

ABSTRACT
In order to investigate the novel function(s) of the herpes simplex virus 1 (HSV-1) immediate early protein ICP22, we screened for ICP22-binding proteins in HSV-1-infected cells. Our results were as follows. (i) Tandem affinity purification of ICP22 from infected cells, coupled with mass spectrometry-based proteomics and subsequent analyses, demonstrates that ICP22 forms a complex(es) with the HSV-1 proteins UL31, UL34, UL47 (or VP13/14), and/or Us3. All these proteins were previously reported to be important for viral egress through the nuclear membrane. (ii) ICP22 colocalizes with UL31 and UL34 at the nuclear membrane in wild-type HSV-1-infected cells. (iii) The UL31-null mutation prevents the targeting of ICP22 to the nuclear membrane. (iv) The ICP22-null mutation resulted in UL31 and UL34 being mislocalized in the endoplasmic reticulum (in addition to the nuclear membrane) and significantly reduced numbers of primary enveloped virions in the perinuclear space, although capsids accumulated in the nuclei. Collectively, these results suggest that (i) ICP22 interacts with HSV-1 regulators of nuclear egress, including UL31, UL34, UL47, and Us3 in HSV-1-infected cells; (ii) UL31 mediates the recruitment and anchorage of ICP22 at the nuclear membrane; and (iii) ICP22 plays a regulatory role in HSV-1 primary envelopment, probably by interacting with and regulating UL31 and UL34. Here we report a previously unknown function for ICP22 in the regulation of HSV-1 nuclear egress.
IMPORTANCE The herpes simplex virus 1 (HSV-1) immediate early protein ICP22 is recognized primarily as a regulator of viral gene expression. In this study, we show that ICP22 interacts with the HSV-1 proteins UL31 and UL34, which play crucial roles at the nuclear membrane in HSV-1 primary envelopment during viral nuclear egress. We also demonstrate that UL31 is required for the targeting of ICP22 to the nuclear membrane and that ICP22 is required for the correct localization of UL31 and/or UL34. Furthermore, we confirm that ICP22 is required for efficient HSV-1 primary envelopment during viral nuclear egress. Thus, we report, for the first time, that ICP22 plays a regulatory role in HSV-1 nuclear egress.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is one of the best-studied members of the Herpesviridae family, causing a variety of diseases such as mucocutaneous diseases, keratitis, skin diseases, and life-threatening encephalitis (1). After HSV-1 entry into the host cell, the nucleocapsid is transported to a nuclear pore, enabling the entry of the viral genome into the nucleus and initiation of viral gene expression (1). HSV-1 encodes >80 different proteins that are expressed in a regulated cascade and fall into three major classes, designated immediate early (IE), early (E), and late (L) (1). Viral DNA replication, capsid assembly, and packaging of replicated viral genomes into capsids take place in the nucleus (1). The nascent progeny nucleocapsids of HSV-1, like those of other herpesviruses, then traverse the inner nuclear membrane (INM) and outer nuclear membrane (ONM) by a unique mechanism. Nucleocapsids acquire a primary envelope by budding through the INM into the perinuclear space between the INM and the ONM (primary envelopment), and enveloped nucleocapsids then fuse with the ONM to release deenveloped nucleocapsids into the cytoplasm (deenvelopment) (2, 3). A heterodimeric complex, designated the nuclear egress complex (NEC), between the HSV-1 proteins UL31 and UL34, conserved in all known herpesviruses, is critical for primary envelopment, the first step in the nuclear egress of herpesvirus nucleocapsids (1–5). It has been reported that, in the absence of UL31 and UL34, nucleocapsids accumulate in the nucleoplasm and that progeny virus intermediates and virions are barely detectable in the perinuclear space, in the cytoplasm, and at the cell surface (5, 6). The HSV-1 UL31/UL34 complex (NEC), and its homologues in other herpesviruses, is thought to facilitate the primary envelopment of nucleocapsids by (i) recruiting host cellular protein kinase C (PKC) isoforms to the nuclear membrane, where they phosphorylate, and thus dissolve, the nuclear lamina, allowing the herpesvirus nucleocapsid access to the INM (2, 3, 7–11); (ii) recruiting nucleocapsids to the INM (2, 3, 12, 13); and (iii) budding of the nucleocapsids into the INM (2, 3, 14, 15). In addition, an HSV-1 serine/threonine protein kinase, Us3, which has been shown to phosphorylate the NEC, is thought to be involved in primary envelopment because it is able to regulate the localization of UL31 and UL34 at the nuclear membrane and to phosphorylate the nuclear lamina (2–4, 7–9, 16, 17). Furthermore, it was recently reported that the NEC forms a complex with Us3 and UL47 (or VP13/14), a major structural protein in the HSV-1 tegument. UL47 plays a regulatory role in primary envelopment, although the precise mechanism by which it acts remains unclear (18). Little is known about the next step in the nuclear egress of nucleocapsids: deenvelopment. The released nucleocapsids acquire a secondary envelope at the cytoplasmic membrane, most likely in the trans-Golgi network (2, 19), and mature virions are then secreted by exocytosis (1).
In the present study, we focused on the role of one HSV-1 IE protein, ICP22, a 420-residue protein encoded by the Us1 gene, conserved in the subfamily Alphaherpesvirinae of the family Herpesviridae. It was previously suggested that ICP22 plays important roles in viral replication and pathogenicity. Mutant viruses lacking ICP22 yield reduced numbers of viral progeny in cell cultures in a cell type-dependent manner (e.g., in human embryonic lung [HEL] fibroblasts but not in Vero cells) and exhibit reduced pathogenicity in mouse models (20). At present, the mechanism(s) by which ICP22 acts in viral replication and pathogenicity is unknown. However, accumulating evidence suggests that ICP22 regulates the expression of specific viral genes (e.g., ICP0 IE and a subset of L genes, including ICP0, UL38, UL41, Us11, gC, UL26, UL26.5, and gJ), probably by interacting with and/or modifying both the viral and cellular proteins involved in viral gene expression, i.e., the cellular proteins RNA polymerase II, TFIID, CDK9, CLOCK, and p53 and the viral proteins ICP4 and ICP27 (21–31). Thus, most research on ICP22 to date has concentrated on its regulatory function(s) in viral gene expression. We note, however, that ICP22 was reported to be required for correct virion composition (32), raising the possibility that ICP22 plays another, yet-to-be-defined role(s) in HSV-1 infection, possibly in HSV-1 virion morphogenesis. To pursue this possibility further, we screened for ICP22-binding proteins by tandem affinity purification of HSV-1-infected HEL cells coupled with mass spectrometry-based proteomics. This screening and subsequent analyses revealed that ICP22 interacted with the HSV-1 nuclear egress regulators UL31, UL34, Us3, and UL47 in HSV-1-infected cells and colocalized with UL31 and UL34 at the nuclear membrane. Studies of the effects of ICP22 and UL31/UL34 on each other showed that UL31 was required for the targeting of ICP22 to the nuclear membrane. Conversely, ICP22 was required for the correct localization of UL31/UL34 at the nuclear membrane. Furthermore, ultrastructural analyses of ICP22-null mutant virus-infected cells demonstrated that ICP22 is required for efficient HSV-1 primary envelopment. These observations suggest that ICP22 facilitates HSV-1 primary envelopment, probably by interacting with and regulating UL31 and UL34. In this paper, we report a novel association between ICP22 and HSV-1 primary envelopment during viral nuclear egress.
MATERIALS AND METHODS
Cells and viruses.
Vero, rabbit skin, and HEL cells were described previously (19, 33). UL31-CV1 cells (a generous gift from J. D. Baines) (34) were cultured in Dulbecco's modified Eagle's medium with 10% fetal calf serum containing 200 μg/ml hygromycin B. The HSV-1(F) wild-type strain; recombinant virus strain YK421 (ΔICP22), an ICP22-null mutant virus in which the entire Us1 open reading frame (ORF) was deleted; recombinant virus strain YK422 (ΔICP22-repair), in which the ICP22-null mutation in YK421 was repaired; recombinant virus strain YK423, in which ICP22 was fused to a Myc epitope–tobacco etch virus (TEV) protease cleavage site–Flag epitope (MEF) tag (MEF-ICP22); recombinant virus strain YK539, in which UL31 was fused to an MEF tag (MEF-UL31); recombinant virus strain YK538, in which UL34 was fused to an MEF tag (MEF-UL34); recombinant virus strain YK511, encoding an enzymatically inactive Us3 mutant in which the lysine at Us3 position 220 was replaced with methionine (Us3K220M); and recombinant virus strain YK513 (Us3K220M-repair), in which the Us3K220M mutation in YK511 was repaired, were described previously (18, 24, 35) (Fig. 1).
FIG 1.

Schematic diagrams of the genome structures of wild-type HSV-1(F) and the relevant domains of the recombinant viruses used in this study. Line 1, wild-type HSV-1(F) genome; line 2, domains encoding viral replication origin S (Ori-S) and the Us1 to Us4 open reading frames (ORFs); line 3, domains of ICP22, Us1.5, and Us3; lines 4 to 8, recombinant viruses with mutations in ICP22 or Us3; line 9, domains encoding the UL30 to UL35 ORFs; line 10, domains of UL34 and UL31; lines 11 to 14, recombinant viruses with mutations in UL34 or UL31.
Plasmids.
The transfer plasmid pBS-UL31-rep, for generating recombinant YK721 (ΔUL31-repair) in which the UL31 deletion in YK720 (ΔUL31) was repaired, was constructed by PCR cloning of the domain containing the UL31 ORF (the 649-bp upstream sequence flanking the UL31 start codon and the 508-bp downstream sequence flanking the UL31 stop codon) from pYEbac102, a full-length infectious clone of HSV-1(F) (33), into pBluescript II KS(+) (Stratagene). A plasmid encoding a fusion protein of glutathione S-transferase (GST) and the N-terminal domain of ICP22 (GST-ICP22N1-90), pGEX-ICP22N1-90, was constructed by cloning Us1 encoding ICP22 codons 1 to 90, amplified by PCR from pBS-ICP22 (24), into pGEX-4T-1 (GE Healthcare) in frame with GST.
Identification of proteins that interact with ICP22.
HEL cells were infected with YK423 (MEF-ICP22) at a multiplicity of infection (MOI) of 3, harvested at 18 h postinfection, and lysed in 0.1% NP-40 buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 50 mM NaF, 0.1% NP-40) containing a protease inhibitor cocktail (Nacalai Tesque). After centrifugation, the supernatants were immunoprecipitated with an anti-Myc monoclonal antibody, and the immunoprecipitates were incubated with AcTEV protease (Invitrogen). After centrifugation, the supernatants were immunoprecipitated with an anti-Flag monoclonal antibody, and the immunoprecipitates were washed three times with 0.1% NP-40 buffer and two times with wash buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 50 mM NaF). Flag elution buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5 mg Flag peptide/ml) was added, and the immunoprecipitates were rotated for 2 h at 4°C. The eluted-protein solution was trypsinized and analyzed by nano-liquid chromatography tandem mass spectrometry (nanoLC-MS/MS), as described previously (36). For this analysis, we used a Q-Star Elite instrument (AB Sciex) coupled with a Dina instrument (KYA Technologies). The MS/MS signals were then analyzed against the human proteins in the RefSeq database (National Center for Biotechnology Information [NCBI]; 38,963 sequences as of 5 July 2010) and the viral proteins in the nonredundant database (NCBI; 754,281 sequences as of 10 June 2011) by using the Mascot algorithm (version 2.2.04; Matrix Science) with the following parameters: variable modifications, oxidation (Met), protein N-terminal acetylation, pyroglutamination (Gln), and phosphorylation (Ser, Thr, and Tyr); maximum missed cleavages, 2; peptide mass tolerance, 100 ppm; and MS/MS tolerance, 0.5 Da. Protein identification was based on the criterion of having at least one MS/MS data signal with a Mascot score that exceeded the threshold (P < 0.05).
Mutagenesis of viral genomes in E. coli and generation of recombinant HSV-1.
A UL31-null mutant virus, YK720 (ΔUL31), in which the UL31 gene was disrupted by replacing UL31 codons 8 to 269 with a foreign gene cassette containing an I-SceI site and a kanamycin resistance gene (Fig. 1), was generated by the Red-mediated mutagenesis procedure using Escherichia coli GS1783 carrying pYEbac102 (33, 35), except that primers 5′-TGTCCCTGGAGCACACCCTGTGTACCTATGTATGACACCGACCCCCATCGAGGATGACGACGATAAGTAGGG-3′ and 5′-GTCGAAGCTGATGTCCCTCATTTTACAATAAATGTCTGCGGCCGACACGGCAACCAATTAACCAATTCTGATTAG-3′ were used and UL31-CV1 cells were transfected with pYEbac102 carrying the UL31-null mutation by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Recombinant virus strain YK721 (ΔUL31-repair), in which the UL31-null mutation in YK720 was repaired (Fig. 1), was generated by cotransfection with pYEbac102 carrying the UL31-null mutation and pBS-UL31-rep into rabbit skin cells, as described previously (33). Plaques were isolated and purified two additional times in Vero cells. Restoration of the original sequence was confirmed by sequencing. In cases where YK720 (ΔUL31) was used, this recombinant virus as well as the control viruses wild-type HSV-1(F) and YK721 (ΔUL31-repair) were propagated and titrated in UL31-CV1 cells.
Production and purification of GST fusion proteins in E. coli.
GST-ICP22N1-90 or GST-ICP22C283-420 fusion proteins were expressed in E. coli that had been transformed with pGEX-ICP22N1-90 or pGEX-ICP22C283-420 (24), respectively, and purified as described previously (37).
Antibodies.
The commercial antibodies used in this study were mouse monoclonal antibodies to Flag (M2; Sigma), Myc (PL14; MBL), and α-tubulin (DM1A; Sigma) and rabbit polyclonal antibodies to calnexin (c4731; Sigma), lamin B1 (ab16048-100; Abcam), Flag (DDDDK; MBL), and VP23 (CAC-CT-HSV-UL18; CosmoBio). Rabbit polyclonal antibodies to UL34, UL31, and Us3; mouse polyclonal antibody to UL31; and chicken polyclonal antibody to UL34 (a generous gift from R. J. Roller) were described previously (4, 18, 35, 38). To generate mouse polyclonal antibodies to ICP22N or ICP22C, BALB/c mice were immunized once with purified GST-ICP22N1-90 or GST-ICP22C283-420 with TiterMax Gold (TiterMax USA, Inc.). Serum from the immunized mouse was used as an anti-ICP22 mouse polyclonal antibody. The anti-ICP22N antibody reacts with ICP22 but not with Us1.5, an amino-terminally truncated form of ICP22 (39), and was used in immunofluorescence studies. The anti-ICP22C antibody was used for immunoblotting.
Antibody analyses.
Immunoprecipitation, immunoblotting, and immunofluorescence were performed as described previously (19, 40).
Electron microscopic analysis.
Vero and HEL cells infected with wild-type HSV-1(F), YK421 (ΔICP22), or YK422 (ΔICP22-repair) at an MOI of 5 for 18 h were examined by ultrathin-section electron microscopy as described previously (38).
RESULTS
Identification of viral and cellular proteins that interact with ICP22 in HSV-1-infected cells.
To identify viral and cellular proteins that interact with ICP22 in HSV-1-infected cells, we used tandem affinity purification of ICP22 fused to an MEF tag with Myc and Flag epitopes and a TEV protease cleavage site (MEF-ICP22). We isolated ICP22-bound proteins from the lysate of HEL cells infected with a recombinant virus, YK423 (MEF-ICP22), encoding MEF-ICP22 (24). Isolated proteins were then identified by mass spectrometry-based proteomic analyses. We identified 19 viral and 237 cellular proteins that coimmunoprecipitated with MEF-ICP22 (data not shown). Notably, these included 70-kDa heat shock cognate protein (hsc70), EBER-associated protein (EAP), subunits of casein kinase II, ICP4, and ICP27, proteins which were previously reported to interact with ICP22 (22, 23, 41, 42). Of these 256 potential ICP22-interacting proteins, we focused on the known viral regulators of HSV-1 primary envelopment, UL31, UL34, and UL47, in order to pursue the possibility that ICP22 is involved in primary envelopment by interacting with these viral proteins (4, 5, 18).
Interaction of ICP22 with UL31, UL34, Us3, and UL47 in HSV-1-infected cells.
The potential ICP22-interacting proteins UL31, UL34, and UL47, identified by mass spectrometry-based proteomics screening as described above, have been reported to form a complex with the HSV-1-encoded serine/threonine protein kinase Us3 (18, 43). To confirm and extend these ICP22 interactions, HEL cells were singly infected with wild-type HSV-1(F), YK423 (MEF-ICP22), YK538 (MEF-UL34) encoding MEF-UL34, or YK539 (MEF-UL31) encoding MEF-UL31; lysed; and immunoprecipitated with anti-Myc antibody. The immunoprecipitates were then analyzed by immunoblotting with antibodies to UL31, UL34, VP23, ICP22, UL47, Us3, and Flag (Fig. 2). We have previously reported that MEF tagging of UL31, UL34, and ICP22 had little effect on viral growth in cell cultures (18, 24). As shown in Fig. 2A, the anti-Myc antibody coprecipitated UL31, UL34, Us3, and UL47 with MEF-ICP22 from lysates of YK423 (MEF-ICP22)-infected HEL cells but did not coprecipitate the capsid protein VP23. In contrast, anti-Myc antibody did not immunoprecipitate any of these viral proteins from lysates of wild-type HSV-1(F)-infected cells (Fig. 2A). These results indicate that ICP22 forms a complex(es) with UL31, UL34, Us3, and/or UL47 in HSV-1-infected cells. Similarly, anti-Myc antibody coprecipitated UL34, Us3, UL47, and ICP22 with MEF-UL31 from lysates of YK539 (MEF-UL31)-infected cells (Fig. 2B) and coprecipitated UL31, UL47, Us3, and ICP22 with MEF-UL34 from lysates of YK538 (MEF-UL34)-infected cells (Fig. 2C). These results indicate that UL31 forms a complex(es) with UL34, Us3, UL47, and/or ICP22 and that UL34 forms a complex(es) with UL31, Us3, UL47, and/or ICP22 in HSV-1-infected cells.
FIG 2.

Interactions between ICP22, UL31, UL34, UL47, and Us3 in HSV-1-infected cells. HEL cells infected with wild-type HSV-1(F) (A to C), YK423 (MEF-ICP22) (A), YK539 (MEF-UL31) (B), or YK538 (MEF-UL34) (C) at an MOI of 5 for 18 h were harvested, immunoprecipitated (IP) with anti-Myc antibody (α-Myc), and analyzed by immunoblotting (IB) with the antibodies indicated. WCE, whole-cell extract.
Colocalization of ICP22 with UL31 and UL34 at the nuclear membrane in HSV-1-infected cells.
To determine where ICP22 interacts with UL31 and UL34 in HSV-1-infected cells, HEL cells infected with wild-type HSV-1(F) or YK423 (MEF-ICP22) were analyzed by immunofluorescence. As reported previously (23), in wild-type HSV-1(F)- or YK423 (MEF-ICP22)-infected HEL cells, ICP22 or MEF-ICP22, respectively, was diffusely localized to the nucleus, in part to discrete nuclear punctate structures (Fig. 3). In addition, ICP22 and MEF-ICP22 were detected at the nuclear rim and colocalized with UL31, UL34, or lamin B1 (an INM marker) in wild-type HSV-1(F)- or YK423 (MEF-ICP22)-infected HEL cells (Fig. 3). These results indicate that ICP22 colocalizes with UL31 and UL34 at the nuclear membrane in HSV-1-infected cells.
FIG 3.
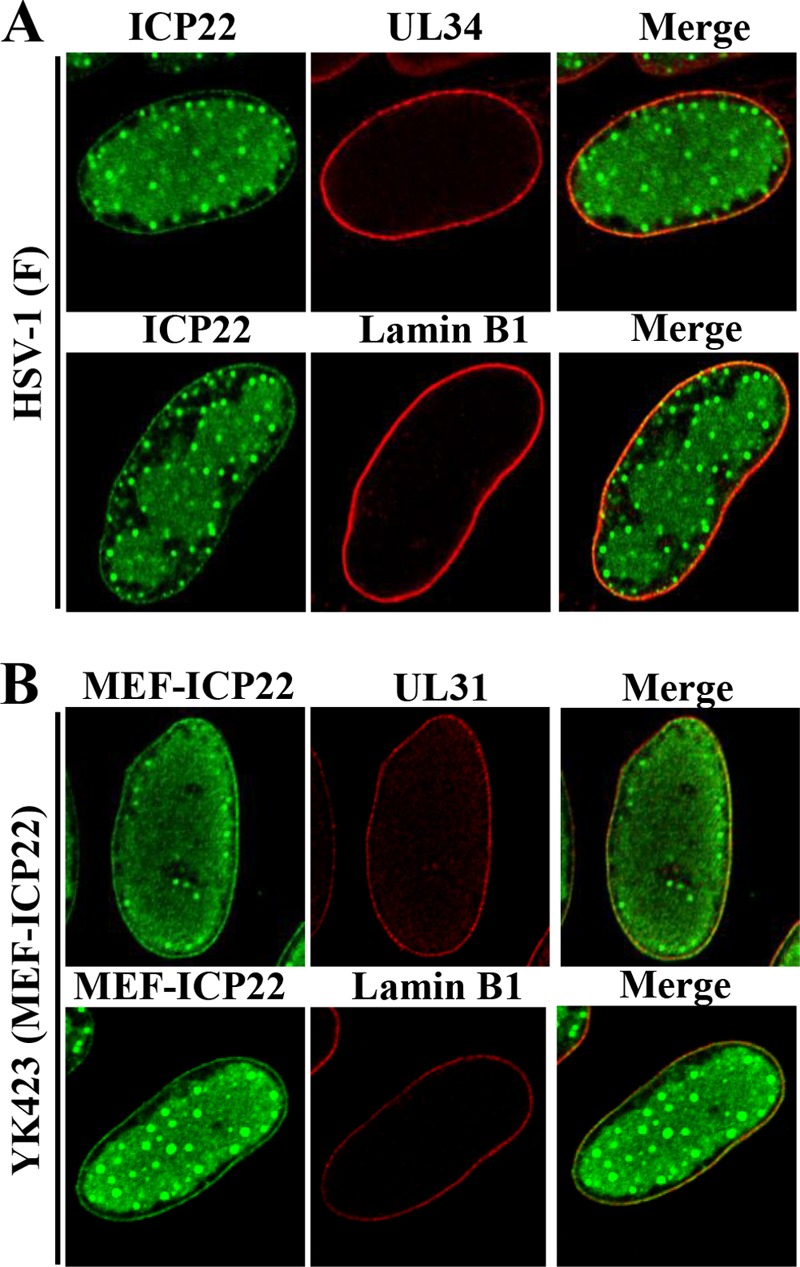
Colocalization of ICP22 with UL31, UL34, or lamin B1 in HSV-1-infected cells. HEL cells infected with wild-type HSV-1(F) (A) or YK423 (MEF-ICP22) (B) at an MOI of 5 for 13 h were fixed, permeabilized, stained with anti-ICP22 and anti-UL34 or anti-lamin B1 (A) or with anti-Flag and anti-UL31 or anti-lamin B1 (B) antibodies, and examined by confocal microscopy.
Effect of Us3 catalytic activity on localization of ICP22 at the nuclear membrane in HSV-1-infected cells.
Us3 has been reported to regulate the localization of UL31 and UL34 at the nuclear membrane in HSV-1-infected cells (4). In the absence of Us3 catalytic activity, UL31 and UL34 were shown to be incorrectly colocalized in punctate structures at the nuclear membrane in HSV-1-infected cells (44). Therefore, we investigated the localization of ICP22 at the nuclear membrane in the absence of US3 kinase activity. As shown in Fig. 3A and 4, in HEL cells infected with wild-type HSV-1(F) or YK513 (Us3K220M-repair), ICP22 and UL34 colocalized at the nuclear membrane with a uniform distribution. In contrast, in HEL cells infected with YK511 (Us3K220M) encoding an enzymatically inactive Us3 mutant, ICP22 colocalized with UL34 and accumulated in punctate structures at the nuclear membrane (Fig. 4).
FIG 4.
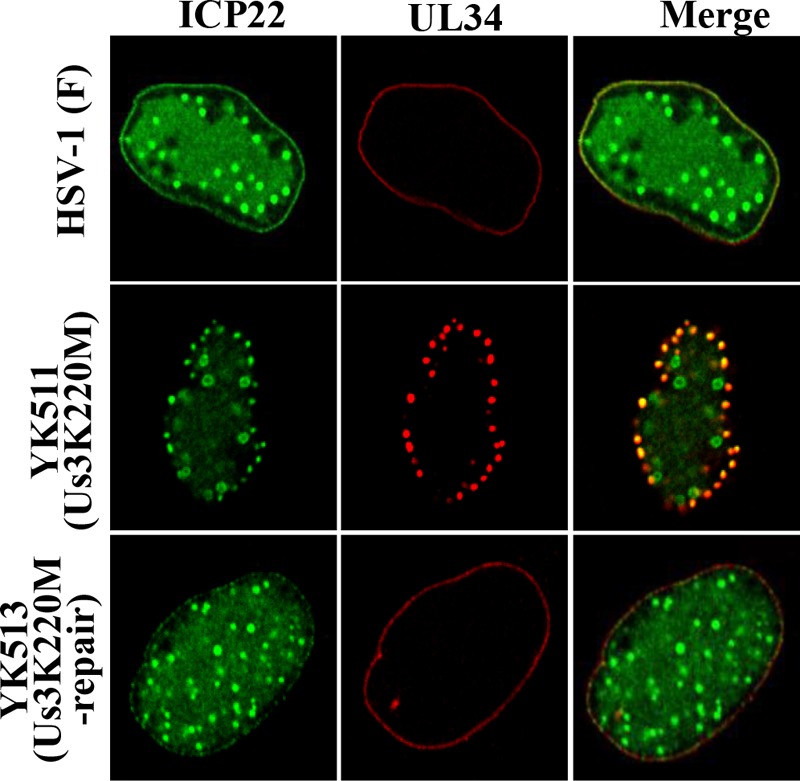
Effect of Us3 kinase activity on localization of ICP22 and UL34 in HSV-1-infected cells. HEL cells infected with wild-type HSV-1(F), YK511 (Us3K220M), or YK513 (Us3K220M-repair) at an MOI of 5 for 13 h were fixed, permeabilized, stained with anti-ICP22 and anti-UL34 antibodies, and examined by confocal microscopy.
Effect of UL31 on localization of ICP22 in HSV-1-infected cells.
To clarify the biological significance of the interaction between ICP22 and the HSV-1 nuclear egress regulators described above (Fig. 2), we examined the effect of UL31, one of the ICP22-binding partners, on ICP22 expression and localization in HSV-1-infected cells. In order to achieve this, we constructed a UL31-null mutant virus, YK720 (ΔUL31), and its repaired virus, YK721 (ΔUL31-repair) (Fig. 1). In agreement with previous reports (6), growth of YK720 (ΔUL31) on Vero cells at an MOI of 5 was severely impaired compared to that of wild-type HSV-1(F) or YK721 (ΔUL31-repair) (Fig. 5A). As shown in Fig. 5B, the levels of expression of ICP22 and UL34 proteins in Vero cells infected with wild-type HSV-1(F), YK720 (ΔUL31), or YK721 (ΔUL31-repair) were similar (Fig. 5A). Comparable results were also obtained with infected HEL cells (data not shown). These results indicate that UL31 had no effect on the expression of ICP22 in HSV-1-infected cells.
FIG 5.
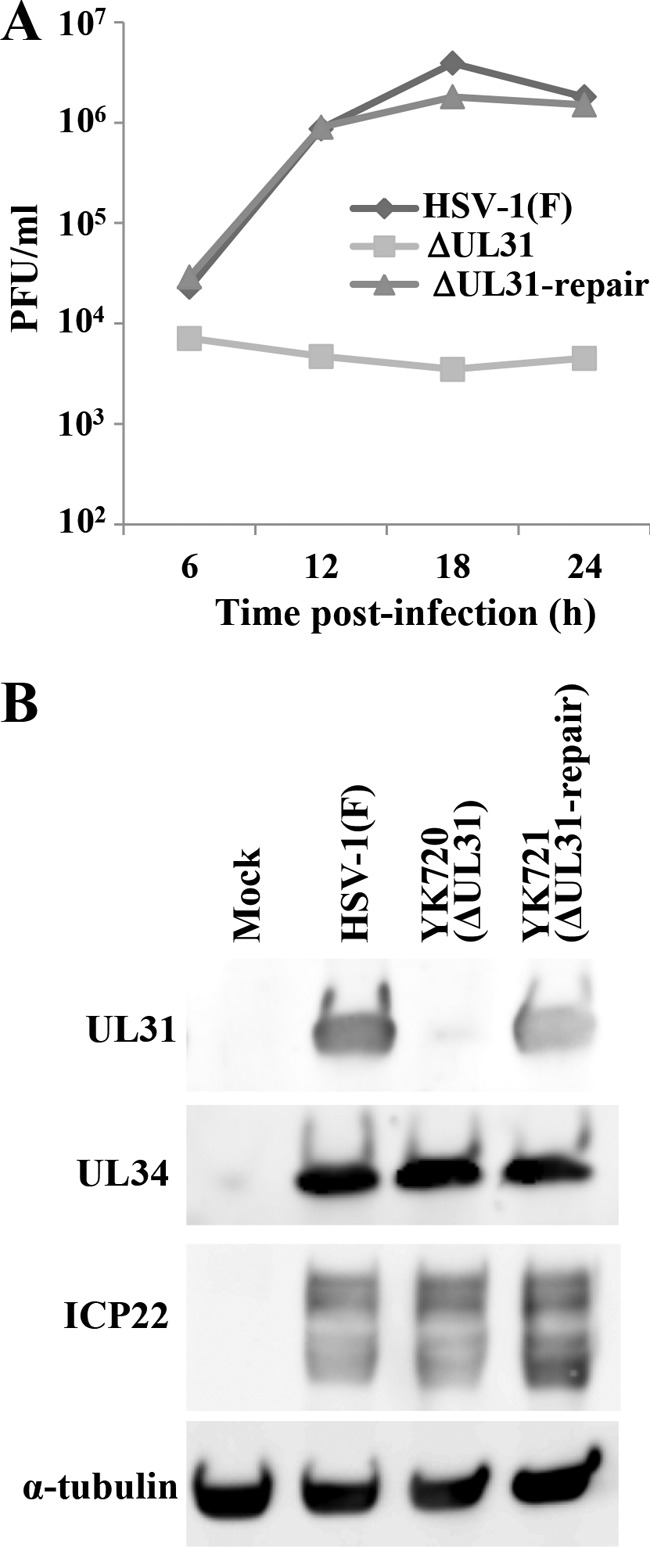
Characterization of YK720 (ΔUL31) and YK721 (ΔUL31-repair). (A) Vero cells were infected with HSV-1(F), YK720 (ΔUL31), or YK721 (ΔUL31-repair) at an MOI of 5. Total virus from the cell culture supernatants and infected cells was harvested at the times indicated and assayed on CV1-UL31 cells. (B) Vero cells mock infected or infected with wild-type HSV-1(F), YK720 (ΔUL31), or YK721 (ΔUL31-repair) at an MOI of 5 for 18 h were analyzed by immunoblotting with antibodies to UL31, UL34, ICP22, and α-tubulin.
Next, to examine the effect of UL31 on the localization of ICP22 in HSV-1-infected cells, HEL cells infected with wild-type HSV-1(F), YK720 (ΔUL31), or YK721 (ΔUL31-repair) were analyzed by immunofluorescence. Consistent with the results shown in Fig. 3, ICP22 was detected at the nuclear membrane and colocalized with UL34 and lamin B1 in cells singly infected with wild-type HSV-1(F) and YK721 (ΔUL31-repair) (Fig. 6A and B). In contrast, ICP22 was not detectable at the nuclear membrane in cells infected with YK720 (ΔUL31) (Fig. 6A and B). Furthermore, UL34 was detectable not only at the nuclear membrane but also in the perinuclear region of the cytoplasm, reminiscent of the endoplasmic reticulum (ER), in cells infected with YK720 (ΔUL31) (Fig. 6A), as previously reported (4). Similar results were also obtained with Vero cells (data not shown). These results indicate that UL31 is required for the targeting of ICP22 to the nuclear membrane in HSV-1-infected cells.
FIG 6.
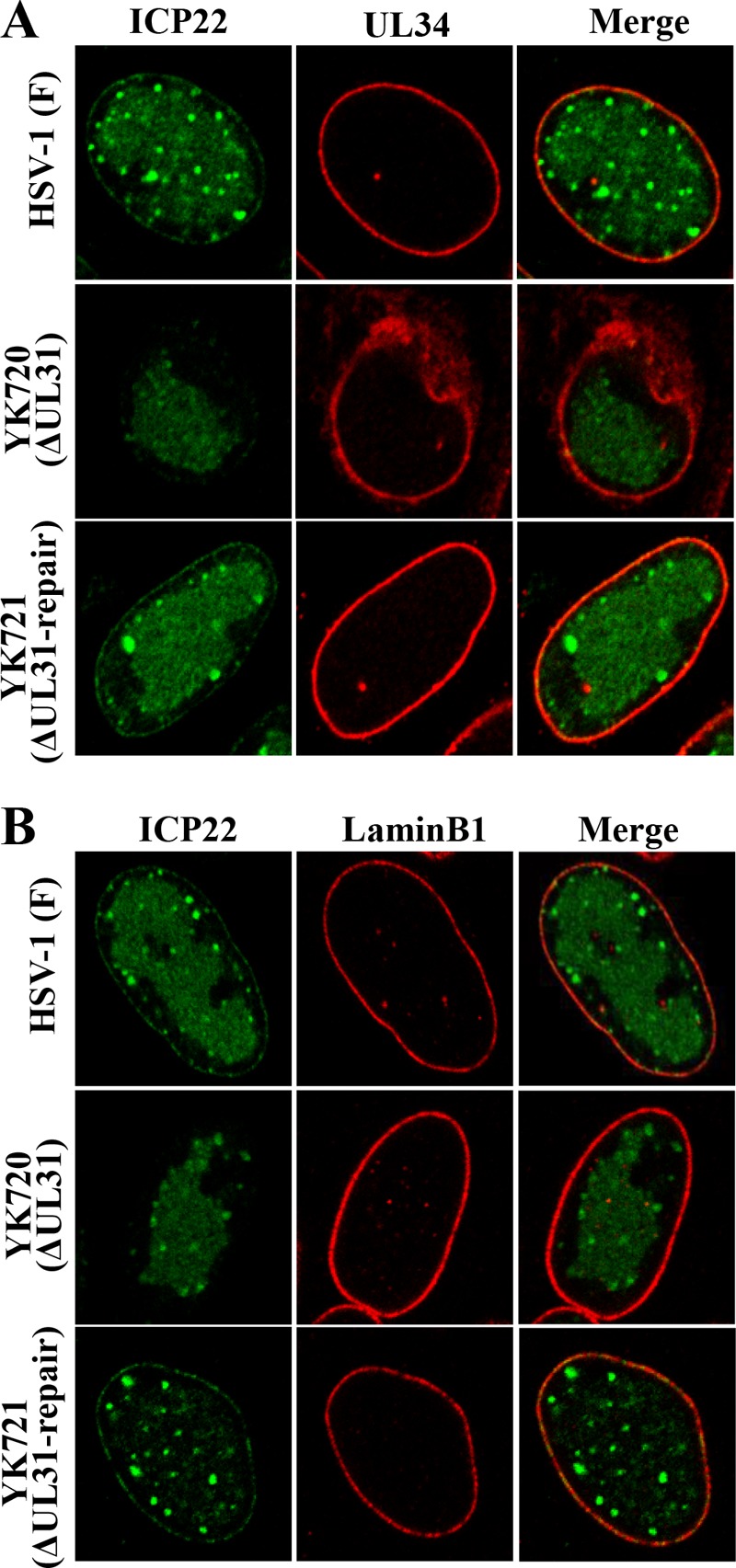
Effect of UL31 on localization of ICP22 in HSV-1-infected cells. HEL cells infected with wild-type HSV-1(F), YK720 (ΔUL31), or YK721 (ΔUL31-repair) at an MOI of 5 for 13 h were fixed, permeabilized, stained with anti-ICP22 and anti-UL34 (A) or with anti-ICP22 and anti-lamin B1 (B) antibodies, and examined by confocal microscopy.
Effect of ICP22 on localization of UL31 and UL34.
We also investigated the effect of ICP22 on the localization of UL31 and UL34 at the nuclear membrane by infecting Vero cells with wild-type HSV-1(F), YK421 (ΔICP22), or YK422 (ΔICP22-repair) and analyzing the infected cells by immunofluorescence. As described above (Fig. 3), UL31 and UL34 were evenly distributed and colocalized around the nuclear rim in Vero cells infected with wild-type HSV-1(F) or YK422 (ΔICP22-repair) (Fig. 7). However, in Vero cells infected with YK421 (ΔICP22), the pattern of localization was markedly different, with UL31 and UL34 being colocalized not only at the nuclear rim but also in a perinuclear region of the cytoplasm (Fig. 7). Since the perinuclear localization of UL31 and UL34 in Vero cells infected with YK421 (ΔICP22) was reminiscent of the ER localization observed in HSV-1-infected cells (45), Vero cells infected with wild-type HSV-1(F), YK421 (ΔICP22), or YK422 (ΔICP22-repair) were stained with antibodies to UL31, UL34, calnexin (an ER marker), or lamin B1. As shown in Fig. 8, UL31 and UL34 colocalized with calnexin in the perinuclear region of the cytoplasm in Vero cells infected with YK421 (ΔICP22). These results indicate that in the absence of ICP22, UL31 and UL34 aberrantly accumulated in both the ER and the nuclear membrane, thus suggesting that ICP22 is an absolute requirement for the correct localization of UL31 and UL34 at the nuclear membrane in HSV-1-infected Vero cells.
FIG 7.
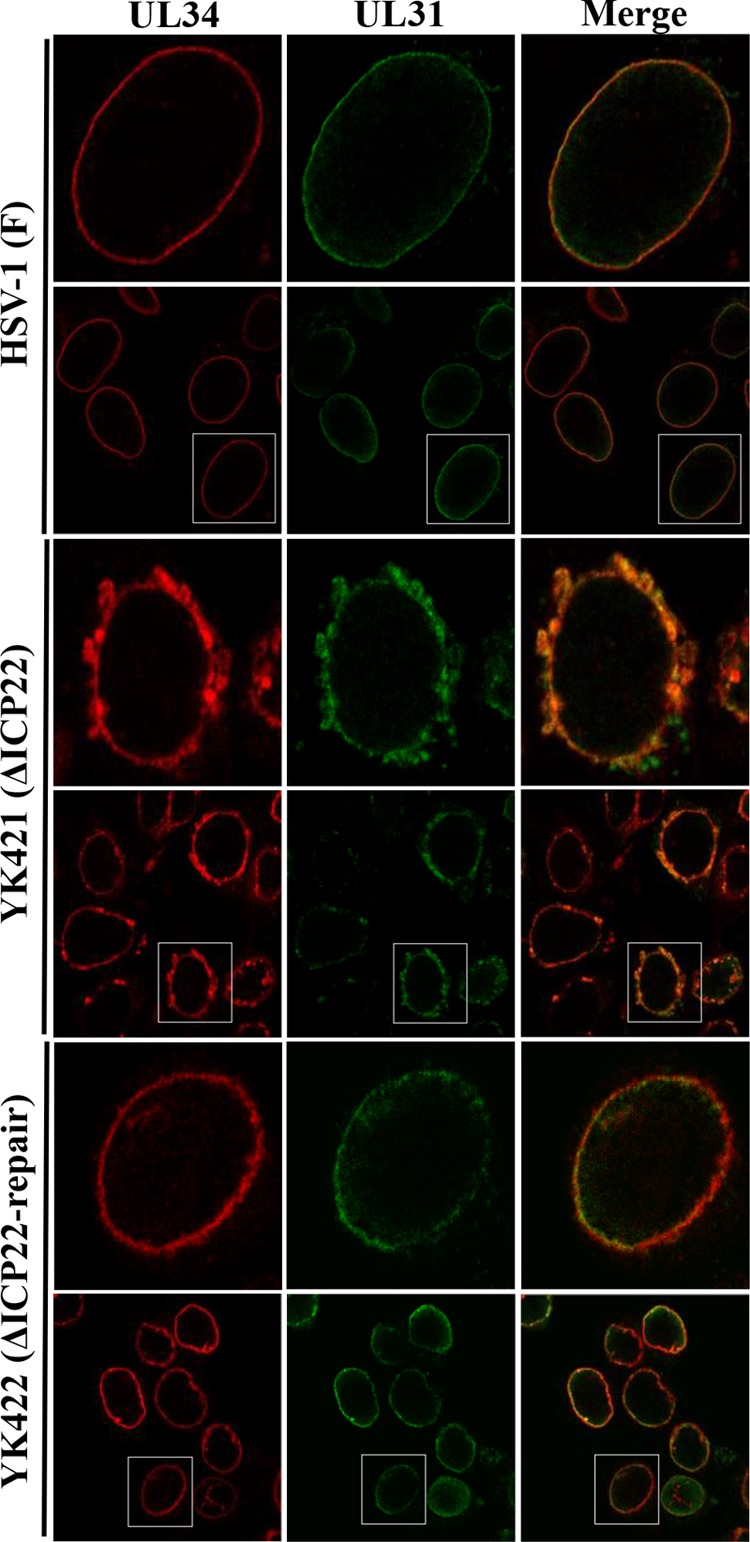
Effect of ICP22 on localization of UL31 and UL34 in HSV-1-infected Vero cells. Vero cells infected with wild-type HSV-1(F), YK421 (ΔICP22), or YK422 (ΔICP22-repair) at an MOI of 5 for 18 h were fixed, permeabilized, stained with anti-UL31 and anti-UL34 antibodies, and examined by confocal microscopy. The top panels are magnified images of insets shown in the bottom panels.
FIG 8.
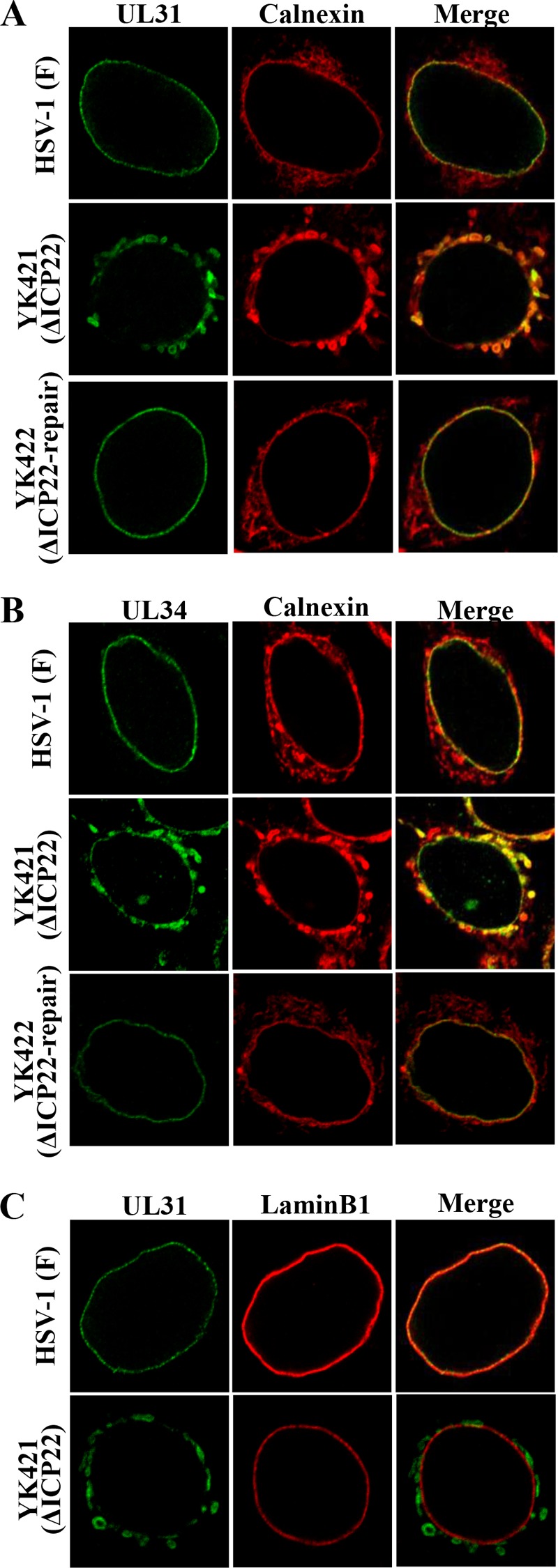
Effect of ICP22 on localization of UL31 and UL34 as well as calnexin and lamin B1 in HSV-1-infected Vero cells. Vero cells infected with wild-type HSV-1(F), YK421 (ΔICP22), or YK422(ΔICP22-repair) at an MOI of 5 for 18 h were fixed; permeabilized; stained with anti-UL31 and anti-calnexin (A), anti-UL34 and anti-calnexin (B), or anti-UL31 and anti-lamin B1 (C) antibodies; and examined by confocal microscopy.
Effect of ICP22 on HSV-1 nuclear egress.
The data described above, showing the interaction of ICP22 with the viral nuclear egress regulators and its regulatory effects on the correct localization of UL31 and UL34 in HSV-1-infected cells, led us to examine whether ICP22 played a role(s) in viral nuclear egress. To this end, we used electron microscopy to investigate the effect of the ICP22-null mutation on viral morphogenesis by quantitating the number of virus particles at different morphogenetic stages in Vero and HEL cells infected with wild-type HSV-1(F), YK421 (ΔICP22), or YK422 (ΔICP22-repair). In HEL cells infected with wild-type HSV-1(F) or YK422 (ΔICP22-repair), 4.4 and 6.6%, respectively, of the total number of virus particles in the perinuclear space were primary enveloped virions (Table 1 and Fig. 9). However, cells infected with YK422 (ΔICP22) had almost zero (0.8%) primary enveloped virions in the perinuclear space, 5.5- and 8.3-fold reductions, respectively (Table 1) (Fig. 9). In contrast, capsids appeared to accumulate in the nucleus in cells infected with YK421 (ΔICP22) (Table 1). While 40.3 and 40.9% of total virus particles were capsids in the nucleus of cells infected with wild-type HSV-1(F) and YK422 (ΔICP22-repair), respectively, the fraction of total virus particles that were capsids in the nucleus in cells infected with YK422 (ΔICP22) increased to 52.8% (Table 1). The differences between the prevalence of these types of virions in YK422 (ΔICP22) and that in wild-type HSV-1(F) or YK422 (ΔICP22-repair) were statistically significant (Table 1). Similar results were also obtained with Vero cells infected with wild-type HSV-1(F), YK545 (ΔUL47), or YK546 (ΔUL47-repair) (Table 2). These results indicate that the ICP22-null mutation resulted in a decrease in the fraction of virus particles that were primary enveloped virions in the perinuclear space and an increase in the fraction that were capsids in the nucleus.
TABLE 1.
Effect of the ICP22-null mutation on distribution of virus particles in infected HEL cells
| Virus | Avg % of virus particles in morphogenetic stage ±SE (no. of virus particles) |
Total no. of particles counted/total no. of cells | ||||
|---|---|---|---|---|---|---|
| Nucleocapsids in the nucleus | Enveloped virions in the perinuclear space | Nucleocapsids in the cytoplasm | Enveloped virions in the cytoplasm | Extracellular enveloped virions | ||
| HSV-1(F) | 40.3 ± 3.5a (2,183) | 4.4 ± 0.7b (227) | 1.8 ± 0.4 (89) | 11.7 ± 2.4 (584) | 41.8 ± 2.9 (2,105) | 5,188/20 |
| YK421 (ΔICP22) | 52.8 ± 3.9 (584) | 0.8 ± 0.5 (8) | 2.2 ± 0.6 (25) | 15.5 ± 2.6 (175) | 28.8 ± 3.8 (329) | 1,121/20 |
| YK422 (ΔICP22-repair) | 40.9 ± 4.0a (1,748) | 6.6 ± 1.3b (256) | 1.7 ± 0.3 (62) | 11.7 ± 1.9 (444) | 39.2 ± 3.9 (1,461) | 3,971/20 |
Statistically significant difference from YK421 (ΔICP22) (P < 0.05).
Statistically significant difference from YK421 (ΔICP22) (P < 0.0005).
FIG 9.
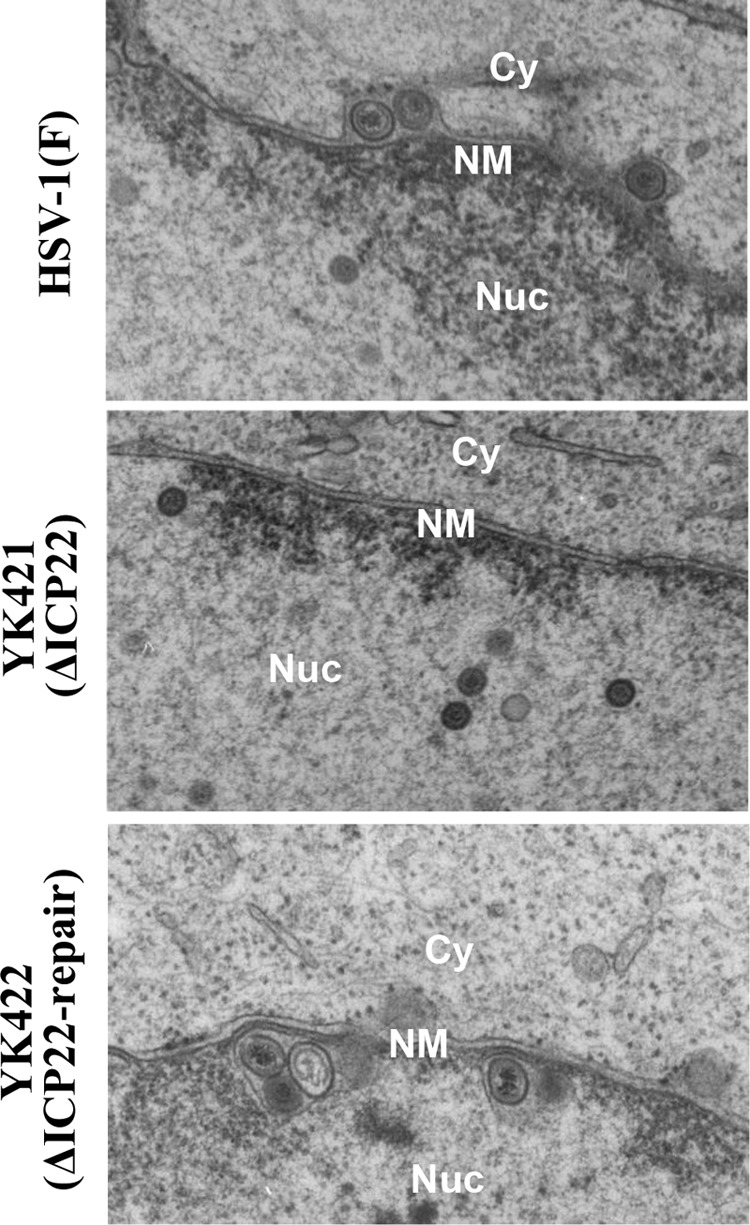
Ultrastructural analysis of the effect of ICP22 on HSV-1 nuclear egress. HEL cells infected with wild-type HSV-1(F), YK421 (ΔICP22), or YK422(ΔICP22-repair) at an MOI of 5 were fixed at 18 h postinfection, embedded, sectioned, stained, and examined by transmission electron microscopy. Nu, nucleus; Cy, cytoplasm; NM, nuclear membrane.
TABLE 2.
Effect of the ICP22-null mutation on distribution of virus particles in infected Vero cells
| Virus | Avg % of virus particles in morphogenetic stage ± SE (no. of virus particles) |
Total no. of particles counted/total no. of cells | ||||
|---|---|---|---|---|---|---|
| Nucleocapsids in the nucleus | Enveloped virions in the perinuclear space | Nucleocapsids in the cytoplasm | Enveloped virions in the cytoplasm | Extracellular enveloped virions | ||
| HSV-1(F) | 42.0 ± 2.9a (1,206) | 16.6 ± 3.0a (516) | 10.7 ± 2.0 (279) | 11.1 ± 1.6 (323) | 19.7 ± 3.7 (539) | 2,863/20 |
| YK421 (ΔICP22) | 60.4 ± 4.1 (1,367) | 7.1 ± 0.9 (140) | 4.6 ± 0.9 (97) | 7.6 ± 1.8 (188) | 20.3 ± 3.2 (391) | 2,183/20 |
| YK422 (ΔICP22-repair) | 47.2 ± 4.3a (871) | 14.8 ± 3.4a (316) | 6.6 ± 1.5 (140) | 10.3 ± 2.4 (208) | 21.0 ± 2.6 (387) | 1,922/20 |
Statistically significant difference from YK421 (ΔICP22) (P < 0.05).
DISCUSSION
The potential interaction of ICP22 with UL31, UL34, and UL47, suggested by tandem affinity purification of MEF-ICP22 expressed in HSV-1-infected HEL cells followed by mass spectrometry-based proteomic analyses and our recent report that the HSV-1 UL31/UL34 complex (NEC) interacts with UL47 and Us3 (18), prompted us to investigate whether ICP22 is also a component of the NEC. We have demonstrated that MEF-ICP22 coimmunoprecipitated with UL31, UL34, Us3, and UL47; MEF-UL31 coimmunoprecipitated with UL34, Us3, UL47, and ICP22; and MEF-UL34 coimmunoprecipitated with UL31, Us3, UL47, and ICP22. Collectively, these results indicate that ICP22 forms a complex(es) with UL31, UL34, Us3, and/or UL47 in HSV-1-infected cells. At present, it remains to be determined whether ICP22, UL31, UL34, Us3, and UL47 form a high-order, multimeric complex in HSV-1-infected cells. However, the reciprocal coimmunoprecipitation experiments described above, combined with previous studies showing coimmunoprecipitation of UL31 and UL34 (4); of Us3 and UL47 (43); and of UL31, UL34, Us3, and UL47 (18), strongly suggest that it does.
The detailed ICP22 localization studies reported here demonstrate that ICP22 localizes not only diffusely in both the nucleoplasm and discrete nuclear punctate structures, as described previously (23), but also to the nuclear membrane, where it colocalizes with UL31 and UL34. Since UL31 and UL34 have been reported to localize predominantly at the nuclear membrane in wild-type HSV-1-infected cells (4), it was probable that the ICP22 interactions with these proteins reported in this study should also take place mainly at the nuclear membrane. In addition, we have shown that ICP22 and UL34 incorrectly colocalize in nuclear membrane punctate structures in cells infected with the Us3 kinase-dead mutant virus strain YK511 (Us3K220M), an observation which we have previously reported for UL31, UL34, and UL47 (18). This result indicates that the localization of ICP22 at the nuclear membrane, like that of UL31, UL34, and UL47 (18, 44), is regulated by Us3 kinase activity and supports the hypothesis that ICP22, UL34, UL31, UL47, and probably Us3 form a complex at the nuclear membrane in HSV-1-infected cells. Thus, it is likely that ICP22 is indeed a novel component of the HSV-1 NEC.
We have shown in this study that UL31 is required for the targeting of ICP22 to the nuclear membrane in HSV-1-infected cells, based on the observation that the absence of UL31 prevented the appearance of ICP22 at the nuclear membrane in HSV-1-infected HEL and Vero cells. It has been reported that the correct localization of the UL31/UL34 complex (NEC) at the nuclear membrane depends on the presence of UL31 and UL34, both individually and bound together (4, 46). Taken together with the data in this study showing that ICP22 interacts with UL31 and UL34, it is reasonable to hypothesize that the UL31/UL34 complex (NEC) recruits and anchors ICP22 at the nuclear membrane by binding to it. In support of this hypothesis, we have demonstrated that the absence of Us3 kinase activity mislocalized ICP22 and UL34 to punctate structures at the nuclear membrane and that ICP22 was not detectable at the nuclear membrane in Vero cells transiently expressing ICP22 alone (data not shown).
We also present data demonstrating that ICP22 is required for the correct localization of UL31 and UL34 at the nuclear membrane in HSV-1-infected Vero cells. The ICP22-null mutation also results in UL31 and UL34 being mislocalized in the ER (in addition to the nuclear membrane) in Vero cells. Notably, the pattern of calnexin staining in Vero cells infected with YK421 (ΔICP22) is different from that in cells infected with wild-type HSV-1(F) or YK422 (ΔICP22-repair), whereas lamin B1 staining is similar. Since the ER appears to collapse into the ONM, these results suggest that ICP22 is required for ER and/or ONM integrity in HSV-1-infected Vero cells. Thus, ICP22 may regulate the localization of UL31 and UL34 by maintaining the correct integrity of the ER and/or ONM in HSV-1-infected cells.
Quantitative electron microscopic analysis of HSV-1-infected Vero and HEL cells showed that the prevalence of primary enveloped virions in the perinuclear space is significantly reduced in the absence of ICP22, whereas that of the nuclear capsids is significantly increased. These results probably reflect an imbalance between the rate of virion delivery into and the rate of exit from the perinuclear space. This suggests that the rate of viral egress from the nucleoplasm decreases in the absence of ICP22 compared to that in the presence of ICP22, but the rate of egress from the perinuclear space in the absence of ICP22 is similar to that in the presence of ICP22. Based on these observations, ICP22 appears to be required for efficient primary envelopment of nucleocapsids in HSV-1 nuclear egress. This hypothesis is supported by our observations that (i) ICP22 interacts with viral regulators for primary envelopment, including UL31, UL34, Us3, and UL47 (4, 5, 18, 47), in HSV-1-infected cells and (ii) ICP22 is required for the correct localization of UL31 and UL34 in Vero cells. It is interesting, but a little confusing, that although the frequencies of virions in the nucleus and in the perinuclear space in cells infected with the ICP22-null mutant virus are significantly increased and reduced, respectively, compared to those in cells infected with wild-type HSV-1 or its repaired virus, the prevalence rates of cytoplasmic and/or extracellular virions do not appear to be obviously different in cells infected with mutant and wild-type HSV-1. As a result of the defect in primary envelopment in cells infected with the ICP22-null mutant virus, one would expect that the frequencies of virions in the cytoplasm and the extracellular space are diminished. There is a possibility that ICP22 negatively regulates an HSV-1 virion maturation step(s) after primary envelopment, probably deenvelopment and/or secondary envelopment. In addition, we cannot completely eliminate the possibility that the increased frequency of virions in the nucleus in cells infected with the ICP22-null mutant virus results from overproduction of nucleocapsids. Further studies to clarify these subjects as well as the more detailed mechanisms by which ICP22 acts in primary envelopment will be needed and are under way in our laboratory.
In summary, we conclude that in HSV-1-infected cells, ICP22 promotes primary envelopment, probably by interacting with and regulating UL31 and UL34. We note, however, that although the prevalence of primary enveloped virions in perinuclear spaces was significantly reduced, enveloped virions in the cytoplasm and on the cell surface were consistently detectable. These observations suggest that ICP22 is not essential for primary envelopment (as are UL31 and UL34) but nevertheless plays a regulatory role in this process. Since ICP22 promotes viral replication in a cell type-dependent manner (20), the activity of ICP22 in regulating primary envelopment appears to play no obvious role in viral replication in some cell types, such as Vero cells. However, the ICP22 activity may be important for HSV-1 pathogenesis in vivo, as reported previously for other HSV-1 regulatory proteins such as Us3, gB, and UL12 (45, 48, 49).
ACKNOWLEDGMENTS
We thank Joel D. Bains and Richard J. Roller for providing UL31-CV1 cells and chicken polyclonal antibody to UL34, respectively. We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers, Grants for Scientific Research, from the Japan Society for the Promotion of Science (JSPS); a contract research fund from the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID); a grant for scientific research on innovative areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan; and a grant from the Takeda Science Foundation.
Footnotes
Published ahead of print 16 April 2014
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1825 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394. 10.1038/nrmicro2559 [DOI] [PubMed] [Google Scholar]
- 3.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell. Microbiol. 15:170–178. 10.1111/cmi.12044 [DOI] [PubMed] [Google Scholar]
- 4.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75:8803–8817. 10.1128/JVI.75.18.8803-8817.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J. Virol. 74:117–129. 10.1128/JVI.74.1.117-129.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang YE, Van Sant C, Krug PW, Sears AE, Roizman B. 1997. The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J. Virol. 71:8307–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bjerke SL, Roller RJ. 2006. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 347:261–276. 10.1016/j.virol.2005.11.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459–6470. 10.1128/JVI.00380-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mou F, Wills EG, Park R, Baines JD. 2008. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J. Virol. 82:8094–8104. 10.1128/JVI.00874-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J. Virol. 80:494–504. 10.1128/JVI.80.1.494-504.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds AE, Liang L, Baines JD. 2004. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes U(L)31 and U(L)34. J. Virol. 78:5564–5575. 10.1128/JVI.78.11.5564-5575.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toropova K, Huffman JB, Homa FL, Conway JF. 2011. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J. Virol. 85:7513–7522. 10.1128/JVI.00837-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang K, Wills EG, Baines JD. 2011. A mutation in UL15 of herpes simplex virus 1 that reduces packaging of cleaved genomes. J. Virol. 85:11972–11980. 10.1128/JVI.00857-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J. 2012. Reconstitution of the Kaposi's sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J. Virol. 86:594–598. 10.1128/JVI.05988-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. U. S. A. 104:7241–7246. 10.1073/pnas.0701757104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 79:9325–9331. 10.1128/JVI.79.14.9325-9331.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purves FC, Spector D, Roizman B. 1991. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J. Virol. 65:5757–5764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J. Virol. 88:4657–4667. 10.1128/JVI.00137-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211. 10.1128/JVI.02681-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sears AE, Halliburton IW, Meignier B, Silver S, Roizman B. 1985. Herpes simplex virus 1 mutant deleted in the alpha 22 gene: growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J. Virol. 55:338–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aubert M, Chen Z, Lang R, Dang CH, Fowler C, Sloan DD, Jerome KR. 2008. The antiapoptotic herpes simplex virus glycoprotein J localizes to multiple cellular organelles and induces reactive oxygen species formation. J. Virol. 82:617–629. 10.1128/JVI.01341-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalamvoki M, Roizman B. 2011. The histone acetyltransferase CLOCK is an essential component of the herpes simplex virus 1 transcriptome that includes TFIID, ICP4, ICP27, and ICP22. J. Virol. 85:9472–9477. 10.1128/JVI.00876-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leopardi R, Ward PL, Ogle WO, Roizman B. 1997. Association of herpes simplex virus regulatory protein ICP22 with transcriptional complexes containing EAP, ICP4, RNA polymerase II, and viral DNA requires posttranslational modification by the U(L)13 protein kinase. J. Virol. 71:1133–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. 2013. Roles of p53 in herpes simplex virus 1 replication. J. Virol. 87:9323–9332. 10.1128/JVI.01581-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng TI, Chang YE, Roizman B. 1997. Infected cell protein 22 of herpes simplex virus 1 regulates the expression of virion host shutoff gene U(L)41. Virology 234:226–234. 10.1006/viro.1997.8659 [DOI] [PubMed] [Google Scholar]
- 26.Ogle WO, Roizman B. 1999. Functional anatomy of herpes simplex virus 1 overlapping genes encoding infected-cell protein 22 and US1.5 protein. J. Virol. 73:4305–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purves FC, Ogle WO, Roizman B. 1993. Processing of the herpes simplex virus regulatory protein alpha 22 mediated by the UL13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc. Natl. Acad. Sci. U. S. A. 90:6701–6705. 10.1073/pnas.90.14.6701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durand LO, Advani SJ, Poon AP, Roizman B. 2005. The carboxyl-terminal domain of RNA polymerase II is phosphorylated by a complex containing cdk9 and infected-cell protein 22 of herpes simplex virus 1. J. Virol. 79:6757–6762. 10.1128/JVI.79.11.6757-6762.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durand LO, Roizman B. 2008. Role of cdk9 in the optimization of expression of the genes regulated by ICP22 of herpes simplex virus 1. J. Virol. 82:10591–10599. 10.1128/JVI.01242-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rice SA, Long MC, Lam V, Schaffer PA, Spencer CA. 1995. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J. Virol. 69:5550–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rice SA, Long MC, Lam V, Spencer CA. 1994. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J. Virol. 68:988–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orlando JS, Balliet JW, Kushnir AS, Astor TL, Kosz-Vnenchak M, Rice SA, Knipe DM, Schaffer PA. 2006. ICP22 is required for wild-type composition and infectivity of herpes simplex virus type 1 virions. J. Virol. 80:9381–9390. 10.1128/JVI.01061-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391. 10.1128/JVI.77.2.1382-1391.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang L, Tanaka M, Kawaguchi Y, Baines JD. 2004. Cell lines that support replication of a novel herpes simplex virus 1 UL31 deletion mutant can properly target UL34 protein to the nuclear rim in the absence of UL31. Virology 329:68–76. 10.1016/j.virol.2004.07.030 [DOI] [PubMed] [Google Scholar]
- 35.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172–6189. 10.1128/JVI.00044-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. 10.1038/nature09420 [DOI] [PubMed] [Google Scholar]
- 37.Kawaguchi Y, Bruni R, Roizman B. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J. Virol. 71:1019–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J. Virol. 83:11624–11634. 10.1128/JVI.00993-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carter KL, Roizman B. 1996. The promoter and transcriptional unit of a novel herpes simplex virus 1 alpha gene are contained in, and encode a protein in frame with, the open reading frame of the alpha 22 gene. J. Virol. 70:172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 71:7328–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bastian TW, Livingston CM, Weller SK, Rice SA. 2010. Herpes simplex virus type 1 immediate-early protein ICP22 is required for VICE domain formation during productive viral infection. J. Virol. 84:2384–2394. 10.1128/JVI.01686-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mitchell C, Blaho JA, Roizman B. 1994. Casein kinase II specifically nucleotidylylates in vitro the amino acid sequence of the protein encoded by the alpha 22 gene of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 91:11864–11868. 10.1073/pnas.91.25.11864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J. Virol. 85:9599–9613. 10.1128/JVI.00845-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78:399–412. 10.1128/JVI.78.1.399-412.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J. Virol. 84:153–162. 10.1128/JVI.01447-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang L, Baines JD. 2005. Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J. Virol. 79:3797–3806. 10.1128/JVI.79.6.3797-3806.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939–8952. 10.1128/JVI.76.17.8939-8952.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J. Virol. 83:5773–5783. 10.1128/JVI.00103-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujii H, Mugitani M, Koyanagi N, Liu Z, Tsuda S, Arii J, Kato A, Kawaguchi Y. 2014. Role of the nuclease activities encoded by herpes simplex virus 1 UL12 in viral replication and neurovirulence. J. Virol. 88:2359–2364. 10.1128/JVI.03621-13 [DOI] [PMC free article] [PubMed] [Google Scholar]