

Abstract
The recent discovery of hantaviruses in shrews and bats in West Africa suggests that other genetically distinct hantaviruses exist in East Africa. Genetic and phylogenetic analyses of newfound hantaviruses, detected in archival tissues from the Geata mouse shrew (Myosorex geata) and Kilimanjaro mouse shrew ( Myosorex zinki) captured in Tanzania, expands the host diversity and geographic distribution of hantaviruses and suggests that ancestral shrews and/or bats may have served as the original mammalian hosts of primordial hantaviruses.
TEXT
While myriad disease-causing RNA viruses were first discovered in sub-Saharan Africa, hantaviruses have been notable exceptions until recently, when Sangassou virus was detected in the African wood mouse (Hylomyscus simus) (1) and Tanganya virus (TGNV) in the Therese's shrew (Crocidura theresae) (2), captured in Guinea. More recently, genetically distinct hantaviruses, designated Azagny virus (AZGV) and Bowé virus (BOWV), have been found in the West African pygmy shrew (Crocidura obscurior) in Côte d'Ivoire (3) and in the Doucet's musk shrew (Crocidura douceti) in Guinea (4), respectively. Here, we report the genetic and phylogenetic analyses of two novel hantaviruses harbored by myosoricine shrews in East Africa.
In accordance with guidelines approved by the American Society of Mammalogists (5), shrews were collected using pitfall traps during faunal surveys of montane forests in Tanzania (6, 7). Frozen liver tissues from 19 Geata mouse shrews (Myosorex geata) (Fig. 1A), 13 Kilimanjaro mouse shrews (Myosorex zinki) (Fig. 1B), 25 Kihaule's mouse shrews (Myosorex kihaulei) (7), 8 climbing shrews (Suncus megalura), 10 Grant's forest shrews (Sylvisorex granti), and 10 Howell's forest shrews (Sylvisorex howelli), captured between August 1995 and August 2002, were analyzed for hantavirus RNA by reverse transcription (RT)-PCR (3, 4). Within minutes after the shrews were sacrificed, tissues were collected and placed in liquid nitrogen and then shipped and stored in liquid nitrogen until testing. The use of archival tissues was exempt from protocol review by the University of Hawaii Institutional Animal Care and Use Committee.
FIG 1.
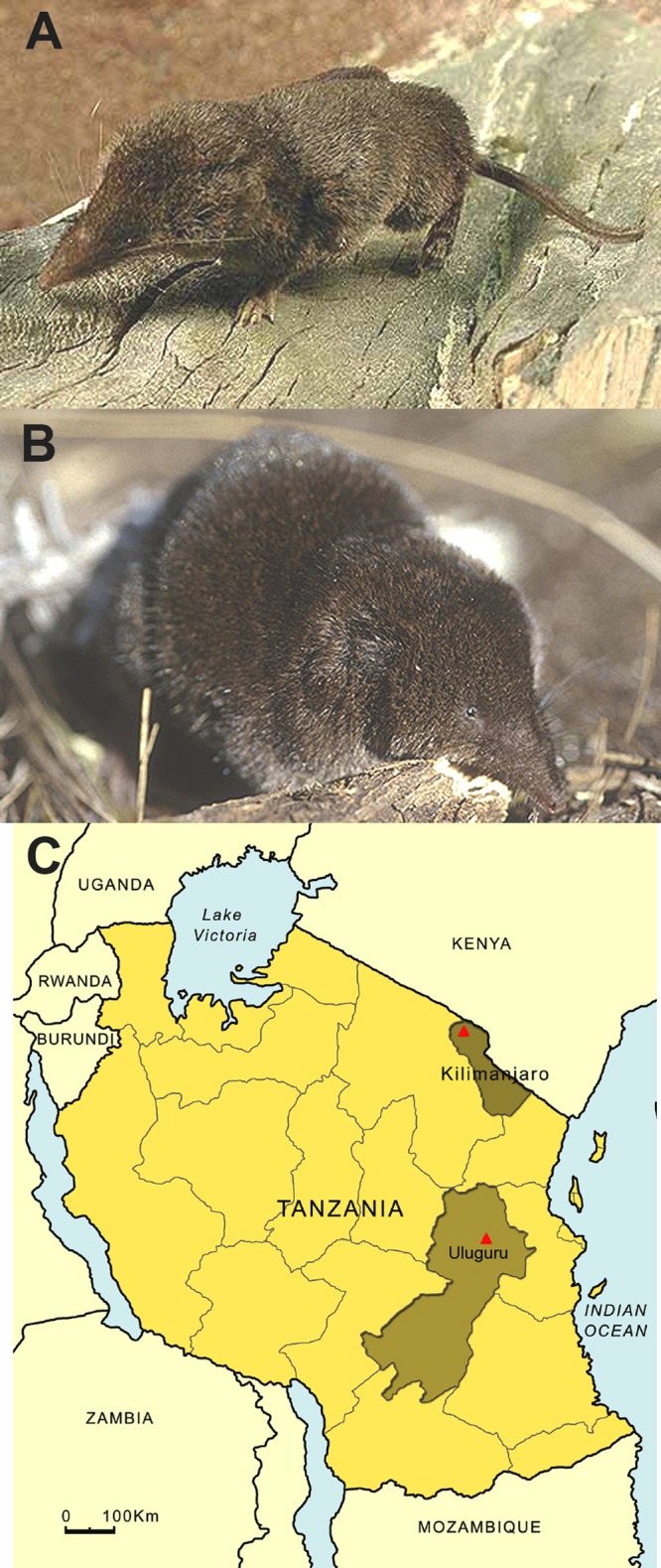
(A) Geata mouse shrew (Myosorex geata); (B) Kilimanjaro mouse shrew (Myosorex zinki); (C) map of Tanzania, showing Uluguru and Kilimanjaro mountains, where hantavirus-infected myosoricine shrews were trapped.
Kilimanjaro virus (KMJV strain FMNH174124) was found in a male Kilimanjaro mouse shrew, captured at 3,475 m elevation in Mt. Kilimanjaro National Park, 13.5 km N and 4 km W of Maua, in the Moshi District, Kilimanjaro Province, on 7 August 2002, and Uluguru virus (ULUV strain FMNH158302) was detected in a male Geata mouse shrew, captured at 1,535 m elevation in Uluguru North Forest Reserve, Uluguru Mountains, 5.1 km W and 2.3 km N of Tegetero, in the Morogoro District, Morogoro Province, on 16 August 1996 (Fig. 1C). Identification of the hantavirus-infected mouse shrews was verified by analysis of the complete 1,140-nucleotide mitochondrial DNA (mtDNA) cytochrome b gene (GenBank JX193701 and JX193702). Despite repeated RT-PCR attempts, hantavirus RNA was not detected in tissues of the other shrew species.
The full-length S and L segments and partial M segment of KMJV and the entire L segment and partial S and M segments of ULUV were sequenced and compared with those of representative rodent- and soricomorph-borne hantaviruses (Table 1). The 1,911-nucleotide S genomic segment of KMJV (GenBank JX193698) contained a single open reading frame (ORF), encoding a 422-amino-acid nucleocapsid (N) protein (nucleotide positions 61 to 1329), and 3′- and 5′-noncoding regions of 60 and 582 nucleotides, respectively. For ULUV, nearly the entire S segment, from nucleotide positions 1 to 1283, was sequenced (GenBank JX193695). Pairwise alignment and comparison of the S segment coding sequences of KJMV and ULUV, using the ClustalW method (8), showed moderately low sequence similarities with other hantaviruses, ranging from 27.3 to 67.8% and 39.9 to 67.8% at the nucleotide and amino acid levels, respectively (Table 1).
TABLE 1.
Sequence similarities of the S, M, and L segments of ULUV strain FMNH158302 in M. geata and KMJV strain FMNH174124 in M. zinki from Tanzania, compared to other rodent- and soricomorph-borne hantavirusesa
| Virus and strain | % sequence similarity with strain: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ULUV FMNH158302 |
KMJV FMNH174124 |
|||||||||||
| S segment |
M segment |
L segment |
S segment |
M segment |
L segment |
|||||||
| 1,187 nt | 395 aa | 1,464 nt | 488 aa | 6,459 nt | 2,152 aa | 1,269 nt | 422 aa | 1,276 nt | 425 aa | 6,447 nt | 2,148 aa | |
| HTNV 76–118 | 48.8 | 46.1 | 40.8 | 38.4 | 65.1 | 61.7 | 49.7 | 50.0 | 46.1 | 43.8 | 63.7 | 62.1 |
| SOOV SOO-1 | 48.3 | 46.3 | 54.2 | 38.4 | 65.3 | 61.3 | 52.8 | 50.5 | 45.2 | 42.6 | 64.5 | 62.3 |
| DOBV Greece | 47.7 | 47.1 | 52.2 | 38.6 | 64.9 | 61.4 | 48.1 | 51.7 | 49.1 | 41.9 | 64.7 | 62.4 |
| SEOV 80–39 | 50.9 | 46.3 | 51.7 | 38.4 | 64.9 | 61.5 | 50.9 | 49.8 | 45.2 | 43.5 | 65.2 | 62.3 |
| SANGV SA14 | 49.2 | 47.1 | 50.8 | 38.6 | 65.2 | 61.8 | 59.6 | 50.5 | 50.3 | 42.6 | 64.0 | 62.6 |
| PUUV Sotkamo | 51.5 | 46.6 | 48.9 | 38.4 | 65.8 | 60.5 | 53.2 | 50.0 | 50.2 | 44.7 | 64.9 | 62.1 |
| TULV 5302v | 48.3 | 47.8 | 49.1 | 41.2 | 63.4 | 59.7 | 49.6 | 49.8 | 49.3 | 44.0 | 64.4 | 61.7 |
| PHV PH-1 | 51.9 | 48.4 | 48.5 | 40.5 | 62.4 | 60.6 | 51.4 | 49.8 | 47.8 | 43.1 | 64.0 | 62.0 |
| SNV NMH10 | 50.5 | 47.1 | 49.2 | 39.8 | 63.6 | 60.9 | 51.8 | 50.7 | 47.6 | 42.8 | 64.5 | 61.6 |
| ANDV Chile9717869 | 48.0 | 47.1 | 48.8 | 38.8 | 64.5 | 60.7 | 51.2 | 50.5 | 46.2 | 42.8 | 64.3 | 61.8 |
| CBNV CBN-3 | 50.7 | 47.1 | 49.1 | 41.2 | 65.1 | 62.1 | 51.1 | 50.5 | 46.8 | 45.2 | 64.5 | 62.5 |
| ARRV MSB73418 | 44.0 | 39.9 | 67.7 | 65.2 | 43.8 | 43.6 | 65.4 | 63.5 | ||||
| JMSV MSB144475 | 49.8 | 45.3 | 43.8 | 33.4 | 64.7 | 63.8 | 50.4 | 50.5 | 44.0 | 34.6 | 65.6 | 63.2 |
| SWSV mp70 | 49.2 | 47.1 | 43.6 | 42.2 | 62.2 | 57.5 | 47.4 | 49.5 | 52.0 | 53.0 | 63.5 | 58.6 |
| KKMV MSB148794 | 50.2 | 46.3 | 42.3 | 34.7 | 65.2 | 63.2 | 48.5 | 48.3 | 38.8 | 28.1 | 65.1 | 63.2 |
| QHSV YN05–284 | 50.4 | 45.8 | 45.5 | 34.2 | 69.6 | 71.1 | 49.3 | 47.9 | 43.8 | 35.3 | 74.4 | 71.1 |
| BOGV 2074 | 52.2 | 40.8 | 67.4 | 70.8 | 44.7 | 36.6 | 67.8 | 70.8 | ||||
| TGNV Tan826 | 27.3 | 44.9 | 71.6 | 71.5 | 27.9 | 45.6 | 70.9 | 68.6 | ||||
| BOWV VN1512 | 47.2 | 46.1 | 50.1 | 39.1 | 64.5 | 60.1 | 50.4 | 48.6 | 50.9 | 43.5 | 65.3 | 61.3 |
| AZGV KBM15 | 37.2 | 46.7 | 49.6 | 40.2 | 65.1 | 61.6 | 45.4 | 50.6 | 54.3 | 46.3 | 65.6 | 62.1 |
| JJUV 10–11 | 48.4 | 45.1 | 53.9 | 42.0 | 64.7 | 60.7 | 50.4 | 47.9 | 43.8 | 43.1 | 66.1 | 61.2 |
| MJNV Cl05–11 | 55.9 | 61.3 | 59.5 | 50.8 | 69.4 | 71.3 | 66.7 | 64.2 | 65.2 | 62.1 | 71.3 | 73.0 |
| TPMV VRC66412 | 53.2 | 62.0 | 61.7 | 50.0 | 69.4 | 72.4 | 67.8 | 67.8 | 65.0 | 63.1 | 69.7 | 73.3 |
| RKPV MSB57412 | 49.5 | 47.8 | 52.5 | 43.2 | 65.0 | 60.6 | 49.8 | 49.8 | 46.5 | 44.7 | 65.4 | 61.5 |
| OXBV Ng1453 | 46.3 | 47.1 | 48.3 | 38.9 | 65.3 | 62.2 | 50.3 | 50.2 | 48.2 | 42.8 | 65.1 | 62.0 |
| ASAV N10 | 50.8 | 45.1 | 49.7 | 41.2 | 67.0 | 62.6 | 46.1 | 47.6 | 49.1 | 44.5 | 66.6 | 63.5 |
| NVAV MSB95703 | 60.6 | 47.6 | 53.4 | 41.9 | 64.1 | 61.4 | 48.9 | 48.8 | 60.8 | 41.9 | 66.1 | 62.4 |
| ULUV FMNH158302 | 71.2 | 73.7 | 59.6 | 47.8 | 70.6 | 74.8 | ||||||
| KMJV FMNH174124 | 71.2 | 73.7 | 59.6 | 47.8 | 70.6 | 74.8 | ||||||
Abbreviations: ANDV, Andes virus; ARRV, Ash River virus; ASAV, Asama virus; AZGV, Azagny virus; BOGV, Boginia virus; BOWV, Bowé virus; CBNV, Cao Bang virus; DOBV, Dobrava virus; HTNV, Hantaan virus; JJUV, Jeju virus; JMSV, Jemez Spring virus; KKMV, Kenkeme virus; KMJV, Kilimanjaro virus; MJNV, Imjin virus; NVAV, Nova virus; OXBV, Oxbow virus; PHV, Prospect Hill virus; PUUV, Puumala virus; QHSV, Qian Hu Shan virus; RKPV, Rockport virus; SANGV, Sangassou virus; SEOV, Seoul virus; SNV, Sin Nombre virus; SOOV, Soochong virus; SWSV, Seewis virus; TGNV, Tanganya virus; TPMV, Thottapalayam virus; TULV, Tula virus; ULUV, Uluguru virus. nt, nucleotides; aa, amino acids.
Partial 1,276- and 1,464-nucleotide regions of the KMJV (GenBank JX193699) and ULUV (GenBank JX193696) M genomic segment, respectively, showed the highly conserved WAASA amino acid motif (amino acid positions 348 to 352 and 359 to 363, respectively). Glycosylation sites, as predicted using NetNlyc 1.0 and Predictprotein (9), showed three potential N-linked glycosylation sites in the Gn glycoprotein for KMJV (positions 4, 51, and 102) and for ULUV (amino acid positions 61, 115, and 225).
The full-length 6,604- and 6,568-nucleotide L segments of KMJV (GenBank JX193700) and ULUV (GenBank JX193697), respectively, encoded a predicted RNA-dependent RNA polymerase (RdRp) of 2,148 and 2,152 amino acids, respectively. The RdRp amino acid sequence similarity, which was highest (72.4% and 73.3%) between KMJV and ULUV (Table 1), exhibited six major conserved motifs (designated premotif A and motifs A, B, C, D, and E), like other hantaviruses (4, 10).
Unrooted phylogenetic trees, based on the coding regions of the S, M, and L segments and generated by maximum likelihood and Bayesian methods, were implemented with the RAxML Blackbox Web server (11) and MrBayes 3.1 (12), under the best-fit GTR+I+Γ model of evolution in MrModeltest v2.3 (13) and jModelTest version 0.1 (14). In trees based on each genomic segment, KMJV and ULUV were distinct from newfound hantaviruses harbored by insectivorous bats, and they shared a common ancestry with Asian crocidurine shrew-borne hantaviruses, namely, Thottapalayam virus (TPMV) (15) and Imjin virus (MJNV) (10) (Fig. 2).
FIG 2.

Phylogenetic trees were generated by the maximum-likelihood and Bayesian methods, using the GTR+I+Γ model of evolution, based on the alignment of the S, M, and L segment sequences of ULUV strain FMNH158302 and KMJV strain FMNH174124. Since tree topologies created using RAxML and MrBayes were very similar, the trees generated by MrBayes are displayed. The phylogenetic positions of ULUV and KMJV are shown in relationship to crocidurine shrew-borne hantaviruses, including Thottapalayam virus (TPMV VRC66412, AY526097, EU001329, EU001330), Imjin virus (MJNV Cl05–11, EF641804, EF641798, EF641806), Azagny virus (AZGV KBM15, JF276226, JF276227, JF276228), Bowé virus (BOWV VN1512, KC631782, KC631783, KC631784), Tanganya virus (TGNV Tan826, EF050455, EF050454), and Jeju virus (JJUV 10-11, HQ834695, HQ834696, HQ834697). Hantaviruses harbored by insectivorous bats included Huangpi virus (HUPV Pa1, JX473273, JX465369), Longquan virus (LQUV Ra10, JX465413, JX465398, JX465379; LQUV Rs168, JX465421, JX465401, JX465387), Magboi virus (MGBV 1209, JN037851), Mouyassué virus (MOYV KB576, JQ287716; MOYV KB577, KJ000540), and Xuan Son virus (XSV F42682, KF704709, KJ000538, KF704714; XSV F44601, KF704712, KJ000539, KF704717). Soricine shrew-borne hantaviruses included Boginia virus (BOGV 2074, JX990966, JX990965), Cao Bang virus (CBNV CBN-3, EF543524, EF543526, EF543525), Ash River virus (ARRV MSB73418, EF650086, EF619961), Jemez Springs virus (JMSV MSB144475, FJ593499, FJ593500, FJ593501), Kenkeme virus (KKMV MSB148794, GQ306148, GQ306149, GQ306150), Qian Hu Shan virus (QHSV YN05-284, GU566023, GU566022, GU566021), and Seewis virus (SWSV mp70, EF636024, EF636025, EF636026). Also shown are mole-borne hantaviruses, including Asama virus (ASAV N10, EU929072, EU929075, EU929078), Nova virus (NVAV MSB95703, FJ539168, HQ840957, FJ593498), Oxbow virus (OXBV Ng1453, FJ539166, FJ539167, FJ593497), and Rockport virus (RKPV MSB57412, HM015223, HM015219, HM015221). Rodent-borne hantaviruses included Hantaan virus (HTNV 76–118, NC_005218, Y00386, NC_005222), Soochong virus (SOOV SOO-1, AY675349, AY675353, DQ562292), Dobrava virus (DOBV Greece, NC_005233, NC_005234, NC_005235), Seoul virus (SEOV 80-39, NC_005236, NC_005237, NC_005238), Sangassou virus (SANV SA14, JQ082300, JQ082301, JQ082302), Puumala virus (PUUV Sotkamo, NC_005224, NC_005223, NC_005225), Prospect Hill virus (PHV PH-1, Z49098, X55129, EF646763), Tula virus (TULV M5302v, NC_005227, NC_005228, NC_005226), Andes virus (ANDV Chile9717869, NC_003466, NC_003467, NC_003468), and Sin Nombre virus (SNV NMH10, NC_005216, NC_005215, NC_005217). The numbers at each node are posterior node probabilities based on 150,000 trees (left) and bootstrap values of 1,000 replicates executed on the RAxML BlackBox Web server (right), respectively. The scale bars indicate nucleotide substitutions per site. Hantavirus taxa are color coded to correspond to the geographic origins of the reservoir hosts: red (Africa); green (Eurasia); blue (Americas).
In the S and L trees, which were based on full-length and/or nearly full-length sequences, KMJV and ULUV were monophyletic, whereas in the M tree, which was based on less than half of the entire M-segment sequence, ULUV was basal to KMJV, TPMV, and MJNV (Fig. 2). The high bootstrap values and posterior node probabilities for ULUV and KMJV in the M segment tree suggested that the disparate topologies based on the M and S/L segments were probably not the result of missing sequences. Instead, this might indicate that the evolutionary history of the M segment differed from that of the S and L segments. Specifically, given that the M segment encodes the envelope glycoprotein, the ULUV M segment may have been under greater selection pressure or the KMJV M segment may have been under negative selection pressure after the two viruses diverged from a shared common ancestor. Alternatively or additionally, genetic recombination or genetic drift may be responsible. The full-length M genomic sequence and future phylogeographic studies may help to explain the discrepant trees.
The Myosorex genus comprises 15 extant species which exist in the forested highlands of central, eastern, and southern Africa (16). As the only endemic mammalian species on Mt. Kilimanjaro, Mysorex zinki is found in forests, heaths, and moorlands and near the edge of the alpine desert, along an elevation gradient, ranging from 2,470 to 4,000 m (17). While Mysorex geata is also endemic in moist montane forests in Tanzania, the true distribution of this species is unclear, particularly at higher altitudes undisturbed by frequent human activities (17).
On the basis of albeit meager fossil records and assuming equally probable and bidirectional exchanges between Eurasia and Africa, the family Soricidae likely originated in Eurasia (18). Shrews of the Myosorex genus are believed to have originated in the tropical forests of central Africa during the Middle Miocene, approximately 12 to 15 million years before the present (16, 18). In reconstructing the biogeographic history of the Soricidae, Dubey and colleagues proposed three equally parsimonious scenarios, based on the premise of two independent origins of the Crocidura genus (18). Viewed within this context, the phylogenetic positions of KMJV and ULUV in relation to TPMV and MJNV, which are hosted by Asian crocidurine shrews, and the clade comprising African crocidurine shrew-borne hantaviruses (BOWV, TGNV, and AZGV) and Jeju virus (JJUV) (19), which is hosted by the Asian lesser white-toothed shrew (Crocidura shantungensis) in Korea, supports a scenario in which the first diversification of the monophyletic Crocidurinae into Crocidurini and Myosoricini tribes occurred in Eurasia rather than Africa.
The discovery of hantaviruses in myosoricine shrew species endemic in Tanzania expands the host diversity and geographic distribution of non-rodent-borne hantaviruses and suggests that other genetically distinct hantaviruses may be widespread elsewhere in Africa. Compared to tissues from rodents, fewer tissues from either shrews or bats in Africa have been examined for hantavirus RNA by RT-PCR. Yet, many more hantaviruses have been found in shrews and bats. That is, of the nine hantaviruses detected thus far in sub-Saharan Africa, five are from shrews (2–4; present study) and two are from insectivorous bats (20, 21), compared to only two from rodents (1, 22). One possible interpretation is that ancestral shrews and/or bats, rather than rodents, may have served as the early mammalian hosts of primordial hantaviruses.
ACKNOWLEDGMENTS
We thank Shannon N. Bennett for helpful suggestions.
This work was supported in part by U.S. Public Health Service grants R01AI075057 and P20GM103516 from the National Institutes of Health, grant DEB-1343517 from the National Science Foundation, and fellowship S13205 from the Japan Society for the Promotion of Science. The services provided by the Genomics Core Facility, funded partially by the Centers of Biomedical Research Excellence program (P30GM103341), are gratefully acknowledged. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 16 April 2014
REFERENCES
- 1.Klempa B, Fichet-Calvet E, Lecompte E, Auste B, Aniskin V, Meisel H, Denys C, Koivogui L, ter Meulen J, Krüger DH. 2006. Hantavirus in African wood mouse, Guinea. Emerg. Infect. Dis. 12:838–840. 10.3201/eid1205.051487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klempa B, Fichet-Calvet E, Lecompte E, Auste B, Aniskin V, Meisel H, Barriere P, Koivogui L, ter Meulen J, Krüger DH. 2007. Novel hantavirus sequences in shrew, Guinea. Emerg. Infect. Dis. 13:520–522. 10.3201/eid1303.061198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang HJ, Kadjo B, Dubey S, Jacquet F, Yanagihara R. 2011. Molecular evolution of Azagny virus, a newfound hantavirus harbored by the West African pygmy shrew (Crocidura obscurior) in Côte d'Ivoire. Virol. J. 8:373. 10.1186/1743-422X-8-373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu SH, Nicolas V, Lalis A, Sathirapongsasuti N, Yanagihara R. 2013. Complete genome sequence analysis and molecular phylogeny of a newfound hantavirus harbored by the Doucet's musk shrew (Crocidura douceti) in Guinea. Infect. Genet. Evol. 20:118–123. 10.1016/j.meegid.2013.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gannon WL, Sykes RS, Animal Care and Use Committee of the American Society of Mammalogists 2007. Guidelines of the American Society of Mammalogists for the use of wild mammals in research. J. Mammal 88:809–823. 10.1644/06-MAMM-F-185R1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stanley WT, Esselstyn JA. 2010. Biogeography and diversity among montane populations of mouse shrew (Soricidae: Myosorex) in Tanzania. Biol. J. Linnean Soc. 100:669–680. 10.1111/j.1095-8312.2010.01448.x [DOI] [Google Scholar]
- 7.Stanley WT, Hutterer 2000. A new species of Myosorex Gray, 1832 (Mammalia: Soricidae) from the Eastern Arc mountains, Tanzania. Bonn. Zool. Beitr. 49:19–29 [Google Scholar]
- 8.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavel Y, von Heijne G. 1990. Sequence differences between glycosylated and nonglycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 3:433–442. 10.1093/protein/3.5.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song J-W, Kang HJ, Gu SH, Moon SS, Bennett SN, Song KJ, Baek LJ, Kim H-C, O'Guinn ML, Chong ST, Klein TA, Yanagihara R. 2009. Characterization of Imjin virus, a newly isolated hantavirus from the Ussuri white-toothed shrew (Crocidura lasiura). J. Virol. 83:6184–6191. 10.1128/JVI.00371-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57:758–771. 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- 12.Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. 10.1093/bioinformatics/btg180 [DOI] [PubMed] [Google Scholar]
- 13.Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818. 10.1093/bioinformatics/14.9.817 [DOI] [PubMed] [Google Scholar]
- 14.Posada D. 2008. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25:1253–1256. 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- 15.Yadav PD, Vincent MJ, Nichol ST. 2007. Thottapalayam virus is genetically distant to the rodent-borne hantaviruses, consistent with its isolation from the Asian house shrew (Suncus murinus). Virol. J. 4:80. 10.1186/1743-422X-4-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willows-Munro S, Matthee CA. 2009. The evolution of the southern African members of the shrew genus Myosorex: understanding the origin and diversification of a morphologically cryptic group. Mol. Phylogenet. Evol. 51:394–398. 10.1016/j.ympev.2009.02.012 [DOI] [PubMed] [Google Scholar]
- 17.Stanley WT, Rogers MA, Hutterer R. 2005. A morphological assessment of Myosorex zinki, an endemic shrew on Mt. Kilimanjaro. Belg. J. Zool. 135(Suppl):141–144 [Google Scholar]
- 18.Dubey S, Salamin N, Ohdachi SD, Barrière P, Vogel P. 2007. Molecular phylogenetics of shrews (Mammalia: Soricidae) reveals timing of transcontinental colonizations. Mol. Phylogenet. Evol. 44:126–137. 10.1016/j.ympev.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 19.Arai S, Gu SH, Baek LJ, Tabara K, Bennett SN, Oh HS, Takada N, Kang HJ, Tanaka-Taya K, Morikawa S, Okabe N, Yanagihara R, Song J-W. 2012. Divergent ancestral lineages of newfound hantaviruses harbored by phylogenetically related crocidurine shrew species in Korea. Virology 424:99–105. 10.1016/j.virol.2011.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sumibcay L, Kadjo B, Gu SH, Kang HJ, Lim BK, Cook JA, Song JW, Yanagihara R. 2012. Divergent lineage of a novel hantavirus in the banana pipistrelle (Neoromicia nanus) in Côte d'Ivoire. Virol. J. 9:34. 10.1186/1743-422X-9-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiss S, Witkowski PT, Auste B, Nowak K, Weber N, Fahr J, Mombouli JV, Wolfe ND, Drexler JF, Drosten C, Klempa B, Leendertz FH, Krüger DH. 2012. Hantavirus in bat, Sierra Leone. Emerg. Infect. Dis. 18:159–161. 10.3201/eid1801.111026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meheretu Y, Cížková D, Těšíková J, Welegerima K, Tomas Z, Kidane D, Girmay K, Schmidt-Chanasit J, Bryja J, Günther S, Bryjová A, Leirs H, Goüy de Bellocq J. 2012. High diversity of RNA viruses in rodents, Ethiopia. Emerg. Infect. Dis. 18:2047–2050. 10.3201/eid1812.120596 [DOI] [PMC free article] [PubMed] [Google Scholar]