

ABSTRACT
Following systemic infection with lymphocytic choriomeningitis virus (LCMV), STAT1 knockout (KO) mice but not wild-type, STAT2 KO, IRF9 KO, or IFNAR KO mice develop lethal disease perpetrated by CD4+ T cells. IRF7 is a key transcriptional activator of type I IFN (IFN-I) during LCMV infection. Here, the role of IRF7 in the lethal host response to LCMV infection in STAT1 KO mice was examined. In contrast to STAT1 KO mice, STAT1/IRF7 double KO (DKO) mice survived LCMV infection with a reduced immune pathology in key organs, such as the liver and spleen. However, similar to STAT1 KO mice, STAT1/IRF7 DKO mice failed to control LCMV replication and spread. LCMV infection in STAT1 KO mice was associated with a significant elevation in the levels of a number of cytokines in serum, including IFN-Is, but this was largely absent in STAT1/IRF7 DKO mice, which had a modest increase in the levels of gamma interferon and CCL2 only. Since IRF7 is known to be a key transcriptional regulator of IFN-I gene expression, the possible role of IFN-I in lethal disease was examined further. STAT1/IFNAR DKO mice, in contrast to STAT1 KO mice, all survived infection with LCMV and exhibited little tissue immune pathology. Additionally, STAT1 KO mice that were deficient for either of the two IFN-I signaling molecules, STAT2 or IRF9, also survived LCMV infection. We conclude that the lethal immune-mediated disease resulting from LCMV infection in STAT1 KO mice is (i) dependent on IRF7-induced IFN-I production and (ii) driven by noncanonical IFN-I signaling via STAT2 and IRF9.
IMPORTANCE Here we report on the basis for the novel, fatal immune-mediated disease of STAT1 KO mice infected with LCMV. Our findings show that, surprisingly, the pathogenesis of this disease is dependent on IRF7-mediated type I interferon production. Moreover, our study identifies noncanonical type I interferon signaling via STAT2 and IRF9 to be essential for the type I IFN-driven fatal disease in LCMV-infected STAT1 KO mice. These results further highlight the significance of noncanonical type I IFN signaling in the pathogenesis of host-mediated injury following viral infection.
INTRODUCTION
Interferons (IFNs) are a family of cytokines that are important in host defense against viral infections, regulating both innate and adaptive antiviral immune responses (reviewed in references 1 and 2). This family consists of the type I IFNs (IFN-Is), which include alpha IFN (IFN-α) and IFN-β; the type II IFN (IFN-II) with a single member, IFN-γ; and the type III IFNs (IFN-IIIs), which consist of IFN-λs. The IFN-Is bind to a common heterodimeric cell surface receptor, termed IFNAR, that triggers the Janus kinase-mediated phosphorylation of the signal transducer and activator of transcription 1 (STAT1) and STAT2. These phosphorylated STATs form a trimolecular complex with interferon regulatory factor 9 (IRF9). This complex, termed interferon-stimulated gene factor 3 (ISGF3), binds to interferon-stimulated response elements (ISREs) to regulate the transcription of IFN-I-regulated genes. The IFN-IIIs bind to a distinct heterodimeric receptor (interleukin-28 receptor α [IL-28Rα]/IL-10Rβ) but, similar to IFN-Is, signal predominantly through the ISGF3 complex. In contrast, binding of IFN-γ to its unique receptor, IFNGR, results in the phosphorylation of STAT1 and the formation of STAT1 homodimers that recognize gamma-activated sequences (GASs) present in the promoter regions of IFN-II-regulated genes.
Interferon regulatory factor 7 (IRF7) is a member of a family of transcription factors containing nine members (IRF1 to IRF9) (reviewed in reference 3). Among the IRFs, IRF3 and IRF7 are crucial in the host response to virus infection, inducing the transcription of the IFN-I genes, including the IFN-β gene as well as a number of IFN-α genes (4–6). Host cell detection of viral infection is dependent on a variety of pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) presented by microbial proteins or nucleic acids (reviewed in references 7 and 8). Recognition of virus-derived single-stranded RNA (ssRNA) or double-stranded RNA by endosomal Toll-like receptor 3 (TLR3), TLR7, TLR8, and TLR9 or the cytoplasmic retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) melanoma differentiation-associated gene 5 (MDA5) and RIG-I results in the activation of IRF3 and/or IRF7, which then activates the production of IFN-Is. The production of IFN-Is occurs as a multiphasic response in which IFN-β and IFN-α4 are initially produced via an IRF3-dependent pathway (4, 5). These bind to the IFN-I receptor (IFNAR) to activate a subset of IFN-stimulated genes (ISGs), including the IRF7 gene. The increase in IRF7 leads to further IFN-α/β production and subsequent transcription of a large set of interferon-stimulated genes that interfere with various steps in the virus replication cycle, inhibiting infection and viral spread (reviewed in references 9 and 10). In addition to its antiviral effects, IFN-Is are also important for the differentiation and maturation of antigen-presenting cells (APCs) and the activation of T and B cells and also promote the development and survival of the CD8+ cytotoxic T cells (CTLs) (for reviews, see references 11 and 12).
Lymphocytic choriomeningitis virus (LCMV) is a bisegmented ssRNA virus belonging to the Arenaviridae and is considered an emerging human pathogen (13). Human transmission has been shown to occur in utero, upon transplantation with infected organs, or upon contact with infected laboratory and wild animals. LCMV infection of adults can lead to the development of fever, malaise, headaches, seizures, and, in some cases, fatal meningitis. LCMV infection in its natural host, the mouse, is a model widely studied for the analysis of virus-host interactions (reviewed in references 14 and 15). In adult wild-type (WT) mice, peripheral infection with LCMV Armstrong (LCMV-Arm) has little clinical impact, with CD8+ T cell-mediated clearance of the virus occurring by 10 to 12 days postinfection. In contrast, intracranial (i.c.) infection of WT mice with LCMV-Arm results in a lethal neurological disease (termed LCM) with characteristic seizures and mononuclear cell infiltrates in the meninges, ependyma, and choroid plexus. LCMV-specific CD8+ T cells produce cytokines and chemokines that mediate extravasation of myelomonocytic cells, including neutrophils and monocytes, which leads to vascular leakage and acute lethality (16). LCMV itself is a relatively noncytopathic virus, as exemplified by the fact that either peripheral or i.c. inoculation of LCMV-Arm in T cell-deficient RAG or SCID mice results in persistent infection without untoward physical effects (14, 15). It was therefore surprising to find that LCMV infection in STAT1 knockout (KO) mice was lethal, whereas similarly infected STAT2 KO, IRF9 KO, IFNGR KO, or STAT1/IFNGR double KO (DKO) mice survived (17). The lethal disease in LCMV-infected STAT1 KO mice is associated with a generalized immune pathology affecting all organs as well as excessive serum levels of a number of proinflammatory cytokines, suggesting that death is due to a destructive antiviral immune process. However, depletion of CD4+ T cells rather than CD8+ T cells protected STAT1 KO mice from lethal disease following LCMV infection.
IFN-Is have a crucial role in the control of LCMV replication and spread. Mice deficient for the IFNAR have enhanced LCMV replication and develop persistent infection following intraperitoneal (i.p.) infection (18, 19). Furthermore, IFNAR-deficient mice (19), mice lacking the IFN-I signaling molecule STAT2 or IRF9 (17), or mice treated with anti-IFN-α/β serum (20) do not develop LCM after i.c. infection with LCMV. Details of the molecular pathway(s) leading to IFN-I induction during LCMV infection have begun to emerge in recent years. Thus, the induction of IFN-Is following LCMV infection is mediated via TLR7/9 or RIG-I/MDA5 (21, 22) and is dependent on IRF7, but not IRF3 (23). While IRF3 is dispensable for the induction of IFN-I during LCMV infection (24), IRF7 is obligatorily required for the production of IFN-α but is only partially required for the production of IFN-β (24–26). Consistent with this role of IRF7, IRF7 KO mice are defective in the initial control of LCMV infection and spread but not the subsequent generation of CD8+ antiviral T cells that promote the clearance of infection (25–27).
In previous work we have shown that the expression of the IRF7 gene is highly upregulated during LCMV infection in mice (28). This IRF7 response to LCMV infection is dependent on IFN-Is, indicating that mutual stimulation of IRF7 and IFN-Is is required for effective antiviral responses. Importantly, the upregulation of IRF7 gene expression during LCMV infection was also observed in the absence of STAT1 but not in the absence of STAT2, suggesting that IRF7 may have a central role in driving the excessive IFN-I production seen in LCMV-infected STAT1 KO mice (29). The aim of this study was 2-fold. First, we sought to determine the role of IRF7 in the regulation of IFN-Is and the development of the lethal host response to LCMV infection in STAT1 KO mice. Second, we wished to elucidate the contribution of IFN-I signaling to the lethal phenotype in LCMV-infected STAT1 KO mice.
MATERIALS AND METHODS
Animals.
STAT1 KO mice (30) were originally provided by Joan Durbin, Nationwide Children's Hospital, Columbus, OH. STAT2 KO mice (31) were provided by Christian Schindler, Columbia University, New York, NY. The IRF7 KO (32) and IRF9 KO (33) mice were obtained from Riken Bioresource Center, Japan. IFNAR KO mice (18) were kindly provided by Michael Frese, Australian National University, Australian Capital Territory, Australia. STAT1 KO double gene-deficient mice with IRF7 KO, STAT2 KO, IRF9 KO, or IFNAR KO were produced by interbreeding, and all genotypes were verified by PCR analysis of tail DNA. All mice used were of the C57BL/6 background and maintained under pathogen-free conditions in the animal facility of the School of Molecular Bioscience, University of Sydney. Ethical approval for the use of all mice in this study was obtained from the University of Sydney Animal Care and Ethics Committee.
LCMV infection.
All mice were aged between 8 and 16 weeks at the time of infection. i.p. infection was performed by injecting 500 PFU of LCMV diluted in 200 μl of phosphate-buffered saline (PBS) with 1% fetal bovine serum (FBS). Sham-infected mice were used as controls and received the same volume of PBS with 1% FBS but without virus. Mice were weighed at the times indicated below, and percent weight change was calculated.
Tissue collection and routine histology.
To determine pathological changes, mice were euthanized at the times indicated below, and the organs (liver, kidney, and spleen) were removed and fixed overnight in ice-cold 4% paraformaldehyde in PBS (pH 7.4). Following paraffin embedding, tissue sections (5 μm) were prepared and stained with hematoxylin-eosin (H&E). Stained sections were examined under a DM4000B bright-field microscope (Leica, Wetzlar, Germany), and images were captured using a Spot Flex camera and Spot (v4.5) software (Diagnostic Instruments).
Multiplex assay for cytokines.
For serum levels of cytokines, mice were anesthetized at the times postinfection indicated below, and blood was collected by cardiac puncture. Blood was clotted at room temperature for 30 min before centrifugation at 1,000 × g for 10 min at 4°C. The serum was removed and stored at −80°C. To determine cytokine levels in serum, a Q-plex array kit (Quansys Biosciences, Logan, UT) for 16 cytokines was used according to the manufacturer's instruction. For each well, 50 μl of serum obtained as described above was used. The plate was scanned with an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE) and analyzed using Q-View software (Quansys Biosciences).
Analysis of serum IFN-I levels. Serum was collected as described above. Commercially available enzyme-linked immunosorbent assay (ELISA) kits were used to measure the levels of IFN-α (PBL Biomedical Laboratories, NJ) and IFN-β (MyBioSource, CA) according to the manufacturers' instructions.
RPA.
Total RNA was isolated from snap-frozen tissue using the TRI Reagent (Sigma-Aldrich, Castle Hill, Australia) according to the manufacturer's instructions. An RNase protection assay (RPA) was performed using IFN-β, IFN-γ, or LCMV nucleoprotein (NP) riboprobes as described previously (20, 34).
LCMV plaque assay.
Vero cells (6 × 105 cells/well) were plated overnight into six-well plates. Tissues were weighed and homogenized in 1 ml ice-cold PBS. Serial dilutions were made with PBS. The medium was removed from the Vero cells, and 200 μl of diluted tissue lysate was added to each well. In addition, a serial dilution from 108 to 100 PFU/ml of LCMV was used as a positive control. After 1 h of incubation at 37°C with 5% CO2, the lysate was removed and 2% carboxymethyl cellulose (Sigma-Aldrich) mixed 1:1 with 2× Dulbecco modified Eagle medium containing 10% FBS was layered on top of the cells. Cells were incubated at 37°C with 5% CO2 for 5 days and then fixed with 10% formalin in PBS for 2 h at room temperature. The cells were washed with H2O and stained with 0.1% (wt/vol) crystal violet in 20% (vol/vol) ethanol for 30 min, rinsed again with H2O, and air dried. The plaques in each well were counted, and the number of PFU per g tissue was calculated.
Statistics analysis.
Data for survival outcome were analyzed by the log-rank test. Other data were evaluated using the two-tailed Mann-Whitney U test. Results are shown as means ± standard errors of the means (SEMs). P values of <0.05 were considered significant.
RESULTS
The absence of IRF7 protects STAT1 KO mice from lethal LCMV infection.
In order to investigate the requirement for IRF7 in the development of lethal disease in STAT1 KO mice following LCMV infection, STAT1/IRF7 DKO mice were generated. STAT1/IRF7 DKO mice were healthy and fertile. Following i.p. inoculation of LCMV, WT and IRF7 KO mice developed mild clinical signs with rough fur and reduced activity from day 5 to day 10 but then recovered (Fig. 1A). WT and IRF7 KO mice did not show a significant loss in body weight (Fig. 1B). Consistent with previous findings in our lab (17), LCMV-infected STAT1 KO mice developed a lethal wasting disease and in accordance with animal welfare requirements were euthanized between days 8 and 10 (Fig. 1A). The disease in the STAT1 KO mice was accompanied by a progressive body weight loss from day 4 postinfection that reached greater than 15% weight loss at the time of euthanasia. In contrast, STAT1/IRF7 DKO mice survived (Fig. 1A) but developed some clinical signs, such as hunched back, rough fur, and reduced activity. Notably, the STAT1/IRF7 DKO mice also developed a significant weight loss from day 7 postinfection, losing up to 15% of their initial body weight (Fig. 1B). The weight loss was sustained and not recovered by day 35 postinfection. In summary, these findings indicate that IRF7 is required for lethal disease following LCMV infection in STAT1 KO mice.
FIG 1.

IRF7 deficiency protects STAT1 KO mice from the development of lethal disease and tissue destruction. WT (n = 8), IRF7 KO (n = 8), STAT1 KO (n = 10), and STAT1/IRF7 DKO (n = 8) mice were infected i.p. with 500 PFU of LCMV, as described in Materials and Methods. (A) Survival outcome. PI, postinfection. For significance (log-rank test): ***, P < 0.001 compared with WT mice. (B) Weight change. The weight of 5 mice for each genotype was monitored for the times shown. The percent weight change was calculated as described in Materials and Methods. Data are shown as means ± SEMs. For significance (Mann-Whitney U test): ^, P < 0.05 for STAT1 KO mice compared with WT mice; #, P < 0.05 for IRF7 KO mice compared with WT mice; *, P < 0.05 for STAT1/IRF7 DKO mice compared with WT mice. Representative histological sections from spleens (C to F) and livers (G to J) removed at day 7 postinfection from WT (n = 3), STAT1 KO (n = 3), IRF7 KO (n = 3), and STAT1/IRF7 DKO (n = 3) mice were processed, and sections were stained with H&E as described in Materials and Methods. Tissue sections from the livers of STAT1 KO, IRF7 KO, and STAT1/IRF7 DKO mice showed the presence of leukocytes (arrows). Magnifications: ×50 (C to F) and ×200 (G to J).
Reduced severity of tissue inflammation and destruction in STAT1 KO mice lacking IRF7.
A hallmark in the lethal wasting disease of LCMV-infected STAT1 KO mice is a significant generalized tissue pathology affecting several organs, including the liver, which is associated with leukocyte infiltration and tissue destruction (17). In these animals, there is also the gross loss of splenic architecture. To determine whether IRF7 deficiency protected STAT1 KO mice from this immune pathology, histological examination was performed on the spleens and livers of WT, STAT1 KO, IRF7 KO, and STAT1/IRF7 DKO mice (Fig. 1C to J). Uninfected mice deficient for STAT1, IRF7, or both showed no differences in tissue pathology compared with uninfected WT mice (data not shown). Similarly, at day 7 postinfection the spleens (Fig. 1C) and livers (Fig. 1G) from WT mice showed no overt pathological changes compared with the spleens and livers from uninfected mice (data not shown). Compared with the spleens of infected WT mice, the spleens of infected STAT1 KO mice showed a complete loss of the follicles and germinal centers (Fig. 1D), while the spleens of infected IRF7 KO mice (Fig. 1E) showed a relatively normal splenic architecture. Although the disruption was not as severe as that in STAT1 KO mice, the splenic organization was also disrupted in LCMV-infected STAT1/IRF7 DKO mice, in which some rudimentary follicles remained but were disrupted (Fig. 1F). In contrast, infected WT mice showed little evidence of leukocyte infiltration into the liver (data not shown), but large numbers of leukocytes were present in the livers of infected STAT1 KO mice (Fig. 1H, arrows), IRF7 KO mice (Fig. 1I, arrows), and STAT1/IRF7 DKO mice (Fig. 1J, arrows). However, the magnitude of the leukocyte infiltration was considerably less in the livers of IRF7 KO and STAT1/IRF7 DKO mice than those of STAT1 KO mice. Furthermore, the livers of infected STAT1 KO mice showed more severe tissue disruption and destruction than those of infected IRF7 and STAT1/IRF7 DKO mice. In summary, these findings show that the survival of LCMV-infected STAT1 KO mice that lacked IRF7 is associated with a reduction in the severity of tissue immune pathology and destruction.
Control of LCMV infection depends crucially on STAT1.
We next compared the role of STAT1 and IRF7 in the initial control of LCMV replication and spread. In order to monitor the levels of LCMV NP RNA in infected mice, RPA was performed on RNA extracted from the livers, kidneys, and spleens of WT, STAT1 KO, IRF7 KO, and STAT1/IRF7 DKO mice (Fig. 2A and B). LCMV NP RNA was not detected in the livers of WT mice but was present at low levels in the kidneys of these animals on day 6 postinfection. In the spleens of WT mice, the viral RNA was present at a low level on day 3 and was undetectable by day 6 postinfection (Fig. 2A and B). In IRF7 KO mice, consistent with our previous finding (25), viral RNA was detectable in the liver, kidney, and spleen on day 3 and the level increased significantly on day 6 postinfection, indicating that IRF7 is important in controlling the initial spread of LCMV (Fig. 2A and B). In contrast, compared with WT or IRF7 KO mice, LCMV NP RNA was present at markedly higher levels (2-fold higher) in the livers and kidneys of STAT1 KO and STAT1/IRF7 DKO mice on day 3 and day 6 postinfection (Fig. 2A and B). On day 3 postinfection, the level of LCMV NP RNA was 2-fold higher in the spleens of STAT1 KO and STAT1/IRF7 DKO mice than in the spleens of WT and IRF7 KO mice, but viral RNA was present at similar levels in the spleens of STAT1 KO, IRF7 KO, and STAT1/IRF7 DKO mice on day 6 postinfection. These findings indicate that in mice lacking STAT1 or both STAT1 and IRF7, there were similar defects in the ability to control viral replication and spread.
FIG 2.

The absence of IRF7 or STAT1 results in impaired control of virus replication and spread following infection with LCMV. (A) Autoradiograph showing LCMV NP RNA levels in the livers, kidneys, and spleens of WT, IRF7 KO, STAT1 KO, and STAT1/IRF7 DKO mice infected i.p. with LCMV, as described in Materials and Methods. Total RNA (10 μg) was analyzed by RPA, as described in Materials and Methods. (B) Quantification of the autoradiographs was performed by densitometry. Values were normalized to the value for the housekeeping gene L32 and are shown as means ± SEMs. For significance (Mann-Whitney U test): *, P < 0.05 compared with uninfected mice; ^, P < 0.05 compared with WT mice at the respective time points; x, P < 0.05 compared with IRF7 KO mice at the respective time points. (C) Plaque assays were performed on tissue lysates from livers and spleens removed at different times from WT, STAT1 KO, IRF7 KO, and STAT1/IRF7 KO mice following i.p. infection with 500 PFU of LCMV. Each symbol represents an individual mouse, with the mean shown as a bar. For significance (Mann-Whitney U test): *, P < 0.05 compared with uninfected mice; ^, P < 0.05 compared with WT mice at the respective time points; x, P < 0.05 compared with IRF7 KO mice at the respective time points.
Plaque assays were performed to determine if increased LCMV NP RNA levels reflected increased infectious virus titers. Infectious LCMV was not detected in the livers of WT mice but was present at a low level in the spleens of WT mice at day 6 postinfection (Fig. 2C). Compared with infected WT mice, a high virus titer was found in the livers and spleens of IRF7 KO mice at day 3 and day 6 postinfection. Furthermore, consistent with the RPA results, the viral load in the livers of STAT1 KO and STAT1/IRF7 DKO mice was several-log-fold higher than that in the livers of IRF7 KO mice (Fig. 2C). In contrast, virus levels in the spleen were similar between STAT1-, IRF7-, and STAT1/IRF7-deficient mice. In comparison with WT mice, the viral load in the spleen was 4 to 8 log units higher in mice that were deficient for STAT1, IRF7, or both. Similar to the RPA results, the plaque assay showed that there was a high level of viral burden in the livers of mice lacking STAT1 or both STAT1 and IRF7 compared with viral burden in the livers of WT and IRF7 KO mice following LCMV infection. However, the plaque assay was unable to distinguish the differences in viral burden in the spleen between these mice. Taken together, the results from the plaque assay and RPA indicated that in the liver and kidney, STAT1 is dominant over IRF7 in the antiviral response.
Reduced cytokine production in LCMV-infected STAT1/IRF7 DKO mice.
Our previous findings showed that the levels of several cytokines in serum are significantly higher in LCMV-infected STAT1 KO mice than similarly infected WT mice at day 3 postinfection (17). In order to investigate whether IRF7 was linked to the production of these cytokines, a Q-plex assay detecting 16 cytokines in parallel was used to analyze serum samples obtained from WT, STAT1 KO, IRF7 KO, or STAT1/IRF7 DKO mice at days 0 and 3 postinfection. Some cytokines (CCL5, granulocyte-macrophage colony-stimulating factor, CCL2, IL-17, IL-2, and IL-1α) were present at very low levels in uninfected WT mice (Fig. 3A), while other cytokines (CCL3, tumor necrosis factor alpha, IFN-γ, IL-12p70, IL-10, IL-6, IL-5, IL-4, IL-3, and IL-1β) were undetectable (Fig. 3A). There were no significant differences in the basal levels of cytokines between WT mice and mice that lacked STAT1, IRF7, or both (Fig. 3A to D). Following LCMV infection, the level of CCL2 increased significantly at day 3 postinfection in WT mice, whereas the levels of the other cytokines showed no significant changes (Fig. 3A), which is consistent with the findings of our previous study (17). However, in infected STAT1 KO mice (Fig. 3B), there was a significant increase in the production of not only CCL2 but also CCL5, IFN-γ, IL-6, and IL-5 compared with that in the uninfected controls and infected WT mice. Furthermore, following LCMV infection, the level of CCL2 was also increased significantly in IRF7 KO mice, whereas the levels of CCL5 and IFN-γ were increased slightly but not significantly (Fig. 3C). Similarly, the levels of these three cytokines were increased in STAT1/IRF7 DKO mice after LCMV infection, but only IFN-γ and CCL2 showed a significant increase (Fig. 3D). In contrast to infected STAT1 KO mice, in infected STAT1/IRF7 DKO mice, neither IL-6 nor IL-5 was increased significantly. In addition, while the level of IFN-γ was significantly increased in infected STAT1/IRF7 DKO mice compared with infected WT and IRF7 KO mice, it was significantly lower (4-fold) than that in infected STAT1 KO mice. Therefore, these results indicate that in LCMV-infected STAT1/IRF7 DKO mice, the systemic cytokine response was, depending on the cytokine involved, either completely or partially ablated compared with that in similarly infected STAT1 KO mice.
FIG 3.

Serum cytokine levels in LCMV-infected mice in the presence and absence of STAT1 or IRF7. A Q-plex assay was performed to analyze the levels of 16 cytokines in sera from WT (A), STAT1 KO (B), IRF7 KO (C), and STAT1/IRF7 DKO (D) mice at day 3 following i.p. infection with 500 PFU, as described in Materials and Methods. Uninfected mice of each genotype served as controls. Experiments were performed using 3 mice at each time point for each genotype and experimental condition. Values are shown as means ± SEMs. GM-CSF, granulocyte-macrophage colony-stimulating factor; TNF-α, tumor necrosis factor alpha. For significance (Mann-Whitney U test): *, P < 0.05 compared with uninfected control mice; ^, P < 0.05 compared with infected WT mice; #, P < 0.05 compared with infected STAT1 mice.
Increased production of IFN-Is and IFN-II in LCMV-infected STAT1 KO mice is dependent on IRF7.
In LCMV infection, the absence of STAT1 is associated with a higher level of IFN-I and IFN-II production than that in WT mice (29). IRF7 is essential for IFN-α production but not for IFN-β and IFN-γ production during LCMV infection, as recently reported by us (25). The expression level of the IFN-β and IFN-γ genes was determined in the liver and spleen of WT, STAT1 KO, IRF7 KO, and STAT1/IRF7 DKO mice at days 0, 3, and 6 following i.p. infection with LCMV (Fig. 4A and B). IFN-β mRNA was undetectable in the livers and spleens of uninfected WT, STAT1 KO, IRF7 KO, and STAT1/IRF7 DKO mice. Following LCMV infection in WT mice, the level of IFN-β mRNA in the liver was below the detection level of the assay at both day 3 and day 6 postinfection. In contrast, in LCMV-infected STAT1 KO mice, the level of IFN-β mRNA was increased significantly in the liver at day 3 and day 6 postinfection. In IRF7 KO mice, the level of IFN-β mRNA in the liver was increased significantly at day 6 only and the level of IFN-β mRNA was similar to that in STAT1 KO mice on day 6 postinfection. Furthermore, IFN-β mRNA was induced in the livers of infected STAT1/IRF7 DKO mice at day 3, and the level further increased at day 6. In the liver, the level of IFN-β mRNA was lower at day 3 but significantly higher at day 6 postinfection in STAT1/IRF7 KO mice than in STAT KO mice.
FIG 4.
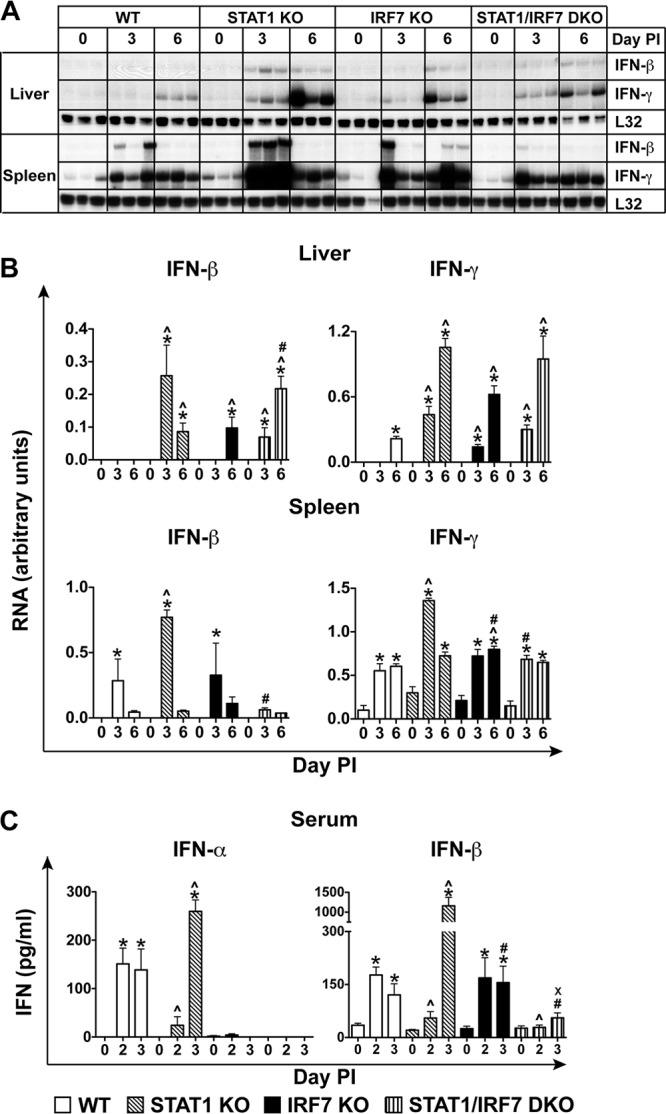
Marked increases in IFN-I and IFN-II levels in STAT1 KO mice but not STAT1/IRF7 DKO mice following LCMV infection. (A) Autoradiograph showing the expression of IFN-β and IFN-γ mRNAs in the livers and spleens of WT, IRF7 KO, STAT1 KO, and STAT1/IRF7 DKO mice infected i.p. with LCMV, as described in Materials and Methods. Total RNA (10 μg) was analyzed by RPA, as described in Materials and Methods. Experiments were performed using three mice at each time point. (B) Quantification of the autoradiographs was performed by densitometry. Values were normalized to the value for the housekeeping gene L32 and are shown as means ± SEMs. (C) IFN-α and IFN-β ELISAs were performed on sera from WT, STAT1 KO, IRF7 KO, and STAT1/IRF7 DKO mice at the times shown following i.p. infection with 500 PFU of LCMV. Experiments were performed using 3 mice at each time point. Values shown are means ± SEMs. For significance (Mann-Whitney U test): *, P < 0.05 compared with uninfected mice; ^, P < 0.05 compared with WT mice at the respective time points; #, P < 0.05 compared with STAT1 KO mice at the respective time points; x, P < 0.05 compared with IRF7 KO mice at the respective time points.
In contrast, in the spleens of LCMV-infected WT mice, the level of IFN-β mRNA was increased significantly at day 3 and deceased to a low level at day 6, whereas in the spleens of STAT1 KO mice, the expression of the IFN-β gene was increased to a significantly higher level than that in WT mice on day 3, but it was also reduced to a low level on day 6 postinfection. In the spleens of IRF7 KO mice, IFN-β mRNA levels were comparable to those observed in the spleens of WT mice and significantly lower than those in the spleens of STAT1 KO mice on day 3 postinfection. Moreover, in the spleens of STAT1/IRF7 DKO mice, IFN-β mRNA was induced at a very low level on day 3 and sustained at a similar level on day 6 postinfection. The level of IFN-β mRNA in the spleens of STAT1/IRF7 DKO mice was significantly lower than that in the spleens of infected STAT1 KO mice. It was also lower than that in the spleens of WT and IRF7 KO mice, but this difference did not reach significance. In all, these findings indicate that following infection with LCMV, expression of the IFN-β gene is exaggerated in the absence of STAT1, a process which is dependent on IRF7 in the spleen but not in the liver.
IFN-γ mRNA was undetectable in the livers of uninfected mice independently of the genotype, but it was present at a low level in the spleens of all mice (Fig. 4A and B). Following i.p. LCMV infection, the IFN-γ mRNA level increased in the livers of WT mice on day 6. In contrast, in the livers of mice deficient for STAT1, IRF7, or both, IFN-γ gene expression was induced at day 3 and was further increased on day 6 postinfection. In the spleens of mice that lacked STAT1, IRF7, or both, IFN-γ mRNA was present at a significantly higher level (2-fold higher) than that in the spleens of WT mice at these times postinfection. Furthermore, IFN-γ mRNA was significantly increased at days 3 and 6 in the spleens of WT, STAT1 KO, IRF7 KO, and STAT1/IRF7 DKO mice following LCMV infection. Of note, on day 3 postinfection, the level of IFN-γ mRNA was significantly higher (2-fold higher) in STAT1 KO mice than WT, IRF7 KO, and STAT1/IRF7 DKO mice. Overall, these findings show that the induction of IFN-γ mRNA in the liver occurs earlier in the mice that are deficient for STAT1, IRF7, or both and also that IRF7 deficiency in STAT1 KO mice is associated with a reduced level of expression of IFN-γ mRNA in the spleen but has no effect in the liver.
To determine the level of systemic IFN-Is in WT, STAT1 KO, IRF7 KO, and STAT1/IRF7 DKO mice during LCMV infection, ELISAs for determination of IFN-α and IFN-β levels of in serum were performed on days 2 and 3 postinfection (Fig. 4C). The level of IFN-α was significantly increased in WT mice on day 2 postinfection and was sustained on day 3 postinfection. In contrast, IFN-α production in STAT1 KO mice, which was only slightly increased on day 2, increased markedly on day 3 postinfection to levels that were significantly (1.5-fold) higher than those in WT mice at the equivalent time postinfection. Furthermore, similar to our previous results (25), IFN-α was not detectable in the sera of mice deficient for IRF7 at either day 2 or day 3 following LCMV infection and was also not detectable in the sera of STAT1/IRF7 DKO mice at these times postinfection.
In the sera of WT mice, IFN-β showed a profile similar to that of IFN-α, with a sustained and significant increase at day 2 and day 3 postinfection (Fig. 4C). In contrast, the level of IFN-β in the sera of STAT1 KO mice was increased only slightly on day 2 postinfection but, similar to the findings for IFN-α, was increased significantly on day 3 postinfection to levels that were significantly higher (7-fold) than those in WT mice at the equivalent time postinfection. In the sera of IRF7 KO mice, IFN-β increased significantly to levels similar to those detected in WT mice on day 2 and day 3 postinfection. Furthermore, the IFN-β level in the sera of STAT1/RF7 DKO mice was slightly increased on day 3 postinfection and was lower than that in WT mice, but the difference did not reach significance. However, in contrast to STAT1 KO and IRF7 KO mice, the level of IFN-β production in STAT1/IRF7 DKO mice was significantly lower. Overall, these findings indicate that following LCMV infection, IFN-I levels are markedly increased in STAT1 KO mice and this amplified level of IFN-I production in the absence of STAT1 is dependent on IRF7.
A lack of IFN-I signaling protects STAT1 KO mice from death after LCMV infection.
Our results showed that following infection with LCMV, STAT1 KO mice exhibited a magnified IFN-I response that was clearly dependent on IRF7. This, together with our observation that STAT1/IRF7 DKO mice did not succumb to lethal disease following LCMV infection, led us to ask whether the lethal disease induced in STAT1 KO mice following LCMV infection was mediated by IFN-I. In order to answer this question, we generated double gene-deficient mice that lacked both STAT1 and IFNAR. These mice developed only mild clinical signs and survived following i.p. infection with LCMV (Fig. 5A). LCMV-infected STAT1/IFNAR DKO mice had a progressive and significant weight loss from day 6 postinfection (Fig. 5B). However, the weight loss was much less than that in STAT1 KO mice. These findings demonstrate that STAT1-independent IFN-I signaling is required for the lethal outcome of LCMV infection in STAT1 KO mice.
FIG 5.

Lethal disease in STAT1 KO mice caused by infection with LCMV is prevented by the absence of IFNAR, STAT2, or IRF9. WT (n = 8), STAT1 KO (n = 8), IFNAR KO (n = 8), STAT1/IFNAR DKO (n = 8), STAT1/STAT2 DKO (n = 8), and STAT1/IRF9 DKO (n = 8) mice were infected i.p. with 500 PFU of LCMV, as described in Materials and Methods. (A) Survival outcome. (B) Weight change. The weight of 5 mice was monitored for the times shown. The percent weight change was calculated as described in Materials and Methods. Data are shown as means ± SEMs. For significance (Mann-Whitney U test): ^, P < 0.05 for STAT1 KO mice compared with WT mice; x, P < 0.05 for IFNAR KO mice compared with WT mice; #, P < 0.05 for STAT1/IFNAR DKO mice compared with WT mice; *, P < 0.05 for STAT1/STAT2 DKO mice compared with WT mice. Spleens (C to F) and livers (G to J) were removed from WT (n = 3), STAT1 KO (n = 3), IFNAR KO (n = 3), and STAT1/IFNAR DKO (n = 3) mice at day 7 postinfection, and sections were stained with H&E as described in Materials and Methods. Tissue sections from the livers of STAT1 KO, IFNAR KO, and STAT1/IFNAR DKO mice showed the presence of leukocytes (arrows). Magnifications: ×50 (C to F) and ×200 (G to J).
Next, we determined whether IFNAR deficiency protected STAT1 KO mice from immune pathology and tissue destruction. Histological examination of the livers and spleens of WT, STAT1 KO, IFNAR KO, and STAT1/IFNAR DKO mice infected i.p. with LCMV was performed (Fig. 5C to J). Uninfected mice deficient for STAT1, IFNAR, or both showed no overt differences in histological appearance compared with uninfected WT mice (data not shown). Following LCMV infection, the spleens of infected STAT1 KO mice (Fig. 5D) showed a dramatic loss of splenic architecture compared with the spleens of infected WT mice (Fig. 5C). However, the spleens of infected IFNAR KO mice (Fig. 5E) and STAT1/IFNAR DKO mice (Fig. 5F) had only a modest disruption of the splenic architecture, with the follicles having less well defined marginal zones but normal-appearing germinal centers. The livers of LCMV-infected WT mice showed no overt abnormalities in histological appearance (Fig. 5G). As expected, large numbers of leukocytes were visible in the livers of infected STAT1 KO mice, and the tissue was disrupted (Fig. 5H). However, in the livers of IFNAR KO mice (Fig. 5I) and STAT1/IFNAR DKO mice (Fig. 5J), the magnitude of leukocyte infiltration was much lower and destruction of the liver tissue was not apparent. Overall, these findings indicate that the lethal disease involving generalized tissue pathology and destruction in LCMV-infected STAT1 KO mice is driven by STAT1-independent IFN-I signaling.
IFN-I-driven lethal disease in LCMV-infected STAT1 KO mice is mediated by STAT2- and IRF9-dependent pathways.
The findings in STAT1/IFNAR KO mice raised the question, what is the nature of the noncanonical IFN-I signaling pathways that mediate lethality in LCMV-infected STAT1 KO mice? In particular, we speculated that STAT2 and IRF9 are perhaps involved in this process. To tackle this issue, additional double gene-deficient mice that lacked STAT1 and either STAT2 or IRF9 were generated. Following i.p. LCMV infection, both STAT1/STAT2 DKO and STAT1/IRF9 DKO mice showed mild clinical signs, but all infected animals survived (Fig. 5A). Additionally, STAT1/STAT2 DKO mice showed significant weight loss from day 6 postinfection (Fig. 5B). The weight loss was not recovered by day 18 postinfection but was considerably less than that of STAT1 KO mice. The weight loss in STAT1/IRF9 DKO mice during LCMV infection was not investigated. In all, these findings demonstrate that in the absence of STAT1, IFN-Is signal through STAT2 and IRF9 following LCMV infection.
DISCUSSION
STAT1 is a crucial signaling molecule for both IFN-I and IFN-II signaling (reviewed in references 35 and 36). We have recently shown that following infection with LCMV, STAT1 KO mice develop an atypical lethal disease which is mediated by CD4+ T cells (17). This lethal disease is not replicated by the loss of signaling by the other canonical IFN-I signaling factors (STAT2 and IRF9), by deficient IFN-γ signaling, or by the combined loss of IFN-I and IFN-γ signaling. The absence of STAT1 during LCMV infection in mice is associated with markedly elevated levels of various cytokines in serum, including IFN-Is and IFN-γ (17, 29). While the development of the lethal disease following LCMV infection in STAT1 KO mice does not require IFN-γ (17), the biological significance of IFN-I signaling in this process was not known but should be considered, since there is now abundant evidence that IFN-I can mediate many biological functions independently of STAT1 (37). IRF7 is a key transcriptional regulator of IFN-I production during LCMV infection (24–27) that, interestingly, is significantly induced in a STAT1-independent fashion in the spleen and central nervous system during LCMV infection (28). Due to these considerations, here we initially studied the role of IRF7 in the lethal host response induced by LCMV in STAT1 KO mice by generating STAT1/IRF7 DKO mice. We showed that IRF7 deficiency in STAT1 KO mice prevented the lethality due to LCMV infection and was associated with the systemic loss of detectable IFN-α and markedly decreased IFN-β. Together with our findings here that STAT1/IFNAR DKO, STAT1/STAT2 DKO, and STAT1/IRF9 DKO mice all survived LCMV infection, this study shows that the lethal immune-mediated disease due to LCMV infection in STAT1 KO mice is dependent on IRF7 and is driven by STAT1-independent IFN-I signaling via STAT2 and IRF9.
The protection against lethal disease afforded by IRF7 deficiency in LCMV-infected STAT1 KO mice correlated well with a general reduction in tissue pathology and the reduced production of a variety of cytokines and chemokines compared with the tissue pathology and the levels of cytokines and chemokines in LCMV-infected STAT1 KO mice. As noted above, LCMV is a noncytopathic virus, which is reflected by the absence of overt tissue injury and disease in immunodeficient mice, even though these animals have very high tissue virus loads (14, 15). Therefore, the reduced tissue damage seen in the STAT1/IRF7 KO mice is related to the overall dampening of the immune response to LCMV infection. However, these mice still developed some clinical signs, such as weight loss, hunched back, and reduced activity, following LCMV infection. Furthermore, these mice showed an inability similar to that of STAT1 KO mice to control LCMV replication and spread, indicating that the antiviral mechanisms that control LCMV in IRF7 KO mice are primarily mediated via STAT1-dependent signaling.
Consistent with our previous findings (17), here we demonstrated that LCMV infection in STAT1 KO mice resulted in high levels of CCL2, IL-5, IL-6, IFN-Is, and IFN-γ in serum, indicative of a cytokine storm. The marked attenuation of this cytokine storm response in the STAT1/IRF7 DKO mice raised the possibility that these cytokines and chemokines, individually or in combination, might play a role in mediating lethal disease in STAT1 KO mice. As noted above, our recent studies showed that IFN-γ is not a crucial mediator in the development of LCMV-induced lethal disease in STAT1 KO mice. However, the involvement of the other cytokines and chemokines whose levels were elevated in the lethal disease in LCMV-infected STAT1 KO mice remains to be determined. Notably, with the exception of IFN-I, IRF7 is not known to regulate the production of the other cytokines and chemokines that were elevated in LCMV-infected STAT1 KO mice. Therefore, the decreased cytokine and chemokine production in LCMV-infected STAT1/IRF7 DKO mice was most likely due to the reduced immune response in these animals rather than the loss of IRF7 per se.
As noted above, IRF7 is a key transcription factor that regulates optimal IFN-I gene expression during LCMV infection. However, while IRF7 is essential for systemic IFN-α production, it is not required for systemic IFN-β production, indicating that the regulation of the IFN-β gene clearly differs from that of the IFN-α genes in LCMV infection (25). In highlighting this point, it is known that for a subset of Toll-like receptors (TLRs), including TLR7 and TLR9, IRF7 associates with MyD88 to induce IFN-α but not IFN-β (38, 39). Nevertheless, the lack of IRF7 in STAT1 KO mice led to a loss of not only systemic IFN-α but also IFN-β, indicating that in the absence of STAT1, IRF7 is required for regulating both IFN-α and IFN-β production following LCMV infection. The mechanism of how the combination of STAT1 and IRF7 regulate IFN-β gene expression during LCMV infection is unknown. It has previously been reported that hematopoietic cells are the principal source of systemic IFN-I production during LCMV infection (17, 27). The significant reduction in IFN-β gene expression in the spleens of LCMV-infected STAT1/IRF7 DKO mice observed here also suggested that a defect in IFN-β production by hematopoietic cells might be the basis for the absence of systemic IFN-β. However, in contrast to the spleen, our results indicated that significant upregulation of the IFN-β gene was maintained in the livers of LCMV-infected STAT1 KO mice, despite the absence of IRF7, suggesting that there are major cell type-specific differences in how the IFN-β gene is regulated in response to LCMV infection. Such differences may reflect the nature of the pattern recognition receptors used by different cells to sense LCMV infection and the subsequent utilization of downstream signaling pathways that ultimately mediate transcriptional activation of the IFN-β gene.
On the basis of the apparent association between IRF7 deficiency, reduced IFN-I levels, and survival of STAT1 KO mice following LCMV infection, we surmised that the underlying driver of the fatal disease in STAT1 KO mice might be STAT1-independent signaling by the IFN-Is. In order to investigate this, STAT1/IFNAR DKO mice that lacked all IFN-I signaling were generated. The results showed unequivocally that these mice were protected against lethal disease after LCMV infection and had less weight loss than STAT1 KO mice and little evidence of tissue immune pathology in comparison with that in STAT1 KO mice. Thus, these findings indicate that the lethal disease in the LCMV-infected STAT1 KO mice is due directly to STAT1-independent IFN-I signaling. We noted that following LCMV infection, STAT1/IRF7 DKO mice exhibited a more severe disease phenotype than STAT1/IFNAR DKO mice. The STAT1/IRF7 DKO mice also had elevated IFN-γ levels, and although these were less than the levels in STAT1 KO mice, the result suggests that there was a low-level antiviral immune response in the STAT1/IRF7 DKO mice. Therefore, we propose that the heightened disease phenotype of STAT1/IRF7 DKO mice compared with that in STAT1/IFNAR KO mice is due to a low level of STAT1-independent IFN-I signaling mediated by residual IFN-β production in these mice. Irrespective of this, our findings demonstrate that STAT1-independent IFN-I signaling is the central driver of the severe tissue pathology and the consequent lethal host response in STAT1 KO mice following LCMV infection.
Our findings point to a noncanonical, STAT1-independent IFN-I signaling pathway(s) as being central in the pathogenesis of the lethal disease in STAT1 KO mice after LCMV infection. The results here also showed that, similar to STAT1/IFNAR DKO mice, STAT1/STAT2 DKO and STAT1/IRF9 DKO mice survived LCMV infection, indicating that the lethal disease mediated by LCMV in STAT1 KO mice is dependent on STAT2 and IRF9. In the context of viral infection, STAT1-independent, STAT2-dependent IFN-I signaling has been reported by a number of studies. For example, IFN-Is can suppress dendritic cell maturation in a STAT1-independent but STAT2-dependent manner, contributing to viral persistence in LCMV CL13 infection (40). Furthermore, STAT2 mediates protective antiviral responses in the absence of STAT1 during cytopathic virus infections, such as vesicular stomatitis virus (VSV) (31) or dengue virus (41) infection, since STAT1/STAT2 DKO mice show a much greater increase in their susceptibility to these infections than mice lacking either gene alone. In contrast to STAT2, STAT1-independent, IRF9-dependent IFN-I signaling has not been reported in the context of antiviral responses, but the findings of our studies presented here suggest that IRF9 is also crucial for the STAT1-independent, STAT2-dependent IFN-I signaling.
STAT2 has a transactivation domain, but it binds DNA poorly (42, 43), while IRF9 appears to be critical for DNA binding and the sequence recognition specificity of the ISGF3 complex (42, 44). STAT2 has been shown to interact with IRF9 in vivo and in vitro and after IFN-α treatment (42, 45–47). Although some studies reported that STAT2-IRF9 complexes are not stable when interacting with DNA (42), other studies have illustrated that STAT2-IRF9 complexes are transcriptionally functional. For example, in HEK293 Tet-On cells that express IRF9 proteins fused with STAT2 transcriptional activation domains, these hybrid proteins are able to increase the expression of ISGs and also exhibit antiviral effects against VSV and herpes simplex virus (45). In addition, recent studies demonstrated that STAT2-IRF9 complexes bind to the ISRE region in the promoter of the RIG-g gene and regulate its expression in STAT1-deficient U3A cells (46). Finally, STAT2 forms homodimers or heterodimers with STAT6 in response to IFN-α (48), and these dimers are capable of binding to IRF9. In all, a role for STAT2 and IRF9 in mediating noncanonical IFN-I signaling in the absence of STAT1 is now clearly strongly supported by the results of our studies presented here, which demonstrate that each of these factors is obligatorily required for the development of lethal disease in STAT1 KO mice following LCMV infection.
In conclusion, the findings from this study indicate that the lethal immune-mediated disease resulting from LCMV infection in STAT1 KO mice is (i) dependent on IRF7-induced IFN-I production and (ii) driven by noncanonical IFN-I signaling via STAT2 and IRF9. Further experiments are required to identify the exact composition of the STAT2-IRF9-containing complexes involved in the STAT1-independent IFN-I signaling pathways. Also, it will be of interest to investigate the mechanism by which STAT1-independent IFN-I signaling contributes to the genesis of the antiviral CD4+ T cell response in STAT1 KO mice. Overall, the findings from this study further highlight the significance of noncanonical IFN-I signaling in the host response to viral infection.
ACKNOWLEDGMENTS
This work was supported in part by a project grant (512407) to I.L.C. from the National Health and Medical Research Council of Australia. W.L. and S.R.J. were the recipients of Australian postgraduate scholarships.
We thank Laura Parker for technical assistance.
Footnotes
Published ahead of print 23 April 2014
REFERENCES
- 1.Le Bon A, Tough DF. 2002. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 14:432–436. 10.1016/S0952-7915(02)00354-0 [DOI] [PubMed] [Google Scholar]
- 2.Pestka S, Krause CD, Walter MR. 2004. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202:8–32. 10.1111/j.0105-2896.2004.00204.x [DOI] [PubMed] [Google Scholar]
- 3.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. 2001. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19:623–655. 10.1146/annurev.immunol.19.1.623 [DOI] [PubMed] [Google Scholar]
- 4.Marie I, Durbin JE, Levy DE. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 17:6660–6669. 10.1093/emboj/17.22.6660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. 1998. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 441:106–110. 10.1016/S0014-5793(98)01514-2 [DOI] [PubMed] [Google Scholar]
- 6.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539–548. 10.1016/S1074-7613(00)00053-4 [DOI] [PubMed] [Google Scholar]
- 7.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137. 10.1038/ni1303 [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi O, Akira S. 2007. Recognition of viruses by innate immunity. Immunol. Rev. 220:214–224. 10.1111/j.1600-065X.2007.00562.x [DOI] [PubMed] [Google Scholar]
- 9.Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8:559–568. 10.1038/nri2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samuel CE. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14:778–809. 10.1128/CMR.14.4.778-809.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alsharifi M, Mullbacher A, Regner M. 2008. Interferon type I responses in primary and secondary infections. Immunol. Cell Biol. 86:239–245. 10.1038/sj.icb.7100159 [DOI] [PubMed] [Google Scholar]
- 12.Seo YJ, Hahm B. 2010. Type I interferon modulates the battle of host immune system against viruses. Adv. Appl. Microbiol. 73:83–101. 10.1016/S0065-2164(10)73004-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tyler KL. 2009. Emerging viral infections of the central nervous system: part 1. Arch. Neurol. 66:939–948. 10.1001/archneurol.2009.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buchmeier MJ, Welsh RM, Dutko FJ, Oldstone MB. 1980. The virology and immunobiology of lymphocytic choriomeningitis virus infection. Adv. Immunol. 30:275–331. 10.1016/S0065-2776(08)60197-2 [DOI] [PubMed] [Google Scholar]
- 15.Kang SS, McGavern DB. 2008. Lymphocytic choriomeningitis infection of the central nervous system. Front. Biosci. 13:4529–4543. 10.2741/3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JV, Kang SS, Dustin ML, McGavern DB. 2009. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 457:191–195. 10.1038/nature07591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofer MJ, Li W, Manders P, Terry R, Lim SL, King NJ, Campbell IL. 2012. Mice deficient in STAT1 but not STAT2 or IRF9 develop a lethal CD4+ T-cell-mediated disease following infection with lymphocytic choriomeningitis virus. J. Virol. 86:6932–6946. 10.1128/JVI.07147-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller U, Steinhoff U, Reis LFL, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. 1994. Functional role of type I and type II interferons in antiviral defense. Science 264:1918–1921. 10.1126/science.8009221 [DOI] [PubMed] [Google Scholar]
- 19.Ou R, Zhou S, Huang L, Moskophidis D. 2001. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal expansion. J. Virol. 75:8407–8423. 10.1128/JVI.75.18.8407-8423.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandberg K, Kemper P, Stalder A, Zhang J, Hobbs MV, Whitton JL, Campbell IL. 1994. Altered tissue distribution of viral replication and T-cell spreading is pivotal in the protection against fatal lymphocytic choriomeningitis in mice after neutralization of IFN-α/β. J. Immunol. 153:220–231 [PubMed] [Google Scholar]
- 21.Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. 2004. Does Toll-like receptor 3 play a biological role in virus infections? Virology 322:231–238. 10.1016/j.virol.2004.01.033 [DOI] [PubMed] [Google Scholar]
- 22.Jung A, Kato H, Kumagai Y, Kumar H, Kawai T, Takeuchi O, Akira S. 2008. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J. Virol. 82:196–206. 10.1128/JVI.01640-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou S, Cerny AM, Zacharia A, Fitzgerald KA, Kurt-Jones EA, Finberg RW. 2010. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 84:9452–9462. 10.1128/JVI.00155-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christensen JE, Fenger C, Issazadeh-Navikas S, Krug A, Liljestrom P, Goriely S, Paludan SR, Finsen B, Christensen JP, Thomsen AR. 2012. Differential impact of interferon regulatory factor 7 in initiation of the type I interferon response in the lymphocytic choriomeningitis virus-infected central nervous system versus the periphery. J. Virol. 86:7384–7392. 10.1128/JVI.07090-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li W, Hofer MJ, Noçon AL, Manders P, Campbell IL. 2013. Interferon regulatory factor 7 (IRF7) is required for the optimal initial control but not subsequent clearance of lymphocytic choriomeningitis virus infection in mice. Virology 439:152–162. 10.1016/j.virol.2013.02.015 [DOI] [PubMed] [Google Scholar]
- 26.Zhou S, Cerny AM, Fitzgerald KA, Kurt-Jones EA, Finberg RW. 2012. Role of interferon regulatory factor 7 in T cell responses during acute lymphocytic choriomeningitis virus infection. J. Virol. 86:11254–11265. 10.1128/JVI.00576-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lang PA, Cervantes-Barragan L, Verschoor A, Navarini AA, Recher M, Pellegrini M, Flatz L, Bergthaler A, Honda K, Ludewig B, Ohashi PS, Lang KS. 2009. Hematopoietic cell-derived interferon controls viral replication and virus-induced disease. Blood 113:1045–1052. 10.1182/blood-2007-10-117861 [DOI] [PubMed] [Google Scholar]
- 28.Ousman SS, Wang J, Campbell IL. 2005. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J. Virol. 79:7514–7527. 10.1128/JVI.79.12.7514-7527.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyagi T, Gil MP, Wang X, Louten J, Chu WM, Biron CA. 2007. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 204:2383–2396. 10.1084/jem.20070401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Durbin JE, Hackenmiller R, Simon MC, Levy DE. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84:443–450. 10.1016/S0092-8674(00)81289-1 [DOI] [PubMed] [Google Scholar]
- 31.Park C, Li S, Cha E, Schindler C. 2000. Immune response in Stat2 knockout mice. Immunity 13:795–804. 10.1016/S1074-7613(00)00077-7 [DOI] [PubMed] [Google Scholar]
- 32.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777. 10.1038/nature03464 [DOI] [PubMed] [Google Scholar]
- 33.Kimura T, Kadokawa Y, Harada H, Matsumoto M, Sato M, Kashiwazaki Y, Tarutani M, Tan RS, Takasugi T, Matsuyama T, Mak TW, Noguchi S, Taniguchi T. 1996. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells 1:115–124. 10.1046/j.1365-2443.1996.08008.x [DOI] [PubMed] [Google Scholar]
- 34.Asensio VC, Kincaid C, Campbell IL. 1999. Chemokines and the inflammatory response to viral infection in the central nervous system with a focus on lymphocytic choriomeningitis virus. J. Neurovirol. 5:65–75. 10.3109/13550289909029747 [DOI] [PubMed] [Google Scholar]
- 35.Goodbourn S, Didcock L, Randall RE. 2000. Interferons: cell signalling, immune modulation, antiviral responses and virus countermeasures. J. Gen. Virol. 81:2341–2364. 10.1099/vir.0.17157-0 [DOI] [PubMed] [Google Scholar]
- 36.Stark GR, Kerr IM, Williams BRG, Silverman RH, Schreiber RD. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227–264. 10.1146/annurev.biochem.67.1.227 [DOI] [PubMed] [Google Scholar]
- 37.van Boxel-Dezaire AH, Rani MR, Stark GR. 2006. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 25:361–372. 10.1016/j.immuni.2006.08.014 [DOI] [PubMed] [Google Scholar]
- 38.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434:1035–1040. 10.1038/nature03547 [DOI] [PubMed] [Google Scholar]
- 39.Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, Takeuchi O, Akira S. 2004. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 5:1061–1068. 10.1038/ni1118 [DOI] [PubMed] [Google Scholar]
- 40.Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. 2005. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity 22:247–257. 10.1016/j.immuni.2005.01.005 [DOI] [PubMed] [Google Scholar]
- 41.Perry ST, Buck MD, Lada SM, Schindler C, Shresta S. 2011. STAT2 mediates innate immunity to dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog. 7:e1001297. 10.1371/journal.ppat.1001297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bluyssen HA, Levy DE. 1997. Stat2 is a transcriptional activator that requires sequence-specific contacts provided by stat1 and p48 for stable interaction with DNA. J. Biol. Chem. 272:4600–4605 [DOI] [PubMed] [Google Scholar]
- 43.Ghislain JJ, Fish EN. 1996. Application of genomic DNA affinity chromatography identifies multiple interferon-alpha-regulated Stat2 complexes. J. Biol. Chem. 271:12408–12413 [DOI] [PubMed] [Google Scholar]
- 44.Bluyssen HA, Muzaffar R, Vlieststra RJ, Van Der Made ACJ, Leung S, Stark GR, Kerr IM, Trapman J, Levy DE. 1995. Combinatorial association and abundance of components of interferon-stimulated gene factor 3 dictate the selectivity of interferon responses. Proc. Natl. Acad. Sci. U. S. A. 92:5645–5649. 10.1073/pnas.92.12.5645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kraus TA, Lau JF, Parisien JP, Horvath CM. 2003. A hybrid IRF9-STAT2 protein recapitulates interferon-stimulated gene expression and antiviral response. J. Biol. Chem. 278:13033–13038. 10.1074/jbc.M212972200 [DOI] [PubMed] [Google Scholar]
- 46.Lou YJ, Pan XR, Jia PM, Li D, Xiao S, Zhang ZL, Chen SJ, Chen Z, Tong JH. 2009. IRF-9/STAT2 functional interaction drives retinoic acid-induced gene G expression independently of STAT1. Cancer Res. 69:3673–3680. 10.1158/0008-5472.CAN-08-4922 [DOI] [PubMed] [Google Scholar]
- 47.Martinez-Moczygemba M, Gutch MJ, French DL, Reich NC. 1997. Distinct STAT structure promotes interaction of STAT2 with the p48 subunit of the interferon-alpha-stimulated transcription factor ISGF3. J. Biol. Chem. 272:20070–20076 [DOI] [PubMed] [Google Scholar]
- 48.Gupta S, Jiang M, Pernis AB. 1999. IFN-alpha activates Stat6 and leads to the formation of Stat2:Stat6 complexes in B cells. J. Immunol. 163:3834–3841 [PubMed] [Google Scholar]