

ABSTRACT
The herpes simplex virus type 1 (HSV-1) UL20 gene encodes a 222-amino-acid nonglycosylated envelope protein which forms a complex with viral glycoprotein K (gK) that functions in virion envelopment, egress, and virus-induced cell fusion. To investigate the role of the carboxyl terminus of the UL20 protein (UL20p) in cytoplasmic virion envelopment, a cadre of mutant viruses was constructed and characterized. The deletion of six amino acids from the carboxyl terminus of UL20p caused an approximately 1-log reduction in infectious virus production compared to that of the wild-type virus. Surprisingly, a phenylalanine-to-alanine replacement at amino acid position 210 caused a gain-of-function phenotype, increasing infectious virus production up to 1 log more than in the wild-type virus. In contrast, the replacement of two membrane-proximal phenylalanines with alanines caused drastic inhibition of infectious virion production and cytoplasmic virion envelopment. Prediction of the membrane topology of UL20p revealed that these two amino acid changes cause retraction of the carboxyl terminus of UL20p from the intracellular space. Confocal microscopy revealed that none of the engineered UL20 mutations affected intracellular transport of UL20p to trans-Golgi network membranes. In addition, a proximity ligation assay showed that none of the UL20 mutations affected UL20p colocalization and potential interactions with the UL37 protein recently found to interact with the gK/UL20 protein complex. Collectively, these studies show that phenylalanine residues within the carboxyl terminus of UL20p are involved in the regulation of cytoplasmic virion envelopment and infectious virus production.
IMPORTANCE We have shown previously that the UL20/gK protein complex serves crucial roles in cytoplasmic virion envelopment and that it interacts with the UL37 tegument protein to facilitate cytoplasmic virion envelopment. In this study, we investigated the role of phenylalanine residues within the carboxyl terminus of UL20p, since aromatic and hydrophobic amino acids are known to be involved in protein-protein interactions through stacking of their aromatic structures. Characterization of mutant viruses carrying phenylalanine (Phe)-to-alanine (Ala) mutations revealed that the two membrane-proximal Phe residues were critical for the proper UL20p membrane topology and efficient virion envelopment and infectious virus production. Surprisingly, a Phe-to-Ala change located approximately in the middle of the UL20p carboxyl terminus substantially enhanced cytoplasmic envelopment and overall production of infectious virions. This work revealed that Phe residues within the UL20p carboxyl terminus are involved in the regulation of cytoplasmic virion envelopment and infectious virus production.
INTRODUCTION
Herpes simplex virus type 1 (HSV-1) is a neurotropic virus that, after replication in epithelial cells, enters into ganglionic neurons via distal axons and establishes latent infection (1–3). HSV-1 is the primary infectious cause of blindness in the western hemisphere, primarily because periodic reactivation causes recurrent keratitis and scarring of the cornea (4, 5). Herpesvirus particles consist of a linear double-stranded DNA genome containing an icosahedral capsid, surrounded by a proteinaceous tegument and, outermost, a cell-derived lipid envelope. The virus encodes at least 12 glycosylated and several nonglycosylated proteins expressed on the viral envelope and in infected cell membranes that are important for the life cycle of the virus, especially during virus entry, intracellular virion morphogenesis, and cell-to-cell spread. HSV-1 virion morphogenesis and egress involves complex mechanisms, which are characterized by sequential morphogenetic steps, including (i) primary envelopment of capsids at the inner nuclear membrane, (ii) de-envelopment of enveloped capsids at the outer nuclear membrane, (iii) acquisition of tegument proteins in the cytoplasm of infected cells, and (iv) re-envelopment of tegumented capsids at the trans-Golgi network (TGN)-derived membranes (6–8). Cytoplasmic virion envelopment is facilitated by multiple interactions among viral glycoproteins embedded in TGN membranes and tegument proteins (8–12).
The UL20 gene encodes a 222-amino-acid nonglycosylated envelope protein, which plays a crucial role in the cytoplasmic virion envelopment and egress of HSV-1. The UL20 gene is highly conserved among alphaherpesviruses, including varicella-zoster virus (13), pseudorabies virus (PRV) (14), and the gammaherpesvirus Marek's disease virus type 2 (15). The UL20 protein (UL20p) has been shown to be important for infectious virion production for both PRV and HSV-1 (16, 17). The UL20 protein forms a functional complex with glycoprotein K (gK), which physically binds with gB and gH to modulate virus-induced cell fusion (18).
Previous studies in our laboratory showed that the deletion of 18 amino acids from the carboxyl terminus of UL20p (UL20 mutant virus 204t) inhibited the intracellular transport and cell surface expression of both gK and UL20p and, also, failed to complement infectious virus production. However, the deletion of either 6 (UL20 216t) or 11 amino acids (UL20 211t) allowed efficient intracellular transport and cell surface expression of both gK and UL20p but failed to complement for infectious virus production (19). Phenylalanine residues are aromatic and hydrophobic amino acids that are known to be involved in protein-protein interactions and have been shown to play an important role in respiratory syncytial virus assembly (20). Therefore, we sought to determine the potential role of phenylalanine residues in the carboxyl terminus of UL20p in cytoplasmic virion envelopment and egress, under the hypothesis that cytoplasmic domains of UL20p may interact with tegument proteins to facilitate cytoplasmic virion envelopment. The UL37 protein is a 120-kDa phosphorylated protein (21) that assumes a predominantly diffuse cytoplasmic distribution in infected cells (22–24) and can be transported to TGN membranes as a complex with the UL36 protein in the absence of capsid formation (25). The UL37 tegument protein specified by HSV-1 and PRV has been shown to play an important role in cytoplasmic virion envelopment, since deletion of the UL37 gene resulted in the accumulation of capsids in the cytoplasm of infected cells (26–28). Recently, we showed that the HSV-1 UL37 protein interacts with gK and UL20p to facilitate cytoplasmic virion envelopment (29).
Here, we show that a UL20 phenylalanine residue at position 210 plays a critical regulatory function in cytoplasmic virion envelopment and infectious virus production, as evidenced by the surprisingly high infectious virus production of the mutant virus containing a Phe-to-Ala substitution at amino acid residue 210, which was 1 log higher than that of wild-type virus. Moreover, two other phenylalanine residues proximal to the membrane of the carboxyl terminus of UL20p are required for proper UL20p membrane topography and function. These phenylalanine-to-alanine mutations did not appreciably alter the colocalization and potential interactions with the UL37 protein, suggesting that they do not play major roles in the overall interactions between the UL37 protein and the gK/UL20 protein complex.
MATERIALS AND METHODS
Cells.
African green monkey kidney (Vero) cells were obtained from the American Type Culture Collection (Rockville, MD), and the Vero-based UL20 protein-complementing FRT cells (Flp-In-CV-1 cells derived from the FLP recombination target [FRT] system) (30), were maintained in Dulbecco's modified Eagle's medium (Gibco-BRL; Grand Island, NY) supplemented with 10% fetal calf serum and antibiotics.
Construction of HSV-1 mutant viruses.
Mutagenesis was accomplished in Escherichia coli cells using the markerless two-step Red recombination mutagenesis system and synthetic oligonucleotides (31, 32) implemented on the bacterial artificial chromosome (BAC) plasmid pYEbac102-VC1 carrying the HSV-1(F) genome with gK and UL20 proteins tagged with V5 and 3× FLAG antigenic epitopes, respectively. Unless otherwise specified, the wild-type virus used in all experiments is the VC-1 strain. HSV-1 mutants UL20ΔPhe and UL20Δ6 were constructed by adding a TAA stop codon in front of the last amino acid or the last 6 amino acids, respectively, from the carboxyl end of UL20p. Mutants UL20F222A, UL20F210A, and UL20F205-206A were created by changing codons for phenylalanine (aromatic amino acid) to alanine (neutral amino acid) at the positions indicated in the names (Table 1). HSV-1 BAC DNAs were purified from 50 ml of overnight bacterial cultures with a Qiagen large-construct kit (Qiagen; Valencia, CA). Using PCR test primers designed to lie outside the target mutation site(s), all mutated DNA regions were sequenced to verify the presence of the desired mutations in BACs. Similarly, viruses recovered from cells transfected with BACs (31) were sequenced to confirm the presence of the desired mutations. The entire genomes of the parental wild-type virus and the UL20F210A mutant viruses were sequenced using the Ion Torrent next-generation sequencing equipment (Life Technologies-Invitrogen; Carlsbad, CA), as we described previously (33). Briefly, total genomic DNA (gDNA) was extracted from the virus-infected Vero cells using the PureLink genomic DNA minikit (Life Technologies-Invitrogen; Carlsbad, CA). High-quality fragment libraries of each virus were prepared from the extracted total gDNAs using the Ion Xpress plus fragment library kit (Life Technologies-Invitrogen; Carlsbad, CA). The fragment libraries were subsequently applied to Ion 316 chips and were sequenced on the Ion Personal Genome Machine system (Life Technologies-Invitrogen; Carlsbad, CA).
TABLE 1.
Names of mutants and amino acid sequences in the wild type and mutants
| Mutanta | Amino acid sequence in: |
|
|---|---|---|
| Wild type | Mutantb | |
| UL20ΔPhe | NAPVAF | NAPVA* |
| UL20Δ6 | RAILNA | RAIL* |
| UL20F222A | NAPVAF | NAPVAA |
| UL20F210A | FFLARF | FFLARA |
| UL20F205-206A | FFLARF | AALARF |
Mutations are described in the text.
An asterisk indicates a stop codon.
SDS-PAGE and Western immunoblot assay.
Western immunoblot analysis was carried out essentially as described previously (34). Confluent monolayers of Vero cells were infected with the indicated viruses at a multiplicity of infection (MOI)/cell of 2. At 24 h postinfection (h.p.i.), cells were washed with PBS and lysed on ice for 30 min in NP-40 lysis buffer (Life Technologies) supplemented with a cocktail of protease inhibitors (Roche, LLC, Basel, Switzerland). The collected samples were mixed with SDS-PAGE sample buffer (NuPAGE) at a 3:1 ratio and were electrophoretically separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (4-to-20% gradient Tris-HEPES-SDS gels; Thermo Scientific). Following electrophoresis, the proteins were transferred onto a nitrocellulose membrane under a constant current. The membrane was blocked in phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST) plus 5% nonfat milk for 1 h at room temperature and was probed with primary mouse anti-FLAG monoclonal antibodies (Abcam; Cambridge, England) at 4°C overnight. Goat anti-mouse secondary antibody conjugated with horseradish peroxidase (HRP) and Immobilon chemiluminescent HRP substrate (Millipore) were used for detection purposes.
Plaque morphologies, replication kinetics, and electron microscopy.
For plaque morphology experiments, confluent monolayers of Vero or FRT cells were infected with the viruses at an MOI/cell of 0.001 and fixed with methanol 48 h.p.i. Immunohistochemistry was performed with primary rabbit anti-HSV antibodies (1:1,000) (Dako) and goat anti-rabbit HRP-labeled antibody (1:1,000) (Dako), and the reactions were developed using the NovaRed substrate (Vector Laboratories). Images were taken with an inverted light microscope (Olympus) using relief contrast. Growth kinetics studies of viruses were performed as we described previously (35, 36). Approximately 90% confluent Vero cells were infected with each virus at 4°C for 1 h at a low or high MOI/cell of 0.2 or 2, respectively. Afterwards, plates were incubated at 37°C for 1 h to allow virus penetration. Any remaining extracellular viruses were inactivated by low-pH treatment using PBS at pH 3, plates were incubated, and infected cells were collected at different time points and stored at 80°C. Viral titers were obtained on FRT cells. The ultrastructural morphology of virions within infected cells was examined by transmission electron microscopy as described previously (18, 31, 35, 37). All flasks with infected cells were incubated at 37°C and 5% CO2 until being processed at 18 h.p.i., and infected cell samples were visualized by transmission electron microscopy.
Quantification of cell-to-cell fusion.
A chemiluminescence-based assay system was used to quantify cellular fusion during viral infection (38–40). Around 80 to 90% confluent Vero cells in 12-well plates were transfected separately with plasmids expressing the T7 polymerase gene or the luciferase gene under the T7 promoter at amounts of 1 μg/well each. Lipofectamine 2000 (Invitrogen) was used for all transfections. The medium was changed 6 h posttransfection, and after 12 h of incubation at 37°C and 5% CO2, cells were detached using trypsin-EDTA. Both populations of cells were washed, further resuspended in complete medium, and mixed in a 1:1 ratio prior to seeding in a 24-well plate. Fifteen hours after seeding, the cells were infected at an MOI/cell of 0.5. At 24 h.p.i., the cells were washed with PBS and lysed with lysis buffer and supernatants were collected. The supernatant was mixed with luciferase assay substrate, and the relative amounts of fusion were obtained by measuring the relative levels of luminescence (relative light units [RLU]) emitted using a TD-20/20 luminometer (Turner Designs).
Quantitative PCR.
Quantitative PCR (qPCR) was utilized to derive the total number of viral genomes within the lysate. Specifically, the primers and probe (6-carboxytetramethylrhodamine [TAMRA]) for the real-time PCR were designed to detect HSV-1 US6 (gD). Lysates were collected at 18 h.p.i., and 200 μl of each suspension was used for the extraction of viral DNA. Viral DNA was extracted using a DNeasy blood and tissue kit (Qiagen, Inc.) according to the manufacturer's instructions. Equal volumes of viral DNA were used for TaqMan PCR analysis. Purified HSV-1 bacterial artificial chromosome (YE102) DNA was used to generate the standard curve.
Confocal microscopy.
Vero cell monolayers grown on an 8-well glass Lab-Tek II chamber slide were infected with the viruses at an MOI/cell of 10. Uninfected cells were used as negative controls. Cells were washed once with PBS and fixed with ice-cold 100% methanol for 10 min at 18 h.p.i. Monolayers were subsequently blocked for 1 h with 2% fetal calf serum in PBS. Further staining was done using mouse monoclonal anti-FLAG IgG1 (Sigma-Aldrich, St. Louis, MO) for recognition of UL20p and TGOLN2 IgG2a (Abnova, Taiwan) for recognition of trans-Golgi network, diluted 1:200 in PBS blocking buffer, followed by Alexa Fluor 555-conjugated goat anti-mouse IgG1 and Alexa Fluor 488-conjugated goat anti-mouse IgG2a secondary antibodies. The nucleus was stained using 4′,6-diamidino-2-phenylindole (DAPI). Specific immunofluorescence was examined using an Olympus FluoView confocal microscope. Image analysis and subcellular scatter plot graphs were generated and analyzed using the Olympus Fluoview FV1000 confocal microscope software interface. Scatterplot graphs with colocalization of two fluorophores within the TGN were generated by specifically defining the regions of interest with a bounding box of equal area based on pixel enumeration for each captured image. To determine an approximate percentage of subcellular colocalization, pixel enumeration and intensity statistics within the Olympus software package were accumulated for each sample image. The percentage of upper-right-quadrant pixels from the total pixels per sample was displayed. Finally, the images were compiled and rendered with Adobe Photoshop.
PLA.
The in situ proximity ligation assay (PLA) for proximity ligation reaction detection was carried out using the appropriate Duolink II In Situ kit components obtained from Sigma-Aldrich, St. Louis, MO. Vero cell monolayers grown on an 8-well glass Lab-Tek II chamber slide were infected with the viruses at an MOI/cell of 10. At 18 h.p.i., the cells were fixed by using methanol for 10 min and blocked using Duolink blocking solution at 37°C in a wet chamber for 2 h. The primary antibodies used were anti-FLAG mouse antibody (Sigma-Aldrich, St. Louis, MO) and anti-UL37 rabbit antibody for the negative controls (mock infection and untagged wild-type F strain) and for the parental wild type, as well as the mutants. The primary antibodies used for the positive control (gK-UL20p interaction) were anti-V5 mouse antibody (Invitrogen Life Technologies) and anti-FLAG rabbit antibody (Sigma-Aldrich, St. Louis, MO). Antibodies were diluted in blocking buffer and incubated in a wet chamber at 4°C overnight. The slides were washed two times with 1× TBS plus 0.05% Tween 20 for 5 min each, and then secondary antibodies (Duolink In Situ PLA probe anti-rabbit plus and Duolink In Situ PLA probe anti-mouse minus) were added and incubated at 37°C for 1 h. Two washes with Duolink In Situ wash buffer A for 5 min each were then followed by the addition of the ligation mixture and incubation in a wet chamber at 37°C for 30 min. After ligation, the slides were washed twice for 2 min using Duolink In Situ wash buffer A before adding amplification reaction mixture for 90 min at 37°C. Texas red-labeled oligonucleotide detection probes (Olink Bioscience) were used. Subsequently, the slides were washed twice with Duolink In Situ wash buffer B and once with 0.1× Duolink In Situ wash buffer B. The slides were mounted with Duolink In Situ mounting medium with DAPI and analyzed in an Olympus FluoView confocal microscope.
RESULTS
Construction and molecular analysis of recombinant viruses.
We have shown previously that deletion of the UL20p carboxyl-terminal six amino acids results in drastic inhibition of cytoplasmic virion envelopment and egress (19). To further investigate the potential role of phenylalanine residues within the carboxyl terminus of the UL20 protein, we constructed a cadre of mutant viruses in which one or more phenylalanine residues were replaced with alanine residues. All mutant viruses were constructed using the markerless two-step Red recombination mutagenesis system implemented on the bacterial artificial chromosome (BAC) plasmid pYEbac102-VC1 carrying the HSV-1(F) genome (see Materials and Methods). This virus expresses gK and UL20p tagged with V5 and 3× FLAG antigenic epitopes, respectively (33, 41). The recombinant mutants included viruses having a deletion of the terminal phenylalanine residue (UL20ΔPhe), deletion of six amino acids from the carboxyl terminus (UL20Δ6), and replacement of one or two internal phenylalanine residues with alanines (UL20F222A, UL20F210A, and UL20F205-206A) (Fig. 1 and Table 1). The deletion mutants were made by the insertion of stop codon TAA in front of the last amino acid for UL20ΔPhe and before the last 6 amino acids for UL20Δ6. Similarly, for the amino acid replacement mutants, codons for phenylalanine (TTT or TTC) were replaced with the codon for alanine (GCA). Virus stocks were prepared after BAC transfection on FRT cells. Mutant viral genomes were characterized using PCR-assisted DNA sequencing, as well as whole-genome sequencing (see Materials and Methods).
FIG 1.

Predicted membrane topology of UL20p and locations of mutations. The amino acid position(s) of the amino acid(s) replacements are indicated, flanked on the left by the targeted phenylalanine(s) and on the right by the changed alanine(s), e.g., UL20F222A. TM, transmembrane region.
Characterization of the ability of UL20 mutant viruses to cause virus-induced cell fusion.
The plaque morphologies of all mutant and parental wild-type viruses were examined on Vero cells (Fig. 2a) and FRT cells (Fig. 2b). As expected, the HSV-1(VC-1) wild-type virus produced the largest virus plaques, while the UL20F205-206A and UL20Δ6 viruses produced smaller viral plaques than the parental wild-type virus on Vero cells. The recombinant viruses UL20ΔPhe and UL20F222A produced syncytial plaques on Vero cells (Fig. 2a). All mutants produced wild-type-like plaques on FRT cells (Vero cells transformed to express the UL20 gene) (Fig. 2b). The extent of virus-induced cell fusion was evaluated using a previously described luciferase assay (see Materials and Methods) (33). The gBΔ28syn mutant HSV-1 virus, which carries a 28-amino-acid deletion of the carboxyl terminus of gB, causes extensive virus-induced cell fusion (34) and was used as a positive control. The relative levels of luminescence were measured for all samples at 24 h h.p.i. Among the mutant viruses, the UL20F222A virus produced the highest extent of fusion, exhibiting RFU levels similar to those of the positive-control gBΔ28syn virus (Fig. 3).
FIG 2.
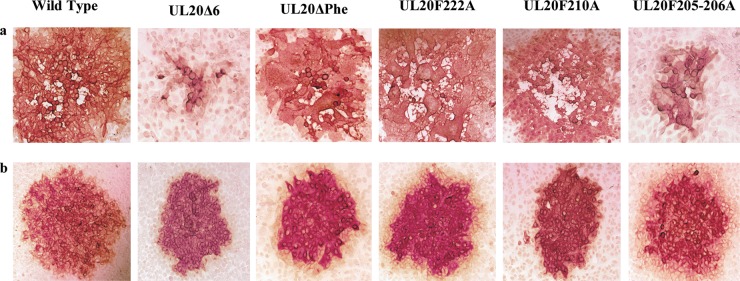
Plaque morphologies of mutant and parental wild-type viruses on Vero (a) or FRT cells (b). Confluent monolayers of Vero cells and FRT cells were infected with an MOI/cell of 0.001 of wild-type and mutant viruses, and viral plaques were detected by immunohistochemistry using polyclonal anti-HSV rabbit antisera at 48 h.p.i., as described in Materials and Methods.
FIG 3.
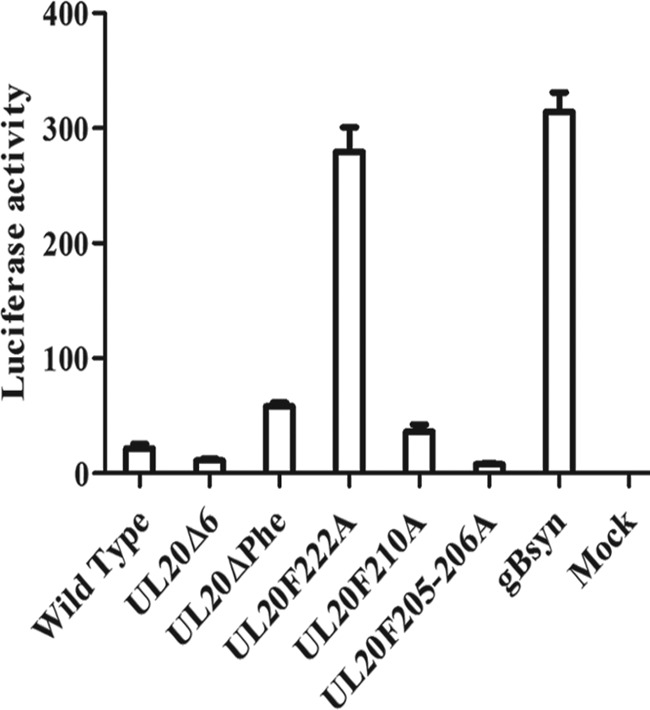
The fusion activity of each virus was quantified by luciferase-based assay (see Materials and Methods). The extent of fusion was assessed at 24 h.p.i. for the wild-type virus and all mutant viruses. Relative luciferase activity readouts are shown, and error bars indicate the standard errors. gBsyn, gBΔ28syn virus.
UL20 protein expression and replication kinetics of mutant viruses.
Western immunoblot analysis revealed that all viruses expressed similar levels of the UL20 protein (Fig. 4). To investigate the role of these mutations in the kinetics of viral replication, replication curves were produced for infections at both a low (MOI/cell of 0.2) (Fig. 5a) and a high (MOI/cell of 2) (Fig. 5b) MOI/cell. At both the high and the low MOI/cell, the UL20F205-206A mutant yielded the lowest end titer and the UL20F210A mutant yielded the highest end titer compared to the end titer of the parental wild-type virus. Importantly, the UL20F210A virus reproducibly produced viral titers approximately 1 log higher than that of the parental wild-type virus at the high MOI. At the low MOI/cell of 0.2, the terminal viral titers of UL20Δ6 and UL20F205-206A were 1 and 2 logs less than the parental wild-type terminal titer, respectively, while the UL20F210A mutant virus produced significantly higher viral titers than the parental wild-type virus. At the MOI/cell of 2, the UL20F205-206A mutant virus titer was more than 1 log less than the parental wild-type virus titer.
FIG 4.

Western immunoblots of UL20 protein (FLAG epitope) in lysates from cells infected with the indicated viruses. Confluent monolayers of Vero cells were infected with the viruses at an MOI/cell of 2. At 24 h.p.i., cell lysates were collected, run on SDS-PAGE gels, and probed with anti-FLAG antibody. Lanes: 1, Vero cell lysate; 2, parental wild-type lysate; 3, UL20Δ6 lysate; 4, UL20ΔPhe lysate; 5, UL20F222A lysate; 6, UL20F210A lysate; 7, UL20F205-206A lysate. Molecular weights (in thousands) are shown at the left.
FIG 5.

Replication kinetics of the parental wild-type and mutant viruses. Confluent Vero cell monolayers were infected with each virus at either a low MOI/cell (0.2) (a) or a high MOI/cell (2.0) (b), and viral titers were obtained by plaque assay on FRT cells at different time points postinfection. HPI, h postinfection; error bars represent standard errors.
Ultrastructural characterization of parental wild-type and mutant viruses.
The ultrastructural phenotypes of all recombinant viruses were compared to that of the wild-type parental virus after infection of Vero cells at an MOI/cell of 3 at 18 h.p.i. (Fig. 6). As expected, the parental wild-type virus did not exhibit any defects in cytoplasmic virion envelopment and egress, as fully enveloped viruses were readily seen outside the infected cells via electron microscopy. In contrast, mutant viruses exhibited various degrees of cytoplasmic defects in virion envelopment, as evidenced by the presence of numerous unenveloped or partially enveloped capsids in the cytoplasm of infected cells (Fig. 6).
FIG 6.

Ultrastructural morphologies of parental wild-type and mutant viruses. Electron micrographs of Vero cells infected with different viruses at an MOI/cell of 3 and processed for electron microscopy at 18 h.p.i. are shown. The extracellular space (e) and cytoplasm (c) are marked. Arrows show viral particles with envelopment defect, magnified in insets.
Characterization of the relative efficiencies of cytoplasmic virion envelopment.
A measure of the relative efficiencies of infectious virion production is indicated by the ratio of total viral genomes produced in infected cells to the number of infectious virions determined by the standard plaque assay. In this regard, The UL20F205-206A mutation caused the most severe defect in the relative efficiency of infectious virion production, followed by the UL20Δ6 mutation (Table 2).
TABLE 2.
Determination of HSV-1 genome-to-PFU ratios
| Virus | Total no. of genomes/PFUa |
|---|---|
| Wild type | 48,000 |
| UL20Δ6 | 538,000 |
| UL20ΔPhe | 170,000 |
| UL20F222A | 166,000 |
| UL20F210A | 38,000 |
| UL20F205-206A | 5,648,000 |
The total number of viral genomes in infected Vero cells at 24 h.p.i. was obtained by qPCR. The total number of intracellular infectious virions was obtained by plaque assay on FRT cells. Ratios reflecting relative efficiency of envelopment and infectious virion production were obtained by dividing the average of the number of viral genomes within the infected cells by the number of PFU.
Intracellular localization of the UL20 mutant proteins and UL20 interactions with the UL37 protein.
The intracellular location of parental wild-type and mutant UL20 proteins was investigated using confocal microscopy. All mutant viruses expressed UL20p that colocalized with the TGN marker, which was monoclonal antibody against trans-Golgi network protein 2 (TGOLN2). These observations were further supported by determination of the approximate percentage of subcellular colocalization using pixel enumeration and intensity statistics as detailed in Materials and Methods (Fig. 7). Recently, we showed that the UL37 protein physically interacted with the gK/UL20 protein complex to facilitate cytoplasmic virion envelopment. To investigate whether the engineered UL20 mutations affected interactions between UL20p and UL37 proteins, we utilized the proximity ligation assay (42, 43) in conjunction with the anti-FLAG monoclonal antibody detecting the UL20-tagged protein and the anti-UL37 polyclonal antibody. The known interaction between the UL20 protein and gK was also tested using the anti-FLAG (UL20) and anti-V5 (gK) antibodies, as a positive PLA control. PLA produced vivid fluorescent signals showing efficient colocalization between the interacting gK and UL20p, as well as between UL20p and UL37, in Vero cells infected with the parental wild-type virus and with all mutant viruses. In contrast, mock-infected cells or cells infected with the HSV-1(F) strain (expresses untagged gK and UL20 proteins) did not exhibit any appreciable fluorescent signals (Fig. 8).
FIG 7.
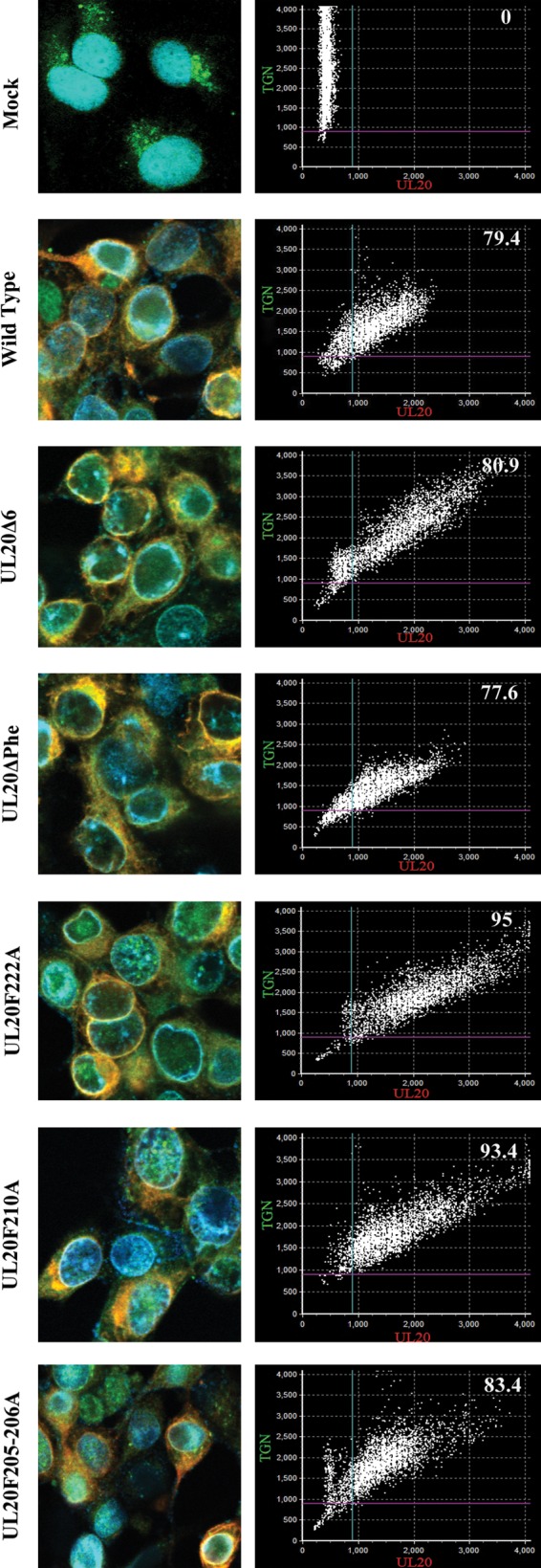
Colocalization of UL20p and TGN in the wild-type virus and mutant viruses. Vero cells were infected with each virus at an MOI/cell of 10, fixed at 18 h.p.i., and stained with anti-FLAG antibody for UL20p (red), TGOLN2 antibody for TGN (green), and DAPI for nucleus (blue). Scatterplot graphs, which depict pixel intensity and distribution and an approximate percentage of subcellular colocalization, are shown for each image.
FIG 8.

Proximity ligation assay to determine UL20p and UL37 interactions. Vero cells were infected with viruses at a high MOI/cell of 10, and PLA was performed at 18 h.p.i. (a to i) Negative-control mock infection (a), negative control with HSV-1(F) strain infection (b), positive control showing gK-UL20p interaction (c), wild type (d), UL20Δ6 (e), UL20ΔPhe (f), UL20F222A (g), UL20F210A (h), and UL20F205-206A (i). DAPI-stained nuclei are shown in blue, and PLA-positive signals generated by the Texas Red dye are shown in red.
DISCUSSION
We have previously shown that the gK/UL20 protein complex serves crucial roles in cytoplasmic virion envelopment (11). Our laboratory and others have shown that the UL37 protein localizes to the TGN membranes and functions in cytoplasmic virion envelopment (44). Recently, we showed that the UL37 protein interacts with the gK/UL20 protein complex and facilitates cytoplasmic virion envelopment and infectious virus production (29). Here, we show that phenylalanine residues located at the carboxyl terminus of the UL20 protein are involved in the regulation of cytoplasmic virion envelopment and infectious virus production.
We have previously reported the use of extensive alanine-scanning mutagenesis of the UL20 protein to delineate domains of the UL20 protein that function in cytoplasmic virion envelopment and virus-induced cell fusion (30). Specifically, we showed that the amino terminus of the UL20 protein functioned in virus-induced cell fusion, while the carboxyl terminus was important for cytoplasmic virion envelopment (30). Of specific interest to the current study was that deletion of the terminal six amino acids of the UL20 protein drastically inhibited cytoplasmic virion envelopment, while it did not affect UL20 protein interactions with gK and virus-induced cell fusion (30). Based on our recent findings that the gK/UL20 protein complex interacts with the UL37 protein, we hypothesized that this interaction may occur between the carboxyl terminus of the UL20 protein and the UL37 protein. These interactions may involve phenylalanine residues, which are known to facilitate protein-protein interactions through residue stacking and hydrophobic interactions. Significantly, the UL20F210A mutant caused more than 1-log-higher replication kinetics and final infectious virus production than the parental wild-type virus on Vero cells at a high MOI/cell. Moreover, the ratio of total viral genomes to PFU was lower than that of the parental wild-type virus, indicating that this mutation increases the efficiency of infectious virus production. The observed increase in the production of infectious virions reflects enhanced cytoplasmic virion envelopment, since it is unlikely that UL20 mutations would affect other aspects of virion assembly, such as capsid assembly in the nucleus. Moreover, the UL20F210A mutant virus produced higher infectious virus titers at a high versus a low MOI/cell compared to the parental wild-type virus, suggesting that enhancement in infectious virus production is dependent on the level of expression of the UL20F210A protein.
The UL20F210A gain-of-function mutation reveals that the phenylalanine residue at position 210 is involved in the regulation of cytoplasmic virion envelopment and infectious virus production. The purpose of this inhibitory mechanism is not immediately apparent. Infectious virus titers and particle-to-PFU ratios are affected by temperature, with optimum virus titers and particle-to-PFU ratios produced at 34°C, presumably because the entire process of virion assembly is more efficient at a reduced assembly speed (K. G. Kousoulas, unpublished observations). Thus, it is possible that the process of cytoplasmic virion envelopment is downregulated by UL20/gK to ensure efficient virion envelopment at 37°C or higher. Alternatively, the replacement of the phenylalanine at position 210 may cause more efficient interaction of UL20p with other viral proteins or cellular proteins involved in cytoplasmic virion envelopment.
The UL20F222A mutant virus caused extensive virus-induced cell fusion on Vero cells, which was completely inhibited on the UL20-complementing FRT cells. This result suggests that the phenylalanine residue at amino acid position 222 is involved in the regulation of membrane fusion, presumably because it alters the interactions of the gK/UL20 protein complex with the fusogenic glycoprotein gB. All mutant viruses produced similar plaques with respect to size and fusogenic characteristics on FRT cells, indicating that wild-type UL20p expression by FRT cells efficiently reverted all mutant phenotypes to that of the parental wild type. Overall, these results suggest that the wild-type UL20p expressed by the FRT cells acts in a dominant manner to reverse the effects of these UL20 mutations, presumably because it interacts more efficiently with gK, gB, and UL37. The reversion of the phenotypic and growth properties of the UL20 mutant viruses indicates the absence of any other spurious mutations within the mutant viral genomes. This conclusion is further supported by PCR-assisted sequencing of targeted genomic areas and sequencing of the parental wild-type and the UL20F201A genomes by next-generation sequencing methodologies, which did not detect any spurious mutations.
Phenylalanine replacement or deletion of carboxyl-terminal regions of the UL20 protein that contained phenylalanine residues caused drastic defects in virion envelopment without affecting colocalization of the UL20 mutant proteins with the UL37 protein, as evidenced by similar PLA-produced fluorescent signals. Of particular interest is the mutagenesis of two membrane-proximal phenylalanine residues to alanines. This double amino acid replacement was predicted to cause retraction of the carboxyl terminus of the UL20 protein so that it would no longer be exposed to intracellular spaces (Fig. 9). The results suggest that the carboxyl terminus of the UL20 protein is not a major contributor to overall interactions with the UL37 protein. Alternatively, mutagenesis of the carboxyl terminus of the UL20 protein may negatively affect other functional aspects of the overall interactions of the gK/UL20 complex, as well as its potential domain-specific affinity to UL37 or other tegument proteins, causing inhibition or enhancement of cytoplasmic virion envelopment and infectious virus production.
FIG 9.
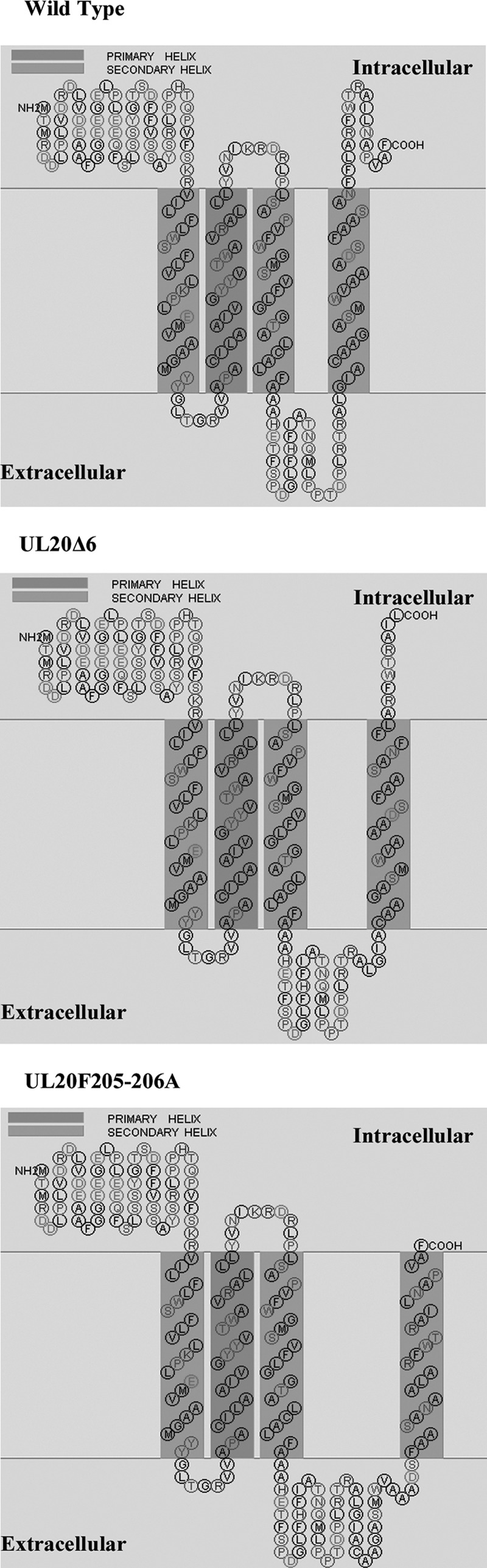
SOSUI program-predicted structures of the wild-type virus and UL20Δ6 and UL20F205-206A mutants. The protein sequences of the viruses were submitted to the SOSUI program, and the predicted structures are shown.
Cytoplasmic virion envelopment has been thought to involve a series of complex interactions between viral glycoproteins embedded in the TGN membranes and tegument proteins found on cytoplasmic capsids or bound to TGN membranes. The results presented here show that cytoplasmic virion envelopment is a dynamic process where interactions can affect cytoplasmic virion envelopment in either a positive or negative manner. These dynamic interactions may provide an advantage for virus replication in certain cells, where the virus may need to control the rate of infectious virus production. The UL20F210A gain-of-function mutation can be incorporated into other mutant herpesviruses engineered for anticancer or gene therapy purposes to enhance their beneficial functions or to alleviate their defects in replication efficiencies and produce higher titers of virus. Further studies are required to explore this gain-of-function behavior of the mutant virus and to investigate whether the mutation has any effect on the pathogenesis of the virus in experimental animal infections.
ACKNOWLEDGMENTS
This work was partially supported by NIH-NIAID grant AI43000 to K.G.K., a Louisiana Board of Regents Governor's Biotechnology Initiative grant to K.G.K., and BioMMED Core Laboratory support by NIH-NIGMS COBRE grant P20GM103458. Also, we acknowledge financial support by the LSU School of Veterinary Medicine to BioMMED.
We thank Yulia Sokolova for technical assistance with electron microscopy and all BioMMED staff for advice and critical reviews of the manuscript.
Footnotes
Published ahead of print 23 April 2014
REFERENCES
- 1.Whitley RJ, Roizman B. 2001. Herpes simplex virus infections. Lancet 357:1513–1518. 10.1016/S0140-6736(00)04638-9 [DOI] [PubMed] [Google Scholar]
- 2.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 6:211–221. 10.1038/nrmicro1794 [DOI] [PubMed] [Google Scholar]
- 3.Bloom DC, Giordani NV, Kwiatkowski DL. 2010. Epigenetic regulation of latent HSV-1 gene expression. Biochim. Biophys. Acta 1799:246–256. 10.1016/j.bbagrm.2009.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liesegang TJ, Melton LJ, III, Daly PJ, Ilstrup DM. 1989. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch. Ophthalmol. 107:1155–1159. 10.1001/archopht.1989.01070020221029 [DOI] [PubMed] [Google Scholar]
- 5.Young RC, Hodge DO, Liesegang TJ, Baratz KH. 2010. Incidence, recurrence, and outcomes of herpes simplex virus eye disease in Olmsted County, Minnesota, 1976-2007: the effect of oral antiviral prophylaxis. Arch. Ophthalmol. 128:1178–1183. 10.1001/archophthalmol.2010.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mettenleiter TC. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537–1547. 10.1128/JVI.76.4.1537-1547.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farnsworth A, Johnson DC. 2006. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J. Virol. 80:3167–3179. 10.1128/JVI.80.7.3167-3179.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211. 10.1128/JVI.02681-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campadelli-Fiume G, Farabegoli F, Di Gaeta S, Roizman B. 1991. Origin of unenveloped capsids in the cytoplasm of cells infected with herpes simplex virus 1. J. Virol. 65:1589–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turcotte S, Letellier J, Lippe R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 79:8847–8860. 10.1128/JVI.79.14.8847-8860.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chouljenko DV, Kim IJ, Chouljenko VN, Subramanian R, Walker JD, Kousoulas KG. 2012. Functional hierarchy of herpes simplex virus 1 viral glycoproteins in cytoplasmic virion envelopment and egress. J. Virol. 86:4262–4270. 10.1128/JVI.06766-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234. 10.1016/j.virusres.2009.03.018 [DOI] [PubMed] [Google Scholar]
- 13.Davison AJ, Scott JE. 1986. The complete DNA sequence of varicella-zoster virus. J. Gen. Virol. 67(Pt 9):1759–1816. 10.1099/0022-1317-67-9-1759 [DOI] [PubMed] [Google Scholar]
- 14.Klupp BG, Kern H, Mettenleiter TC. 1992. The virulence-determining genomic BamHI fragment 4 of pseudorabies virus contains genes corresponding to the UL15 (partial), UL18, UL19, UL20, and UL21 genes of herpes simplex virus and a putative origin of replication. Virology 191:900–908. 10.1016/0042-6822(92)90265-Q [DOI] [PubMed] [Google Scholar]
- 15.Hatama S, Jang HK, Izumiya Y, Cai JS, Tsushima Y, Kato K, Miyazawa T, Kai C, Takahashi E, Mikami T. 1999. Identification and DNA sequence analysis of the Marek's disease virus serotype 2 genes homologous to the herpes simplex virus type 1 UL20 and UL21. J. Vet. Med. Sci. 61:587–593. 10.1292/jvms.61.587 [DOI] [PubMed] [Google Scholar]
- 16.Baines JD, Ward PL, Campadelli-Fiume G, Roizman B. 1991. The UL20 gene of herpes simplex virus 1 encodes a function necessary for viral egress. J. Virol. 65:6414–6424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. 1997. The UL20 gene product of pseudorabies virus functions in virus egress. J. Virol. 71:5639–5646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foster TP, Melancon JM, Baines JD, Kousoulas KG. 2004. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 78:5347–5357. 10.1128/JVI.78.10.5347-5357.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melancon JM, Fulmer PA, Kousoulas KG. 2007. The herpes simplex virus UL20 protein functions in glycoprotein K (gK) intracellular transport and virus-induced cell fusion are independent of UL20 functions in cytoplasmic virion envelopment. Virol. J. 4:120. 10.1186/1743-422X-4-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaikh FY, Cox RG, Lifland AW, Hotard AL, Williams JV, Moore ML, Santangelo PJ, Crowe JE., Jr 2012. A critical phenylalanine residue in the respiratory syncytial virus fusion protein cytoplasmic tail mediates assembly of internal viral proteins into viral filaments and particles. mBio 3:e00270–11. 10.1128/mBio.00270-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albright AG, Jenkins FJ. 1993. The herpes simplex virus UL37 protein is phosphorylated in infected cells. J. Virol. 67:4842–4847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLauchlan J. 1997. The abundance of the herpes simplex virus type 1 UL37 tegument protein in virus particles is closely controlled. J. Gen. Virol. 78(Pt 1):189–194 [DOI] [PubMed] [Google Scholar]
- 23.Schmitz JB, Albright AG, Kinchington PR, Jenkins FJ. 1995. The UL37 protein of herpes simplex virus type 1 is associated with the tegument of purified virions. Virology 206:1055–1065. 10.1006/viro.1995.1028 [DOI] [PubMed] [Google Scholar]
- 24.McLauchlan J, Liefkens K, Stow ND. 1994. The herpes simplex virus type 1 UL37 gene product is a component of virus particles. J. Gen. Virol. 75(Pt 8):2047–2052. 10.1099/0022-1317-75-8-2047 [DOI] [PubMed] [Google Scholar]
- 25.Desai P, Sexton GL, Huang E, Person S. 2008. Localization of herpes simplex virus type 1 UL37 in the Golgi complex requires UL36 but not capsid structures. J. Virol. 82:11354–11361. 10.1128/JVI.00956-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desai P, Sexton GL, McCaffery JM, Person S. 2001. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 75:10259–10271. 10.1128/JVI.75.21.10259-10271.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leege T, Granzow H, Fuchs W, Klupp BG, Mettenleiter TC. 2009. Phenotypic similarities and differences between UL37-deleted pseudorabies virus and herpes simplex virus type 1. J. Gen. Virol. 90:1560–1568. 10.1099/vir.0.010322-0 [DOI] [PubMed] [Google Scholar]
- 28.Klupp BG, Granzow H, Mundt E, Mettenleiter TC. 2001. Pseudorabies virus UL37 gene product is involved in secondary envelopment. J. Virol. 75:8927–8936. 10.1128/JVI.75.19.8927-8936.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jambunathan N, Chouljenko DV, Desai P, Charles A-S, Subramanian R, Chouljenko VN, Kousoulas KG. 5 March 2014. The herpes simplex virus type-1 UL37 protein interacts with viral glycoprotein gK and the membrane protein UL20 and functions in cytoplasmic virion envelopment. J. Virol. 10.1128/JVI.00278-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melancon JM, Foster TP, Kousoulas KG. 2004. Genetic analysis of the herpes simplex virus type 1 UL20 protein domains involved in cytoplasmic virion envelopment and virus-induced cell fusion. J. Virol. 78:7329–7343. 10.1128/JVI.78.14.7329-7343.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee HC, Chouljenko VN, Chouljenko DV, Boudreaux MJ, Kousoulas KG. 2009. The herpes simplex virus type 1 glycoprotein D (gD) cytoplasmic terminus and full-length gE are not essential and do not function in a redundant manner for cytoplasmic virion envelopment and egress. J. Virol. 83:6115–6124. 10.1128/JVI.00128-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 33.Kim IJ, Chouljenko VN, Walker JD, Kousoulas KG. 2013. Herpes simplex virus 1 glycoprotein M and the membrane-associated protein UL11 are required for virus-induced cell fusion and efficient virus entry. J. Virol. 87:8029–8037. 10.1128/JVI.01181-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chouljenko VN, Iyer AV, Chowdhury S, Chouljenko DV, Kousoulas KG. 2009. The amino terminus of herpes simplex virus type 1 glycoprotein K (gK) modulates gB-mediated virus-induced cell fusion and virion egress. J. Virol. 83:12301–12313. 10.1128/JVI.01329-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foster TP, Kousoulas KG. 1999. Genetic analysis of the role of herpes simplex virus type 1 glycoprotein K in infectious virus production and egress. J. Virol. 73:8457–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foster TP, Melancon JM, Kousoulas KG. 2001. An alpha-helical domain within the carboxyl terminus of herpes simplex virus type 1 (HSV-1) glycoprotein B (gB) is associated with cell fusion and resistance to heparin inhibition of cell fusion. Virology 287:18–29. 10.1006/viro.2001.1004 [DOI] [PubMed] [Google Scholar]
- 37.Melancon JM, Luna RE, Foster TP, Kousoulas KG. 2005. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 79:299–313. 10.1128/JVI.79.1.299-313.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McShane MP, Longnecker R. 2005. Analysis of fusion using a virus-free cell fusion assay. Methods Mol. Biol. 292:187–196. 10.1385/1-59259-848-X:187 [DOI] [PubMed] [Google Scholar]
- 39.Okuma K, Nakamura M, Nakano S, Niho Y, Matsuura Y. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235–244. 10.1006/viro.1998.9530 [DOI] [PubMed] [Google Scholar]
- 40.Pogue-Geile KL, Lee GT, Shapira SK, Spear PG. 1984. Fine mapping of mutations in the fusion-inducing MP strain of herpes simplex virus type 1. Virology 136:100–109. 10.1016/0042-6822(84)90251-4 [DOI] [PubMed] [Google Scholar]
- 41.Jambunathan N, Chowdhury S, Subramanian R, Chouljenko VN, Walker JD, Kousoulas KG. 2011. Site-specific proteolytic cleavage of the amino terminus of herpes simplex virus glycoprotein K on virion particles inhibits virus entry. J. Virol. 85:12910–12918. 10.1128/JVI.06268-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jarvius M, Paulsson J, Weibrecht I, Leuchowius KJ, Andersson AC, Wahlby C, Gullberg M, Botling J, Sjoblom T, Markova B, Ostman A, Landegren U, Soderberg O. 2007. In situ detection of phosphorylated platelet-derived growth factor receptor beta using a generalized proximity ligation method. Mol. Cell. Proteomics 6:1500–1509. 10.1074/mcp.M700166-MCP200 [DOI] [PubMed] [Google Scholar]
- 43.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. 2006. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3:995–1000. 10.1038/nmeth947 [DOI] [PubMed] [Google Scholar]
- 44.Pasdeloup D, Beilstein F, Roberts AP, McElwee M, McNab D, Rixon FJ. 2010. Inner tegument protein pUL37 of herpes simplex virus type 1 is involved in directing capsids to the trans-Golgi network for envelopment. J. Gen. Virol. 91:2145–2151. 10.1099/vir.0.022053-0 [DOI] [PMC free article] [PubMed] [Google Scholar]