

ABSTRACT
The coxsackievirus and adenovirus receptor (CAR) is a cell contact protein with an important role in virus uptake. Its extracellular immunoglobulin domains mediate the binding to coxsackievirus and adenovirus as well as homophilic and heterophilic interactions between cells. The cytoplasmic tail links CAR to the cytoskeleton and intracellular signaling cascades. In the heart, CAR is crucial for embryonic development, electrophysiology, and coxsackievirus B infection. Noncardiac functions are less well understood, in part due to the lack of suitable animal models. Here, we generated a transgenic mouse that rescued the otherwise embryonic-lethal CAR knockout (KO) phenotype by expressing chicken CAR exclusively in the heart. Using this rescue model, we addressed interspecies differences in coxsackievirus uptake and noncardiac functions of CAR. Survival of the noncardiac CAR KO (ncKO) mouse indicates an essential role for CAR in the developing heart but not in other tissues. In adult animals, cardiac activity was normal, suggesting that chicken CAR can replace the physiological functions of mouse CAR in the cardiomyocyte. However, chicken CAR did not mediate virus entry in vivo, so that hearts expressing chicken instead of mouse CAR were protected from infection and myocarditis. Comparison of sequence homology and modeling of the D1 domain indicate differences between mammalian and chicken CAR that relate to the sites important for virus binding but not those involved in homodimerization. Thus, CAR-directed anticoxsackievirus therapy with only minor adverse effects in noncardiac tissue could be further improved by selectively targeting the virus-host interaction while maintaining cardiac function.
IMPORTANCE Coxsackievirus B3 (CVB3) is one of the most common human pathogens causing myocarditis. Its receptor, the coxsackievirus and adenovirus receptor (CAR), not only mediates virus uptake but also relates to cytoskeletal organization and intracellular signaling. Animals without CAR die prenatally with major cardiac malformations. In the adult heart, CAR is important for virus entry and electrical conduction, but its nonmuscle functions are largely unknown. Here, we show that chicken CAR expression exclusively in the heart can rescue the otherwise embryonic-lethal CAR knockout phenotype but does not support CVB3 infection of adult cardiomyocytes. Our findings have implications for the evolution of virus-host versus physiological interactions involving CAR and could help to improve future coxsackievirus-directed therapies inhibiting virus replication while maintaining CAR's cellular functions.
INTRODUCTION
Coxsackieviruses belong to the Enterovirus genus in the family of Picornaviridae and are important human pathogens, which cause a plethora of infectious diseases such as myocarditis, meningitis, and pancreatitis (1). They are one of the most common causes of myocarditis, with up to 33% of patients positive for coxsackievirus group B (CVB) (2). The classification of coxsackieviruses is based on the systemic versus localized pathology in the mouse, which is the experimental model of choice to study the CVB3 serotype (3, 4). In addition to mouse and human, other species are also infected by CVB3, which has been reported in diverse primate species with myocarditis (5–7). Infections with other CVBs have been reported in primates and dogs (8, 9). Infections of the pig have been attributed to the swine vesicular disease (SVD) virus—an adaptation of the human CVB5 to the swine (10).
The coxsackievirus and adenovirus receptor (CAR) was identified in 1997 (11, 12) and binds group B coxsackieviruses and adenoviruses of the subgenera A and C to F, as well as the SVD virus (12–14). CAR is crucial for CVB3 uptake as documented by protection from infection of heart or pancreas in the respective tissue-specific knockouts (3, 4). Virus attachment is mediated by binding of CAR's N-terminal immunoglobulin (Ig) domain to the canyon of the virus, where the viral capsid proteins VP1 to -3 interact with CAR (15). This interaction is conserved in the family Picornaviridae (16–18). CVB3s use the coreceptor decay accelerating factor (DAF) for cell attachment (19), which binds adjacent to the CAR interaction sites on the virus surface (20). DAF alone is not sufficient for virus infection (21). It has been proposed to facilitate the presentation of virions to CAR and contribute to systemic infections, helping the virus to cross epithelial cell barriers (22).
CAR is a type I transmembrane protein localized in the tight junctions of epithelial cells and in the intercalated discs between cardiomyocytes (23, 24). It consists of an N-terminal signal peptide, two extracellular Ig domains, a transmembrane domain, and an intracellular tail. Because of its two extracellular Ig-like domains, CAR belongs to the immunoglobulin superfamily (IgSF) (11). The N-terminal Ig domain is important for virus binding but also for homophilic interactions or interaction with other IgSF proteins (15, 25–27). The cytoplasmic tail contains a PDZ-binding motif (28) that links to the cytoskeleton and intracellular signaling cascades (24, 29, 30).
To analyze the role of CAR in vivo, several knockout (KO) animals have been generated (31). Conventional CAR KO animals die between embryonic day E11.5 and E13.5 (32–35). The embryos suffer from defects in cardiac development, hemorrhage, and edema. Their myofilaments are disorganized and less compact than those of wild-type (WT) controls. To circumvent the embryonic-lethal phenotype and analyze the function of CAR in the adult heart, several conditional KO animals have been generated (33, 35, 36). The adult KOs revealed a role for CAR in the electrical signal propagation between atrium and ventricle (35–37). Together, this indicates an essential function of CAR in both the embryonic and the adult heart. Deletion of CAR in noncardiac tissues did not result in severe phenotypes (3, 37).
To analyze possible noncardiac functions of CAR and its role in mediating and sustaining coxsackievirus infections, we generated a novel animal model that expresses CAR exclusively in cardiomyocytes but not in other cells. With Gallus gallus CAR (chCAR) under the control of the heart-specific MLC-2 promoter, we were able to rescue the embryonic-lethal phenotype of the conventional CAR KO. CVB3 infection studies of the noncardiac CAR KO (ncKO) mouse demonstrate the protection from pathology throughout the body, including the heart, where mouse CAR (mCAR) is replaced with chCAR. Sequence alignment and modeling of the protein structure indicated crucial differences in CAR-CVB3 interaction between species. The respective amino acids were less well conserved than were the amino acids involved in homodimerization and cluster differentially within the Ig domain.
MATERIALS AND METHODS
Animal procedures.
All experiments involving animals were carried out following the Guide for the Care and Use of Laboratory Animals of the German Animal Welfare Act, and protocols were approved by the Committee on the Ethics of Animal Experiments of Berlin state authorities and the regional board of Tübingen.
In brief, we generated a transgenic mouse (TG) that expresses the full-length CAR protein from Gallus gallus, including all annotated exons (chCAR), under the control of the heart-specific MLC-2 promoter, in addition to the wild-type mouse CAR. Animals with high expression levels of chCAR as determined by TaqMan analysis died early postnatally, while lower levels were compatible with life. These animals were mated with heterozygous CAR knockout mice (38) for two generations to obtain rescue animals, which are homozygously deficient for mouse CAR and express chicken CAR only in the cardiac ventricle. The colony was maintained by breeding animals with and without the transgene to ensure heterozygosity for the transgene and to reduce potential side effects from integration of the transgene.
For echocardiography or electrocardiogram (ECG) analysis, mice were anesthetized with 1.5% isoflurane. For echocardiography, M and B modes were measured with the Vevo 2100 system (Visual Sonics) and the MS400 transducer. Data acquisition and analysis with the Vevo 2100 software, including the trace ejection fraction (EF) (%), have been described previously (39). ECG standard intervals were measured in six-limb leads. The ECG was recorded with the PL3508 PowerLab 8/35 data acquisition system (AD Instruments). Data analysis was performed with the LabChart 7.3 software (AD Instruments). For body composition analysis by magnetic resonance (MR) technology, we used the LF90II body composition analyzer (Bruker). The Evans blue (EB) extravasation assay was performed by injecting 4 ml of 2% (wt/vol) EB dye solution/kg of body weight (BW) intravenously. Two hours later, the animals were sacrificed. The brains and kidneys were dissected and photographed. Ten-milligram sections of the organs were then homogenized in phosphate-buffered saline (PBS), mixed with the same volumes of 60% trichloroacetic acid, and centrifuged (10,000 rpm, 20 min). Absorbance of the supernatants was measured at 620 nm.
Electron microscopy.
Mice were killed by cervical dislocation and perfused with 4% paraformaldehyde (PFA) in phosphate buffer. Left ventricular tissue was postfixed with 2.5% glutaraldehyde in phosphate buffer overnight. Tissues were washed with cacodylate buffer, treated with 1% osmium tetroxide (pH 2.5) for 2 h, and dehydrated in a graded ethanol series. Tissues were embedded in Epon resin, cut in 70-nm sections, and contrasted with uranyl and lead citrate. Pictures were taken with a Zeiss 910 electron microscope and a charge-coupled device (CCD) camera (Proscan).
Virus inoculation.
The CVB3 Nancy strain (40) was grown and propagated in Vero cells. Stock virus was prepared by freezing and thawing three times and purified by sucrose gradient centrifugation as described previously (41). The same stock was used to infect mice by intraperitoneal injection with 1 × 105 PFU or intravenously with 1 × 106 PFU of purified CVB3. Animals were analyzed 4 or 10 days postinjection (dpi) with virus.
Cell culture.
Cells were cultured in Dulbecco modified Eagle medium (DMEM; Lonza) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin and incubated at 37°C with 5% CO2.
Transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For one cavity of a 6-well plate, 2.6 μg plasmid DNA was used. Protein expression was analyzed 48 h after transfection.
Transfected CHO cells were infected with CVB3 at a multiplicity of infection (MOI) of 0.1 for 30 min at 37°C. Subsequently, cells were washed twice with PBS. For RNA preparation, cells were lysed 4 h postinfection with 1 ml TRIzol. For plaque assay, cells with supernatant were harvested 24 h postinfection and lysed by three cycles of freezing and thawing.
Plaque assay.
Plaque assays were carried out as described previously (42). Briefly, HeLa cells were cultured in six-well culture plates as confluent monolayers at a density of 1 × 106 cells/well. After 24 h, medium was removed and cells were overlaid with 1 ml of diluted supernatant harvested from infected CHO cells and then incubated at 37°C for 30 min. After removal of the supernatant, cells were overlaid with 2 ml of agar containing Eagle's minimal essential medium (MEM). Three days later, the cells were stained with 0.025% neutral red in PBS. Virus titers were determined by counting plaques 3 h after staining. Data shown represent the results of two independent plaque assays, each performed in duplicate.
Immunofluorescence analysis.
H9C2 cells transfected with the chCAR expression construct were fixed with 4% PFA in PBS and stained according to the published protocol (43). Primary antibody was a rabbit anti-chCAR antibody targeting the extracellular domain of chCAR (a generous gift from Fritz Rathjen [26]). As secondary antibody, we used an anti-rabbit IgG Cy3-conjugated antibody raised in goat (Invitrogen). Image acquisition was performed on a Leica SPE microscope, and Image analysis was done in ImageJ (44).
Tissue preparation.
Samples of tissues were either quick-frozen in liquid nitrogen for the subsequent preparation of RNA or fixed for 12 h in phosphate-buffered (pH 7.2) 4% paraformaldehyde and embedded in paraffin for histology, immunohistology, and in situ hybridization.
Western blotting.
Protein preparation, SDS-PAGE, and Western blotting have been previously described (44). Primary antibodies used against chCAR (a generous gift from Fritz Rathjen; 1:500) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (mouse monoclonal; Calbiochem; 1:5,000) and secondary antibody conjugated with horseradish peroxidase (HRP; GE Healthcare Life Sciences; 1:5,000) were used according to the manufacturer's instructions.
Histopathology.
Histological analysis was performed on deparaffinized 5-μm-thick tissue sections that were stained with Masson's trichrome or hematoxylin and eosin (H&E) to assess cellular injury and inflammation (45). Immunohistochemistry using the CVB3-specific VP1 antibody (1:400) provided by Mediagnost (Reutlingen, Germany) was used to follow CVB3 replication. Subsequently, the sections were processed with the MaxHomo Mouse on Mouse Polymer HRP detection kit (MaxVision, Washington, USA) followed by HistoGreen substrate (Linaris, Dossenheim, Germany). The tissue sections were counterstained with hematoxylin. We evaluated cellular damage comprising cell necrosis, inflammation, and scarring as published previously (46), using a Zeiss Axioskop 40 microscope, with the following grading system: 0, no cellular damage and/or inflammatory infiltrates; 1, small foci of tissue damage and/or inflammatory cells; 2, larger foci of damage and/or >100 inflammatory cells; 3, larger foci with an area of ≤10% of a cross-section involved; 4, >10 to 30% of a cross-section involved; 5, up to 100% of a cross-section involved.
CVB3 RNA in situ hybridization.
For localization and quantification of CVB3 RNA in paraffinized tissue sections, we used radioactive in situ hybridization. At the indicated time points, tissue samples were fixed in 4% paraformaldehyde-0.1 mol/liter sodium phosphate buffer (pH 7.2) and embedded in paraffin for detection of viral RNA with a 35S-labeled enterovirus-specific RNA probe as previously described (41, 45). For quantification of CVB3 RNA in high-power fields (×200), we used the following scoring system: 0, no positive cells; 1, a few small foci with positive cells; 2, several foci with >100 positive cells; 3, ≤10% of the tissue containing positive cells; 4, 10 to 30% of the tissue containing positive cells; 5, up to 100% of the tissue containing positive cells. The same score was used for detection of VP1-positive cells using a Zeiss Axioskop 40 microscope, also reflecting virus replication at the cellular level.
Expression analysis.
RNA from the indicated tissues was amplified with specific TaqMan probes (sequence available on request). The gene expression assays for RANTES and beta interferon 1 (IFN-β1) as well as the 18S RNA as a housekeeping gene were purchased from Applied Biosystems (Foster City, CA). Data acquisition and analysis have been previously described (47).
Multiplex bead immunoassay.
To measure the serum concentration of RANTES, we used the BioPlex Pro mouse cytokine assay (Bio-Rad) according to the manufacturer's instructions. The fluorescence signal was detected using the Luminex 200 system (Millipore). Concentration was determined using a standard curve. All samples were run in triplicate.
Coxsackievirus B3 IgG serum ELISA.
To determine the concentration of CVB3-specific antibodies, the Serion enzyme-linked immunosorbent assay (ELISA) classic coxsackievirus IgG kit (Virion Serion) was used according to the manufacturer's instructions. Serum samples were initially diluted 1:200. The secondary antibody was replaced by an alkaline phosphatase-coupled anti-mouse IgG antibody (Invitrogen). The signal at 450 nm was measured with the F200 Pro microplate reader (Tecan). All samples were run in triplicate.
Statistical analysis.
For statistical analysis, GraphPad Prism software 5.0 (San Diego, CA) was used. Results are expressed as means ± standard errors of the means (SEM). To compare two groups, the Student t test was performed. Analysis of more than 2 groups was performed by one-way analysis of variance (ANOVA). Asterisks in the figures indicate statistically significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Homology modeling of Gallus gallus CAR D1 domain.
The three-dimensional structure of the CAR D1 domain amino acids (aa) 19 to 144 (UniProtKB accession number P78310) from Homo sapiens (PDB identifier [ID] 1RSF chain A) was used as the template for homology modeling. The homology modeling of the Gallus gallus CXADR D1 domain amino acids 20 to 139 (UniProtKB accession number F1NSL7) was performed using the ESyPred3D homology modeling program (48). The resulting three-dimensional structure was analyzed and compared using the Discovery Studio Visualizer 3.1 (Accelrys).
RESULTS
Heart-specific expression of CAR rescues the embryonic-lethal CAR KO phenotype.
CAR is essential for embryonic development, as knockouts die in midgestation with cardiac malformations and hemorrhage (32–34). To test if survival exclusively depends on CAR expression in the heart and to potentially circumvent the embryonic-lethal KO phenotype, we used a genetic rescue approach with cardiac tissue-specific expression of chCAR in the CAR KO mouse. We inserted the coding region of chicken CAR into the MLC-2 expression vector and validated the construct by restriction analysis and sequencing of the open reading frame (Fig. 1A). The MLC-2 promoter restricts expression to the heart, and its perinatal regulation matches that of CAR with high expression in the embryo and reduced levels after birth (49). To confirm protein expression, the rat cardiomyoblast cell line H9C2 was transfected with the MLC-2 chCAR expression construct followed by immunofluorescence staining with an anti-chCAR antibody (Fig. 1B). After validation of protein expression and proper localization of chCAR at the plasma membrane, we used pronuclear microinjection of the linearized construct to obtain transgenic (TG) animals. Founders with high expression levels of the chCAR transgene died within the first weeks of age (data not shown). Surviving transgenic animals were backcrossed to the CAR KO mice for two generations to obtain noncardiac CAR KO animals (ncKO), which survive and do not display an obvious phenotype (Fig. 1C and D). To validate the ncKO model, we determined CAR protein expression by Western blotting (Fig. 1E) and CAR mRNA levels by reverse transcription-quantitative PCR (qRT-PCR) with specific primers discriminating between mouse and chicken CAR. RNA from several tissues was isolated from 10-week-old mice, which express CAR at low levels, but it was clearly distinct from the background signal in ncKO animals (Fig. 1F). Expression of the transgenic chCAR was restricted to the heart of the ncKO mice (Fig. 1G), with no activity of the MLC-2 promoter in other tissues. The transgenic animals as well as the ncKO mice developed normally. Their weight and the body composition of fat, free water, and muscle tissue did not differ from those of wild-type (WT) animals (Table 1). Because of the importance of CAR in cardiac development and function, the hearts of the ncKO mice and the parental transgenic strain underwent additional analysis. The heart-weight-to-body-weight ratio of these animals was normal (Fig. 1H), as was the histology of adult hearts (Fig. 1I). There were no signs of hypertrophy, dilation, or changes in tissue composition. The ultrastructure as determined by electron microscopy was unremarkable in the rescue animals, in contrast to the conventional (34) or cardiac (35) CAR KO mice. The myofilament structure, mitochondria, and intercalated discs appeared normal in all genotypes (Fig. 1J). Cardiac function, as determined by echocardiography, was unchanged in TG or ncKO animals (Table 2). The electrical activity of the heart that was affected in the cardiac CAR KO with atrioventricular (AV) block (35–37) was normal in both strains (Table 3). Thus, chCAR completely restores the cardiac function of mCAR in the rescue animals. Furthermore, there were no obvious phenotypes observed caused by the deletion of CAR in other tissues studied, as the animals developed normally and had a normal body composition and an intact blood-brain barrier (Fig. 1K to M).
FIG 1.

Generation and basic characterization of the noncardiac CAR knockout mouse (ncKO). (A) The CAR expression construct contains the MLC-2 promoter, β-globin intron 2, chicken CAR cDNA (chCAR), and the simian virus 40 polyadenylation sequence (SV40pA). (B) Proper expression and localization were validated by immunofluorescence of chCAR (green) in H9C2 cells. Nuclei are stained in blue (4′,6-diamidino-2-phenylindole [DAPI]). Bar, 20 μm. (C) Breeding scheme. The MLC promoter drives expression of chCAR exclusively in the heart (a). Mouse CAR is expressed from the wild-type alleles. The heterozygous knockout contains a recombined CAR allele with a residual lox side after excision of exon 1 (triangle; b). Homozygous CAR knockout mice with the chCAR transgene (k) are available after two generations. (D) Pedigree of the heart-specific CAR rescue animals with the parental (P), F1, and F2 generations. Genotypes of animals (a to k) are provided below. The transgenic mouse expresses chicken CAR (chCAR) under the control of the heart-specific MLC-2 promoter (a). These animals were mated with heterozygote CAR-deficient mice carrying the recombined mouse CAR allele (CAR rec; b). Heterozygous offspring with (e) and without (f) the transgene were mated to obtain the noncardiac CAR KO animals (k). (E) In transgenic animals, chicken CAR (chCAR) protein is expressed and localizes to the intercalated disc. (F) In wild-type tissues, mouse CAR mRNA is expressed at normal levels as determined by TaqMan analysis, while expression in noncardiac knockout mice (ncKO) is at the detection limit (Fc, fold change). (G) The chCAR transgene is exclusively expressed in the hearts of ncKO animals. (H) Heart-weight-to-body-weight ratio (HW/BW) was normal in transgenic and ncKO animals. (I) Masson's trichrome staining of paraffin sections from hearts of 10-week-old wild-type (WT), chCAR transgenic (TG), and noncardiac CAR KO (ncKO) mice. There were no differences in heart morphology and tissue composition. Bar, 1 mm. (J) Ultrathin sections of hearts from 10-week-old WT, transgenic, and ncKO mice were analyzed by electron microscopy. The left panel shows the intercalated discs; the right panel shows the myofilament structure with mitochondria. There were no differences in cardiac ultrastructure. Bar, 2 μm. (K and L) CAR WT and ncKO mice were injected intravenously with 2% Evans blue (EB) dye to test the blood-brain barrier function. The brains (K) of both WT and ncKO mice showed no signs of dye leakage, as opposed to WT and ncKO kidneys (L), which stained dark blue. (M) Absorbance measurement of extravasated EB confirmed that ncKO mice had no increase of the dye content in the brain compared to WT mice. The amount of EB extracted from the brains of both WT and ncKO animals was significantly smaller than the amount extracted from the kidneys (P < 0.001). WT, n = 4; ncKO, n = 4. Bar, 1 cm.
TABLE 1.
Body composition analysis of transgenic and ncKO animals
| Characteristic | Value for mouse type: |
||
|---|---|---|---|
| WT (n = 6) | TG (n = 6) | ncKO (n = 6) | |
| Wt (g) | 23.00 ± 4.22 | 24.07 ± 4.79 | 20.93 ± 4.72 |
| Fat (%) | 15.19 ± 1.08 | 14.43 ± 1.01 | 16.95 ± 3.41 |
| Free water (%) | 6.43 ± 0.42 | 6.75 ± 0.74 | 6.67 ± 0.56 |
| Muscle (%) | 75.96 ± 1.18 | 76.51 ± 1.46 | 74.16 ± 3.97 |
TABLE 2.
Echocardiography of transgenic and ncKO animals
| Characteristica | Value for mouse type: |
||
|---|---|---|---|
| WT (n = 8) | TG (n = 5) | ncKO (n = 6) | |
| BW (g) | 21.84 ± 3.05 | 25.14 ± 3.84 | 20.92 ± 4.64 |
| HW (mg) | 99.16 ± 11.06 | 116.17 ± 23.94 | 106.45 ± 25.95 |
| HW/BW (mg/g) | 4.57 ± 0.41 | 4.63 ± 0.50 | 5.15 ± 1.01 |
| LVD diastole (mm) | 4.16 ± 0.18 | 4.13 ± 0.52 | 3.99 ± 0.45 |
| LVD systole (mm) | 3.09 ± 0.23 | 3.06 ± 0.29 | 2.99 ± 0.41 |
| FS (%) | 25.64 ± 4.46 | 26.24 ± 2.32 | 25.12 ± 4.57 |
| EF (%) | 50.92 ± 7.61 | 51.12 ± 4.71 | 48.96 ± 6.71 |
BW, body weight; HW, heart weight; LVD, left ventricular diameter; FS, fractional shortening; EF, ejection fraction.
TABLE 3.
ECG of transgenic and ncKO animals
| Characteristic | Value for mouse type: |
||
|---|---|---|---|
| WT (n = 6) | TG (n = 6) | ncKO (n = 6) | |
| Interval (ms) | |||
| PR | 39.70 ± 1.14 | 41.22 ± 1.36 | 39.49 ± 3.08 |
| QRS | 13.26 ± 1.55 | 11.74 ± 1.65 | 11.32 ± 1.45 |
| QT | 25.66 ± 1.79 | 26.05 ± 2.14 | 25.70 ± 0.91 |
| Amplitude (mV) | |||
| P | 0.08 ± 0.02 | 0.09 ± 0.02 | 0.11 ± 0.02 |
| Q | −0.05 ± 0.03 | −0.06 ± 0.04 | −0.08 ± 0.08 |
| R | 1.55 ± 0.34 | 1.53 ± 0.35 | 1.60 ± 0.37 |
| S | −0.45 ± 0.15 | −0.33 ± 0.27 | −0.49 ± 0.26 |
The CAR ncKO mouse is protected from CVB3 infection.
The relevance of CAR and its coreceptors for virus entry and CVB3-associated pathology has recently been addressed using loss-of-function mutants in heart and pancreas (3, 4). To facilitate the differential analysis of multiple organs and to determine how CAR influences virus spread within the organism, we infected WT and ncKO animals intraperitoneally with the CVB3 Nancy strain. Ten days after infection, tissues were isolated and analyzed for virus and associated pathology, such as signs of inflammation. Of the noncardiac tissues analyzed, we found necrotic lesions in histological sections of the pancreas of WT but not ncKO animals (Fig. 2A). The area of necrosis and inflammation with infiltration of mononuclear cells was used to assign an arbitrary score from 0 (no damage) to 5 (30 to 100% affected area). For WT pancreas, the inflammation score was 4.70 ± 0.48 (mean ± standard deviation [SD]). Spleen, liver, kidney, and lung appeared normal in both genotypes (score 0). In situ hybridization with a probe specific for viral plus-strand RNA confirmed replicating virus in the exocrine pancreas of WT but not of ncKO animals (Fig. 2B). The presence of viral RNA was scored from 0 (no positive cells) to 5 (30 to 100% positive cells). In the pancreas of wild-type mice, the infection score was 4.20 ± 0.42 (mean ± SD). There was no viral RNA as a sign of replicating virus in ncKO animals (score 0).
FIG 2.
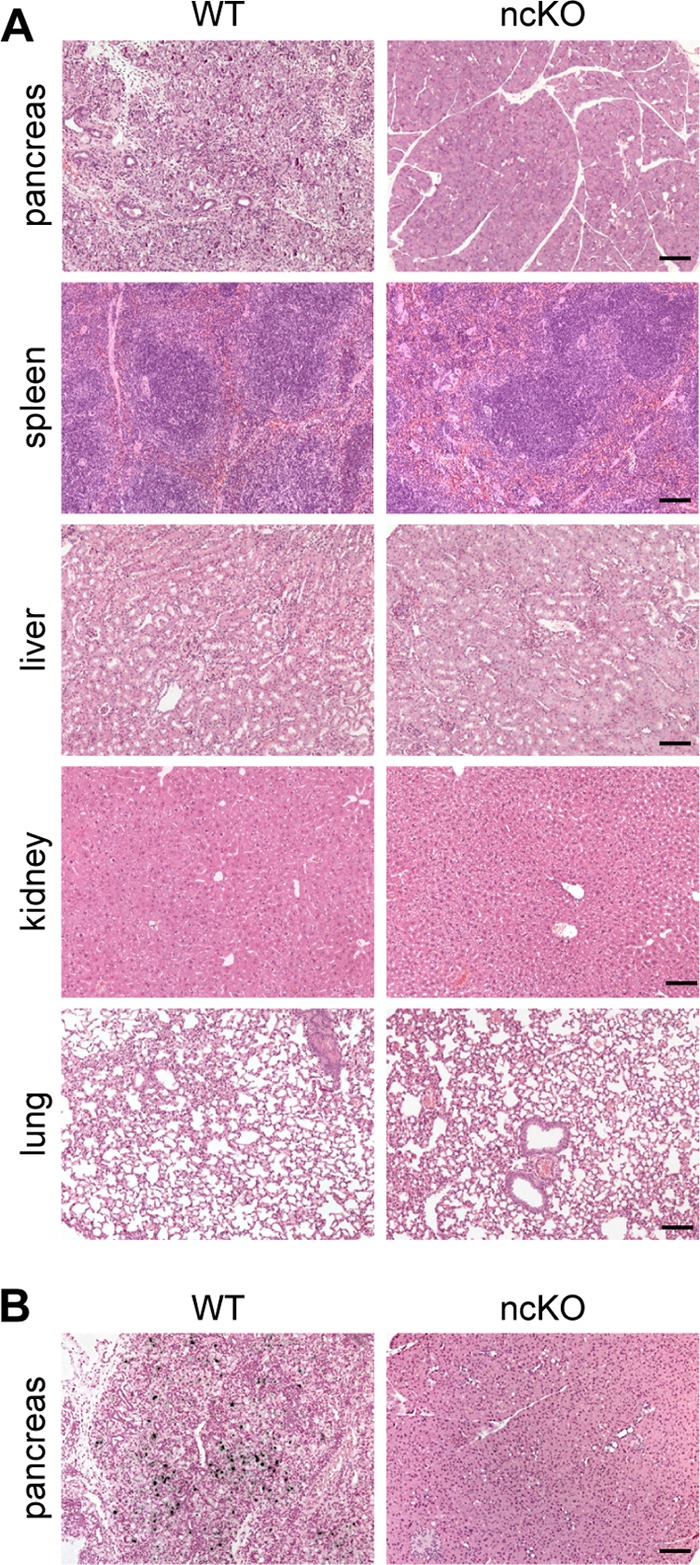
In ncKO mice, noncardiac tissues are protected from CVB3 infection. CAR WT and ncKO animals were inoculated intraperitoneally with 1 × 105 PFU of the CVB3 Nancy strain and analyzed after 10 days by H&E staining and in situ hybridization on paraffin sections. (A) H&E staining reveals necrosis and inflammation with infiltration of mononuclear cells in WT pancreas, which is not detected in the ncKO animals. The morphology of spleen, liver, kidney, and lung was normal. There was no sign of infection in any ncKO tissue analyzed. (B) CVB3 infection was documented by the presence of viral RNA in WT pancreas. There was no viral RNA as a sign of replicating virus in ncKO animals. WT, n = 10; ncKO, n = 10. Bar, 50 μm.
In a more global approach to virus infection, we analyzed the immune response in animals exposed to CVB3. Intraperitoneal infection of ncKO mice did not result in the induction of inflammation markers. CVB3-specific IgG levels were increased from 4 through 10 days postinfection (dpi) in wild-type animals, while ncKO animals did not produce a specific immune response. In the serum of ncKO mice at 4 or 10 dpi, there was no induction of the inflammation marker “regulated on activation, normal T-cell expressed and secreted” (RANTES). In contrast, WT animals had high levels of serum RANTES in the acute phase of the infection at 4 dpi, which decreased but were still elevated in the subacute phase at 10 dpi (Fig. 3).
FIG 3.

The immune response to CVB3 is not induced in ncKO mice. Serum of CVB3-inoculated WT and ncKO animals was collected 4 and 10 days after infection (dpi). (A) Specific antibody production was determined using a CVB3 serum ELISA. At 4 dpi, CVB3-specific IgGs are present in WT animals and increase through day 10 postinfection. No CVB3-specific antibodies were detected in ncKO animals. (B) In the acute phase of infection, the inflammation marker RANTES was upregulated more than 5-fold in the serum of WT but not ncKO animals. At 10 dpi, serum levels of RANTES are decreased in WT mice but still elevated compared to those in ncKO mice. *, P < 0.05; ***, P < 0.005. Uninfected: WT, n = 3; ncKO, n = 6; 4 dpi: WT, n = 4; ncKO, n = 3; 10 dpi: WT, n = 6; ncKO, n = 8.
As there was no prominent immune response to virus load in the ncKO mouse, we investigated if CVB3 infection was blocked not only in noncardiac cells but also in cardiomyocytes of the rescue animals. We analyzed hearts from ncKO mice in the acute phase of infection and found that hearts expressing chCAR instead of mCAR were protected from CVB3 infection. Comparing WT versus ncKO hearts at 4 and 10 dpi, we found signs of infiltration, viral CVB3 RNA, and VP1 protein only in the wild-type mice, not in ncKO mice (Fig. 4A to D). Quantification was based on the affected area and/or number of positive cells with infiltration based on histology (WT, 1.30 ± 0.48, versus ncKO, 0.0 ± 0.0), in situ hybridization (WT, 1.20 ± 0.42, versus ncKO, 0.0 ± 0.0), and presence of capsid protein VP1 (WT, 0.8 ± 0.42, versus ncKO, 0.0 ± 0.0).
FIG 4.

Replacing mouse with chicken CAR prevents CVB3 entry and the associated cardiac pathology. Analysis of virus uptake and the associated immune response in hearts expressing mouse versus chicken CAR (CAR WT and ncKO animals). (A to D) H&E staining documents the infiltration with mononuclear cells (A) and replicating virus detected by in situ hybridization of wild-type but not ncKO hearts 10 days (B) after intraperitoneal infection with CVB3. Viral RNA detected by qRT-PCR (C) as well as the viral capsid protein VP1 (D) was present only in the hearts of WT but not ncKO mice at 4 dpi. (E) The inflammation markers RANTES, TNF-α, IFN-β1, IL-6, and IL-10 were induced only in the hearts of WT and not ncKO animals as determined by TaqMan analysis. *, P < 0.05; **, P < 0.01; ***, P < 0.001. WT, n = 4; ncKO, n = 3. Bars, 50 μm.
We also tested the expression of the cytokines RANTES, tumor necrosis factor alpha (TNF-α), beta interferon 1 (IFN-β1), interleukin 6 (IL-6), and interleukin 10 (IL-10) in infected heart tissue samples. In contrast to WT animals, there was no induction in ncKO mice (Fig. 4E).
To determine if the route of delivery in the ncKO mouse affected virus-induced cardiac pathology, we performed a second set of infections delivering the virus intravenously to ensure that the virus reached the heart. Viral RNA was present in wild-type hearts (infection score, 1.25 ± 0.50) and pancreas (infection score, 4.25 ± 0.50). Again, there were no histological signs of infection and no viral RNA in the hearts of infected ncKO animals (Fig. 5A). Intravenous delivery resulted in a slight induction of CVB3-specific IgGs, but there was still a significant difference from WT animals with replicating virus (Fig. 5B). The slight induction in the ncKO mouse might be caused by circulating virus in the blood and does not necessarily indicate a viral infection. Serum levels of RANTES were increased only in WT animals (Fig. 5C). Taken together, the protection from virus infection of noncardiac and cardiac tissue when mouse CAR is replaced by chicken CAR indicates an important role for CAR in mediating virus uptake throughout the body and suggests a species-specific interaction of CVB3 with its receptor.
FIG 5.

Protection from CVB3-associated pathology is independent of the delivery route. CAR WT and ncKO animals were inoculated intravenously with 1 × 106 PFU of the CVB3 Nancy strain. (A) At 10 dpi, viral RNA was present in heart and pancreas of WT but not ncKO animals as detected by in situ hybridization of paraffin sections. (B) CVB3-specific antibodies were detected in both WT and ncKO animals by using a coxsackievirus IgG-specific ELISA. The immune response was significantly reduced in ncKO animals. (C) Serum levels of RANTES at 10 dpi were increased only in WT animals. *, P < 0.05; ***, P < 0.001. WT, n = 4; ncKO, n = 5. Bar, 50 μm.
CVB3 infection is species dependent.
As chCAR restored the physiological functions of mCAR but did not mediate CVB3 infection, we investigated differences in virus uptake in a cell culture system. Native CHO cells were not infected with the CVB3 Nancy strain but with the PD strain, which entered cells independently of CAR (50) and resulted in high virus titers (Fig. 6A). CHO cells, which do not express CAR and thus cannot be infected by the CVB3 Nancy strain (51), were transfected with mouse CAR (mCAR) or chicken CAR (chCAR) and subsequently infected with CVB3. Transfection of CHO cells with mouse CAR resulted in high levels of infection and virus replication verified by a plaque assay. Nontransfected CHO cells and cells transfected with chicken CAR were not infected (Fig. 6B). To validate these findings, RNA from transfected cells 4 h postinfection was isolated and analyzed by qRT-PCR. Viral RNA was detected only in cells expressing mCAR and not in cells expressing chicken or no CAR (Fig. 6C). Nontransfected CHO cells were CAR negative, and only the transfected CAR species was expressed in the cells (Fig. 6D and E).
FIG 6.

Species-specific variation in CAR affects CVB3 infection. CAR-negative CHO cells were transfected with CAR from mouse (mCAR) and chicken (chCAR), infected with CVB3, and analyzed using a plaque assay. (A) The CVB3 Nancy strain does not replicate in CAR-negative CHO cells, whereas the CVB3 PD strain infects cells independently of CAR. (B) Replicating virus arose only from cells transfected with mCAR. Untransfected CHO cells or cells transfected with chCAR did not contain virus. (C) At 4 h postinfection, CVB3 RNA was present only in mCAR-transfected cells and not in those expressing chCAR. (D and E) TaqMan analysis confirmed the species-specific CAR expression. *, P < 0.05; ***, P < 0.001.
To relate these functional differences to differences in the amino acid compositions of chicken versus mouse CAR, we performed sequence alignments and modeled the interaction site of chicken CAR. As the N-terminal Ig domain is most important for virus binding as well as the interaction with other IgSF proteins, and as the structures had been solved for human and mouse, we focused our analysis on that region (Fig. 7). The published binding sites of CVB3 to CAR (15) comprise 27 amino acids (aa), of which 24 are identical between mouse and human (89% homology). The differentiating amino acids are located close to the D2 domain of CAR, suggesting only minor relevance of this region for virus binding. In contrast, the homology of CVB3 binding amino acids between mouse and chicken is 63% (17 identical aa). Modeling of the chicken CAR D1 domain localizes changed amino acids also to the upper part of the D1 domain. The published interaction sites between two CAR molecules (25, 26) are highly conserved between mouse and human (100%) and mouse and chicken (80%). The differential amino acids are not clustered but distributed throughout the D1 domain. These species-specific differences might explain why chCAR can preserve the cellular function of CAR but not CVB3 replication.
FIG 7.

Cellular and viral CAR binding sites differ between species. Sequence analysis and modeling of the N-terminal Ig domain of human, mouse, and chicken CAR. Published binding sites of the CAR-CVB3 interaction or CAR-CAR are displayed on a dark gray background. Homology to the published sites is indicated by the light gray background. (A) Alignment of the first D1 domain shows the conservation of amino acids within the binding sites for CVB3 between Homo sapiens, Mus musculus, and G. gallus. Three amino acids are exchanged between human and mouse, and 7 additional amino acids are exchanged in chicken (indicated in red). An asterisk indicates a position with a single, fully conserved residue. A colon indicates conservation between groups of strongly similar properties. A period indicates conservation between groups of weakly similar properties. (B) The structures of the human and mouse Ig domains have been solved, and the G. gallus D1 domain was modeled by using human D1 domain as a template. Nonconserved amino acids are indicated in red. The additional amino acid exchanges in G. gallus mainly affect the upper part of the molecule. (C) The mutations affecting amino acids in the chicken Ig domain (red) do not cluster within the structure. (D) The amino acids involved in CAR dimerization (blue) are well conserved between species, with complete identity between mouse and human and 4 additional mutations in chicken.
With the generation of our novel ncKO mouse, we were able to show that the essential function of CAR in vivo is related to cardiac development. The species-specific differences in virus uptake versus cardiac function became apparent as chCAR restored normal heart development but did not mediate virus infection. This can be explained by differential conservation of virus binding sites versus sites involved in cellular interactions.
DISCUSSION
CAR is a multifunctional protein that not only mediates uptake of type B coxsackieviruses and adenoviruses but also is required for critical cellular functions such as cell contact formation and signal transduction. Thus, CAR-deficient mice die early in embryonic development (32–34). In this study, we show that the expression of chicken CAR in the mouse heart is sufficient to rescue the embryonic-lethal CAR KO phenotype as it adopts the physiological functions of mouse CAR. Surprisingly, it does not restore susceptibility to coxsackievirus infection.
To study the cellular function of CAR, various KO mice have been generated. The conventional KO of CAR exon 1 or 2 leads to cardiac malformations and embryonic lethality between day E11.5 and E13.5. Survival of cardiac tissue-specific knockout mice depends on the promoter driving the Cre transgene and the targeted exon, ranging from complete lethality in the MHCcre-driven KO of CAR exon 1 or the TnTcre-driven KO of CAR exon 2 (33, 36) to survival of 20% or 100% of the embryos in the MHCcre-driven CAR exon 2 KO (32–34). The inducible KO of CAR in the adult heart is compatible with life but results in an electrical phenotype with impaired cardiac function (35–37). Together, these mouse models indicate an important role of CAR in development and function of the heart.
Here, we used a rescue approach to evaluate the role of CAR in extracardiac tissues and study species-specific differences in virus uptake versus cardiac physiology. We used the MLC-2 promoter, which mimics the physiological timing of CAR expression (32, 49, 52, 53), to express chicken CAR exclusively in the mouse heart. This approach was associated with a dose-dependent lethality, as mice expressing high levels of the transgene died within the first weeks after birth while founders with lower levels of chicken CAR expression survived. This is consistent with reported observations after a 6-fold overexpression of CAR in the heart (54). These animals develop cardiomyopathy and cardiac hypertrophy and die within their first month of life. In contrast, our surviving transgenic mice with low expression of the chCAR transgene and proper developmental regulation through the MLC-2 promoter do not show any obvious abnormalities.
The chCAR transgene rescued the embryonic lethality of the homozygous CAR KO mice, resulting in normal cardiac function and development of the ncKO animals (32–34). Thus, expression of CAR exclusively in the heart is sufficient for the survival of CAR KO embryos.
Postnatal development of the ncKO mice was normal, including body weight, body composition, heart weight, and cardiac morphology. The spectrum of cardiac pathology in CAR KO mice ranges from enlarged mitochondria over thin myofilament bundles (34) to disrupted connections of the myofilaments to the intercalated discs (35). In contrast, there were no obvious morphological, ultrastructural, or functional changes in our ncKO animals, including mechanics and electrical activity, as determined by echocardiography and ECG. The electrical conduction defect with severe AV block, the hallmark of the adult CAR knockout heart (35, 36), was completely restored with the chicken transgene. Indeed, normal cardiac development and function in the ncKO mouse indicate that chCAR can replace the physiological function of the endogenous CAR in the mouse heart.
The survival of the ncKO mouse suggests that CAR is not a critical component in the development of extracardiac tissues. The phenotype of the tamoxifen-inducible ubiquitous CAR knockout (55) with tamoxifen injection at E12.5 resulted in death on day E18.5. The embryos suffered from subcutaneous edema and hemorrhages, and the presence of erythrocytes in lymphatic vessels had been attributed to a potential contribution of CAR to the separation between blood and lymphatic vessels (55). Our analysis of the ncKO mouse, which did not reproduce this phenotype, would suggest that the hemorrhage is more likely secondary to the cardiac phenotype and not a vascular defect.
The expression of CAR in male germ cells and Sertoli cells suggests a function for CAR in male fertility (56, 57). As our animals breed and produce normal litters, an essential function in male fertility can be excluded. These findings are consistent with the inducible ubiquitous downregulation of CAR in adult mice that has no effect on metabolism, fertility, or behavior (37). The localization of CAR to tight junctions implies a role for CAR in epithelial barrier function and tissue homeostasis (58). Accordingly, we investigated the blood-brain barrier in ncKO mice and did not find an increased permeability, indicating an intact barrier function. The lack of an obvious extracardiac phenotype in the ncKO mouse under baseline conditions does not exclude a role for CAR in noncardiac tissues, and we anticipate that future appropriate challenges will help develop the ncCAR KO as a suitable tool to study the role of CAR in the lung endothelium or kidney or in the migration of neutrophils (59). Conveniently, the robust rescue avoids the necessity to induce CAR deficiency and provides a complete loss of CAR in all cells from early embryonic development.
Challenging wild-type mice with CVB3 resulted in infection, virus replication, and the expected pathology, predominantly in heart and pancreas as the prime target tissues (3, 4). The ncKO mouse was protected from virus-induced pathology. Unexpectedly, this involved not only noncardiac tissues but also the heart, which did not show any signs of infection despite the cardiac expression of chCAR. In hearts of infected rescue animals, we did not detect viral RNA by in situ hybridization or TaqMan analysis, nor did we detect CVB3 capsid protein VP1. There were no signs of histopathology and no induction of an immune response in infected ncKO animals. A similar protection of the pancreas or heart has been reported in the respective tissue-specific CAR KO mice with reduced virus load, inflammation, and tissue damage compared to those of WT mice (3, 4). The absence of viral replication in heart or pancreas as primary organs of CVB3 replication was not associated with reduced virus spread or replication in other organs (3, 4).
To exclude a compartmentalization effect where the loss of CAR in noncardiac tissue prevents virus spread, we monitored cardiac infection after intravenous versus intraperitoneal delivery of CVB3. Neither route of virus administration resulted in cardiac damage or viral replication. After intravenous application, we detected CVB3-specific antibodies in the serum of infected rescue animals, although at lower levels than those in wild-type mice. The production of antibodies likely results from circulating virus in the blood and not from the infection, as the induction of an immune response by nonreplicating virus capsids has previously been described (60).
With mounting evidence that chCAR does not mediate virus entry in vivo, we used a cell culture-based system to eliminate the complicating effects of virus spread and immune response. In CAR-negative CHO cells transfected with mouse or chicken CAR, we compared virus replication levels via plaque assays and found that unlike mouse CAR, chCAR does not mediate CVB3 infection. CVB3 has been reported to infect several species, including humans and other primates (5–7) and mice and dogs (8, 9). Infections of nonmammalian species have not been reported, but transfection of CAR-negative CHO cells with zebrafish CAR allows CVB3 infection (61). Chicken CAR did not mediate virus uptake into CHO cells, possibly related to the diversity of amino acid sequences, with a homology between zebrafish and chicken CAR of only 51%. While amino acid conservation of CAR is high between mammals, there is a considerable disparity with nonmammalian species, specifically between sites involved in cellular versus virus-host interactions. Specifically, the CVB3 binding sites are highly conserved between human and mouse but not between human and chicken. Modeling of the protein structure localizes the 10 nonconserved amino acids between human and chicken throughout the D1 region. In contrast, the 3 aa differentiating human and mouse localize in D1 close to the D2 domain, which is not as accessible for interactions as is the distal region of the molecule. Amino acids of the N-terminal Ig domain that are involved in the physiological interaction between CAR molecules or CAR and other members of the IgSF and the cytoplasmic tail with its PDZ consensus site (GSIV) are conserved between human, mouse, and chicken (15). This suggests that amino acids involved in CAR's physiological interactions underlie a higher selection pressure and are conserved between species, in contrast to amino acids that relate to the interaction with pathogens. Thus, CAR-dependent cell contacts and signaling pathways can be sustained by chCAR, but CVB3 infection cannot.
In summary, expression of chCAR in the heart of CAR knockout mice has allowed us to rescue the embryonic-lethal KO phenotype and address interspecies differences in virus-host interactions. Replacing mouse with chicken CAR protects animals from CVB3 infection but maintains normal cardiomyocyte morphology and function. Thus, the cardiac rescue survives and does not display any obvious noncardiac phenotypes. Accordingly, we expect only minor nonmuscle side effects from CAR-directed therapeutic strategies and added safety for cardiac antiviral strategies that exploit species-specific differences in CAR physiology versus pathology.
ACKNOWLEDGMENTS
We thank Janine Fröhlich, Mathias Pippow, and Sandra Bundschuh for technical assistance. We thank the transgenic core facility at the MDC for excellent support with generation of the chCAR transgenic mouse. We thank Fritz Rathjen for the chCAR plasmid and the antibody, Michael Bader for the MLC-2 promoter plasmid, Michaela Schmidtke for CVB3-PD, and Bettina Purfürst for support with the electron microscopy data acquisition.
This research was funded by the German Research Foundation (DFG; SFB-TR/19 and GO865/9-1); F. Freiberg received a TransCard Ph.D. fellowship.
Footnotes
Published ahead of print 16 April 2014
REFERENCES
- 1.Modlin JF, Rotbart HA. 1997. Group B coxsackie disease in children. Curr. Top. Microbiol. Immunol. 223:53–80 [DOI] [PubMed] [Google Scholar]
- 2.Kühl U, Pauschinger M, Noutsias M, Seeberg B, Bock T, Lassner D, Poller W, Kandolf R, Schultheiss H-P. 2005. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 111:887–893. 10.1161/01.CIR.0000155616.07901.35 [DOI] [PubMed] [Google Scholar]
- 3.Kallewaard NL, Zhang L, Chen J-W, Guttenberg M, Sanchez MD, Bergelson JM. 2009. Tissue-specific deletion of the coxsackievirus and adenovirus receptor protects mice from virus-induced pancreatitis and myocarditis. Cell Host Microbe 6:91–98. 10.1016/j.chom.2009.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi Y, Chen C, Lisewski U, Wrackmeyer U, Radke M, Westermann D, Sauter M, Tschöpe C, Poller W, Klingel K, Gotthardt M. 2009. Cardiac deletion of the Coxsackievirus-adenovirus receptor abolishes Coxsackievirus B3 infection and prevents myocarditis in vivo. J. Am. Coll. Cardiol. 53:1219–1226. 10.1016/j.jacc.2008.10.064 [DOI] [PubMed] [Google Scholar]
- 5.He W, Lu H, Song D, Zhao K, Gai X, Wang X, Chen Q, Gao F. 2009. The evidence of Coxsackievirus B3 induced myocarditis as the cause of death in a Sichuan snub-nosed monkey (Rhinopithecus roxellana). J. Med. Primatol. 38:192–198. 10.1111/j.1600-0684.2008.00336.x [DOI] [PubMed] [Google Scholar]
- 6.Han T, He W, Song D, Zhao K, Wu C, Gao F, Lu H, Gai X, Wang X, Li F, Ji C, Lin X. 2012. Experimental SSM-CVB3 infection in macaques. Exp. Mol. Pathol. 92:131–139. 10.1016/j.yexmp.2011.10.008 [DOI] [PubMed] [Google Scholar]
- 7.Paque RE, Gauntt CJ, Nealon TJ. 1981. Assessment of cell-mediated immunity against coxsackievirus B3-induced myocarditis in a primate model (Papio papio). Infect. Immun. 31:470–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly ME, Soike K, Ahmed K, Iatropoulos MJ. 1978. Coxsackievirus in an infant chimpanzee. J. Med. Primatol. 7:119–121 [DOI] [PubMed] [Google Scholar]
- 9.Lundgren DL, Hobbs CH, Clapper WE. 1971. Experimental infection of beagle dogs with Coxsackievirus type B1. Am. J. Vet. Res. 32:609–613 [PubMed] [Google Scholar]
- 10.Verdaguer N, Jimenez-Clavero MA, Fita I, Ley V. 2003. Structure of swine vesicular disease virus: mapping of changes occurring during adaptation of human coxsackie B5 virus to infect swine. J. Virol. 77:9780–9789. 10.1128/JVI.77.18.9780-9789.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomko RP, Xu R, Philipson L. 1997. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. U. S. A. 94:3352–3356. 10.1073/pnas.94.7.3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. 1997. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320–1323. 10.1126/science.275.5304.1320 [DOI] [PubMed] [Google Scholar]
- 13.Martino TA, Petric M, Weingartl H, Bergelson JM, Opavsky MA, Richardson CD, Modlin JF, Finberg RW, Kain KC, Willis N, Gauntt CJ, Liu PP. 2000. The coxsackie-adenovirus receptor (CAR) is used by reference strains and clinical isolates representing all six serotypes of coxsackievirus group B and by swine vesicular disease virus. Virology 271:99–108. 10.1006/viro.2000.0324 [DOI] [PubMed] [Google Scholar]
- 14.Roelvink PW, Lizonova A, Lee JG, Li Y, Bergelson JM, Finberg RW, Brough DE, Kovesdi I, Wickham TJ. 1998. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J. Virol. 72:7909–7915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He Y, Chipman PR, Howitt J, Bator CM, Whitt MA, Baker TS, Kuhn RJ, Anderson CW, Freimuth P, Rossmann MG. 2001. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat. Struct. Biol. 8:874–878. 10.1038/nsb1001-874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belnap DM, McDermott BM, Jr, Filman DJ, Cheng N, Trus BL, Zuccola HJ, Racaniello VR, Hogle JM, Steven AC. 2000. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. U. S. A. 97:73–78. 10.1073/pnas.97.1.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolatkar PR, Bella J, Olson NH, Bator CM, Baker TS, Rossmann MG. 1999. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 18:6249–6259. 10.1093/emboj/18.22.6249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olson NH, Kolatkar PR, Oliveira MA, Cheng RH, Greve JM, McClelland A, Baker TS, Rossmann MG. 1993. Structure of a human rhinovirus complexed with its receptor molecule. Proc. Natl. Acad. Sci. U. S. A. 90:507–511. 10.1073/pnas.90.2.507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shafren DR, Bates RC, Agrez MV, Herd RL, Burns GF, Barry RD. 1995. Coxsackieviruses B1, B3, and B5 use decay accelerating factor as a receptor for cell attachment. J. Virol. 69:3873–3877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoder JD, Cifuente JO, Pan J, Bergelson JM, Hafenstein S. 2012. The crystal structure of a coxsackievirus B3-RD variant and a refined 9-angstrom cryo-electron microscopy reconstruction of the virus complexed with decay-accelerating factor (DAF) provide a new footprint of DAF on the virus surface. J. Virol. 86:12571–12581. 10.1128/JVI.01592-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shafren DR, Williams DT, Barry RD. 1997. A decay-accelerating factor-binding strain of coxsackievirus B3 requires the coxsackievirus-adenovirus receptor protein to mediate lytic infection of rhabdomyosarcoma cells. J. Virol. 71:9844–9848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selinka H-C, Wolde A, Sauter M, Kandolf R, Klingel K. 2004. Virus-receptor interactions of coxsackie B viruses and their putative influence on cardiotropism. Med. Microbiol. Immunol. 193:127–131. 10.1007/s00430-003-0193-y [DOI] [PubMed] [Google Scholar]
- 23.Honda T, Saitoh H, Masuko M, Katagiri-Abe T, Tominaga K, Kozakai I, Kobayashi K, Kumanishi T, Watanabe YG, Odani S, Kuwano R. 2000. The coxsackievirus-adenovirus receptor protein as a cell adhesion molecule in the developing mouse brain. Brain Res. Mol. Brain Res. 77:19–28. 10.1016/S0169-328X(00)00036-X [DOI] [PubMed] [Google Scholar]
- 24.Cohen CJ, Shieh JT, Pickles RJ, Okegawa T, Hsieh JT, Bergelson JM. 2001. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. U. S. A. 98:15191–15196. 10.1073/pnas.261452898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Raaij MJ, Chouin E, van der Zandt H, Bergelson JM, Cusack S. 2000. Dimeric structure of the coxsackievirus and adenovirus receptor D1 domain at 1.7 A resolution. Structure 8:1147–1155. 10.1016/S0969-2126(00)00528-1 [DOI] [PubMed] [Google Scholar]
- 26.Patzke C, Max KE, Behlke J, Schreiber J, Schmidt H, Dorner AA, Kröger S, Henning M, Otto A, Heinemann U, Rathjen FG. 2010. The coxsackievirus-adenovirus receptor reveals complex homophilic and heterophilic interactions on neural cells. J. Neurosci. 30:2897–2910. 10.1523/JNEUROSCI.5725-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verdino P, Witherden DA, Havran WL, Wilson IA. 2010. The molecular interaction of CAR and JAML recruits the central cell signal transducer PI3K. Science 329:1210–1214. 10.1126/science.1187996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Excoffon KJDA, Hruska-Hageman A, Klotz M, Traver GL, Zabner J. 2004. A role for the PDZ-binding domain of the coxsackie B virus and adenovirus receptor (CAR) in cell adhesion and growth. J. Cell Sci. 117:4401–4409. 10.1242/jcs.01300 [DOI] [PubMed] [Google Scholar]
- 29.Sollerbrant K, Raschperger E, Mirza M, Engstrom U, Philipson L, Ljungdahl PO, Pettersson RF. 2003. The Coxsackievirus and adenovirus receptor (CAR) forms a complex with the PDZ domain-containing protein ligand-of-numb protein-X (LNX). J. Biol. Chem. 278:7439–7444. 10.1074/jbc.M205927200 [DOI] [PubMed] [Google Scholar]
- 30.Coyne CB, Voelker T, Pichla SL, Bergelson JM. 2004. The coxsackievirus and adenovirus receptor interacts with the multi-PDZ domain protein-1 (MUPP-1) within the tight junction. J. Biol. Chem. 279:48079–48084. 10.1074/jbc.M409061200 [DOI] [PubMed] [Google Scholar]
- 31.Fischer R, Poller W, Schultheiss H-P, Gotthardt M. 2009. CAR-diology—a virus receptor in the healthy and diseased heart. J. Mol. Med. (Berl.) 87:879–884. 10.1007/s00109-009-0489-5 [DOI] [PubMed] [Google Scholar]
- 32.Asher DR, Cerny AM, Weiler SR, Horner JW, Keeler ML, Neptune MA, Jones SN, Bronson RT, Depinho RA, Finberg RW. 2005. Coxsackievirus and adenovirus receptor is essential for cardiomyocyte development. Genesis 42:77–85. 10.1002/gene.20127 [DOI] [PubMed] [Google Scholar]
- 33.Chen JW, Zhou B, Yu QC, Shin SJ, Jiao K, Schneider MD, Baldwin HS, Bergelson JM. 2006. Cardiomyocyte-specific deletion of the coxsackievirus and adenovirus receptor results in hyperplasia of the embryonic left ventricle and abnormalities of sinuatrial valves. Circ. Res. 98:923–930. 10.1161/01.RES.0000218041.41932.e3 [DOI] [PubMed] [Google Scholar]
- 34.Dorner AA, Wegmann F, Butz S, Wolburg-Buchholz K, Wolburg H, Mack A, Nasdala I, August B, Westermann J, Rathjen FG, Vestweber D. 2005. Coxsackievirus-adenovirus receptor (CAR) is essential for early embryonic cardiac development. J. Cell Sci. 118:3509–3521. 10.1242/jcs.02476 [DOI] [PubMed] [Google Scholar]
- 35.Lim B-K, Xiong D, Dorner A, Youn T-J, Yung A, Liu TI, Gu Y, Dalton ND, Wright AT, Evans SM, Chen J, Peterson KL, McCulloch AD, Yajima T, Knowlton KU. 2008. Coxsackievirus and adenovirus receptor (CAR) mediates atrioventricular-node function and connexin 45 localization in the murine heart. J. Clin. Invest. 118:2758–2770. 10.1172/JCI34777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lisewski U, Shi Y, Wrackmeyer U, Fischer R, Chen C, Schirdewan A, Jüttner R, Rathjen F, Poller W, Radke MH, Gotthardt M. 2008. The tight junction protein CAR regulates cardiac conduction and cell-cell communication. J. Exp. Med. 205:2369–2379. 10.1084/jem.20080897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pazirandeh A, Sultana T, Mirza M, Rozell B, Hultenby K, Wallis K, Vennström B, Davis B, Arner A, Heuchel R, Löhr M, Philipson L, Sollerbrant K. 2011. Multiple phenotypes in adult mice following inactivation of the Coxsackievirus and Adenovirus Receptor (Car) gene. PLoS One 6:e20203. 10.1371/journal.pone.0020203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marsman RFJ, Bezzina CR, Freiberg F, Verkerk AO, Adriaens ME, Podliesna S, Chen C, Purfürst B, Spallek B, Koopmann TT, Baczko I, Dos Remedios CG, George AL, Jr, Bishopric NH, Lodder EM, de Bakker JMT, Fischer R, Coronel R, Wilde AAM, Gotthardt M, Remme CA. 2014. Coxsackie and adenovirus receptor is a modifier of cardiac conduction and arrhythmia vulnerability in the setting of myocardial ischemia. J. Am. Coll. Cardiol. 63:549–559. 10.1016/j.jacc.2013.10.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, King NM, Yu Q, Tschöpe C, McNabb M, Larson DF, Labeit S, Gotthardt M. 2009. Truncation of titin's elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circ. Res. 105:557–564. 10.1161/CIRCRESAHA.109.200964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kandolf R, Hofschneider PH. 1985. Molecular cloning of the genome of a cardiotropic Coxsackie B3 virus: full-length reverse-transcribed recombinant cDNA generates infectious virus in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 82:4818–4822. 10.1073/pnas.82.14.4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gruhle S, Sauter M, Szalay G, Ettischer N, Kandolf R, Klingel K. 2012. The prostacyclin agonist iloprost aggravates fibrosis and enhances viral replication in enteroviral myocarditis by modulation of ERK signaling and increase of iNOS expression. Basic Res. Cardiol. 107:287. 10.1007/s00395-012-0287-z [DOI] [PubMed] [Google Scholar]
- 42.Fechner H, Sipo I, Westermann D, Pinkert S, Wang X, Suckau L, Kurreck J, Zeichhardt H, Müller O, Vetter R, Erdmann V, Tschope C, Poller W. 2008. Cardiac-targeted RNA interference mediated by an AAV9 vector improves cardiac function in coxsackievirus B3 cardiomyopathy. J. Mol. Med. (Berl.) 86:987–997. 10.1007/s00109-008-0363-x [DOI] [PubMed] [Google Scholar]
- 43.Weinert S, Bergmann N, Luo X, Erdmann B, Gotthardt M. 2006. M line-deficient titin causes cardiac lethality through impaired maturation of the sarcomere. J. Cell Biol. 173:559–570. 10.1083/jcb.200601014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Da Silva Lopes K, Pietas A, Radke MH, Gotthardt M. 2011. Titin visualization in real time reveals an unexpected level of mobility within and between sarcomeres. J. Cell Biol. 193:785–798. 10.1083/jcb.201010099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R. 1992. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc. Natl. Acad. Sci. U. S. A. 89:314–318. 10.1073/pnas.89.1.314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szalay G, Sauter M, Hald J, Weinzierl A, Kandolf R, Klingel K. 2006. Sustained nitric oxide synthesis contributes to immunopathology in ongoing myocarditis attributable to interleukin-10 disorders. Am. J. Pathol. 169:2085–2093. 10.2353/ajpath.2006.060350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lisewski U, Shi Y, Wrackmeyer U, Fischer R, Chen C, Schirdewan A, Jüttner R, Rathjen F, Poller W, Radke MH, Gotthardt M. 2008. The tight junction protein CAR regulates cardiac conduction and cell-cell communication. J. Exp. Med. 205:2369–2379. 10.1084/jem.20080897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lambert C, Léonard N, De Bolle X, Depiereux E. 2002. ESyPred3D: prediction of proteins 3D structures. Bioinformatics 18:1250–1256. 10.1093/bioinformatics/18.9.1250 [DOI] [PubMed] [Google Scholar]
- 49.Franz WM, Breves D, Klingel K, Brem G, Hofschneider PH, Kandolf R. 1993. Heart-specific targeting of firefly luciferase by the myosin light chain-2 promoter and developmental regulation in transgenic mice. Circ. Res. 73:629–638. 10.1161/01.RES.73.4.629 [DOI] [PubMed] [Google Scholar]
- 50.Schmidtke M, Selinka HC, Heim A, Jahn B, Tonew M, Kandolf R, Stelzner A, Zell R. 2000. Attachment of coxsackievirus B3 variants to various cell lines: mapping of phenotypic differences to capsid protein VP1. Virology 275:77–88. 10.1006/viro.2000.0485 [DOI] [PubMed] [Google Scholar]
- 51.Kramer B, Huber M, Kern C, Klingel K, Kandolf R, Selinka HC. 1997. Chinese hamster ovary cells are non-permissive towards infection with coxsackievirus B3 despite functional virus-receptor interactions. Virus Res. 48:149–156. 10.1016/S0168-1702(96)01438-4 [DOI] [PubMed] [Google Scholar]
- 52.Fechner H, Noutsias M, Tschoepe C, Hinze K, Wang X, Escher F, Pauschinger M, Dekkers D, Vetter R, Paul M, Lamers J, Schultheiss HP, Poller W. 2003. Induction of coxsackievirus-adenovirus-receptor expression during myocardial tissue formation and remodeling: identification of a cell-to-cell contact-dependent regulatory mechanism. Circulation 107:876–882. 10.1161/01.CIR.0000050150.27478.C5 [DOI] [PubMed] [Google Scholar]
- 53.Kashimura T, Kodama M, Hotta Y, Hosoya J, Yoshida K, Ozawa T, Watanabe R, Okura Y, Kato K, Hanawa H, Kuwano R, Aizawa Y. 2004. Spatiotemporal changes of coxsackievirus and adenovirus receptor in rat hearts during postnatal development and in cultured cardiomyocytes of neonatal rat. Virchows Arch. 444:283–292. 10.1007/s00428-003-0925-9 [DOI] [PubMed] [Google Scholar]
- 54.Caruso L, Yuen S, Smith J, Husain M, Opavsky MA. 2010. Cardiomyocyte-targeted overexpression of the coxsackie-adenovirus receptor causes a cardiomyopathy in association with beta-catenin signaling. J. Mol. Cell. Cardiol. 48:1194–1205. 10.1016/j.yjmcc.2010.01.022 [DOI] [PubMed] [Google Scholar]
- 55.Mirza M, Pang M-F, Zaini MA, Haiko P, Tammela T, Alitalo K, Philipson L, Fuxe J, Sollerbrant K. 2012. Essential role of the coxsackie- and adenovirus receptor (CAR) in development of the lymphatic system in mice. PLoS One 7:e37523. 10.1371/journal.pone.0037523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mirza M, Hreinsson J, Strand ML, Hovatta O, Soder O, Philipson L, Pettersson RF, Sollerbrant K. 2006. Coxsackievirus and adenovirus receptor (CAR) is expressed in male germ cells and forms a complex with the differentiation factor JAM-C in mouse testis. Exp. Cell Res. 312:817–830. 10.1016/j.yexcr.2005.11.030 [DOI] [PubMed] [Google Scholar]
- 57.Mirza M, Petersen C, Nordqvist K, Sollerbrant K. 2007. Coxsackievirus and adenovirus receptor is up-regulated in migratory germ cells during passage of the blood-testis barrier. Endocrinology 148:5459–5469. 10.1210/en.2007-0359 [DOI] [PubMed] [Google Scholar]
- 58.Raschperger E, Thyberg J, Pettersson S, Philipson L, Fuxe J, Pettersson RF. 2006. The coxsackie- and adenovirus receptor (CAR) is an in vivo marker for epithelial tight junctions, with a potential role in regulating permeability and tissue homeostasis. Exp. Cell Res. 312:1566–1580. 10.1016/j.yexcr.2006.01.025 [DOI] [PubMed] [Google Scholar]
- 59.Zen K, Liu Y, McCall IC, Wu T, Lee W, Babbin BA, Nusrat A, Parkos CA. 2005. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Mol. Biol. Cell 16:2694–2703. 10.1091/mbc.E05-01-0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang L, Parham NJ, Zhang F, Aasa-Chapman M, Gould EA, Zhang H. 2012. Vaccination with coxsackievirus B3 virus-like particles elicits humoral immune response and protects mice against myocarditis. Vaccine 30:2301–2308. 10.1016/j.vaccine.2012.01.061 [DOI] [PubMed] [Google Scholar]
- 61.Petrella J, Cohen CJ, Gaetz J, Bergelson JM. 2002. A zebrafish coxsackievirus and adenovirus receptor homologue interacts with coxsackie B virus and adenovirus. J. Virol. 76:10503–10506. 10.1128/JVI.76.20.10503-10506.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]