS5A hyperphosphorylation has also been reported in the case of JFH-1 or JFH-1-based recombinant virus (25, 58–60). Consistent with a previous report (27), we found that CKI-α depletion inhibited the replication of the genotype 1b subgenomic replicon LucNeo#2, which carries the adaptive S2204R mutation in NS5A (38, 39) (Fig. 4C). Our transient-replication assay with the JFH-1 subgenomic replicon showed a slight but significant reduction in replication following CKI-α depletion (Fig. 4B). However, CKI-α silencing did not affect RNA replication in SGR-JFH1/LucNeo cells, where the JFH-1 subgenomic replicon stably replicates (Fig. 4C). Thus, the involvement of CKI-α in HCV RNA replication might be genotype or isolate dependent. We observed a difference in the replication capacity following CKI-α knockdown between transient and stable replication of JFH-1 replicons (Fig. 4B and C). A moderate reduction of replication was also detected when the JFH-1 genome carrying the S225A or S232A mutation was transiently transfected (Fig. 7B). One may infer that CKI-α is involved in the initiation of viral RNA replication rather than in its maintenance.
Our intra- and extracellular infectivity assays following CKI-α depletion suggested that CKI-α primarily targets virion assembly in the HCV life cycle (Fig. 4D to F), although a slight but nonnegligible negative effect of CKI-α knockdown on viral replication was observed (Fig. 4B). It is accepted that the assembly of HCV particles requires recruitment of NS proteins, including NS5A as well as structural proteins, to cytoplasmic membrane structures around LDs, leading to an interaction between NS5A and the core, which is important for efficient encapsidation of the viral genome (7, 8, 56). To understand how CKI-α is involved in virion assembly, we performed a subcellular fractionation assay and immunofluorescence confocal microscopy. Our subcellular fractionation assay clearly showed that hyperphosphorylated NS5A, p58, is mainly localized in low-density membrane fractions, while hypophosphorylated NS5A, p56, prefers high-density fractions. NS5A abundance in lighter fractions was decreased following CKI-α depletion (Fig. 5A). These results are supported by microscopic analyses that demonstrated that CKI-α silencing reduced colocalization of NS5A with LDs and the core (Fig. 5D and E). We tried to confirm the interaction of NS5A and core in HCVcc-infected cells that had been transfected with CKI-α siRNA or irrelevant siRNA. However, the interaction was not observed under this condition, presumably because immunoprecipitated NS5A and/or core was not abundant enough to assess their coimmunoprecipitation in siRNA-transfected cells. Alternatively, we assessed the interaction in Huh-7 cells coexpressing core and NS5A, although no p58 form (no functional NS5A) was detected in this setting. We clearly detected the interaction of NS5A and core in this experiment and found that CKI-α depletion had no significant effect on this interaction (data not shown). Taken together with the results of confocal microscopy (Fig. 5E), this finding might suggest that both (i) phosphorylation of serine residues in the C terminus of NS5A, which is involved in the generation of basally phosphorylated NS5A, as shown previously (8), and (ii) CKI-α-mediated hyperphosphorylation, in which serine residues in the LCS I region are mainly involved, are important for an efficient interaction between NS5A and core in HCV replicating cells. Collectively, CKI-α-mediated hyperphosphorylation of NS5A may contribute to an increase in the local concentration of NS5A at low-density membrane structures around LDs rather than in facilitating the physical interaction of NS5A and core. The relationship between the phosphorylation status of NS5A and its localization on cellular membranes has been previously reported. Miyanari et al. showed that mutated NS5A expressed from JFH-1 variants (JFH1AAA99 and JFH1AAA102 in their report), whose p58/p56 ratios were lower than that of wild-type virus, was not recruited to LDs (56). Qiu et al. fractionated lysates from replicon cells and demonstrated that a substantial amount of hyperphosphorylated NS5A was detected in lighter fractions. Treatment with an NS5A inhibitor, BMS-790052, reduced hyperphosphorylation of NS5A and concomitantly decreased the overall amount of NS5A in low-density membrane fractions (62). These findings raise questions about the regulatory mechanism(s) of the subcellular localization of NS5A, especially at low-density membrane structures. The above-mentioned NS5A mutants, JFH1AAA99 and JFH1AAA102, have triple alanine substitutions for the APK sequence at aa 99 to 101 and the PPT sequence at aa 102 to 104 in NS5A, respectively, but neither is likely to be a CKI recognition site. In addition, the NS5A inhibitor has no inhibitory effect on CKI activity (62). Thus, it appears that two or more kinds of serine/threonine-specific protein kinases, including CKI-α, participate in NS5A phosphorylation that is important for the regulation of its subcellular distribution. It is tempting to speculate that host factors involved in membrane trafficking or lipogenesis possibly interact with NS5A in a phosphorylation-dependent manner and facilitate recruitment of NS5A to low-density membrane structures surrounding LDs. Although further study is needed to validate this speculation, NS5A-interacting factors, such as VAP-A and diacylglycerol acyltransferase-1 (19, 63, 64), might be candidates for involvement in the regulation of this process.
To identify potential phospho-acceptor sites for CKI-α, a phospho-proteome analysis was carried out with NS5A isolated from HCVcc-infected cell lines with and without CKI-α knockdown (Fig. 6). Three kinds of phosphopeptides were identified out of a total of 629 peptides after peptide selection, although fine mapping of the phosphorylation sites was not completely successful. Among them, only the relative frequency of phosphopeptide 1 (GSPPSEASSSVSQLSAPSLR) was decreased after CKI-α knockdown, suggesting the possibility that peptide 1 contains amino acids phosphorylated by CKI-α. Serine residues in peptide 1, which are well conserved among HCV genotypes, matched the consensus sequences for CKI-α-mediated phosphorylation (57). In contrast, a threonine residue in peptide 2 is not conserved and does not match the consensus sequences for phosphorylation by CKI-α. The frequency of phosphopeptide 3 was unchanged with and without CKI-α knockdown (0.1% versus 0.1%). Further mutagenesis analyses targeting the peptide 1 region suggested that S225 and S232 are possible CKI-α phosphorylation sites involved in NS5A hyperphosphorylation and infectious virus production, because alanine substitution for either of these serine residues reproduced the viral phenotype after CKI-α knockdown more accurately than the other mutants within the peptide 1 region (Fig. 7B to E). The S229A and S235A mutations severely impaired viral replication, suggesting that phosphorylation at S229 and S235 is essential for efficient viral replication (Fig. 7B). However, the HCV protein expression assay using vaccinia virus expressing the T7 RNA polymerase revealed that phosphorylation of these residues is not involved in NS5A hyperphosphorylation (Fig. 7E). In the case of genotype 1 isolates, NS5A mutations that reduce hyperphosphorylation enhanced viral RNA replication (17, 18). However, this was not the case for the S225A or S232A mutation in genotype 2a. In addition, S229A and S235A mutations in genotype 1b (Con1) markedly enhance viral replication (18), but the same mutations are lethal in genotype 2a. S232 has been shown to be a potential phosphorylation site for CKI-α by a peptide-based kinase assay in vitro (28). However, the present study is the first to demonstrate the significance of phosphorylation at S225 and S232 in infectious virus production. The sequence coverage of NS5A by our mass spectrometry analysis was less than 60% and was especially low in domain III of NS5A (Fig. 6B). We cannot exclude the possibility of the presence of additional CKI-α phosphorylation sites in NS5A.
Recently, two cellular kinases involved in the regulation of NS5A phosphorylation have been identified. PI4K-IIIα is essential for HCV replication (65–70) and catalyzes the synthesis of phosphatidylinositol 4-phosphate accumulating in HCV replicating cells through its enzymatic activation resulting from an interaction with NS5A (71, 72). PI4K-IIIα directly interacts with the C-terminal end of NS5A domain I, and NS5A–PI4K-IIIα binding is essential for viral replication. Its depletion resulted in a relative increase of p58 abundance, while overexpression of enzymatically active PI4K-IIIα increased the relative abundance of p56 (73). Plk1 has been shown to play a role in viral replication through hyperphosphorylation of NS5A (25). Plk1 was coimmunoprecipitated with NS5A, and knockdown of Plk1 or treatment with a specific inhibitor decreased both NS5A hyperphosphorylation and HCV replication. Since the recognition sites for CKI-α, PI4K-IIIα, and Plk1 have been assumed to be spatially close to each other (25, 28, 73), it is interesting to analyze their interactive actions in regard to regulation of NS5A phosphorylation.
The exquisite balance between the two different phosphorylated forms of NS5A has been proposed to regulate the HCV life cycle; basally phosphorylated p56 abundance is hypothetically involved in viral RNA replication, and hyperphosphorylated p58 is required for virion assembly (9, 17, 18, 73, 74). However, this hypothesis has not yet been fully proven. Our results here provide strong evidence supporting the involvement of NS5A hyperphosphorylation in viral assembly and that of CKI-α in mediating this process. These results not only contribute to a better understanding of the regulatory details of the HCV life cycle but also illuminate targets for potential antiviral strategies.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to F. V. Chisari for kindly providing Huh7.5.1 cells, C. M. Rice for providing anti-NS5A mouse monoclonal antibody (9E10), K. Watashi and K. Shimotohno for providing LucNeo#2 cells, T. Pietschmann for providing the expression plasmid carrying the JFH-1 envelope glycoprotein gene, and Y. Matsuura for providing the plasmid expressing the VSV-G envelope glycoprotein. We thank D. Akazawa for help in preparing HCVpp, the Michael Hooker Microscopy Facility of the University of North Carolina for assistance with confocal microscopy, M. Sasaki, N. Sugiyama, K. Goto, and T. Date for their technical assistance, and T. Mizoguchi for secretarial assistance.
This work was supported in part by grants-in-aid from the Ministry of Health, Labor, and Welfare of Japan and the Ministry of Education, Culture, Sports, Science, and Technology, Japan, by Research on Health Sciences Focusing on Drug Innovation from the Japan Health Sciences Foundation, and by a grant (R01-AI095690) from the U.S. National Institutes of Health.
Footnotes
Published ahead of print 23 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03170-13.
REFERENCES
- 1.Shepard CW, Finelli L, Alter MJ. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5:558–567. 10.1016/S1473-3099(05)70216-4 [DOI] [PubMed] [Google Scholar]
- 2.Alter MJ. 2007. Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 13:2436–2441 http://www.wjgnet.com/1007-9327/13/2436.asp [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ, Weiner AJ, Bradley DW, Kuo G, Houghton M. 1991. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 88:2451–2455. 10.1073/pnas.88.6.2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki T, Ishii K, Aizaki H, Wakita T. 2007. Hepatitis C viral life cycle. Adv. Drug Deliv. Rev. 59:1200–1212. 10.1016/j.addr.2007.04.014 [DOI] [PubMed] [Google Scholar]
- 5.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. 10.1126/science.285.5424.110 [DOI] [PubMed] [Google Scholar]
- 6.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76:5974–5984. 10.1128/JVI.76.12.5974-5984.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 4:e1000035. 10.1371/journal.ppat.1000035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, Suzuki T. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J. Virol. 82:7964–7976. 10.1128/JVI.00826-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4:e1000032. 10.1371/journal.ppat.1000032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi ST, Lee KJ, Aizaki H, Hwang SB, Lai MM. 2003. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 77:4160–4168. 10.1128/JVI.77.7.4160-4168.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyanari Y, Hijikata M, Yamaji M, Hosaka M, Takahashi H, Shimotohno K. 2003. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 278:50301–50308. 10.1074/jbc.M305684200 [DOI] [PubMed] [Google Scholar]
- 12.Tellinghuisen TL, Marcotrigiano J, Rice CM. 2005. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435:374–379. 10.1038/nature03580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang L, Hwang J, Sharma SD, Hargittai MR, Chen Y, Arnold JJ, Raney KD, Cameron CE. 2005. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J. Biol. Chem. 280:36417–36428. 10.1074/jbc.M508175200 [DOI] [PubMed] [Google Scholar]
- 14.Reed KE, Xu J, Rice CM. 1997. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J. Virol. 71:7187–7197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanji Y, Kaneko T, Satoh S, Shimotohno K. 1995. Phosphorylation of hepatitis C virus-encoded nonstructural protein NS5A. J. Virol. 69:3980–3986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaneko T, Tanji Y, Satoh S, Hijikata M, Asabe S, Kimura K, Shimotohno K. 1994. Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem. Biophys. Res. Commun. 205:320–326. 10.1006/bbrc.1994.2667 [DOI] [PubMed] [Google Scholar]
- 17.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974. 10.1126/science.290.5498.1972 [DOI] [PubMed] [Google Scholar]
- 18.Appel N, Pietschmann T, Bartenschlager R. 2005. Mutational analysis of hepatitis C virus nonstructural protein 5A: potential role of differential phosphorylation in RNA replication and identification of a genetically flexible domain. J. Virol. 79:3187–3194. 10.1128/JVI.79.5.3187-3194.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans MJ, Rice CM, Goff SP. 2004. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc. Natl. Acad. Sci. U. S. A. 101:13038–13043. 10.1073/pnas.0405152101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katze MG, Kwieciszewski B, Goodlett DR, Blakely CM, Neddermann P, Tan SL, Aebersold R. 2000. Ser(2194) is a highly conserved major phosphorylation site of the hepatitis C virus nonstructural protein NS5A. Virology 278:501–513. 10.1006/viro.2000.0662 [DOI] [PubMed] [Google Scholar]
- 21.Reed KE, Rice CM. 1999. Identification of the major phosphorylation site of the hepatitis C virus H strain NS5A protein as serine 2321. J. Biol. Chem. 274:28011–28018 [DOI] [PubMed] [Google Scholar]
- 22.Nordle Gilliver A, Griffin S, Harris M. 2010. Identification of a novel phosphorylation site in hepatitis C virus NS5A. J. Gen. Virol. 91:2428–2432. 10.1099/vir.0.023614-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemay KL, Treadaway J, Angulo I, Tellinghuisen TL. 2013. A hepatitis C virus NS5A phosphorylation site that regulates RNA replication. J. Virol. 87:1255–1260. 10.1128/JVI.02154-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neddermann P, Quintavalle M, Di Pietro C, Clementi A, Cerretani M, Altamura S, Bartholomew L, De Francesco R. 2004. Reduction of hepatitis C virus NS5A hyperphosphorylation by selective inhibition of cellular kinases activates viral RNA replication in cell culture. J. Virol. 78:13306–13314. 10.1128/JVI.78.23.13306-13314.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen YC, Su WC, Huang JY, Chao TC, Jeng KS, Machida K, Lai MM. 2010. Polo-like kinase 1 is involved in hepatitis C virus replication by hyperphosphorylating NS5A. J. Virol. 84:7983–7993. 10.1128/JVI.00068-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coito C, Diamond DL, Neddermann P, Korth MJ, Katze MG. 2004. High-throughput screening of the yeast kinome: identification of human serine/threonine protein kinases that phosphorylate the hepatitis C virus NS5A protein. J. Virol. 78:3502–3513. 10.1128/JVI.78.7.3502-3513.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quintavalle M, Sambucini S, Di Pietro C, De Francesco R, Neddermann P. 2006. The alpha isoform of protein kinase CKI is responsible for hepatitis C virus NS5A hyperphosphorylation. J. Virol. 80:11305–11312. 10.1128/JVI.01465-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quintavalle M, Sambucini S, Summa V, Orsatti L, Talamo F, De Francesco R, Neddermann P. 2007. Hepatitis C virus NS5A is a direct substrate of casein kinase I-alpha, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J. Biol. Chem. 282:5536–5544. 10.1074/jbc.M610486200 [DOI] [PubMed] [Google Scholar]
- 29.Kato T, Date T, Miyamoto M, Sugiyama M, Tanaka Y, Orito E, Ohno T, Sugihara K, Hasegawa I, Fujiwara K, Ito K, Ozasa A, Mizokami M, Wakita T. 2005. Detection of anti-hepatitis C virus effects of interferon and ribavirin by a sensitive replicon system. J. Clin. Microbiol. 43:5679–5684. 10.1128/JCM.43.11.5679-5684.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796. 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phan T, Beran RK, Peters C, Lorenz IC, Lindenbach BD. 2009. Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1–E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 83:8379–8395. 10.1128/JVI.00891-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO. 2005. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol. Ther. 11:435–443. 10.1016/j.ymthe.2004.10.016 [DOI] [PubMed] [Google Scholar]
- 33.Ryan MD, King AM, Thomas GP. 1991. Cleavage of foot-and-mouth disease virus polyprotein is mediated by residues located within a 19 amino acid sequence. J. Gen. Virol. 72:2727–2732. 10.1099/0022-1317-72-11-2727 [DOI] [PubMed] [Google Scholar]
- 34.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199. 10.1016/0378-1119(91)90434-D [DOI] [PubMed] [Google Scholar]
- 35.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299. 10.1073/pnas.0503596102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akazawa D, Date T, Morikawa K, Murayama A, Miyamoto M, Kaga M, Barth H, Baumert TF, Dubuisson J, Wakita T. 2007. CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J. Virol. 81:5036–5045. 10.1128/JVI.01573-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. 2003. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 125:1808–1817. 10.1053/j.gastro.2003.09.023 [DOI] [PubMed] [Google Scholar]
- 38.Murata T, Ohshima T, Yamaji M, Hosaka M, Miyanari Y, Hijikata M, Shimotohno K. 2005. Suppression of hepatitis C virus replicon by TGF-beta. Virology 331:407–417. 10.1016/j.virol.2004.10.036 [DOI] [PubMed] [Google Scholar]
- 39.Goto K, Watashi K, Murata T, Hishiki T, Hijikata M, Shimotohno K. 2006. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem. Biophys. Res. Commun. 343:879–884. 10.1016/j.bbrc.2006.03.059 [DOI] [PubMed] [Google Scholar]
- 40.Murayama A, Sugiyama N, Yoshimura S, Ishihara-Sugano M, Masaki T, Kim S, Wakita T, Mishiro S, Kato T. 2012. A subclone of HuH-7 with enhanced intracellular hepatitis C virus production and evasion of virus related-cell cycle arrest. PLoS One 7:e52697. 10.1371/journal.pone.0052697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tadokoro D, Takahama S, Shimizu K, Hayashi S, Endo Y, Sawasaki T. 2010. Characterization of a caspase-3-substrate kinome using an N- and C-terminally tagged protein kinase library produced by a cell-free system. Cell Death Dis. 1:e89. 10.1038/cddis.2010.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawasaki T, Gouda MD, Kawasaki T, Tsuboi T, Tozawa Y, Takai K, Endo Y. 2005. The wheat germ cell-free expression system: methods for high-throughput materialization of genetic information. Methods Mol. Biol. 310:131–144. 10.1007/978-1-59259-948-6_10 [DOI] [PubMed] [Google Scholar]
- 43.Sawasaki T, Ogasawara T, Morishita R, Endo Y. 2002. A cell-free protein synthesis system for high-throughput proteomics. Proc. Natl. Acad. Sci. U. S. A. 99:14652–14657. 10.1073/pnas.232580399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sawasaki T, Kamura N, Matsunaga S, Saeki M, Tsuchimochi M, Morishita R, Endo Y. 2008. Arabidopsis HY5 protein functions as a DNA-binding tag for purification and functional immobilization of proteins on agarose/DNA microplate. FEBS Lett. 582:221–228. 10.1016/j.febslet.2007.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1–E2 envelope protein complexes. J. Exp. Med. 197:633–642. 10.1084/jem.20021756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masaki T, Suzuki R, Saeed M, Mori K, Matsuda M, Aizaki H, Ishii K, Maki N, Miyamura T, Matsuura Y, Wakita T, Suzuki T. 2010. Production of infectious hepatitis C virus by using RNA polymerase I-mediated transcription. J. Virol. 84:5824–5835. 10.1128/JVI.02397-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masaki T, Matsuura T, Ohkawa K, Miyamura T, Okazaki I, Watanabe T, Suzuki T. 2006. All-trans retinoic acid down-regulates human albumin gene expression through the induction of C/EBPβ-LIP. Biochem. J. 397:345–353. 10.1042/BJ20051863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwahori T, Matsuura T, Maehashi H, Sugo K, Saito M, Hosokawa M, Chiba K, Masaki T, Aizaki H, Ohkawa K, Suzuki T. 2003. CYP3A4 inducible model for in vitro analysis of human drug metabolism using a bioartificial liver. Hepatology 37:665–673. 10.1053/jhep.2003.50094 [DOI] [PubMed] [Google Scholar]
- 49.Li Q, Lau A, Morris TJ, Guo L, Fordyce CB, Stanley EF. 2004. A syntaxin 1, Gα(o), and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J. Neurosci. 24:4070–4081. 10.1523/JNEUROSCI.0346-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim J, Lee D, Choe J. 1999. Hepatitis C virus NS5A protein is phosphorylated by casein kinase II. Biochem. Biophys. Res. Commun. 257:777–781. 10.1006/bbrc.1999.0460 [DOI] [PubMed] [Google Scholar]
- 51.Ide Y, Tanimoto A, Sasaguri Y, Padmanabhan R. 1997. Hepatitis C virus NS5A protein is phosphorylated in vitro by a stably bound protein kinase from HeLa cells and by cAMP-dependent protein kinase A-alpha catalytic subunit. Gene 201:151–158. 10.1016/S0378-1119(97)00440-X [DOI] [PubMed] [Google Scholar]
- 52.Benga WJ, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, Schuster C. 2010. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51:43–53. 10.1002/hep.23278 [DOI] [PubMed] [Google Scholar]
- 53.Farquhar MJ, Harris HJ, Diskar M, Jones S, Mee CJ, Nielsen SU, Brimacombe CL, Molina S, Toms GL, Maurel P, Howl J, Herberg FW, van Ijzendoorn SC, Balfe P, McKeating JA. 2008. Protein kinase A-dependent step(s) in hepatitis C virus entry and infectivity. J. Virol. 82:8797–8811. 10.1128/JVI.00592-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- 55.Matsumura T, Kato T, Sugiyama N, Tasaka-Fujita M, Murayama A, Masaki T, Wakita T, Imawari M. 2012. 25-Hydroxyvitamin D3 suppresses hepatitis C virus production. Hepatology 56:1231–1239. 10.1002/hep.25763 [DOI] [PubMed] [Google Scholar]
- 56.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097. 10.1038/ncb1631 [DOI] [PubMed] [Google Scholar]
- 57.Ubersax JA, Ferrell JE., Jr 2007. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 8:530–541. 10.1038/nrm2203 [DOI] [PubMed] [Google Scholar]
- 58.Ross-Thriepland D, Harris M. 2014. Insights into the complexity and functionality of hepatitis C virus NS5A phosphorylation. J. Virol. 88:1421–1432. 10.1128/JVI.03017-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fridell RA, Valera L, Qiu D, Kirk MJ, Wang C, Gao M. 2013. Intragenic complementation of hepatitis C virus NS5A RNA replication-defective alleles. J. Virol. 87:2320–2329. 10.1128/JVI.02861-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fridell RA, Qiu D, Valera L, Wang C, Rose RE, Gao M. 2011. Distinct functions of NS5A in hepatitis C virus RNA replication uncovered by studies with the NS5A inhibitor BMS-790052. J. Virol. 85:7312–7320. 10.1128/JVI.00253-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim S, Welsch C, Yi M, Lemon SM. 2011. Regulation of the production of infectious genotype 1a hepatitis C virus by NS5A domain III. J. Virol. 85:6645–6656. 10.1128/JVI.02156-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qiu D, Lemm JA, O'Boyle DR, II, Sun JH, Nower PT, Nguyen V, Hamann LG, Snyder LB, Deon DH, Ruediger E, Meanwell NA, Belema M, Gao M, Fridell RA. 2011. The effects of NS5A inhibitors on NS5A phosphorylation, polyprotein processing and localization. J. Gen. Virol. 92:2502–2511. 10.1099/vir.0.034801-0 [DOI] [PubMed] [Google Scholar]
- 63.Camus G, Herker E, Modi AA, Haas JT, Ramage HR, Farese RV, Jr, Ott M. 2013. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J. Biol. Chem. 288:9915–9923. 10.1074/jbc.M112.434910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gao L, Aizaki H, He JW, Lai MM. 2004. Interactions between viral nonstructural proteins and host protein hVAP-33 mediate the formation of hepatitis C virus RNA replication complex on lipid raft. J. Virol. 78:3480–3488. 10.1128/JVI.78.7.3480-3488.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. 2009. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:7577–7582. 10.1073/pnas.0902693106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vaillancourt FH, Pilote L, Cartier M, Lippens J, Liuzzi M, Bethell RC, Cordingley MG, Kukolj G. 2009. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 387:5–10. 10.1016/j.virol.2009.02.039 [DOI] [PubMed] [Google Scholar]
- 67.Borawski J, Troke P, Puyang X, Gibaja V, Zhao S, Mickanin C, Leighton-Davies J, Wilson CJ, Myer V, Cornellataracido I, Baryza J, Tallarico J, Joberty G, Bantscheff M, Schirle M, Bouwmeester T, Mathy JE, Lin K, Compton T, Labow M, Wiedmann B, Gaither LA. 2009. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 83:10058–10074. 10.1128/JVI.02418-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, Chung RT. 2009. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host. Microbe 5:298–307. 10.1016/j.chom.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, Elledge SJ. 2009. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. U. S. A. 106:16410–16415. 10.1073/pnas.0907439106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trotard M, Lepere-Douard C, Regeard M, Piquet-Pellorce C, Lavillette D, Cosset FL, Gripon P, Le Seyec J. 2009. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 23:3780–3789. 10.1096/fj.09-131920 [DOI] [PubMed] [Google Scholar]
- 71.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, Poenisch M, Blankenburg H, Hiet MS, Longerich T, Diehl S, Ramirez F, Balla T, Rohr K, Kaul A, Buhler S, Pepperkok R, Lengauer T, Albrecht M, Eils R, Schirmacher P, Lohmann V, Bartenschlager R. 2011. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9:32–45. 10.1016/j.chom.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berger KL, Kelly SM, Jordan TX, Tartell MA, Randall G. 2011. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 85:8870–8883. 10.1128/JVI.00059-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reiss S, Harak C, Romero-Brey I, Radujkovic D, Klein R, Ruggieri A, Rebhan I, Bartenschlager R, Lohmann V. 2013. The lipid kinase phosphatidylinositol-4 kinase III alpha regulates the phosphorylation status of hepatitis C virus NS5A. PLoS Pathog. 9:e1003359. 10.1371/journal.ppat.1003359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pietschmann T, Zayas M, Meuleman P, Long G, Appel N, Koutsoudakis G, Kallis S, Leroux-Roels G, Lohmann V, Bartenschlager R. 2009. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 5:e1000475. 10.1371/journal.ppat.1000475 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.