

ABSTRACT
Kaposi's sarcoma-associated herpesvirus (KSHV) typically displays two different phases in its life cycle, the default latent phase and the lytic phase. There is a short period of lytic gene expression in the early stage of KSHV primary infection. The factors involved in the shutdown process of lytic gene expression are poorly identified. It has been shown that the latency-associated nuclear antigen (LANA) encoded by KSHV plays an important role in the establishment of viral latency. In screening, we identified a host protein, Krüppel-associated box domain-associated protein 1 (KAP1), that bound to LANA. We validated the interaction between LANA and KAP1 in vivo and in vitro, as well as their colocalization in the nucleus. We mapped out that LANA interacted with both the N- and C-terminal domains of KAP1. Based on the interface of LANA-KAP1 interaction determined, we proved that LANA recruited KAP1 to the RTA promoter region of the KSHV genome. We revealed that KAP1 was involved in transcriptional repression by LANA. We found multiple cooccupation sites of LANA and KAP1 on the whole KSHV genome by chromatin immunoprecipitation for sequencing (ChIP-seq) and demonstrated that LANA-recruited KAP1 played a critical role in the shutdown of lytic gene expression during the early stage of KSHV primary infection. Taken together, our data suggest that LANA interacts with KAP1 and represses lytic gene expression to facilitate the establishment of KSHV latency.
IMPORTANCE Our study revealed the mechanism of transcriptional repression by LANA during KSHV primary infection, providing new insights into the process of KSHV latency establishment.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also called human herpesvirus 8, belongs to the subfamily Gammaherpesviridae. It is the most recently identified oncogenic DNA herpesvirus. KSHV is related to Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease (MCD), which cause severe illness in AIDS patients (1–3). As a member of the herpesvirus family, KSHV typically displays two different phases in its life cycle, the default latent phase and the lytic phase (4, 5). No infectious viral particles are produced, and only limited gene expression is detected during the default latent phase (6, 7). The virus can be reactivated from the latent phase with specific stimuli, such as valproic acid, butyrate, or hypoxia (8–10). Replication transcriptional activator (RTA), the switch protein encoded by KSHV, is able to trigger the reactivation; however, its expression is tightly restricted during the latent phase (11–13). The establishment of KSHV latency is a key step for viral infection. There is a short period of lytic gene expression at the early stage of KSHV primary infection (14–16). Lytic gene expression begins early and is then quickly shut down in the human foreskin fibroblast, primary human dermal microvascular endothelial cell, and primary human umbilical vein endothelial cell infection systems (14, 15). The process of lytic gene expression shutdown remains largely unknown, and the factors involved have not been fully identified. It has been shown that the latency-associated nuclear antigen (LANA) encoded by KSHV plays an important role in the establishment of KSHV latency (13, 17–20).
The LANA protein is essential for the replication and persistence of the viral episome during latent infection (21–23). Deleting LANA from the KSHV genome results in the loss of the viral episome and enhanced lytic gene expression (18, 19). At the early stage of KSHV primary infection, lytic gene expression is shut down, along with increasing LANA expression (14, 20). LANA can inhibit RTA expression by repressing its promoter (13, 17). The same phenomenon is also seen in rhesus monkey rhadinovirus, which suggests a conserved mechanism of viral latency control in the herpesvirus family (24). However, the mechanism by which LANA represses gene expression remains less clear. It has been reported that LANA can accumulate heterochromatin components on the terminal repeat (TR) of the KSHV genome to repress viral gene expression and recruit Dnmt3a or other repressors to silence host gene expression (25–27), but how LANA represses the transcriptional activity of the RTA promoter remains largely unknown. Both the N- and C-terminal domains of LANA can mediate repression of gene expression (28, 29). In an artificial system, it has been shown that the N-terminal domain can interact with the mSin3A complex to repress gene expression (30), yet little is known about the C-terminal domain of LANA.
In screening, we identified a host protein, Krüppel-associated box domain-associated protein 1 (KAP1), that binds to LANA. KAP1 is also known as tripartite-motif-containing protein 28 (TRIM28) and transcription intermediary factor 1β (TIF1β) (31). KAP1 functions as a transcriptional repressor and can change epigenetic state by recruiting a histone deacetylase (HDAC) and methyltransferase complex (31–33). KAP1 can be recruited to the genome as a scaffold protein by KRAB (Krüppel-associated box) or non-KRAB zinc finger protein (31). The repression mediated by the KRAB-KAP1 complex can exert a long-range effect on the genome by spreading H3K9me3 and heterochromatin protein 1β (HP1β) (34). Also, the KRAB-KAP1 complex can mediate the repression of episomal gene expression of adeno-associated viral and nonintegrated lentiviral vectors, which implies a functional role in the herpesvirus family (35). A previous study has demonstrated that KAP1 regulates KSHV latency by association with transcriptional promoters in the viral genome, which is modulated by the viral protein kinase (vPK) (36). However, how KAP1 is recruited to the KSHV genome is completely unknown.
In this study, we found and validated direct interaction between LANA and KAP1, as well as their colocalization in the nucleus. We mapped out that LANA can interact with both the N- and C-terminal domains of KAP1. Based on the interface determined, we proved that LANA can recruit KAP1 to the KSHV genome to repress gene expression. We found multiple cooccupation sites of LANA and KAP1 on the whole KSHV genome and demonstrated that LANA-recruited KAP1 plays a critical role in the shutdown of lytic gene expression during the early stage of KSHV primary infection. Our results indicate that LANA interacts with the KAP1 protein and represses lytic gene expression to facilitate the establishment of KSHV latency.
MATERIALS AND METHODS
Cells, plasmids, and antibodies.
KSHV-positive B lymphoma cell lines (BCBL1, JSC-1, and BC-3) were maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and antibiotics (penicillin and streptomycin). Human embryonic kidney (HEK) 293T and HeLa cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS and antibiotics (penicillin and streptomycin). The HeLa-shKAP1 stable cell line was a kind gift from Chuangui Wang (East China Normal University, Shanghai, China). All cell lines were grown at 37°C supplemented with 5% CO2.
A DNA construct expressing Strep-Flag-tagged LANA was generated by cloning full-length LANA into a modified pCDH-puro vector, which was introduced into a fragment encoding tandem Strep-tag II and Flag epitopes at the BamHI site. A plasmid encoding Flag-tagged KAP1 was a kind gift from Xiajun (John) Li (Black Family Stem Cell Institute, New York, NY, USA). Plasmid pCAGGS-HA-LANA was described previously (37). The LANA truncated constructs glutathione S-transferase (GST)-LANA-N (amino acids [aa] 1–340) and GST-LANA-C (aa 1022–1162) were described previously (38). GST-fused full-length KAP1 and its truncated constructs (ΔRBCC, ΔPB, RBCC, M, and PB) were obtained by cloning the corresponding fragments into the 5′ EcoRI and 3′ XhoI sites of pGEX-4T-1 vector. Hemagglutinin (HA)-tagged KAP1 truncated constructs (ΔRBCC, ΔPB, and M) were constructed by cloning the corresponding fragments into the pCMV-HA vector in frame at the 5′ EcoRI and 3′ XhoI sites. All primers are listed in Table 1. The luciferase reporter plasmid with the RTA promoter (∼3 kb), pGL2-RTAp, was described previously (13). Plasmid pGL3p-TR was generated by subcloning the TR fragment from plasmid pBSpuroA3 (a kind gift from Subhash C. Verma, University of Nevada, Reno, NV, USA) into the pGL3p vector at the NotI site.
TABLE 1.
Primers for PCR amplification and analysis

Anti-HA and anti-Flag M2 monoclonal antibodies were purchased from Sigma. Anti-KAP1 rabbit monoclonal antibody (no. 4124) was purchased from Cell Signaling Technology. Anti-KAP1 mouse monoclonal antibody (chromatin immunoprecipitation [ChIP] grade; ab22553) was purchased from Abcam. Anti-acetyl-histone H3 (anti-AcH3) rabbit polyclonal antibody (no. 06-599) was purchased from Merck Millipore. Anti-LANA mouse monoclonal antibody produced by 1B5 hybridoma was made in our laboratory (antigen source for immunization, LANA aa 900 to 1162).
TAP-MS.
Tandem affinity purification-mass spectrometry (TAP-MS) was used to identify LANA protein complexes. The procedures were described previously (39). Strep-Flag-tagged LANA was overexpressed in 293T cells. Specifically, Strep-Tactin Sepharose (IBA) and Flag M2 Sepharose (Sigma) were utilized to purify native protein complexes.
Immunoprecipitation (IP) and Western blotting.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF]) for 15 min. The cells were centrifuged at 12,000 × g at 4°C for 30 min to remove cell debris. Five percent of the cell lysates were kept as inputs, and the remainder were precleared with protein A- or protein G-coupled Sepharose (Life Technologies) for 2 h at 4°C and then immunoprecipitated with the corresponding antibodies for 6 to 12 h. For the Strep-tagged protein, the cell lysates were directly incubated with Strep-Tactin Sepharose for 6 to 10 h. The immunoprecipitates were washed four times with RIPA buffer and then boiled in SDS loading buffer for Western blot analysis. For Western blotting, protein samples were analyzed by SDS-PAGE and transferred onto nitrocellulose membranes, followed by blocking and probing with the indicated antibodies for detection.
Protein purification and in vitro binding assay (GST pulldown).
Escherichia coli strain BL21(DE3) expressing GST or GST fusion proteins was grown in Luria broth (LB) medium at 37°C to exponential phase and cultured overnight at 16°C after induction with isopropyl-thiogalactopyranoside. Cells were harvested and resuspended in ice-cold phosphate-buffered saline (PBS), followed by sonication lysis (Sonics; cycle, 3-s on/5-s off pulses; amplitude, 35%). The cell lysates were centrifuged at 12,000 × g to obtain the supernatant, which was combined with Sepharose 4B-glutathione resin (GE Healthcare) for affinity purification, according to the manufacturer's instructions. In vitro-translated biotin-labeled proteins produced by a TNT coupled transcription/translation system (Promega) or nuclear extracts were incubated with purified GST fusion protein-bound beads in RIPA buffer for 6 to 8 h. After washing with RIPA buffer four times, the pulled-down products were analyzed with a Transcend chemiluminescent translation detection system (Promega) or by Western blotting.
Immunofluorescence assay (IFA).
Cells were fixed with 4% paraformaldehyde for 30 min, followed by permeabilization with 0.1% Triton X-100 for 10 min. After blocking with 5% normal goat serum (Life Technologies) for 1 h, the cells were incubated with anti-LANA mouse monoclonal antibody and anti-KAP1 rabbit monoclonal antibody at a 1:50 dilution for 1 h and then further stained with goat anti-mouse IgG conjugated with Alexa Fluor 488 (Life Technologies) and goat anti-rabbit IgG conjugated with Alexa Fluor 555 (Life Technologies) at a 1:1,000 dilution for 1 h. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma). Coverslips were mounted with anti-fade mounting medium (Beyotime) and photographed using a digital camera and software (Leica).
ChIP.
The ChIP protocol was adapted from the Rockland website with some modifications. Cells were cross-linked in the medium with 1% formaldehyde for 30 min (LANA and KAP1) or 5 min (acetyl-histone H3) at room temperature and quenched with 0.125 M glycine. After cross-linking, the cells were washed with PBS twice and resuspended in 1 ml buffer A (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.2% Triton X-100, 1 mM dithiothreitol, 0.5 mM EDTA, 0.2 mM PMSF) for 10 min at 4°C. The extracted nuclei were pelleted by centrifugation at 1,300 × g for 5 min. The nuclei were lysed in SDS lysis buffer (50 mM HEPES, 1 mM EDTA, 1% SDS, 1 mM PMSF) for 10 min on ice. The lysates were subjected to sonication to obtain 200- to 500-bp fragments of DNA (Sonics; cycle, 2 6-s pulses; amplitude, 30 to 35%) and then centrifuged at 12,000 × g at 4°C for 10 min to obtain the supernatants. The supernatants were diluted 1:10 with RIPA buffer. Samples were precleared with pretreated protein A or G beads (1 mg/ml bovine serum albumin [BSA], 1 mg/ml sperm DNA, 20% beads) for 2 h at 4°C. A small fraction of the supernatants were kept as input, and the remainder were divided into groups according to the experiment. The aliquots were incubated with pretreated protein A or G beads and the corresponding antibody overnight at 4°C. After extensive washing with RIPA buffer, wash buffer (20 mM Tris-HCl, pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 1 mM PMSF) and TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) (four times each), the beads were resuspended in TE buffer. The resuspended beads were subjected to RNase A and proteinase K digestion, and the cross-linking was reversed at 65°C for 8 to 10 h. DNA was recycled with a DNA purification kit (Tiangen).
RNA interference.
KAP1 knockdown was achieved with short hairpin RNA (shRNA) (target sequence, GCGTCCTGGCACTAACTCA), as previously described (40). The knockdown effect of KAP1 cannot be detected at the protein level in a transient-transfection assay (we tested at least five reported shRNAs, and none of them worked); thus, the stable HeLa-shKAP1 cell line (a kind gift from Chuangui Wang) was utilized in this study.
Luciferase assay.
All the procedures were described in the manual of the luciferase assay system (Promega). A dual luciferase reporter assay was not used because LANA may affect the expression of the control plasmid (29). The luciferase reporter plasmid was transfected into cells, along with the plasmid expressing HA-LANA, and the total amount of DNA was normalized with empty vector in the transfection. Cells were harvested at 36 h posttransfection for the luciferase assay.
KSHV virion purification and primary infection.
KSHV virions were acquired by the induction of iSLK.219 cells with doxycycline as previously described (41). The primary infection of HeLa-Ctrl and HeLa-shKAP1 cells was achieved by centrifugation at 1,250 × g at 30°C for 2 h after adding concentrated virus to the medium.
qPCR.
Quantitative PCR (qPCR) was used to determine the relative quantities of RNA (cDNA) and DNA. Total RNA was extracted from harvested cells using TRIzol reagent (Life Technologies), and cDNA was obtained by reverse transcription with a genomic DNA (gDNA) eraser RT kit (TaKaRa). Total DNA (host and viral genomes) was extracted from harvested cells using the genome DNA extraction kit (Life Technologies). qPCR was performed with a SYBR green Real-Time PCR master mix kit (Toyobo). Reaction mixtures contained 5 μl master mix plus Rox passive reference dye, 1 μM each primer, and 4 μl diluted sample. All primers are listed in Table 1. The program set on the 7900HT sequence detection system (Life Technologies) was 95°C for 5 min, followed by 40 cycles at 95°C for 15 s, 58°C for 20 s, and 72°C for 30 s. Values for the relative quantification were calculated by the ΔΔCT method. Melting curve analysis was performed to verify the specificity of the products, and each sample was tested in triplicate.
ChIP-seq.
The procedures for ChIP for sequencing (ChIP-seq) were similar to those for ChIP described above, except for the pretreated beads, to which sperm DNA was not added. More cells were needed for ChIP-seq, and 108 BC-3 cells were harvested for the experiment. The lysates were sonicated to obtain DNA fragments of ∼200 bp. The enriched DNA was subjected to library preparation for high-throughput sequencing. The ChIP-seq library was prepared according to the Illumina instructions. The size selection range for the library was 100 to 400 bp.
Bioinformatics analysis of ChIP-seq data.
The ChIP-seq data were aligned with the human genome (hg19) and the KSHV genome (HQ404500 plus 35 copies of the TR [U75699.1]) using Bowtie2 (42); only one mismatch was allowed. The output files were subjected to peak calling with MACS (model-based analysis of ChIP-seq), as described previously (43). The data aligned to the human genome were analyzed to obtain a model, because the KSHV genome is too small to build a model for analysis. All the parameters were default, except that the parameter of the fold range was changed to 8 to 30. The default P value cutoff for peak detection was 10−5. However, the d values (distances between the modes of the Watson and Crick peaks) in the calculated models of LANA and KAP1 were both <60 bp, so the analysis was done without the MACS model (−nomodel), according to the protocol (43). The parameters were customized to analyze data aligned to the KSHV genome. Because of the smaller size of the KSHV genome, the IgG group was not used as a control for its strong bias, and the parameters were set as follows: --bw (bandwidth) = 100, --nomodel (without the MACS model), --shiftsize (shift tags by an arbitrary number) = 50, --nolambda (use of a global lambda). P value cutoffs at 10−3 and 10−5 were both analyzed. The results were visualized with IGV software (44). For data validation, we chose the more reliable results (P < 10−5).
RESULTS
LANA forms a complex with KAP1 in vivo and in vitro.
LANA works like a scaffold protein in the nucleus, with many protein-protein interactions (38, 45). To study the function of the LANA protein systematically, we used TAP-MS to identify proteins interacting with LANA. Strep-Flag-tagged LANA was expressed in HEK 293T cells. The cell lysates were subjected to affinity purification with streptavidin beads, followed by IP with Flag M2 beads, and the purified eluates were analyzed by MS. A few novel proteins were identified by peptide correlation with the International Protein Index (IPI) database. The results are summarized in Table 2. KAP1 (an ∼110-kDa band was observed) (Fig. 1A) was one of the identified binding partners of LANA. The interaction between LANA and KAP1 was first confirmed by coimmunoprecipitation (co-IP). Flag-tagged KAP1 was transfected into HEK 293T cells, along with Strep-Flag-tagged LANA. After affinity purification with streptavidin beads, the purified proteins, along with input samples, were detected by Western blotting with anti-Flag and anti-KAP1 antibodies. KAP1 was coimmunoprecipitated with LANA (Fig. 1B). The reverse co-IP experiment was also used to test the interaction. HA-tagged LANA and Flag-tagged KAP1 were utilized. After IP with Flag M2 beads, the immunoprecipitated proteins, along with input samples, were detected by Western blotting with anti-Flag and anti-HA antibodies. LANA was coimmunoprecipitated with KAP1 (Fig. 1C). The interaction between endogenous LANA and KAP1 was verified in JSC-1 cells (Fig. 1D). To determine whether this interaction was direct, we investigated the interaction by in vitro binding assay. In vitro-translated biotin-KAP1 was incubated with purified GST, GST-fused LANA-N (aa 1 to 340), and GST-fused LANA-C (aa 1022 to 1162) beads. KAP1 bound to the C-terminal domain of LANA instead of the N-terminal domain (Fig. 1E). Taken together, these results demonstrate that LANA forms a complex with KAP1 in vivo and in vitro.
TABLE 2.

Previously reported LANA-interacting protein.
FIG 1.
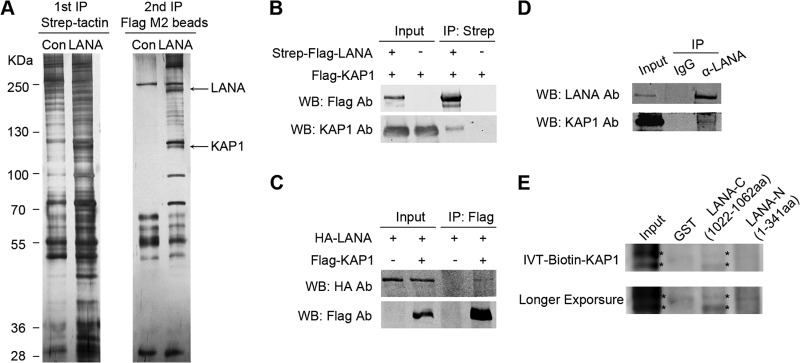
LANA forms a complex with KAP1 in vivo and in vitro. (A) TAP. A plasmid expressing Strep-Flag-tagged LANA was transfected into HEK 293T cells. The equivalent empty vector was transfected as a control (Con). Cell lysates were subjected to affinity purification with streptavidin beads, followed by IP with Flag M2 beads. The purified eluates were resolved by SDS-PAGE and visualized by silver staining. The bands corresponding to LANA and KAP1 are indicated. (B and C) Co-IP of LANA and KAP1 in HEK 293T cells. (B) Flag-tagged KAP1 was transfected into cells, along with Strep-Flag-tagged LANA or empty-vector controls. After affinity purification with streptavidin beads, the purified proteins, along with input samples, were detected by Western blotting (WB) with anti-Flag and anti-KAP1 antibodies (Ab). (C) HA-tagged LANA was transfected into cells, along with Flag-tagged KAP1or empty-vector controls. After IP with Flag M2 beads, the purified proteins, along with input samples, were detected by Western blotting with anti-HA and anti-Flag antibodies. (D) Co-IP of endogenous LANA and KAP1 in JSC-1 cells. JSC-1 cell lysates were subjected to IP with anti-LANA (α-LANA) antibody or mouse IgG controls. Purified proteins, along with input samples, were detected by Western blotting with anti-LANA and anti-KAP1 antibodies. (E) In vitro interaction between LANA and KAP1. Purified GST, GST-fused LANA-N (aa 1 to 340), and GST-fused LANA-C (aa 1022 to 1162) beads were incubated with equivalent in vitro-translated (IVT) biotin-KAP1, and pulled-down proteins were subjected to Transcend chemiluminescent translation detection. The asterisks indicate the specific bands of IVT biotin-KAP1.
LANA is colocalized with KAP1 in the nucleus.
To address whether LANA and KAP1 were in the same nuclear compartment of naturally KSHV-infected cells, we performed an immunofluorescence assay in BCBL-1 and JSC-1 cells. BCBL-1 and JSC-1 cells were fixed and probed with mouse antibody against LANA and rabbit antibody against KAP1, followed by incubation with appropriate secondary antibodies. LANA was distributed in a nuclear punctate pattern, and KAP1 was abundantly diffused in the nucleus (Fig. 2). LANA colocalized with KAP1 in the nuclei of BCBL-1 (Fig. 2, top) and JSC-1 (bottom) cells.
FIG 2.

LANA colocalizes with KAP1 in the nucleus. BCBL-1 (top row) and JSC-1 (bottom row) cells were fixed and probed with mouse antibody against LANA and rabbit antibody against KAP1, followed by incubation with goat anti-mouse IgG conjugated with Alexa Fluor 488 (green) and goat anti-rabbit IgG conjugated with Alexa Fluor 555 (red). Significant colocalized dot signals are indicated by arrowheads. Magnification: oil lens, ×63; zoom, ×2. Magnified views of the boxed areas are shown in the insets.
LANA interacts with both N- and C-terminal domains of KAP1.
KAP1 is a member of the TRIM protein family, with typical structural features. It has a ring finger, B-box, zinc finger, coiled-coil (RBCC) domain at its N terminus; a plant homeodomain and bromodomain (PB) at its C terminus; and a domain for HP1 binding in the middle (31). To explore the function of the interaction between LANA and KAP1, we determined the interface of LANA-KAP1 interaction by GST pulldown and co-IP. A series of GST-fused, truncated KAP1 constructs were generated in the GST pulldown assay (Fig. 3A and B). The purified, GST-fused, truncated KAP1 beads were incubated with cell lysates containing HA-tagged LANA. The pulled-down proteins and input samples were detected by Western blotting with anti-HA antibody. As shown in Fig. 3C, HA-tagged LANA was pulled down by KAP1-FL, KAP1-ΔRBCC, KAP1-ΔPB, KAP1-PB, and KAP1-RBCC, but not GST and KAP1-M; thus, we concluded that LANA interacted with both the N- and C-terminal domains of KAP1. This conclusion was supported by co-IP, and only HA-tagged KAP1-ΔRBCC and KAP1-ΔPB were coimmunoprecipitated with Strep-Flag-tagged LANA, but not KAP1-M (Fig. 3D).
FIG 3.

LANA interacts with both N- and C-terminal domains of KAP1. (A) Domains of KAP1 and truncated constructs used in this study. (B) Purified GST-fused KAP1 truncated constructs for GST pulldown assay. Purified GST-fused KAP1 truncated constructs were subjected to SDS-PAGE and Coomassie blue staining. (C) GST pulldown assay. Purified GST and GST-fused KAP1 truncated constructs were incubated with equivalent cell lysates containing HA-tagged LANA, and the pulled-down proteins were subjected to Western blotting with anti-LANA antibody. (D) Co-IP of LANA and KAP1 truncated constructs in HEK 293T cells. HA-tagged KAP1 truncated constructs were transfected into HEK 293T cells, along with Strep-Flag-tagged LANA. After affinity purification with streptavidin beads, the purified proteins, along with input samples, were detected by Western blotting with anti-Flag and anti-HA antibodies. The asterisks indicate the major bands.
We established that LANA interacted with the N- and C-terminal domains of KAP1. KAP1 does not bind directly to DNA, and the RBCC domain is mainly responsible for its recruitment (31). Transcriptional repression by LANA of the KSHV and cellular genomes has been reported (13, 17, 24–30). Combined with the repressive role of KAP1 and a previous report that KAP1 is associated with the KSHV genome (36), we hypothesized that LANA might recruit KAP1 to the KSHV genome to repress gene expression.
LANA recruits KAP1 to the RTA promoter region.
LANA strongly binds to the TR region and has relatively weak binding at other regions of the KSHV genome, such as the RTA promoter (46–48). To determine whether LANA recruited KAP1 to the KSHV genome, we examined the binding of LANA and KAP1 at the TR and RTA promoter regions by ChIP assay. We performed ChIP with anti-LANA and anti-KAP1 antibodies, as well as the control IgG in KSHV-positive BCBL-1 and JSC-1 cell lines. The purified DNA eluate was quantitated by qPCR and gel analysis, and the results are shown in Fig. 4A (BCBL-1) and B (JSC-1). LANA bound specifically to the TR region at a high level, while it had relatively weak binding in the RTA promoter region. KAP1 bound to the TR region more weakly than LANA and bound weakly to the RTA promoter. KAP1 strongly bound to the 3′ end of the ZNF554 exon, which was a positive control. LANA binds to the RTA promoter through interaction with recombination signal binding protein-Jκ (RBP-Jκ), specificity protein 1 (Sp1), and histone H2A/B (17, 49, 50). To explore further the specific binding sites of KAP1 at the RTA promoter, we designed eight pairs of primers at the RTA promoter to determine the binding (Fig. 4C). Both LANA and KAP1 showed comprehensive binding throughout the RTA promoter (Fig. 4D). These results demonstrate that LANA and KAP1 cooccupy the TR and RTA promoter regions of the KSHV genome.
FIG 4.

LANA recruits KAP1 to the RTA promoter region. (A and B) ChIP assays of LANA and KAP1 in BCBL-1 (A) and JSC-1 (B) cells. The immunoprecipitated DNA in the ChIP assays was subjected to qPCR (left) and DNA gel analysis (right). Binding of LANA and KAP1 at the RTA promoter and TR regions was determined using corresponding primers. The ZNF554 region was detected as a positive control for KAP1 binding. ChIP-qPCR data were normalized by the percent input method (signals obtained from ChIP were divided by signals obtained from an input sample). The data are presented as means ± standard deviations (SD). (C) Illustration of the primer sets at the RTA promoter used in the ChIP assays. (D) Binding of LANA and KAP1 at the RTA promoter region as determined by the primers shown in panel C. ChIP-qPCR data were normalized by the percent input method. (E) ChIP assays of KAP1 in the presence (+) or absence (−) of LANA expression. Plasmid pGL2-RTAp containing the RTA promoter or pGL3p-TR containing the TR fragment was transfected into cells with LANA expression plasmid or empty-vector controls. Cells were collected for ChIP assay at 36 h posttransfection. ChIP-qPCR data were normalized by the fold enrichment method (ChIP signals were divided by the IgG signals). The data are presented as means and SD.
To demonstrate the role of LANA in KAP1 recruitment, that is, whether the recruitment of KAP1 to the TR and RTA promoter regions was dependent on LANA, we determined the binding of KAP1 in the presence or absence of LANA expression. Plasmid pGL2-RTAp, containing the RTA promoter, or pGL3p-TR, containing a TR fragment, was transfected into cells with or without the LANA expression plasmid. Cells were collected for ChIP assay at 36 h posttransfection. Figure 4E shows that enrichment of KAP1 at the RTA promoter region was detected only in cells with LANA expression. However, we did not obtain the enrichment signal for KAP1 in the TR region, even in the presence of LANA. Therefore, our results indicate that LANA recruits KAP1 specifically to the RTA promoter region.
KAP1 is involved in transcriptional repression by LANA.
Although we proved that LANA recruits KAP1 to the RTA promoter region, we still did not know whether the binding of KAP1 is functional, so we determined whether KAP1 was involved in transcriptional repression by LANA. We performed a luciferase reporter assay in HeLa-Ctrl and HeLa-shKAP1 stable cell lines. Expression of KAP1 was reduced significantly by shRNA in HeLa-shKAP1 cells (Fig. 5A). The reporter plasmid pGL2-RTAp, which contained the RTA promoter, was transfected into HeLa-Ctrl and HeLa-shKAP1 cells, along with HA-LANA expression plasmid. Figure 5B, top, shows that LANA repressed transcriptional activity of the RTA promoter in HeLa-Ctrl cells in a dose-dependent manner. In contrast, transcriptional repression by LANA was abrogated in HeLa-shKAP1 cells (Fig. 5B, bottom). Therefore, we concluded that KAP1 is involved in transcriptional repression by LANA. To confirm the result, we also knocked down KAP1 in BCBL-1 cells. The expression of RTA was elevated when KAP1 was knocked down, as shown in Fig. 5C. However, very few virions were detected in the supernatants of KAP1 knockdown BCBL-1 cells (data not shown). Knocked-down KAP1 alone did not reactivate KSHV, suggesting other restriction factors are involved in the maintenance of KSHV latency. Another explanation for this result is that the effect of KAP1 knockdown by shRNA in BCBL-1 cells may not be strong enough for us to see an increase in virus release in the supernatant.
FIG 5.
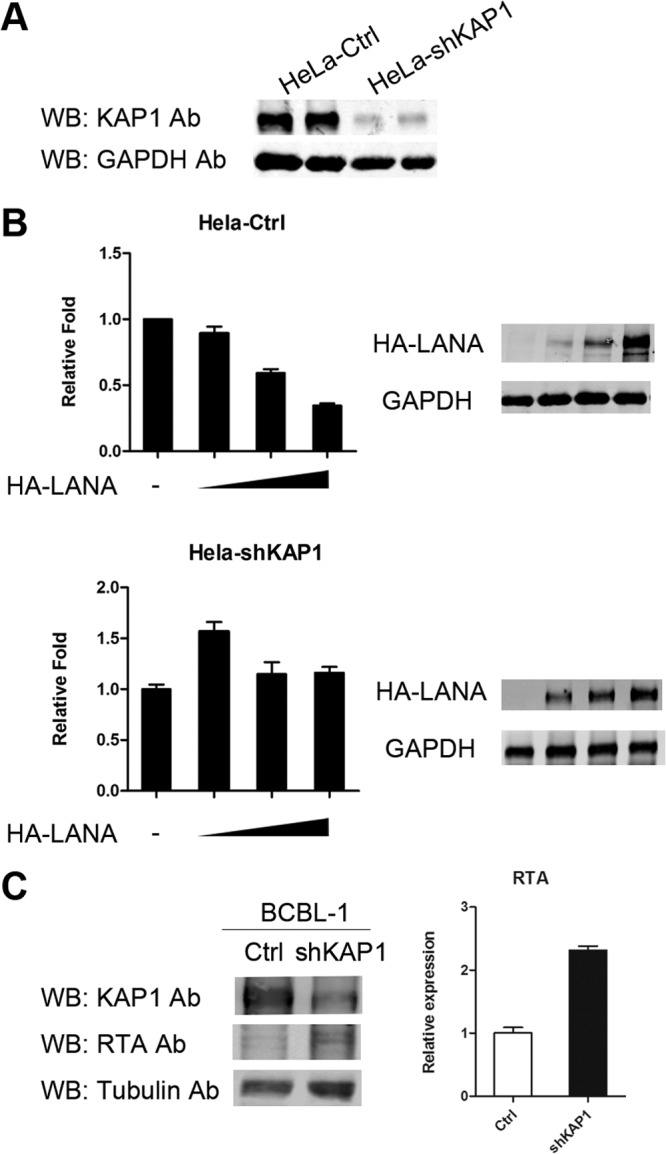
KAP1 is involved in transcriptional repression by LANA. (A) Knocked-down KAP1 in HeLa-shKAP1 cells. (B) Luciferase reporter gene assay in HeLa-Ctrl and HeLa-shKAP1 cells. pGL2-RTAp reporter plasmid (0.2 μg) was transfected into HeLa-Ctrl cells, along with 0, 0.5, 1, or 2 μg pCAGGS-HA-LANA and HeLa-shKAP1 cells and 0, 1, 2, or 4 μg pCAGGS-HA-LANA due to lower LANA expression in HeLa-shKAP1 cells. The total transfected DNA was normalized with the pCAGGS empty vector. Promoter activity is presented as the fold relative to the control (without LANA expression). The data are presented as means and SD. Multiple independent experiments were performed in triplicate. (C) Knocked-down KAP1 in BCBL-1 cells. The expression of RTA was detected by Western blotting (left) and qPCR (right).
LANA and KAP1 have multiple cooccupation sites on the KSHV genome.
To determine whether LANA recruited KAP1 to other regions of the KSHV genome, we mapped all the genome-wide binding sites of LANA and KAP1 by ChIP-seq. The ChIP-enriched DNA with anti-LANA and anti-KAP1 antibodies in the KSHV-positive cell line BC-3 was analyzed by deep sequencing. Sequence reads for each sample were mapped to the KSHV genome (HQ404500 plus 35 copies of the TR) and human genome (hg19) using Bowtie2, and then the aligned file was subjected to peak calling using MACS. The peak models of LANA and KAP1 built by MACS based on the human genome alignment indicated that both LANA and KAP1 had broad peaks (Fig. 6A); thus, we applied the −nomodel parameter to MACS analysis. The peaks of LANA and KAP1 binding (P < 10−3) on the whole KSHV genome are shown in Fig. 6B. The first ∼28-kb region of the KSHV genome failed to show an enrichment signal for LANA and KAP1 due to possible deletion or large sequence variation in the KSHV genome of BC-3 cells, which was mentioned previously (the KSHV genome in BC-3 cells is smaller than in BC1 and BC2 cells by Southern blotting) (51). The most significant peaks of LANA binding are grouped in the TR region, and smaller peaks are scattered throughout the genome, which is consistent with previous results (46). The annotated information on LANA peaks (P < 10−5) is shown in Table 3. The peaks of KAP1 binding identified on the viral genome were small. The enrichment signal at the TR region, as determined by real-time PCR, was not detected for the alignment of 35 copies of the TR. The annotated information on KAP1 peaks (P < 10−5) is shown in Table 4. The peaks of KAP1 covered regions of lytic genes, as well as latent genes, and most of them coincided with peaks of LANA (Fig. 6C), which implies that LANA may be important for KAP1 recruitment to the KSHV genome, or vice versa. The incompletely overlapping peaks of LANA and KAP1 indicated that KAP1 could also be recruited to the KSHV genome by an alternative mechanism. To validate the identified peaks, we determined the binding by qPCR. Figure 6D shows that LANA and KAP1 cooccupied the newly identified regions. The ChIP-seq demonstrated that LANA and KAP1 had multiple cooccupation sites on the KSHV genome, indicating that LANA-recruited KAP1 functions widely on the genome to control gene expression. Additionally, we validated the possible deletion or large sequence variation in the KSHV genome of BC-3 cells by PCR (Fig. 6E).
FIG 6.

LANA and KAP1 had multiple cooccupation sites on the KSHV genome. (A) Peak models of LANA and KAP1 built by MACS based on the human genome alignment. (B) Illustration of LANA and KAP1 binding sites on the whole KSHV genome. The ChIP-seq data on LANA and KAP1 were aligned to the KSHV genome (HQ404500 plus 35 copies of the TR [U75699.1]) and subjected to peak calling with MACS (P < 10−3). The output files (peaks, summits, and wigs) were visualized in IGV software. The identified peaks are shown in the peak diagrams. The summit diagrams show the sites with the highest scores in the peaks. The wig diagrams show the general binding information for the whole KSHV genome. The region marked by the asterisk covers ∼28 kb of the KSHV genome that failed to show the enrichment signal of LANA and KAP1, due to possible deletion or large sequence variation in the KSHV genome of BC-3 cells. (C) Diagram of regions occupied by LANA (red) and KAP1 (blue) (P < 10−5). (D) Validation of ChIP-seq results by qPCR. ChIP-qPCR data were normalized by fold enrichment (ChIP signals were divided by the IgG signals). GAPDH was a negative control. The data are presented as means and SD. (E) Validation of possible deletion or large sequence variation in the KSHV genome of BC-3 cells by PCR.
TABLE 3.
LANA peaks on the KSHV genome (P < 10−5)

Peaks (start to end) were annotated to genes in the KSHV genome (HQ404500) that could be promoters, coding sequences, or untranslated regions.
Previously reported LANA peak on KSHV genome by ChIP-seq.
Previously reported LANA peak on KSHV genome by ChIP-on-chip.
TABLE 4.
KAP1 peaks on the KSHV genome (P < 10−5)

Peaks (start to end) were annotated to genes in the KSHV genome (HQ404500) that could be promoters, coding sequences, or untranslated regions.
LANA-recruited KAP1 is crucial for establishment of KSHV latency.
To determine whether LANA-recruited KAP1 was involved in establishment of KSHV latency, we observed the effect of KAP1 knockdown on viral gene expression and the change in early epigenetic modification during KSHV primary infection. HeLa-Ctrl and HeLa-shKAP1 cells were de novo infected with rKSHV.219 at the same multiplicity of infection, and lytic gene expression was determined (Fig. 7A). To ensure the correctness of the gene expression analysis, we examined the relative KSHV genome copy numbers in HeLa-Ctrl and HeLa-shKAP1 cells at 24 h postinfection. The KSHV genome copy numbers in HeLa-Ctrl and HeLa-shKAP1 cells were similar, as assayed by both K2 and LANA primer sets (Fig. 7B). Expression of a limited number of lytic genes can be detected during KSHV primary infection (14). We determined the expression of the RTA, K2, and K15 genes. The results of the lytic gene expression at each time point are shown in Fig. 7C. As expected, RTA expression was highest at the beginning (as early as 2 h postinfection) but soon decreased. It was almost completely stopped at 24 h postinfection in HeLa-Ctrl cells, although it remained high in HeLa-shKAP1 cells, even at 72 h postinfection. RTA expression was significantly higher in HeLa-shKAP1 cells at each time point. Similar to that of the RTA gene, K2 gene expression was also higher in HeLa-shKAP1 cells and remained at a high level at 72 h postinfection. The expression pattern of the K15 gene differed from that of the RTA and K2 genes, which is consistent with a previous study (14). It showed a slight increase over time until 48 h postinfection. K15 expression achieved a more robust peak at 48 h postinfection in HeLa-shKAP1 cells and was not shut down at 72 h postinfection. The effect of KAP1 knockdown on viral gene expression during KSHV de novo infection indicates that KAP1 is involved in the shutdown of lytic genes. KAP1 represses gene expression via epigenetic change of histone acetylation or methylation (31–33). The epigenetic modification of the RTA promoter is mainly subject to the regulation of histone acetylation (17, 52), and therefore, we determined the AcH3 level in the RTA promoter region at the early stages of KSHV primary infection (Fig. 7D). The AcH3 level was significantly upregulated in HeLa-shKAP1 cells at 48 h postinfection. The upregulated AcH3 level disrupted shutdown of RTA gene expression. The elevated AcH3 level in HeLa-shKAP1 cells also validated the higher RTA expression. Therefore, we conclude that LANA-recruited KAP1 is crucial for the rapid establishment of KSHV latency.
FIG 7.

LANA-recruited KAP1 is crucial for establishment of KSHV latency. (A) Illustration of de novo KSHV infection in HeLa-Ctrl and HeLa-shKAP1 cells. (B) Relative KSHV genome DNA copy numbers in cells. Cells were collected for DNA extraction at 24 h postinfection, and the relative quantity was determined by qPCR. Two primer sets were used. The data were normalized against GAPDH. The data are presented as means and SD. (C) Kinetics of lytic gene expression during primary infection. Cells were collected for RNA extraction at the indicated time points and reverse transcribed to cDNA. The relative quantity was determined by qPCR. The data were normalized against GAPDH. The data are presented as means ± SD. Shown is a representative result of de novo infection experiments. The complete de novo infection experiments were done twice independently and showed the same tendency. For some individual time points, the results were repeated at least three times. (D) ChIP assay of AcH3 at the RTA promoter region. Cells were collected for AcH3 ChIP assay at 48 h postinfection. ChIP-qPCR data were normalized by the percent input method (signals obtained from ChIP were divided by signals obtained from an input sample). The data are presented as means and SD.
DISCUSSION
The establishment of KSHV latency is a rapid and efficient process during primary infection. Lytic gene expression is shut down, while latent gene expression is sustained, which implies a process of differential gene expression. The mechanism of this process remains largely unclear, but previous studies on the LANA protein provide some clues. The LANA protein is crucial for the establishment of viral latency, and thus, insight from LANA sheds light on the process. In this study, we showed that LANA interacted with the KAP1 protein and repressed lytic gene expression to facilitate the establishment of KSHV latency.
Multiple proteins have been shown to interact with LANA (26, 30, 38, 45, 48–50, 53–69). In our TAP-MS screening, a few novel proteins that were related to transcriptional regulation and posttranslational modification were identified, such as KAP1, NCOR2, retinoblastoma-associated protein (RBBP), SMAD, USP7, and SUMO3. Previously reported LANA-interacting proteins, like BRD2 (54), SIN3A (30), PARP1 (67), PRKDC (DNA-PK) (68), XRCC6 (KU70) (68), and KDM3A (48), were also found in this study (Table 2). We validated the interaction between LANA and KAP1 in vivo and in vitro. We found that KAP1 interacted with the C-terminal domain of LANA directly. We mapped out that LANA interacted with both the N- and C-terminal domains of KAP1. Recent studies by different groups have resolved the structure of the C-terminal domain of LANA and have demonstrated that the C-terminal domain works in an oligomeric state (70–72). The N-terminal domain of KAP1 (RBCC) has been reported to function as a homo-oligomer, which is recruited by the KRAB domain to the genome (73). Thus, our results imply that KAP1 is recruited by the oligomeric LANA to the genome. Coincidentally, when this paper was in preparation, Cai et al. reported that LANA does not directly interact with KAP1 but with sumoylated KAP1 through the SUMO-interacting motif (SIM) in the N-terminal domain of the LANA protein (74). However, our results demonstrated that LANA directly interacted with both the N- and C-terminal domains of KAP1. We think that these conflicting results were caused by the different in vitro binding assays. We also explored the possible function of the interaction between LANA and the C-terminal domain of KAP1, but the result was negative, as LANA did not affect the sumoylation of KAP1 (data not shown).
In the ChIP assay, we demonstrated the cooccupation by LANA and KAP1 at the RTA promoter and in the TR region of the KSHV genome. However, the binding of LANA and KAP1 in the RTA promoter region was weak, which is likely due to their indirect binding to DNA. The binding of LANA in the TR region was stronger than the binding of KAP1, which suggests a weak interaction between LANA and KAP1 in the TR region. This is possibly true, because DNA binding may promote higher oligomerization of LANA (70), which partially disrupts the interaction between LANA and KAP1. The recruitment assay demonstrated that LANA recruited KAP1 to the RTA promoter region, but not the TR region (Fig. 4E). KAP1 did not bind to the TR region in transiently transfected cells, which was different from the observation in PEL cell lines. The transient transfection could be regarded as a process mimicking KSHV primary infection, and thus, the result indicated that KAP1 bound to the RTA promoter, but not the TR region, at the early stage of KSHV primary infection. The binding of KAP1 in the TR region might be a later event after the establishment of KSHV latency. We unexpectedly demonstrated that both LANA and KAP1 showed comprehensive binding in the RTA promoter region, which implies that the repressive role of LANA may not be limited to one or two sites in the RTA promoter region. This result also verified the recent finding that LANA and KAP1 can cooccupy the HIF-1α binding sites at the RTA promoter (74).
We revealed that KAP1 was involved in transcriptional repression by LANA in a reporter gene assay, which is consistent with the result in the recent publication by Cai et al. (74). The expression level of transfected LANA was lower in HeLa-shKAP1 cells, which suggests that KAP1 stabilizes LANA in the nucleus. KAP1 is an abundant protein expressed in the nucleus, with a possible long half-life (difficult to knock down by transient RNA interference). It has also been shown that LANA has a long half-life, exceeding 24 h (75). KAP1 is a SUMO intramolecular E3 ligase, and LANA is subject to sumoylation modification (74, 76–78). Thus, it is reasonable to speculate that KAP1 is responsible for the sumoylation of LANA and that the degradation of LANA may be regulated by its sumoylation.
We identified multiple cooccupation sites of LANA and KAP1 on the whole KSHV genome by ChIP-seq. In the previous study, ChIP-seq and ChIP-on-chip methods were both applied to study the binding sites of LANA on the KSHV genome (46–48). Besides the strong binding sites of LANA in the TR region, only a few binding sites (K7, vIRF2, open reading frame 73 [ORF73], and K15) on the KSHV genome were reported across studies. The ChIP-seq data on LANA in BC-3 cells were consistent with previous ChIP-seq data in BCBL-1 cells (Table 3), except that more peaks were identified in this study, such as the well-studied RTA promoter region (17, 50). Most of the identified KAP1 peaks coincided with LANA peaks, implying that LANA may be important for the recruitment of KAP1 to the KSHV genome. A previous study only selectively detected the binding of KAP1 in the promoter regions of the KSHV genome (36). However, we found that the cooccupation sites of LANA and KAP1 were not limited to the promoter regions. KAP1 can repress transcriptional activity, not only in the promoter regions, but also in the coding sequence regions, which produces an advantage for LANA in controlling gene expression (31, 79). We found cooccupation by LANA and KAP1 in the latent gene regions. Latent gene expression should be maintained at an appropriate level to evade immune surveillance, and thus, the interaction between LANA and KAP1 may also regulate the expression of latent genes.
Our results demonstrated that LANA-recruited KAP1 is crucial for KSHV primary infection. Lytic gene expression is enhanced and cannot be shut down in HeLa-shKAP1 cells in time. The enhanced expression can be detected at an early time point, suggesting that KAP1 is involved in an immediate-early step during primary infection. However, knockdown of KAP1 does not change the expression pattern of viral genes. Expression of the K15 gene in HeLa-shKAP1 cells remains a slowly increasing process at the beginning, implying that another mechanism is responsible for selective lytic gene expression. KAP1 represses gene expression by changing the epigenetic state of the genome. It can recruit both the histone methyltransferase and deacetylase complexes (32, 33). Here, we found upregulation of the AcH3 level in the RTA promoter region in HeLa-shKAP1 cells, which suggests that KAP1 utilizes the deacetylase complex to repress gene expression. Indeed, many of the identified host proteins interacting with LANA, along with KAP1, are members of the deacetylase complex, such as HDAC1, HDAC2, and RBBP. Previous studies have demonstrated that transcriptional repression by LANA can be interrupted by deacetylase inhibitors (9, 17, 24). Therefore, the interaction between LANA and KAP1 could be important for the latent KSHV genome to maintain a hypoacetylated state.
In conclusion, our study revealed the mechanism of transcriptional repression by LANA during KSHV primary infection, providing new insights into the process of KSHV latency establishment.
ACKNOWLEDGMENTS
This work was supported by grants from the key project of the Natural Science Foundation of China (81230037) and the National Basic Research Program of China (2011CB504805 and 2012CB519002) to K.L. and the Science Foundation of China for the Youth (81101478) to Y.G.
We thank Chuangui Wang (East China Normal University, Shanghai, China) and Subhash C. Verma (University of Nevada, Reno, NV) for their plasmids and cell lines. We acknowledge the Omics Core, CAS-MPG Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, for ChIP-seq service.
Footnotes
Published ahead of print 16 April 2014
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191. 10.1056/NEJM199505043321802 [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 4.Boshoff C, Chang Y. 2001. Kaposi's sarcoma-associated herpesvirus: a new DNA tumor virus. Annu. Rev. Med. 52:453–470. 10.1146/annurev.med.52.1.453 [DOI] [PubMed] [Google Scholar]
- 5.Ye F, Lei X, Gao SJ. 2011. Mechanisms of Kaposi's sarcoma-associated herpesvirus latency and reactivation. Adv. Virol. 2011:193860. 10.1155/2011/193860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dittmer DP. 2003. Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 63:2010–2015 http://cancerres.aacrjournals.org/content/63/9/2010.long [PubMed] [Google Scholar]
- 8.Shaw RN, Arbiser JL, Offermann MK. 2000. Valproic acid induces human herpesvirus 8 lytic gene expression in BCBL-1 cells. AIDS 14:899–902. 10.1097/00002030-200005050-00021 [DOI] [PubMed] [Google Scholar]
- 9.Yu Y, Black JB, Goldsmith CS, Browning PJ, Bhalla K, Offermann MK. 1999. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J. Gen. Virol. 80:83–90 [DOI] [PubMed] [Google Scholar]
- 10.Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R. 2001. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 97:3244–3250. 10.1182/blood.V97.10.3244 [DOI] [PubMed] [Google Scholar]
- 11.Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312. 10.1006/viro.1998.9486 [DOI] [PubMed] [Google Scholar]
- 12.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871. 10.1073/pnas.95.18.10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lan K, Kuppers DA, Verma SC, Robertson ES. 2004. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J. Virol. 78:6585–6594. 10.1128/JVI.78.12.6585-6594.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnan HH, Naranatt PP, Smith MS, Zeng L, Bloomer C, Chandran B. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620. 10.1128/JVI.78.7.3601-3620.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoo SM, Zhou FC, Ye FC, Pan HY, Gao SJ. 2005. Early and sustained expression of latent and host modulating genes in coordinated transcriptional program of KSHV productive primary infection of human primary endothelial cells. Virology 343:47–64. 10.1016/j.virol.2005.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao SJ, Deng JH, Zhou FC. 2003. Productive lytic replication of a recombinant Kaposi's sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 77:9738–9749. 10.1128/JVI.77.18.9738-9749.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu F, Day L, Gao SJ, Lieberman PM. 2006. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J. Virol. 80:5273–5282. 10.1128/JVI.02541-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye FC, Zhou FC, Yoo SM, Xie JP, Browning PJ, Gao SJ. 2004. Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J. Virol. 78:11121–11129. 10.1128/JVI.78.20.11121-11129.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q, Zhou F, Ye F, Gao SJ. 2008. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 379:234–244. 10.1016/j.virol.2008.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lan K, Kuppers DA, Verma SC, Sharma N, Murakami M, Robertson ES. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J. Virol. 79:7453–7465. 10.1128/JVI.79.12.7453-7465.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644. 10.1126/science.284.5414.641 [DOI] [PubMed] [Google Scholar]
- 22.Hu J, Garber AC, Renne R. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 76:11677–11687. 10.1128/JVI.76.22.11677-11687.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Si H, Verma SC, Lampson MA, Cai Q, Robertson ES. 2008. Kaposi's sarcoma-associated herpesvirus-encoded LANA can interact with the nuclear mitotic apparatus protein to regulate genome maintenance and segregation. J. Virol. 82:6734–6746. 10.1128/JVI.00342-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeWire SM, Damania B. 2005. The latency-associated nuclear antigen of rhesus monkey rhadinovirus inhibits viral replication through repression of Orf50/Rta transcriptional activation. J. Virol. 79:3127–3138. 10.1128/JVI.79.5.3127-3138.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakakibara S, Ueda K, Nishimura K, Do E, Ohsaki E, Okuno T, Yamanishi K. 2004. Accumulation of heterochromatin components on the terminal repeat sequence of Kaposi's sarcoma-associated herpesvirus mediated by the latency-associated nuclear antigen. J. Virol. 78:7299–7310. 10.1128/JVI.78.14.7299-7310.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shamay M, Krithivas A, Zhang J, Hayward SD. 2006. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi's sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. U. S. A. 103:14554–14559. 10.1073/pnas.0604469103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Bartolo DL, Cannon M, Liu YF, Renne R, Chadburn A, Boshoff C, Cesarman E. 2008. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood 111:4731–4740. 10.1182/blood-2007-09-110544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwam DR, Luciano RL, Mahajan SS, Wong L, Wilson AC. 2000. Carboxy terminus of human herpesvirus 8 latency-associated nuclear antigen mediates dimerization, transcriptional repression, and targeting to nuclear bodies. J. Virol. 74:8532–8540. 10.1128/JVI.74.18.8532-8540.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garber AC, Shu MA, Hu J, Renne R. 2001. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 75:7882–7892. 10.1128/JVI.75.17.7882-7892.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krithivas A, Young DB, Liao G, Greene D, Hayward SD. 2000. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 74:9637–9645. 10.1128/JVI.74.20.9637-9645.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iyengar S, Farnham PJ. 2011. KAP1 protein: an enigmatic master regulator of the genome. J. Biol. Chem. 286:26267–26276. 10.1074/jbc.R111.252569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schultz DC, Friedman JR, Rauscher FJ., III 2001. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 15:428–443. 10.1101/gad.869501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., III 2002. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16:919–932. 10.1101/gad.973302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groner AC, Meylan S, Ciuffi A, Zangger N, Ambrosini G, Denervaud N, Bucher P, Trono D. 2010. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 6:e1000869. 10.1371/journal.pgen.1000869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barde I, Laurenti E, Verp S, Groner AC, Towne C, Padrun V, Aebischer P, Trumpp A, Trono D. 2009. Regulation of episomal gene expression by KRAB/KAP1-mediated histone modifications. J. Virol. 83:5574–5580. 10.1128/JVI.00001-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang PC, Fitzgerald LD, Van Geelen A, Izumiya Y, Ellison TJ, Wang DH, Ann DK, Luciw PA, Kung HJ. 2009. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi's sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 69:5681–5689. 10.1158/0008-5472.CAN-08-4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin Y, He Z, Liang D, Zhang Q, Zhang H, Deng Q, Robertson ES, Lan K. 2012. Carboxyl-terminal amino acids 1052 to 1082 of the latency-associated nuclear antigen (LANA) interact with RBP-Jkappa and are responsible for LANA-mediated RTA repression. J. Virol. 86:4956–4969. 10.1128/JVI.06788-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaul R, Verma SC, Robertson ES. 2007. Protein complexes associated with the Kaposi's sarcoma-associated herpesvirus-encoded LANA. Virology 364:317–329. 10.1016/j.virol.2007.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gloeckner CJ, Boldt K, Ueffing M. 2009. Strep/FLAG tandem affinity purification (SF-TAP) to study protein interactions. Curr. Protoc. Protein Sci. Chapter 19:Unit 19.20. 10.1002/0471140864.ps1920s57 [DOI] [PubMed] [Google Scholar]
- 40.Hu C, Zhang S, Gao X, Gao X, Xu X, Lv Y, Zhang Y, Zhu Z, Zhang C, Li Q, Wong J, Cui Y, Zhang W, Ma L, Wang C. 2012. Roles of Kruppel-associated box (KRAB)-associated co-repressor KAP1 Ser-473 phosphorylation in DNA damage response. J. Biol. Chem. 287:18937–18952. 10.1074/jbc.M111.313262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174:12–21. 10.1016/j.jviromet.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9:R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol. 29:24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shamay M, Liu J, Li R, Liao G, Shen L, Greenway M, Hu S, Zhu J, Xie Z, Ambinder RF, Qian J, Zhu H, Hayward SD. 2012. A protein array screen for Kaposi's sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J. Virol. 86:5179–5191. 10.1128/JVI.00169-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu F, Tsai K, Chen HS, Wikramasinghe P, Davuluri RV, Showe L, Domsic J, Marmorstein R, Lieberman PM. 2012. Identification of host-chromosome binding sites and candidate gene targets for Kaposi's sarcoma-associated herpesvirus LANA. J. Virol. 86:5752–5762. 10.1128/JVI.07216-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Campbell M, Chang PC, Huerta S, Izumiya C, Davis R, Tepper CG, Kim KY, Shevchenko B, Wang DH, Jung JU, Luciw PA, Kung HJ, Izumiya Y. 2012. Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J. Biol. Chem. 287:5806–5818. 10.1074/jbc.M111.289496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim KY, Huerta SB, Izumiya C, Wang DH, Martinez A, Shevchenko B, Kung HJ, Campbell M, Izumiya Y. 2013. Kaposi's sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen regulates the KSHV epigenome by association with the histone demethylase KDM3A. J. Virol. 87:6782–6793. 10.1128/JVI.00011-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861. 10.1126/science.1120541 [DOI] [PubMed] [Google Scholar]
- 50.Lan K, Kuppers DA, Robertson ES. 2005. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J. Virol. 79:3468–3478. 10.1128/JVI.79.6.3468-3478.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arvanitakis L, Mesri EA, Nador RG, Said JW, Asch AS, Knowles DM, Cesarman E. 1996. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 88:2648–2654 [PubMed] [Google Scholar]
- 52.Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, Jung JU. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 6:e1001013. 10.1371/journal.ppat.1001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76:11596–11604. 10.1128/JVI.76.22.11596-11604.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Platt GM, Simpson GR, Mittnacht S, Schulz TF. 1999. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 73:9789–9795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe A, Higuchi M, Fukushi M, Ohsawa T, Takahashi M, Oie M, Fujii M. 2007. A novel KRAB-Zinc finger protein interacts with latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus and activates transcription via terminal repeat sequences. Virus Genes 34:127–136. 10.1007/s11262-006-0048-x [DOI] [PubMed] [Google Scholar]
- 56.Chen W, Dittmer DP. 2011. Ribosomal protein S6 interacts with the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 85:9495–9505. 10.1128/JVI.02620-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paudel N, Sadagopan S, Balasubramanian S, Chandran B. 2012. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen and angiogenin interact with common host proteins, including annexin A2, which is essential for survival of latently infected cells. J. Virol. 86:1589–1607. 10.1128/JVI.05754-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.An J, Sun Y, Rettig MB. 2004. Transcriptional coactivation of c-Jun by the KSHV-encoded LANA. Blood 103:222–228. 10.1182/blood-2003-05-1538 [DOI] [PubMed] [Google Scholar]
- 59.Cai Q, Lan K, Verma SC, Si H, Lin D, Robertson ES. 2006. Kaposi's sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 alpha to upregulate RTA expression during hypoxia: latency control under low oxygen conditions. J. Virol. 80:7965–7975. 10.1128/JVI.00689-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kusano S, Eizuru Y. 2010. Human I-mfa domain proteins specifically interact with KSHV LANA and affect its regulation of Wnt signaling-dependent transcription. Biochem. Biophys. Res. Commun. 396:608–613. 10.1016/j.bbrc.2010.04.111 [DOI] [PubMed] [Google Scholar]
- 61.Lim C, Gwack Y, Hwang S, Kim S, Choe J. 2001. The transcriptional activity of cAMP response element-binding protein-binding protein is modulated by the latency associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 276:31016–31022. 10.1074/jbc.M102431200 [DOI] [PubMed] [Google Scholar]
- 62.Liu J, Martin HJ, Liao G, Hayward SD. 2007. The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J. Virol. 81:10451–10459. 10.1128/JVI.00804-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muromoto R, Okabe K, Fujimuro M, Sugiyama K, Yokosawa H, Seya T, Matsuda T. 2006. Physical and functional interactions between STAT3 and Kaposi's sarcoma-associated herpesvirus-encoded LANA. FEBS Lett. 580:93–98. 10.1016/j.febslet.2005.11.057 [DOI] [PubMed] [Google Scholar]
- 64.Radkov SA, Kellam P, Boshoff C. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6:1121–1127. 10.1038/80459 [DOI] [PubMed] [Google Scholar]
- 65.Roupelieva M, Griffiths SJ, Kremmer E, Meisterernst M, Viejo-Borbolla A, Schulz T, Haas J. 2010. Kaposi's sarcoma-associated herpesvirus Lana-1 is a major activator of the serum response element and mitogen-activated protein kinase pathways via interactions with the Mediator complex. J. Gen. Virol. 91:1138–1149. 10.1099/vir.0.017715-0 [DOI] [PubMed] [Google Scholar]
- 66.Verma SC, Borah S, Robertson ES. 2004. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J. Virol. 78:10348–10359. 10.1128/JVI.78.19.10348-10359.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ohsaki E, Ueda K, Sakakibara S, Do E, Yada K, Yamanishi K. 2004. Poly(ADP-ribose) polymerase 1 binds to Kaposi's sarcoma-associated herpesvirus (KSHV) terminal repeat sequence and modulates KSHV replication in latency. J. Virol. 78:9936–9946. 10.1128/JVI.78.18.9936-9946.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cha S, Lim C, Lee JY, Song YJ, Park J, Choe J, Seo T. 2010. DNA-PK/Ku complex binds to latency-associated nuclear antigen and negatively regulates Kaposi's sarcoma-associated herpesvirus latent replication. Biochem. Biophys. Res. Commun. 394:934–939. 10.1016/j.bbrc.2010.03.086 [DOI] [PubMed] [Google Scholar]
- 69.Fujimuro M, Wu FY, ApRhys C, Kajumbula H, Young DB, Hayward GS, Hayward SD. 2003. A novel viral mechanism for dysregulation of beta-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat. Med. 9:300–306. 10.1038/nm829 [DOI] [PubMed] [Google Scholar]
- 70.Domsic JF, Chen HS, Lu F, Marmorstein R, Lieberman PM. 2013. Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLoS Pathog. 9:e1003672. 10.1371/journal.ppat.1003672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hellert J, Weidner-Glunde M, Krausze J, Richter U, Adler H, Fedorov R, Pietrek M, Ruckert J, Ritter C, Schulz TF, Luhrs T. 2013. A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLoS Pathog. 9:e1003640. 10.1371/journal.ppat.1003640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Correia B, Cerqueira SA, Beauchemin C, Pires de Miranda M, Li S, Ponnusamy R, Rodrigues L, Schneider TR, Carrondo MA, Kaye KM, Simas JP, McVey CE. 2013. Crystal structure of the gamma-2 herpesvirus LANA DNA binding domain identifies charged surface residues which impact viral latency. PLoS Pathog. 9:e1003673. 10.1371/journal.ppat.1003673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peng H, Begg GE, Schultz DC, Friedman JR, Jensen DE, Speicher DW, Rauscher FJ., III 2000. Reconstitution of the KRAB-KAP-1 repressor complex: a model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein-protein interactions. J. Mol. Biol. 295:1139–1162. 10.1006/jmbi.1999.3402 [DOI] [PubMed] [Google Scholar]
- 74.Cai Q, Cai S, Zhu C, Verma SC, Choi JY, Robertson ES. 2013. A unique SUMO-2-interacting motif within LANA is essential for KSHV latency. PLoS Pathog. 9:e1003750. 10.1371/journal.ppat.1003750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen W, Hilton IB, Staudt MR, Burd CE, Dittmer DP. 2010. Distinct p53, p53:LANA, and LANA complexes in Kaposi's sarcoma-associated herpesvirus lymphomas. J. Virol. 84:3898–3908. 10.1128/JVI.01321-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou MM, Rauscher FJ., III 2007. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 28:823–837. 10.1016/j.molcel.2007.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zeng L, Yap KL, Ivanov AV, Wang X, Mujtaba S, Plotnikova O, Rauscher FJ, III, Zhou MM. 2008. Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat. Struct. Mol. Biol. 15:626–633. 10.1038/nsmb.1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Campbell M, Izumiya Y. 2012. Post-translational modifications of Kaposi's sarcoma-associated herpesvirus regulatory proteins—SUMO and KSHV. Front. Microbiol. 3:31. 10.3389/fmicb.2012.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Iyengar S, Ivanov AV, Jin VX, Rauscher FJ, III, Farnham PJ. 2011. Functional analysis of KAP1 genomic recruitment. Mol. Cell. Biol. 31:1833–1847. 10.1128/MCB.01331-10 [DOI] [PMC free article] [PubMed] [Google Scholar]