

ABSTRACT
Broadly targeted cellular immune responses are thought to be important for controlling replication of human and simian immunodeficiency viruses (HIV and SIV). However, eliciting such responses by vaccination is complicated by immunodominance, the preferential targeting of only a few of the many possible epitopes of a given antigen. This phenomenon may be due to the coexpression of dominant and subdominant epitopes by the same antigen-presenting cell and may be overcome by distributing these sequences among several different vaccine constructs. Accordingly, we tested whether vaccinating rhesus macaques with “minigenes” encoding fragments of Gag, Vif, and Nef resulted in broadened cellular responses capable of controlling SIV replication. We delivered these minigenes through combinations of recombinant Mycobacterium bovis BCG (rBCG), electroporated recombinant DNA (rDNA) along with an interleukin-12 (IL-12)-expressing plasmid (EP rDNA plus pIL-12), yellow fever vaccine virus 17D (rYF17D), and recombinant adenovirus serotype 5 (rAd5). Although priming with EP rDNA plus pIL-12 increased the breadth of vaccine-induced T-cell responses, this effect was likely due to the improved antigen delivery afforded by electroporation rather than modulation of immunodominance. Indeed, Mamu-A*01+ vaccinees mounted CD8+ T cells directed against only one subdominant epitope, regardless of the vaccination regimen. After challenge with SIVmac239, vaccine efficacy was limited to a modest reduction in set point in some of the groups and did not correlate with standard T-cell measurements. These findings suggest that broad T-cell responses elicited by conventional vectors may not be sufficient to substantially contain AIDS virus replication.
IMPORTANCE Immunodominance poses a major obstacle to the generation of broadly targeted, HIV-specific cellular responses by vaccination. Here we attempted to circumvent this phenomenon and thereby broaden the repertoire of SIV-specific cellular responses by vaccinating rhesus macaques with minigenes encoding fragments of Gag, Vif, and Nef. In contrast to previous mouse studies, this strategy appeared to minimally affect monkey CD8+ T-cell immundominance hierarchies, as seen by the detection of only one subdominant epitope in Mamu-A*01+ vaccinees. This finding underscores the difficulty of inducing subdominant CD8+ T cells by vaccination and demonstrates that strategies other than gene fragmentation may be required to significantly alter immunodominance in primates. Although some of the regimens tested here were extremely immunogenic, vaccine efficacy was limited to a modest reduction in set point viremia after challenge with SIVmac239. No correlates of protection were identified. These results reinforce the notion that vaccine immunogenicity does not predict control of AIDS virus replication.
INTRODUCTION
The HIV epidemic continues to afflict millions of people worldwide. In spite of advances in prevention strategies and the scale-up of antiretroviral therapy access in the last decade, the World Health Organization still recorded more than 6,000 new HIV infections daily in 2012 (1). Given these statistics and the lack of resources of those countries with the highest number of cases, a prophylactic vaccine is probably the best long-term solution to halt the spread of HIV.
Unfortunately, developing an effective AIDS vaccine has been exceedingly difficult, as evidenced by the disappointing results of most human efficacy trials conducted to date (2–6). Although a modest reduction in the rate of HIV acquisition was reported in the RV144 study (7), this effect was short-lived and will have to be improved in order to significantly impact the size of the pandemic in high-risk areas. The induction of broadly reactive neutralizing antibodies remains a long-sought goal of the HIV vaccine field (8). However, the complexity and variability of the HIV Env glycoprotein have frustrated attempts to engender this type of response (9). Cellular immune responses have also been the focus of many vaccine approaches, since extensive correlative data from HIV-infected patients indicate that virus-specific T cells play a key role in suppressing viral replication and delaying progression to AIDS (10–13). Most convincingly, experimental depletion of CD8+ lymphocytes in simian immunodeficiency virus (SIV)-infected rhesus macaques leads to a brisk rise in viremia (14, 15), implicating these cells in virologic control. Furthermore, for many T-cell-based vaccine regimens, significant reductions in plasma virus concentrations in macaques challenged with pathogenic strains of SIV have been reported (16–20). Collectively, these studies support the premise that cellular immunity would improve the efficacy of HIV vaccines.
Exploiting the potential of cellular immunity in HIV vaccine strategies has not been straightforward, due to our limited understanding of the immunological properties of protective T-cell responses (21). It is still not clear, for instance, which effector functions vaccine-elicited T cells must perform in order to inhibit viral production in vivo. The differentiation state and phenotype of efficacious anti-HIV T cells also need further characterization. Knowledge of these features would provide important clues for the development of HIV immunization strategies, since the amount and duration of antigenic stimulation can directly impact the quality of memory T cells (22). Additionally, the choice of immunogens for HIV vaccines remains an open question since control of viral replication may be affected, depending on which viral proteins are targeted by the immune system (12, 23). Although Gag has been a preferred antigen for the induction of cellular immunity, mounting evidence suggests that Vif and Nef might be useful targets as well (24–27). Clarifying these issues would provide a rational basis for the design of new vaccine approaches aimed at generating cellular immunity against HIV.
It is widely accepted that HIV vaccines must elicit broad cellular responses to cope with the diversity of circulating isolates (28–30). However, the T-cell repertoire is shaped by host-intrinsic immunodominance hierarchies that often favor the expansion of T cells targeting a few “dominant” epitopes while other potentially immunogenic (i.e., “subdominant”) sequences are ignored (31). Interestingly, this phenomenon can be modulated by adjusting immunization regimens so that dominant and subdominant epitopes are not expressed in the same antigen-presenting cell (APC). For example, mouse experiments have shown that inactivating or removing dominant epitopes from immunogens allows the host to respond more effectively to subdominant epitopes (32–34). Moreover, isolating epitopes in separate DNA vaccine constructs has been shown to induce equivalent frequencies of T-cell responses directed against dominant and subdominant epitopes (32, 33, 35). Along these lines, concomitant vaccination with multiple DNA plasmids encoding fragments of a viral protein can broaden the repertoire of virus-specific T-cell responses compared to responses to vectors expressing the full-length antigens (36, 37). Since this latter strategy is more applicable to HIV vaccine design, we explored it in the present study.
Here we tested the hypothesis that broad T-cell responses elicited by gag, vif, and nef minigene vaccine constructs can control AIDS virus replication. Six minigenes covered parts of the Gag polyprotein, two inserts expressed the amino and carboxyl halves of Vif, and one gene fragment covered the central region of Nef. Each minigene was inserted into individual constructs of the following vector platforms: recombinant Mycobacterium bovis BCG (rBCG), electroporated recombinant DNA (rDNA), yellow fever vaccine virus 17D (rYF17D), and recombinant adenovirus serotype 5 (rAd5). To maximize the immunogenicity of the rDNA vectors, we delivered them by electroporation in the presence of an IL-12-carrying plasmid (EP rDNA plus pIL-12). Macaques in group 1 were immunized with EP rDNA plus pIL-12/rAd5. Animals in group 2 received all vectors in the following combination: rBCG, EP rDNA plus pIL-12, rYF17D, and rAd5. Macaques in group 3 were vaccinated with rYF17D and rAd5, while those in group 4 received rAd5 only. After performing a comprehensive evaluation of the immunogenicity of these vaccine regimens, we assessed their efficacy by challenging all animals with repeated intrarectal inoculations of SIVmac239.
MATERIALS AND METHODS
Research animals.
The 39 animals used in this study were Indian rhesus macaques (Macaca mulatta) obtained from the Wisconsin National Primate Research Center (WNPRC). They were cared for in accordance with the guidelines of the Weatherall Report under a protocol approved by the University of Wisconsin Graduate School Animal Care and Use Committee. Vaccinations and SIV challenges were performed under anesthesia, and all efforts were made to minimize suffering. The major histocompatibility complex (MHC) class I genotype of each animal was determined by sequence-specific primer PCR (SSP-PCR) analysis (Table 1) (38). Based on these results, we included two Mamu-A*01+ macaques in each of groups 1 to 4 to facilitate monitoring of vaccine-induced CD8+ T cells. Since Mamu-A*01 expression has been linked to control of SIVmac239 replication (39, 40), we balanced this effect by including four animals that were positive for this allele in group 5, the control group. Furthermore, none of the animals in this study expressed the MHC class I alleles Mamu-B*08 or Mamu-B*17, which have been associated with elite control of SIV infection (41, 42). Since combinations of tripartite motif 5 (TRIM5) alleles can restrict replication of certain SIV strains (43, 44), we determined the TRIM5 genotypes of these animals by sequencing genomic DNA or by SSP-PCR, as described previously (44).
TABLE 1.
Animal characteristics
| Group | Animal ID | Age (yrs)a | Gender | MHC class I | TRIM5 |
|---|---|---|---|---|---|
| Group 1 | r03084 | 6 | Female | Mamu-A*01 | TFP/TFP |
| r05061 | 5 | Female | Mamu-A*01 | TFP/CypA | |
| r02081 | 8 | Female | TFP/CypA | ||
| r04062 | 6 | Male | Q/TFP | ||
| rh2001 | 13 | Male | Q/Q | ||
| rhAS05 | 16 | Male | Q/TFP | ||
| r98006 | 12 | Male | TFP/CypA | ||
| r03128 | 6 | Female | Q/CypA | ||
| Group 2 | rh2036 | 10 | Male | Mamu-A*01 | Q/TFP |
| r03043 | 7 | Female | Mamu-A*01 | TFP/TFP | |
| r01105 | 8 | Female | Q/TFP | ||
| r03018 | 7 | Male | TFP/CypA | ||
| r99088 | 10 | Male | Q/CypA | ||
| r98025 | 12 | Female | Q/TFP | ||
| r03050 | 7 | Female | Q/TFP | ||
| r01051 | 8 | Female | Q/TFP | ||
| Group 3 | r98036 | 12 | Female | Mamu-A*01 | Q/Q |
| rh2034 | 11 | Male | Mamu-A*01 | Q/Q | |
| r02122 | 8 | Female | TFP/CypA | ||
| r03071 | 7 | Female | TFP/CypA | ||
| r04025 | 7 | Female | Q/CypA | ||
| r04036 | 6 | Female | Q/TFP | ||
| rh2044 | 15 | Female | TFP/TFP | ||
| rh2231 | 15 | Female | TFP/CypA | ||
| Group 4 | r03021 | 7 | Female | Mamu-A*01 | TFP/TFP |
| r03115 | 7 | Female | Mamu-A*01 | TFP/TFP | |
| r04064 | 6 | Male | Q/TFP | ||
| rh1969 | 14 | Female | TFP/TFP | ||
| r01060 | 9 | Male | Q/TFP | ||
| r04022 | 6 | Female | Q/TFP | ||
| r04101 | 5 | Female | Q/TFP | ||
| Group 5 | r03116 | 7 | Male | Mamu-A*01 | TFP/TFP |
| rh2306 | 8 | Male | Mamu-A*01 | TFP/TFP | |
| r02108 | 9 | Male | Mamu-A*01 | Q/CypA | |
| r03141 | 7 | Male | Mamu-A*01 | Q/TFP | |
| r02076 | 9 | Female | Q/TFP | ||
| rh2284 | 7 | Male | Q/CypA | ||
| r03103 | 7 | Male | Q/TFP | ||
| r03111 | 7 | Female | Q/TFP |
Animal age at beginning of the study.
Vector platforms and immunogens.
The nine SIV minigenes employed here encoded the regions of SIVmac239 Gag, Vif, and Nef depicted in Fig. 1A. These sequences were inserted into three vector platforms: rDNA, rYF17D, and rAd5. The rDNA constructs consisted of nine pCMVkan plasmids (45), each carrying one of the SIV minigenes shown in Fig. 1A. Expression of these gene fragments was under the control of the human cytomegalovirus (CMV) promoter and the bovine growth hormone polyadenylation signal. Except for the two first rDNA vaccinations of group 1, these rDNA vectors were codelivered with the AG157 plasmid, which encodes the two subunits of rhesus IL-12 expressed from two separate transcription units (46). We refer to this plasmid as pIL-12 here. Aldevron (Fargo, ND) produced the large-scale plasmid batches used for rDNA vaccinations. The nine rAd5 vectors used in these studies contained the same optimized SIV minigenes and were made by Viraquest, Inc., by using the RAPAd method (47). Of note, the sequences inserted in the rDNA and rAd5 vectors were codon optimized for high expression in mammalian cells.
FIG 1.

SIV minigenes and vaccination scheme. (A) Relative sizes and amino acid regions encoded by the SIVmac239 gag, vif, and nef minigenes. The six gag minigenes covered amino acids 6 to 52, 44 to 84, and 76 to 123 of matrix (MA), 142 to 186, 178 to 258, and 250 to 415 of capsid (CA), p2, and nucleocapsid (NC). Two vif minigenes encoded the amino (aa 1 to 110) and carboxyl (aa 102 to 214) halves of the Vif protein, while the central part of Nef (aa 45 to 210) was covered by one gene fragment. (B) We delivered the SIV minigenes through four vaccine regimens comprised of combinations of three vector platforms (rBCG, EP rDNA plus pIL-12, and rYF17D) followed by a common rAd5 boost. Each SIV minigene was inserted into an individual vector, yielding a total of nine constructs for each vector platform. The rBCG vectors were an exception, as they contained smaller SIV minigenes and thus required 14 different constructs to cover the same regions of Gag, Vif, and Nef described above. At week 14 to 25 after the rAd5 boost, we began challenging the animals intrarectally with 800 TCID50 of SIVmac239. Animals were inoculated every week, and viral loads were assessed 7 days after each challenge. If an animal was not productively infected after a given virus exposure, it was rechallenged the following week. Since several animals remained uninfected after eight inoculations with 800 TCID50, we increased the dose of the inoculum according to the information provided in Table 2.
The nine live-attenuated rYF17D viruses employed were generated as described previously (48). In sum, SIV minigenes with a codon usage matching that of the YF17D virus were inserted into the junction of the genes encoding E and NS1 in the YF17D backbone. We generated viable viruses by transfecting in vitro-generated, full-length rYF17D genomic mRNA into Vero cells, after which virus present in the supernatant of these cultures was propagated in Vero cell monolayers. We confirmed the presence of all SIV inserts in their corresponding rYF17D viruses by reverse transcription-PCR analysis of the E/NS1 intergenic region and by nucleotide sequencing.
The rBCG constructs contained SIV inserts that were slightly different than those mentioned above; instead of nine SIV minigenes, a total of 14 codon-optimized sequences covered the following amino acid regions of the SIVmac239 Gag, Vif, and Nef proteins: gag-1 (amino acids [aa] 6 to 52), gag-2 (aa 44 to 84), gag-3 (aa 76 to 123), gag-4 (aa 142 to 186), gag-5 (aa 178 to 227), gag-6 (aa 239 to 358), vif-1 (aa 1 to 60), vif-2 (aa 51 to 110), vif-3 (aa 102 to 160), vif-4 (aa 151 to 214), nef-1 (aa 45 to 96), nef-2 (aa 88 to 139), nef-3 (aa 131 to 180), and nef-4 (aa 172 to 210). Each of these gene fragments was inserted into individual rBCG vectors.
Vaccinations.
In order to maximize the dispersal of our SIV inserts among different lymph nodes and thereby avoid immunodominance (32, 35), we delivered each of the nine rDNA, rYF17D, and rAd5 constructs to separate anatomical locations. These sites were rotated in subsequent immunizations so that each location did not receive vectors encoding the same SIV minigene twice. The sequence and timing of these vaccinations are shown in Fig. 1B.
For the rDNA vaccinations, we prepared individual syringes containing 1.0 mg of each rDNA and two different amounts of pIL-12, depending on the vaccination group. We used phosphate-buffered saline (PBS) to adjust the volume of these syringes to 0.5 ml. Macaques in group 1 did not receive pIL-12 at the first and second EP rDNA vaccinations. At the third occasion, we added 0.4 mg of pIL-12 to each syringe containing the rDNA constructs, yielding a total of 3.2 mg of pIL-12. For group 2, 0.08 mg of pIL-12 was codelivered with each of the rDNA plasmids, yielding a total of 0.64 mg of pIL-12 given at each EP rDNA vaccination. The TriGrid in vivo electroporation system developed by Ichor Medical Systems, Inc. (San Diego, CA) was used to administer the rDNA constructs intramuscularly, using standard 1-ml syringes with a 22-gauge needle. The rDNA constructs were injected into the right and left legs (tibialis), right and left thighs (lower and upper lateralis), and right and left forearms (brachialis), yielding a total of eight injection sites. We combined the rDNA plasmids carrying the gag (aa 6 to 52) and nef (aa 45 to 210) minigenes and codelivered them during the EP vaccinations. Macaques in group 4 were vaccinated three times with a single dose of empty DNA (1.0 mg; no pIL-12) delivered to a separate muscle on each occasion.
Vaccinees in groups 2 and 3 were vaccinated with 2.0 × 105 PFU of each rYF17D construct. Each construct was diluted in PBS to a final volume of 1.0 ml and injected subcutaneously (using 27-gauge needles) into one of the following anatomical sites: right and left shoulders, right and left forearms, right and left thighs, right and left calves, and abdomen. Macaques in group 4 received a single dose of the parental YF17D vaccine (2.0 × 105 PFU) in the right forearm.
All animals in groups 1 to 4 were vaccinated with 1011 viral particles of each of the nine rAd5 vectors carrying the SIV minigenes. These constructs were diluted in 0.5 ml of PBS and delivered intramuscularly using 25-gauge needles. The same sites were used as for the EP rDNA vaccinations, with the addition of the left shoulder.
Macaques in group 2 were vaccinated once with rBCG. As described above, the SIV inserts expressed by the rBCG vectors were broken down into additional fragments, yielding a total of 14 individual constructs. We resuspended 2.0 × 105 CFU of each rBCG vector in 100 μl of PBS and injected the mixtures intradermally into nine different sites: right and left calves, right and left thighs, right and left arms, right and left shoulders, and abdomen.
SIVmac239 challenges.
We challenged the animals in this study by using repeated, intrarectal (i.r.) inoculations of SIVmac239 of 800 50% tissue culture infective doses (TCID50) per exposure, corresponding to 6.52 × 106 viral RNA (vRNA) copies. We exposed the macaques to SIV every week and assessed plasma viral loads 7 days after each inoculation to determine if they had acquired SIV infection. If viral loads were ≥1,000 vRNA copies/ml of plasma, animals were considered to be infected and were not rechallenged. If viral loads were <1,000 vRNA copies/ml of plasma, we obtained an additional measurement at day 9 or 10 to ensure productive infection. If viral loads were positive at this second measurement, we did not rechallenge the animals; however, if they were negative, we resumed the i.r. inoculations at day 14. After eight exposures with the initial dose of 800 TCID50 per challenge, several animals remained uninfected, and so we increased the challenge inoculum as shown in Table 2.
TABLE 2.
Dose and number of intrarectal challenges with SIVmac239
| Group | Animal ID no. | No. of challenges received by dose group |
Total no. of challenges | |||
|---|---|---|---|---|---|---|
| 800 TCID50 | 4,000 TCID50 | 5,000 TCID50 | 50,000 TCID50 | |||
| Group 1 | r03084 | 5 | 5 | |||
| r05061 | 8 | 1 | 9 | |||
| r02081 | 6 | 6 | ||||
| r04062 | 8 | 3 | 11 | |||
| rh2001 | 8 | 6 | 4 | 2 | 20 | |
| rhAS05 | 8 | 2 | 10 | |||
| r98006 | 3 | 3 | ||||
| r03128 | 8 | 3 | 11 | |||
| Median | 9.5 | |||||
| Group 2 | rh2036 | 4 | 4 | |||
| r03043 | 8 | 2 | 10 | |||
| r01105 | 4 | 4 | ||||
| r03018 | 8 | 2 | 10 | |||
| r99088 | 2 | 2 | ||||
| r98025 | 2 | 2 | ||||
| r03050 | 8 | 5 | 13 | |||
| r01051 | 4 | 4 | ||||
| Median | 4 | |||||
| Group 3 | r98036 | 3 | 3 | |||
| rh2034 | 7 | 7 | ||||
| r02122 | 8 | 4 | 12 | |||
| r03071 | 5 | 5 | ||||
| r04036 | 7 | 7 | ||||
| rh2231 | 4 | 4 | ||||
| r04025 | 6 | 6 | ||||
| rh2044 | 8 | 5 | 13 | |||
| Median | 6.5 | |||||
| Group 4 | r03021 | 8 | 3 | 11 | ||
| r03115 | 8 | 2 | 10 | |||
| r04064 | 7 | 7 | ||||
| rh1969 | 8 | 3 | 11 | |||
| r01060 | 3 | 3 | ||||
| r04022 | 4 | 4 | ||||
| r04101 | 4 | 4 | ||||
| Median | 7 | |||||
| Group 5 | r03116 | 2 | 2 | |||
| r03103 | 3 | 3 | ||||
| r02076 | 8 | 6 | 2 | 16 | ||
| r02108 | 6 | 6 | ||||
| rh2306 | 8 | 2 | 10 | |||
| r03111 | 8 | 6 | 4 | 4 | 22 | |
| r03141 | 3 | 3 | ||||
| rh2284 | 8 | 2 | 10 | |||
| Median | 8 | |||||
IFN-γ enzyme-linked immunosorbent spot (ELISPOT) assays.
We isolated peripheral blood mononuclear cells (PBMC) from EDTA-treated blood by using Ficoll-Paque Plus (GE Health Sciences) density centrifugation. We used PBMC or PBMC depleted of CD8+ cells (CD8− PBMC) by magnetic bead separation (Miltenyi Biotec, Auburn, CA), directly in precoated ELISpotPLUS kits (Mabtech Inc., Mariemont, OH) for the detection of monkey gamma interferon (IFN-γ) according to the manufacturer's protocols. Briefly, 105 PBMC or CD8− PBMC were used per well and incubated for 14 to 18 h at 37°C in 5.0% CO2. As a positive control, 5.0 μg/ml of concanavalin A (Sigma Chemical, St. Louis, MO) was added to the cells, and a set of negative control wells of medium only was also included on each plate. Peptide pools of 10 15-mers overlapping by 11 amino acids spanning the regions of SIVmac239 Gag (n = 11 pools), Vif (n = 5 pools), and Nef (n = 4 pools) covered by the minigenes were used at a concentration of 1.0 μM. Reactivity to each protein is reported as the sum of responses to these pools. The concentration of minimal optimal peptides corresponding to SIVmac239 epitopes was 10.0 μM. All SIV peptide pools were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
Test wells were run in duplicates, while control wells were run with replicates of 2, 4, or 6, depending on the assay. Assay results are shown as spot-forming cells (SFC) per 106 PBMC or CD8− PBMC. Responses comprising <50 SFC per 106 cells were considered negative and were not tested statistically. Positive responses were determined by using a one-tailed t test and an alpha level of 0.05, where the null hypothesis was that the background level would be greater than or equal to the treatment level. If determined to be positive statistically, the values were reported as the average of the test wells minus the average of all negative control wells.
ICS assay.
We performed a multiparameter intracellular cytokine staining (ICS) assay by incubating freshly isolated PBMC from rhesus macaques with pools of Gag, Vif, and Nef peptides, as previously described (49). Samples were acquired using FACS DIVA, version 6.2, on a special order research product (SORP) BD LSR II apparatus equipped with a 50-mW 405-nm violet, a 100-mW 488-nm blue, and a 50-mW 640-nm red laser, and results were analyzed by using FlowJo 9.4 (TreeStar, Inc.). Analysis and presentation of distributions were performed with Microsoft Excel and GraphPad Prism. In the prechallenge assays, we incubated PBMC with the same pools of Gag, Vif, and Nef peptides employed in the IFN-γ ELISPOT assays (see above). Our staining panel included antibodies directed against CD4 (peridinin chorophyll protein [PerCP]-Cy5.5 conjugated; clone L200; BD Biosciences), CD8 (Pacific Blue; clone RPA-T8; BD Biosciences), CD14 (electron-coupled dye [ECD]; clone RMO52; Beckman Coulter), CD19 (ECD; clone JA.119; Beckman Coulter), IFN-γ (fluorescein isothiocyanate; clone 4S.B3; BD Biosciences), tumor necrosis factor alpha (TNF-α; Alexa Fluor 700; MAb11; BD Biosciences), CD107a (phycoerythrin [PE]; clone H4A3; BD Biosciences), and IL-2 (allophycocyanin; clone MQ1-17H12; BD Biosciences). In the postchallenge assays, our antigen stimuli consisted of four pools of peptides spanning (i) amino acids 1 to 291 and (ii) 281 to 510 of Gag; (iii) the Vif open reading frame (ORF); and (iv) the Nef ORF. Our staining panel included antibodies directed against CD4 (PerCP Cy5.5; clone L200; BD Biosciences), CD8 (AmCyan; clone SK1; BD Biosciences), CD14 (ECD; clone RMO52; Beckman Coulter), CD19 (ECD; clone JA.119; Beckman Coulter), IFN-γ (Alexa Fluor 700; clone B27; BD Biosciences), TNF-α (V450; clone Mab11; BD Horizon), CD107a (PE; clone H4A3; BD Biosciences), and IL-2 (allophycocyanin; clone MQ1-17H12; BD Biosciences). The ICS data are reported as sums of CD4+ and CD8+ lymphocytes reacting against all Gag, Vif, and Nef pools.
For data analysis, we first gated on forward scatter height (FSC-H) versus forward scatter area (FSC-A) to remove doublets (see Fig. S1 in the supplemental material). Subsequently, we gated on the lymphocyte population and then created a dump channel by excluding CD14+ CD19+ events. At this stage, we separated lymphocyte subsets based on their expression of either CD4 or CD8 (excluding those expressing both markers) and conducted our functional analyses within these two compartments. After making gates for each function (IFN-γ, CD107a, TNF-α, and IL-2), we used the Boolean gate platform to generate a full array of possible combinations, equating to 16 response patterns when testing four functions (24 = 16). We used three criteria to determine positivity of responses: (i) background-subtracted responses had to be at least 2-fold higher than the background itself; (ii) Boolean gates for each response pattern had to contain ≥10 events; (iii) response patterns had to be greater than matched values obtained in ICS assays using PBMC from three SIV-naive Indian rhesus macaques stimulated under the same conditions.
Viral load measurements.
Viral loads were measured from EDTA anticoagulated plasma by using a previously described protocol (50). The limit of reliable quantification as performed on an input volume of 0.5 ml of plasma is 30 copy equivalents of SIV genomic RNA per ml.
Statistical analyses.
We utilized the IFN-γ ELISPOT data for comparisons of SIV-specific T-cell responses among the four vaccinated groups. We carried out pairwise permutation tests in which 10,000 permutation samples were generated by swapping the group labels. P values were obtained according to the two-sample t statistic. To confirm the results obtained by the permutation tests, we also performed Mann-Whitney tests and obtained similar P values. To maintain the type I error rate at no higher than α, we applied the Bonferroni-Holm correction to these comparisons and obtained a corrected threshold of 0.0015.
We defined each animal's peak viral load as the highest viral load measurement during the first 4 weeks of infection. Set point viremia was determined as the geometric mean of viral loads measured within weeks 6 to 12 postinfection. To determine vaccine efficacy, we compared viral loads from each of groups 1 to 4 with that of group 5, the control group. We performed pairwise comparisons of the groups by using permutation tests. For each test, 10,000 permutations samples were generated by permuting the vaccination group labels. We obtained P values by using the customary two-sample t statistic. The threshold of significance for the viral load comparisons was 0.0026 according to the Bonferroni-Holm adjustment.
To determine whether vaccination affected the rate at which animals acquired SIV infection, we used Kaplan-Meier survival analyses to test whether vaccinees in each of groups 1 to 4 and control animals in group 5 differed in the number of challenges required until infection. All comparisons yielded P values of >0.3, according to log rank tests. For our correlates analysis, we compared nine immunological variables with each animal's set point viral loads by using the Spearman rank correlation test (see Table 3, below). We obtained P values from 1,000 bootstrap resamples from the observed pairs of values.
TABLE 3.
Correlations between nine immunological variables measured in vaccinees in groups 1 to 4 and their corresponding set point viral loads
| Immunological variable | Time point | Correlation coefficient (r) | P value |
|---|---|---|---|
| Total magnitude of IFN-γ-producing cells measured by ELISPOT assay with CD8− PBMCa | Day 7 post-rAd5 boost | −0.21 | 0.56 |
| Total magnitude of IFN-γ-producing cells measured by ELISPOT assay with whole PBMC | Day 14 post-rAd5 boost | −0.16 | 0.54 |
| Total magnitude of IFN-γ-producing cells measured by ELISPOT assay with whole PBMC | Day of first SIV challenge | −0.11 | 0.68 |
| Total magnitude of IFN-γ-producing cells measured by ELISPOT assay with whole PBMC | Day 14 post-SIV infection | 0.02 | 0.9 |
| Total breadth of T-cell responsesb | Day 21 post-rAd5 boost | −0.13 | 0.6 |
| Total magnitude of polyfunctional CD4+ T-cell responsesc | Day 7 post-rAd5 boost | −0.28 | 0.49 |
| Total magnitude of polyfunctional CD8+ T-cell responses | Day 14 post-rAd5 boost | 0.04 | 0.87 |
| Total magnitude of polyfunctional CD4+ T-cell responses | Day 14 post-SIV infection | −0.36 | 0.51 |
| Total magnitude of polyfunctional CD8+ T-cell responses | Day 14 post-SIV infection | −0.05 | 0.84 |
For comparisons of the total magnitude of IFN-γ-producing cells measured in the ELISPOT assays, we used the sum of SFC/106 PBMC (or CD8− PBMC) values measured in wells stimulated with Gag, Vif, or Nef peptides at the indicated time points in the correlation analysis.
For comparisons of the total breadth of T-cell responses, we used the sum of Gag-, Vif-, and Nef-specific T-cell responses mapped in each vaccinee at day 21 post-rAd5 boost for the correlation analysis.
For comparisons of the total magnitude of polyfunctional T-cell responses, we used the total frequency of Gag-, Vif-, and Nef-specific CD4+ or CD8+ T cells performing a minimum of three immunological functions for the correlation analysis.
RESULTS
Vaccination regimens.
Previous studies have shown that dividing dominant and subdominant epitopes among different rDNA vectors during vaccination can overcome natural immunodominance hierarchies and result in equivalent CD8+ T-cell responses to both types of epitopes (32, 35–37). Here, we applied this strategy to a T-cell-based SIV vaccine in an attempt to broaden the repertoire of vaccine-elicited T-cell responses. We hypothesized that vaccination with minigenes encoding fragments of the SIVmac239 Gag, Vif, and Nef proteins would generate broadly targeted T-cell responses capable of controlling viral replication after mucosal challenge with SIVmac239. To test this hypothesis, we designed a total of nine minigenes, six of which covered parts of the matrix (MA), capsid (CA), p2, and nucleocapsid (NC) of the Gag polyprotein (Fig. 1A). Two inserts covered the entire Vif ORF, while one sequence encoded the central part of Nef (Fig. 1A). We delivered these minigenes by using four vector platforms: rBCG, EP rDNA plus pIL-12, rYF17D, and rAd5. Each minigene was inserted into a separate vaccine vector, yielding a total of nine constructs for each vector platform. The only exception was rBCG, for which we had 14 different constructs (6 for Gag, 4 for Vif, and 4 for Nef; see Materials and Methods). In order to maximize the dispersal of antigens among different lymph nodes, we administered each vaccine vector to a separate anatomical site and rotated these locations in subsequent immunizations so that constructs encoding fragments of the same protein were not administered at the same site twice.
We immunized a total of 31 Indian rhesus macaques with four different heterologous prime-boost regimens comprising combinations of the vaccine vectors mentioned above. Animals in group 1 (n = 8) received three EP rDNA primes, with pIL-12 given only at the third immunization (Fig. 1B). Of note, animals in this group experienced lymphopenia in the weeks that followed the third administration of EP rDNA plus pIL-12, which was likely caused by the high dose of pIL-12 (3.2 mg) delivered on that occasion (51). To ensure that the animals had sufficient time to recover, we administered the rAd5 boost 26 weeks later (Fig. 1B). Macaques in group 2 (n = 8) were vaccinated with all four vectors, given in the following order: rBCG, EP rDNA plus pIL-12, rYF17D, and rAd5 (Fig. 1B). Importantly, the dose of pIL-12 delivered to group 2 vaccinees was 5-fold lower (0.64 mg) than that employed in group 1 and did not cause any observable adverse events. Group 3 animals (n = 8) received the same rYF17D prime and rAd5 boost reported recently (27).
The fact that macaques in groups 1 to 3 were boosted with rAd5 might have masked the contributions of their priming immunizations to their overall SIV-specific T-cell response, since rAd5 is highly immunogenic in nonhuman primates (52). To control for this, macaques in group 4 (n = 7) were primed with “empty” versions of the DNA (without pIL-12) and YF17D constructs described above, and then animals were boosted with the same rAd5 vectors given to groups 1 to 3 (Fig. 1B). This way, rAd5 was the sole source of Gag-, Vif-, and Nef-specific T-cell responses in the group 4 animals, providing a reference for assessing the immunogenicity and efficacy of the vaccine regimens employed in groups 1 to 3. Macaques in group 5 (n = 8) did not receive any vaccination regimen and served as controls for the present study (Fig. 1B).
Immunogenicity of vaccination regimens.
We assessed the immunogenicity of the four vaccine regimens by carrying out IFN-γ ELISPOT and ICS assays at several time points following the rAd5 boost, including the time of challenge. The IFN-γ ELISPOT assays, performed on PBMC and CD8− PBMC, provided a quantitative readout for comparing cellular responses among the groups (Fig. 2; see also Fig. 4, below). To complement this analysis, we set up ICS assays at the same time points to characterize the functional profiles of vaccine-induced CD4+ and CD8+ T cells (Fig. 3). The immunogenicity and efficacy data for each of groups 1, 2, 3, 4, and 5 are color coded with blue, green, beige, black, and red symbols, respectively.
FIG 2.

Immunogenicity of vaccine regimens at days 7 and 14 post-rAd5 boost. We carried out IFN-γ ELISPOT assays on PBMC and CD8-depleted (CD8− PBMC) at days 7 (A and C) and 14 (B and D) after the rAd5 boost by using peptide pools spanning the regions of SIVmac239 Gag, Vif, and Nef covered by the nine minigenes. The magnitude of each animal's vaccine-induced T-cell response to Gag, Vif, and Nef is shown in panels A and B. Response values were calculated as described in Materials and Methods. The total SIV-specific T-cell response for each animal comprises the sum of IFN-γ-producing cells in PBMC or CD8− PBMC that were stimulated by the Gag, Vif, and Nef peptide pools at day 7 (C) or 14 (D) post-rAd5 boost. We compared the total magnitudes of these responses among the four groups by using pairwise permutation tests. The threshold of significance for these comparisons was 0.0015 according to the Bonferroni-Holm adjustment. Groups 1, 2, 3, and 4 are color coded in blue, green, beige, and black, respectively. Lines represent medians, and each symbol corresponds to one vaccinee.
FIG 4.

Magnitude of vaccine-induced T-cell responses at the time of challenge. We carried out IFN-γ ELISPOT assays on PBMC and CD8-depleted PBMC on the day of each group's first intrarectal challenge. For groups 1, 2, 3, and 4, this time point occurred 14, 25, 22, and 19 weeks after the rAd5 boost, respectively. Response values were calculated as described in Materials and Methods. We compared the total magnitude of SIV-specific T-cell responses among the four groups by using pairwise permutation tests. The threshold of significance for these comparisons was 0.0015 according to the Bonferroni-Holm adjustment. Groups 1, 2, 3, and 4 are color coded in blue, green, beige, and black, respectively. Lines represent medians, and each symbol corresponds to one vaccinee.
FIG 3.

Functional profile of vaccine-induced T-cell responses. We carried out multiparameter ICS assays to determine the ability of SIV-specific T cells induced by the four vaccine regimens to degranulate (i.e., CD107a expression) and produce cytokines (IFN-γ, TNF-α, and IL-2). To characterize vaccine-elicited CD4+ and CD8+ T cells at their peak expansion, we performed this analysis at days 7 and 14 after the rAd5 boost, respectively. Response magnitudes for each animal were calculated by adding the percentages of Gag-, Vif-, and Nef-specific CD4+ or CD8+ lymphocytes producing each combination of functions tested. Panels on the left correspond to CD4+ T-cell responses in group 1 (A), group 2 (C), group 3 (E), and group 4 (G). Panels on the right correspond to CD8+ T-cell responses in group 1 (B), group 2 (D), group 3 (F), and group 4 (H). Results for groups 1, 2, 3, and 4 are color coded in blue, green, beige, and black, respectively.
The rAd5 boost resulted in vigorous expansion of Gag- and Vif-specific cellular responses in groups 1 and 2 (Fig. 2A and B). Nef was also targeted, but at lower frequencies (Fig. 2). Notably, vaccinees in both groups mounted impressive CD4+ T-cell responses at day 7 post-rAd5 boost (Fig. 2A), with total medians of spot-forming cells exceeding 6,000 SFC/106 CD8− PBMC (Fig. 2C). In addition to IFN-γ, these SIV-specific CD4+ T cells were also capable of elaborating TNF-α and IL-2, but not CD107a (Fig. 3A and C). By day 14 post-rAd5 boost, CD4+ T-cell responses had contracted to less than half of their peak levels recorded at day 7 (Fig. 2B and D), while the number of SFC/106 PBMC remained high (Fig. 2B). This implied that SIV-specific CD8+ T lymphocytes predominated at this time point. Indeed, contemporary ICS measurements confirmed this pattern and revealed that production of IFN-γ either alone or in conjunction with CD107a were the main functional signatures of vaccine-induced CD8+ T cells on this occasion (Fig. 3B and D and data not shown). Importantly, vaccinees in groups 1 and 2 maintained readily detectable SIV-specific T-cell responses until the time of challenge (total medians, >950 SFC/106 PBMC), with CD4+ T cells still comprising a substantial fraction of their total response (Fig. 4A). Of note, the total magnitude of cellular responses in these two groups was statistically indistinguishable at all prechallenge time points analyzed (Fig. 2C and D and 4B), in spite of substantial differences in the vaccinations that preceded the rAd5 boost.
Vaccine-induced T-cell responses in groups 3 and 4 were of lower frequencies than those detected in groups 1 and 2 but conformed to a similar pattern; Gag and Vif were preferentially targeted, and the peak expansion of CD4+ and CD8+ T-cell responses occurred at days 7 and 14 post-rAd5 boost, respectively (Fig. 2A and B). Concomitant ICS assays demonstrated that SIV-specific CD4+ and CD8+ T lymphocytes in groups 3 and 4 were infrequent and less heterogeneous than their counterparts in groups 1 and 2 (Fig. 3E to H). Not surprisingly, statistical comparisons of the total magnitude of cellular responses measured in the four groups confirmed the superior immunogenicity of the group 1 and group 2 regimens compared to those given to groups 3 and 4 (Fig. 2C and D and 4B). This analysis also revealed that the total frequencies of SIV-specific T cells in groups 3 and 4 were similar, except for a modest increase in the number of IFN-γ-producing cells measured in PBMC from group 3 animals at day 14 post-rAd5 boost (P = 0.0446) (Fig. 2D). Of note, this difference was not significant after correcting for multiple comparisons. The similarity in the total sizes of the group 3 and group 4 responses suggests that the rYF17D vaccination did not effectively prime SIV-specific T cells in the group 3 animals. Indeed, only a few group 3 vaccinees had measurable SIV-specific T cells at day 14 post-rYF17D vaccination (see Fig. S2D in the supplemental material). This finding is consistent with a recent study conducted in our laboratory that showed that rYF17D vectors are poorly immunogenic in rhesus macaques (49). As for the persistence of these responses, we detected low frequencies of vaccine-elicited T cells in both group 3 and group 4 animals at the time of challenge (Fig. 4A and B).
We determined the total breadth of vaccine-induced T-cell responses in all animals at day 21 post-rAd5 boost (Fig. 5). To do that, we performed IFN-γ ELISPOT in PBMC using deconvoluted peptide pools for which positive responses were observed at days 7 and/or 14 after the rAd5 boost. This allowed us to map T-cell responses down to single 15-mers, depending on blood availability. Remarkably, vaccinees in groups 1 and 2 recognized a total median of 23 and 22 epitopes, respectively, which were concentrated in Gag, followed by Vif and Nef (Fig. 5A). These values were significantly greater than the total number of responses targeted by macaques in group 3 (median, 14 epitopes) and group 4 (median, 9 epitopes) (Fig. 5A and B). Of note, the majority of Gag-specific T-cell responses elicited in groups 1 to 4 targeted the parts of CA, p2, and NC encoded by the gag (aa 178 to 258) and gag (aa 250 to 415) minigenes (see Fig. S3 in the supplemental material). Their larger sizes and the fact that many CD4+ and CD8+ T-cell epitopes are present within this region of the Gag polyprotein likely contributed to their superior immunogenicity compared to the smaller gag (aa 6 to 52), gag (aa 44 to 84), gag (aa 76 to 123), and gag (aa 142 to 186) minigenes (53, 54). The two vif minigenes were equally immunogenic in all groups, while most Nef-specific T cells were directed against the central part of the nef minigene (see Fig. S3 in the supplemental material).
FIG 5.
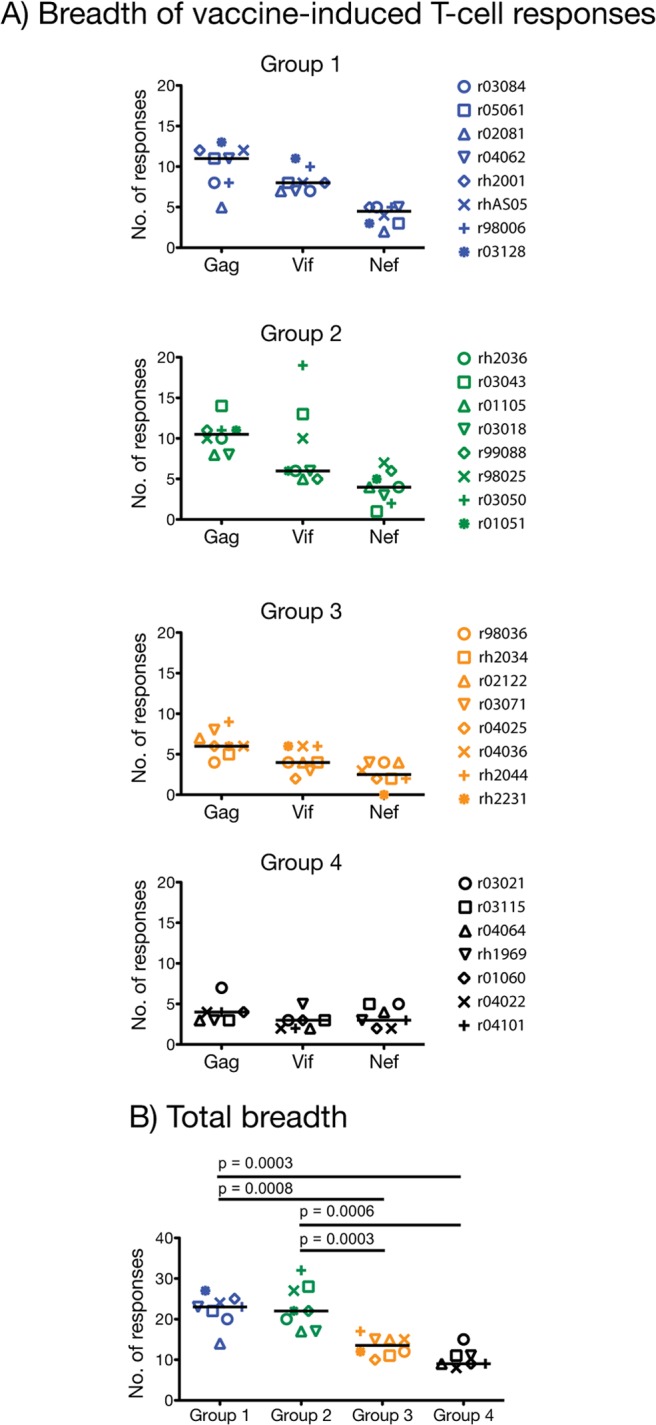
Breadth of vaccine-induced T-cell responses. At day 21 post-rAd5 boost, we carried out IFN-γ ELISPOT assays on PBMC using deconvoluted Gag, Vif, and Nef peptide pools for which a positive IFN-γ ELISPOT response was observed at previous time points. Since the 15-mers that comprise each peptide pool overlap by 11 amino acids, we avoided overestimating the breadth of T-cell responses by considering a minimum of two consecutive positive peptides as one response. (A) Number of unique responses in Gag, Vif, and Nef recognized by vaccinees in groups 1 to 4. (B) The total breadth was calculated by adding the number of responses directed against Gag, Vif, and Nef measured within each animal. The resulting number was used for comparing the breadth of vaccine-induced T-cell responses among groups 1 to 4. We used pairwise permutation tests for this analysis, and the threshold of significance for these comparisons was 0.0015 according to the Bonferroni-Holm adjustment. Groups 1, 2, 3, and 4 are color coded in blue, green, beige, and black, respectively. Lines represent medians, and each symbol denotes one vaccinee.
The exceptional breadth of responses measured in the group 1 and group 2 animals prompted us to characterize the contribution of CD4+ and CD8+ T cells to their repertoire of vaccine-elicited cellular responses. Although the approach mentioned above yielded high sensitivity for mapping individual T-cell responses in each animal, we could not discriminate between the two subsets in the IFN-γ ELISPOT assays performed on whole PBMC. We addressed this issue by calculating the total number of pools of peptides recognized by CD4+ and CD8+ T cells in the ICS assays performed at days 7 and 14 post-rAd5 boost, respectively. Each one of these pools contained 10 15-mers overlapping by 11 amino acids spanning the regions of Gag (n = 11 pools), Vif (n = 5 pools), and Nef (n = 4 pools) covered by the minigenes. We found that, on average, vaccine-elicited CD4+ T cells in groups 1 and 2 recognized 70% of these pools, compared to 23% and 14% for groups 3 and 4, respectively (data not shown). This difference was less pronounced in the CD8+ T-cell compartment; while vaccinees in groups 1 and 2 mounted CD8+ T cells against 42% of the pools of Gag, Vif, and Nef peptides, this number reached 37% for group 3 and 26% for group 4 (data not shown).
The high frequency CD4+ T cells engendered in groups 1 and 2 stemmed from priming those animals with rDNA delivered by electroporation, which has been shown to dramatically improve the immunogenicity of gene-based vaccines (55–60). Indeed, electroporation is highly efficient at transfecting cells in vivo and can increase gene expression levels by 100- to 1,000-fold above levels for intramuscular injection of rDNA without electrical stimulation (61). Along these lines, we detected sizeable SIV-specific T-cell responses in the majority of vaccinees in groups 1 and 2 after the last EP rDNA plus pIL-12 and prior to the rAd5 boost (see Fig. S2A to C in the supplemental material). Thus, priming with EP rDNA resulted in robust expansion of broad, high-frequency, and multifunctional cellular responses after the rAd5 boost in the group 1 and group 2 vaccinees. Most of these vaccine-induced cellular responses were comprised of CD4+ T cells, although CD8+ T lymphocytes contributed a sizable fraction as well.
Vaccine-induced CD8+ T-cell responses to Mamu-A*01-restricted dominant and subdominant epitopes in groups 1 to 4.
The gene fragmentation strategy employed here attempted to circumvent immunodominance by dispersing dominant and subdominant epitopes among different APCs in order to broaden the T-cell repertoire induced by vaccination. Previous studies in the murine model have shown that this approach induces equivalent responses to both types of epitopes (32, 35–37). Using this rationale, we investigated whether the immunization regimens employed in groups 1 to 4 affected the immunodominance hierarchy of known SIV-specific CD8+ T-cell epitopes. For this analysis, we focused on the two Mamu-A*01+ macaques present in each group, since this MHC class I molecule binds many SIVmac239-derived peptides with defined immunodominance patterns (62, 63). We used IFN-γ ELISPOT assay to assess reactivity against the immunodominant Gag181–189CM9 epitope as well as several other subdominant epitopes in Gag (Gag149–157LW9, Gag254–262QI9, and Gag372–379LF8) and Vif (Vif100–109VL10 and Vif144–152QA9) (Fig. 6). Nef-derived peptides capable of binding to Mamu-A*01 at physiologically relevant 50% inhibitory concentrations have not been described. We found that all Mamu-A*01+ vaccinees in groups 1 to 4 mounted CD8+ T-cell responses to Gag181–189CM9 and Vif100–109VL10, though over a wide spectrum of frequencies (Fig. 6). Responses to the other subdominant epitopes were either undetectable or at borderline levels, as was the case with rh2036's response to Gag372–379LF8 at day 14 post-rAd5 boost (Fig. 6B). Strikingly, vaccine-elicited CD8+ T cells to the immunodominant Gag181–189CM9 epitope were either below or close to the threshold of positivity of our IFN-γ ELISPOT assay (50 SFC/106 PBMC) in the group 1 vaccinees (Fig. 6A). In contrast, these animals had easily detectable Vif100–109VL10-specific CD8+ T cells after the rAd5 boost, which outnumbered the Gag181–189CM9 response at all time points analyzed (Fig. 6A). In macaques in groups 2 to 4, these responses conformed to a different immunodominance pattern whereby both Gag181–189CM9- and Vif100–109VL10-specific CD8+ T cells peaked at similar frequencies but the expansion kinetics of the latter cells appeared protracted compared to the Gag-directed response (Fig. 6B to D).
FIG 6.

Vaccine-induced CD8+ T-cell responses to dominant and subdominant epitopes restricted by Mamu-A*01. We determined whether Mamu-A*01+ vaccinees in groups 1 to 4 developed CD8+ T-cell responses against dominant (Gag181–189CM9) and subdominant (Gag149–157LW9, Gag254–262QI9, Gag372–379LF8, Vif100–109VL10, and Vif144–152QA9) epitopes after vaccination. We assessed reactivity against these peptides via IFN-γ ELISPOT assays performed at days 7 (left column) and 14 (middle column) after the rAd5 boost. We also repeated these measurements on the day of the first SIV challenge (right column) which, for groups 1, 2, 3, and 4, corresponded to week 14, 25, 22, and 19 after the rAd5 boost, respectively. Bar graphs indicate the magnitude of IFN-γ-producing cells (in SFC/106 PBMC) of macaques in group 1 (A), group 2 (B), group 3 (C), and group 4 (D). The dotted line represents the threshold of positivity of our IFN-γ assays (50 SFC/106 PBMC). Groups 1, 2, 3, and 4 are color coded in blue, green, beige, and black, respectively.
With only two Mamu-A*01+ macaques in each group, it is difficult to conclude whether these data reflect bona fide changes to the immundominance ranks of the Gag181–189CM9 and Vif100–109VL10 epitopes or are simply due to biological variability among the animals. However, the fact that the immunodominance hierarchy adopted by these responses in the two group 1 animals was the opposite of what is typically seen during primary SIVmac239 infection of Mamu-A*01+ macaques suggests that the former scenario is more likely.
Mucosal challenge with SIVmac239.
In order to approximate clinically relevant human exposures to HIV, we challenged all animals intrarectally with repeated inoculations of SIVmac239 sufficient to infect only a fraction of challenged animals per occasion (64). These challenges began after various intervals after the rAd5 boost that ranged from 14 to 25 weeks (Fig. 1B). We determined the initial dose of our inoculum based on a previous in vivo titration study conducted in our laboratory and on the results of a recent efficacy trial (60), which utilized the same SIVmac239 stock used here. In that study, Winstone et al. reported that all macaques in their control group became infected after seven intrarectal challenges with 800 TCID50 (60). We therefore chose this dose as our starting point.
After a series of eight weekly challenges with 800 TCID50, three vaccinees in group 1, five in group 2, six in group 3, four in group 4, and four control animals in group 5 became productively infected (Table 2). Given that several macaques remained uninfected after this first challenge phase, we increased the dose of the SIVmac239 inoculum to 4,000 TCID50 for the next 6 weeks. During this time, all vaccinees in groups 1 to 4 (except for rh2001 in group 1) and two control animals acquired SIV infection (Table 2). Notably, two control macaques, r02076 and r03111, remained uninfected after this second round of challenges. We then began a third series of challenges with 5,000 TCID50 for the next 4 weeks, which was sufficient to infect r02076 after two exposures (Table 2). Nevertheless, rh2001 and r03111 resisted these four challenges with 5,000 TCID50. These animals finally became infected after we increased the dose of the SIVmac239 inoculum to 50,000 TCID50, which infected rh2001 and r03111 after two and four exposures, respectively (Table 2). Of note, none of the vaccination regimens tested in the present study affected acquisition of SIVmac239 infection (Fig. 7).
FIG 7.
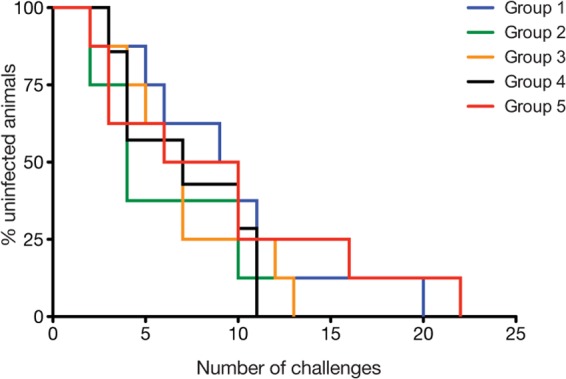
Kaplan-Meier rate of infection after repeated challenges with escalating doses of SIVmac239. This graphical representation of the data presented in Table 2 shows the percentage of uninfected vaccinees in each of groups 1 to 4 or control animals in group 5 after each intrarectal inoculation with SIVmac239. Animals were inoculated with 800 TCID50 in challenges 1 to 8, 4,000 TCID50 in challenges 9 to 14, 5,000 TCID50 in challenges 15 to 18, and 50,000 TCID50 in challenges 19 to 22. The rate at which vaccinees in groups 1 to 4 acquired SIV infection was not significantly different from that of control animals in group 5, as determined by the log rank test (P > 0.3).
To assess vaccine efficacy, we compared viral loads between vaccinees in groups 1 to 4 and the control animals in group 5 during the acute and chronic phases of infection. Our significance threshold for these comparisons was 0.0026, as established by the Bonferroni-Holm correction for multiple testing. SIVmac239 replicated vigorously in the group 5 macaques, reaching a median peak viral load of 1.2 × 107 vRNA copies/ml of plasma and then plateauing at 6.9 × 105 vRNA copies/ml (Fig. 8E to H). The majority of vaccinated macaques also experienced high levels of viral replication in the acute phase (Fig. 8A to D), with median peak viral loads ranging from 4.4 × 106 vRNA copies/ml in group 4 to 7.1 × 106 vRNA copies/ml in group 2 (Fig. 8G). The similarity of these peak viremia values among the groups was unexpected, given the marked differences in cellular immunity induced by the four immunization regimens (Fig. 2C and D). Of note, two vaccinees controlled acute-phase viremia to less than 105 vRNA copies/ml; rh2001 in group 1 and rh1969 in group 4 had peak viral loads of 1.4 × 104 and 4.6 × 104 vRNA copies/ml, respectively. Nevertheless, in spite of these two outliers, median peak viral loads in groups 1 to 4 were statistically indistinguishable from that of group 5 (Fig. 8G).
FIG 8.

Plasma virus concentrations after SIVmac239 infection. Vaccinees in group 1 (A), group 2 (B), group 3 (C), group 4 (D), and the control animals in group 5 (E) were infected after various intrarectal exposures to SIVmac239 (Table 2). Viral loads were log-transformed and correspond to the number of vRNA copies/ml of plasma. The solid and dashed lines in graphs A to F are for reference only and indicate a viral load of 106 and 103 vRNA copies/ml, respectively. (F) Median viral loads for groups 1 to 5 for each time point up to week 12 postinfection, the last follow-up measurement. (G) Median peak viral loads for groups 1 to 5. (H) Median set point viral loads for groups 1 to 5. We determined the set point viremia for each animal by calculating the geometric mean of viral loads measured within weeks 6 to 12 postinfection. We compared peak and set point viral loads among gropus 1 to 5 by using pairwise permutation tests. The threshold of significance for these comparisons was 0.0026 according to the Bonferroni-Holm adjustment.
Encouragingly, several vaccinees controlled viral replication in the chronic phase. This effect was most evident in group 1, with three macaques (rh2001, r05061, and r98006) experiencing viral loads of less than 103 vRNA copies/ml at week 12 postinfection (Fig. 8A). Overall, the median set point viral load for this group was 5.7 × 104 vRNA copies/ml, corresponding to a significant 1-log reduction in virus production compared to group 5 (P = 0.002) (Fig. 8H). Macaques in group 2 performed similarly and lowered set point viremia to a median of 7.5 × 104 vRNA copies/ml (P = 0.006) (Fig. 8H). Although the P value for this comparison was slightly above our significance threshold, the difference in set points between groups 2 and 5 appears biologically significant, considering the distribution of viral loads among animals within each group (Fig. 8B and E). As for group 3, chronic-phase viremia in those animals plateaued at a median of 1.9 × 105 vRNA copies/ml and was not statistically distinct from that of the control group (Fig. 8C, F, and H). Strikingly, group 4 had the lowest median set point of all vaccinated groups (1.6 × 104 vRNA copies/ml), and two of its animals (r04022 and r03115) had plasma viral loads below 103 vRNA copies/ml at weeks 8 and 12 postinfection (Fig. 8D and H). This outcome was quite surprising, given how unremarkable the cellular immune responses induced in the group 4 animals were. However, perhaps because of the smaller sample size of this group, the median set point viral load in group 4 was not statistically distinct from that of group 5 at the Bonferroni-Holm-corrected level (P = 0.018) (Fig. 8H).
We also evaluated whether combinations of TRIM5 alleles expressed by vaccinees in groups 1 to 4 impacted the outcome of this trial. The TRIM5 gene encodes a cytoplasmic protein (TRIM5α) that interacts with the HIV/SIV capsid early after infection and restricts viral replication (65). Multiple TRIM5 variants have been identified in rhesus macaques and can be grouped into three allelic classes: TRIM5CypA, TRIM5TFP, and TRIM5Q (43). Previous studies have shown that macaques expressing combinations of these alleles, particularly the TRIM5TFP/CypA and TRIM5TFP/TFP genotypes, tend to experience lower levels of viral replication than animals that are homozygous for TRIM5Q after infection with SIVsmE543-3 (43). In the present study, vaccinees that were positive for restrictive TRIM5 genotypes (TRIM5TFP/CypA and TRIM5TFP/TFP) had similar viral loads as those expressing more permissive (TRIM5Q/Q) and moderately restrictive (TRIM5Q/TFP and TRIM5Q/CypA) allele combinations (Table 1; Fig. 9A and B). Furthermore, expression of restrictive TRIM5 genotypes was not linked to the exceptionally high number of i.r. challenges required to infect the group 1 vaccinee rh2001 (TRIM5Q/Q) and the group 5 macaques r02076 (TRIM5Q/TFP) and r03111 (TRIM5Q/TFP). These results are consistent with previous reports showing that SIVmac239 is refractory to the antiviral effects of TRIM5α (43, 44).
FIG 9.
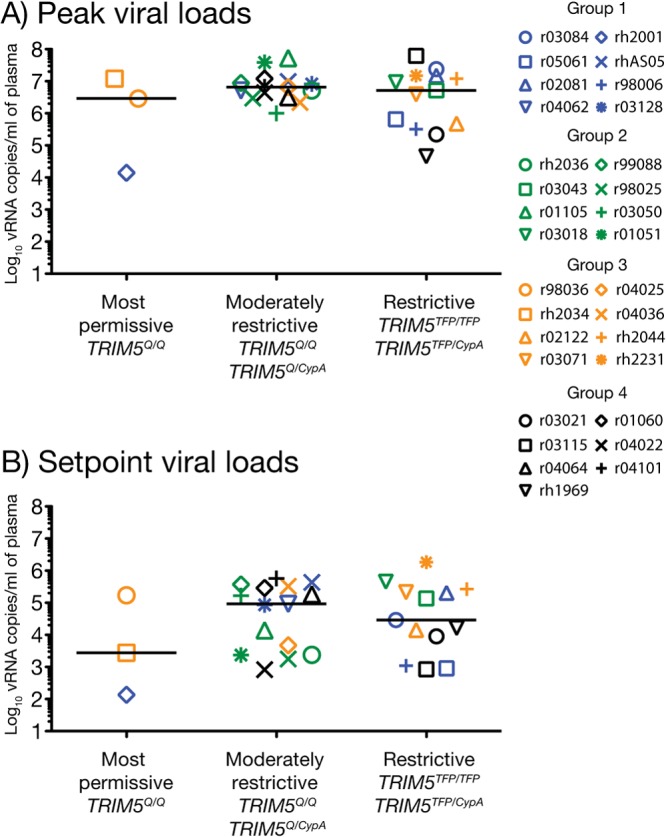
TRIM5 genotype did not affect peak or set point viremia among vaccinees in groups 1 to 4. The graphs show comparisons of peak (A) and set point (B) viral loads among vaccinees expressing TRIM5 alleles that are permissive (TRIM5Q/Q), moderately restrictive (TRIM5Q/TFP and TRIM5Q/CypA), or restrictive (TRIM5TFP/CypA and TRIM5TFP/TFP) for the replication of certain SIV strains. Viral loads were log transformed and correspond to the number of vRNA copies/ml of plasma. Groups 1, 2, 3, and 4 are color coded in blue, green, beige, and black, respectively. Lines represent medians, and each symbol denotes one vaccinee.
In sum, none of the vaccination regimens employed in groups 1 to 4 delayed acquisition of SIV infection or affected peak viremia. Vaccine-induced T-cell responses in groups 1 and 2 were partially effective in the chronic phase, affording a modest 1-log reduction in set point viral loads. Unexpectedly, macaques in group 4 also controlled viral replication at this stage, but the 1.6-log reduction in median set point achieved in this group was not significant after correcting for multiple comparisons. Even though the immunogenicity of the group 1 and group 2 regimens surpassed that obtained in group 4 by all T-cell measurements employed here (i.e., magnitude of IFN-γ-producing T cells, breadth, and ability to perform multiple immunological functions), these groups lowered viral loads to nearly the same extent. The similarity in these outcomes underscores our limited understanding of the T-cell correlates of virologic control of immunodeficiency virus infection.
Anamnestic responses and correlates analysis.
We evaluated anamnestic cellular responses by performing IFN-γ ELISPOT and ICS assays at day 14 postinfection. We found that vaccine-induced T-cell responses in all groups underwent robust expansion after infection, reaching levels that surpassed those measured in the vaccine phase (Fig. 2 and 10). The magnitude of these responses was considerably higher in PBMC than in CD8− PBMC, indicating a predominance of CD8+ T cells at this stage (Fig. 10A and B). Similar to the pattern observed during the vaccinations, Gag and Vif were preferentially targeted by the majority of the animals (Fig. 10A). Interestingly, the total frequency of SIV-specific T cells in groups 1 and 2 no longer outnumbered those in groups 3 and 4; instead, all groups had largely equivalent levels of anamnestic cellular responses after infection, except for a mild increase in the magnitude of the group 2 response in PBMC compared to that in groups 1 and 3 (Fig. 10B).
FIG 10.

Magnitude of anamnestic T-cell responses at day 14 post-SIV infection. We carried out IFN-γ ELISPOT assays on PBMC and CD8− PBMC at day 14 after SIV infection of each vaccinee in groups 1 to 4. The SFC/106 PBMC (or CD8− PBMC) values for each vaccine-encoded antigen (A) and the overall SIV-specific response (B) were calculated as described in the legend for Fig. 2. We compared the total magnitudes of SIV-specific T-cell responses among the four groups by using pairwise permutation tests. The threshold of significance for these comparisons was 0.0015 according to the Bonferroni-Holm adjustment. Groups 1, 2, 3, and 4 are color coded in blue, green, beige, and black, respectively. Lines represent medians, and each symbol denotes one vaccinee.
The ICS analysis confirmed that CD8+ T cells greatly outnumbered CD4+ T cells at this time point, likely due to the depletion of the latter subset caused by the high levels of viral replication that vaccinees experienced in the acute phase (Fig. 11). The functional profile of anamnestic CD8+ T-cell responses was similar among the four groups and resembled the phenotype observed during the vaccine phase, with the majority of the cells producing two or more functions (Fig. 11B, D, F, and H). One exception was that several animals in groups 1 to 4 mounted CD8+ T cells that stained positive for CD107a alone after infection; these cells were not detected after the rAd5 boost. As for CD4+ T cells, two functional signatures distinguished prechallenge and postchallenge responses. First, while IL-2-producing CD4+ T cells were easily detected after the rAd5 boost, they nearly disappeared following SIV infection (Fig. 11A, C, E, and G). This phenomenon might be due to a preferential depletion of this subtype, as IL-2-producing CD4+ T cells have been shown to be highly susceptible to lentivirus infection (66). Second, cytotoxic (CD107a+) CD4+ T cells, which were not detected during the vaccine phase, expanded to considerable levels in several group 1 and group 2 vaccinees after infection (Fig. 11A and C). This phenotype was typically accompanied by the simultaneous production of IFN-γ and TNF-α. Indeed, IFN-γ+ TNF-α+ CD107a+ CD4+ T cells were detected in half of the group 2 vaccinees at day 14 postinfection (Fig. 11C). Of note, a few investigators have reported cytolytic CD4+ T cells in HIV-1-infected patients, and these responses have recently been linked to improved control of HIV replication and delayed progression to AIDS (67–69). However, the magnitude of SIV-specific CD4+ T cells that stained positive for CD107a did not correlate with either peak or set point viral loads in the present study (data not shown).
FIG 11.

Functional profile of anamnestic T-cell responses. We carried out multiparameter ICS assays at day 14 postinfection to determine the ability of anamnestic SIV-specific T cells to degranulate (i.e., CD107a expression) and produce cytokines (IFN-γ, TNF-α, and IL-2). The antigen stimuli for these assays consisted of peptide mixtures spanning (i) amino acids 1 to 291 and (ii) 281 to 510 of the Gag polyprotein, (iii) the Vif ORF, and (iv) the Nef ORF. Each symbol denotes one vaccinee and corresponds to the total frequency of Gag-, Vif-, and Nef-specific T cells capable of producing each combination of functions tested. Panels on the left correspond to CD4+ T-cell responses in group 1 (A), group 2 (C), group 3 (E), and group 4 (G). Panels on the right correspond to CD8+ T-cell responses in group 1 (B), group 2 (D), group 3 (F), and group 4 (H). Groups 1, 2, 3, and 4 are color coded in blue, green, beige, and black, respectively.
Since vaccination may have altered the immunodominance hierarchy of the Gag181–189CM9 and Vif100–109VL10 epitopes in some of the Mamu-A*01+ vaccinees, we examined the expansion kinetics of vaccine-induced CD8+ T-cell responses to these peptides after infection. As a reference, we carried out this analysis early after infection of the four unvaccinated Mamu-A*01+ macaques in group 5 and confirmed that CD8+ T cells directed against Gag181–189CM9 dominated over those specific for Vif100–109VL10 at all time points investigated (Fig. 12A). At weeks 2 and 3 postinfection, all Mamu-A*01+ vacccinees mounted recall CD8+ T-cell responses to Gag181–189CM9, albeit over a wide range of frequencies (Fig. 12B to E). In the group 2 macaques, for instance, responses against this immunodominant epitope exceeded 10,000 SFC/106 PBMC (Fig. 12C), while r03084 in group 1 had a peak Gag181–189CM9-specific CD8+ T-cell response of <2,000 SFC/106 PBMC (Fig. 12B). Animal r05061 in group 1 and the group 4 vaccinee r03115 also responded less robustly to this epitope (Fig. 12B and E). The fact that r03084, r05061, and r03115 had Gag181–189CM9-specific CD8+ T cells below the limits of detection at the time of challenge might explain their lower anamnestic responses against this epitope (Fig. 6A and D). Interestingly, CD8+ T cells to the Vif100–109VL10 epitope adopted two patterns of expansion after infection: they either matched or surpassed the size of their contemporaneous Gag181–189CM9-specific response, as seen in both group 1 animals (Fig. 12B), the group 2 macaque rh2036 (Fig. 12C), rh2034 in group 3 (Fig. 12D), and r03021 in group 4 (Fig. 12E), or underwent limited proliferation, reverting to their typical subdominant status (e.g., r03043, r98036, and r03115 in groups 2, 3, and 4 respectively) (Fig. 12C to E). The magnitude of these responses was also related to their frequency before infection, as the former animals had the highest numbers of Vif100–109VL10-specific CD8+ T cells at the time of the first SIV challenge (Fig. 6). These results demonstrate that vaccine-induced CD8+ T cells directed against a subdominant epitope can expand to high levels after SIV infection. However, the magnitude of the anamnestic Vif100–109VL10-specific CD8+ T-cell response did not seem to affect acute-phase viremia in the present study; except for r05061 (group 1) and r03021 (group 4), the remaining vaccinees mounting high frequencies of these Vif-specific CD8+ T cells experienced peak viral loads that exceeded 106 vRNA copies/ml (Fig. 8G). Furthermore, the magnitude of vaccine-induced Vif100–109VL10-specific CD8+ T cells measured at the day of the first challenge did not correlate with set point viral loads in Mamu-A*01+ vaccinees (see Fig. S4A in the supplemental material).
FIG 12.

Magnitude of Gag181–189CM9- and Vif100–109VL10-specific CD8+ T cells in Mamu-A*01+ macaques after SIV infection. We measured the frequencies of CD8+ T cells directed against the Gag181–189CM9 and Vif100–109VL10 epitopes by carrying out IFN-γ ELISPOT assays on PBMC from Mamu-A*01+ macaques at several time points after infection. To confirm the immunodominant and subdominant ranks of the Gag181–189CM9 and Vif100–109VL10 epitopes, respectively, we performed this analysis on the four unvaccinated Mamu-A*01+ animals in group 5 at weeks 2 and 3 after infection of r02108 and rh2306, weeks 4 and 6 after infection of r03116, and weeks 6 and 8 after infection of r03141 (A). For vaccinees in group 1 (B), group 2 (C), group 3 (D), and group 4 (E), we measured the frequencies of anamnestic responses against these epitopes at weeks 2 and 3 postinfection. Bar graphs indicate the magnitude of IFN-γ-producing cells (in SFC/106 PBMC). Groups 1, 2, 3, and 4 are color coded in blue, green, beige, and black, respectively.
Lastly, we carried out a correlates analysis in an attempt to understand the basis of the partial virologic control reported here. Since none of the vaccine regimens significantly reduced the acute-phase viremia compared to the control group, we focused on set point viral loads only. For this analysis, we selected nine immunological variables that were measured either in the vaccine phase or after the animals became infected (day 14 postinfection). These variables included the total magnitude and breadth of vaccine-induced T-cell responses, as determined by IFN-γ ELISPOT assays on PBMC and CD8− PBMC, and the percentage of polyfunctional CD4+ and CD8+ T-cell responses, defined as cells staining positive for at least three functions. Of note, we could not analyze whether the number of CD4+ or CD8+ T-cell responses elicited by vaccination was associated with virologic control, because our approach to determine T-cell breadth was based on IFN-γ production by PBMC in ELISPOT assays. Strikingly, none of the immunological parameters mentioned above correlated with set point viremia (Table 3). Moreover, the breadth of Gag-, Vif-, and Nef-specific T-cell responses measured after vaccination and the total number of responses recalled during the acute phase of infection were not associated with set point viral loads either (see Fig. S4B to E). Together, these results demonstrate that neither the breadth nor magnitude nor polyfunctionality of vaccine-induced, SIV-specific T-cell responses dictated the outcome of this trial.
DISCUSSION
In this study, we attempted to overcome immunodominance and ultimately improve the breadth of a T-cell-based SIV vaccine by delivering immunogens as minigenes rather than full-length ORFs. Since coexpression of dominant and subdominant epitopes within the same APC appears to be important for immunodominance (70, 71), dividing these epitopes by gene fragmentation has been shown to induce broadly targeted cellular immunity in mice (32, 35–37). We designed nine minigenes encoding fragments of the SIVmac239 Gag, Vif, and Nef proteins and delivered them to rhesus macaques by using combinations of rBCG, EP rDNA plus pIL-12, and rYF17D vectors followed by a common rAd5 boost. The group 1 (EP rDNA plus pIL-12/rAd5) and group 2 (rBCG/EP rDNA plus pIL-12/rYF17D/rAd5) regimens achieved the best immunogenicity and induced broad and abundant cellular immune responses. The group 3 (rYF17D/rAd5) and group 4 (rAd5) vaccine modalities also elicited SIV-specific T-cell responses, but their levels were significantly lower. The superior immunogenicity of the group 1 and group 2 regimens stemmed from immunizing these animals with rDNA delivered by electroporation, an extremely effective way to engender T-cell immunity (61). Notably, EP rDNA has been shown to elicit broad and high-frequency cellular immune responses in humans, even without the benefit of a heterologous viral vector boost (59). Moreover, the ability of EP rDNA to induce T-cell responses can be further increased by the coadministration of molecular adjuvants, such as the IL-12-expressing plasmid used here (46, 55, 60). These observations corroborate the efficiency of electroporated rDNA immunizations to prime cellular immune responses.
Of note, we did not formally address whether the gene fragmentation strategy employed here improved the overall breadth of vaccine-induced T-cell responses compared to delivering the Gag, Vif, and Nef immunogens as whole ORFs. This would require a head-to-head comparison of the immunogenicities of each of the four vaccine regimens tested in groups 1 to 4 with those achieved by the same vector combinations encoding full-length Gag, Vif, and Nef, a prohibitively expensive experiment. However, inferences can be made from past studies conducted in our laboratory that used similar vaccine modalities encoding full-length, codon-optimized SIVmac239 antigens. For example, rDNA priming (delivered by conventional intramuscular injection) followed by a rAd5 boost encoding the entire SIVmac239 proteome (except for Env) elicited a median of 8 Gag-specific T-cell responses (20). Critically, the number of these responses was calculated according to the same approach we used to determine the T-cell breadth in the present study and, for this comparison, we only considered epitopes lying within the same regions covered by the six Gag minigenes. We found that this number was significantly lower than the median of 11 epitopes recognized by the group 1 and group 2 vaccinees in the current study (P < 0.05). An important caveat of this analysis is the difference in the way the rDNA vectors were delivered in the two trials, which makes it difficult to conclude whether the superior breadth achieved by the group 1 and group 2 regimens was due to the gene fragmentation strategy and/or the fact that the rDNA constructs were administered via electroporation. In this regard, we carried out a more straightforward analysis by comparing the number of Gag-specific T-cell responses induced in the group 4 animals with that achieved by another rAd5-based vaccine reported recently (49). While the Gag immunogen was delivered to group 4 macaques in fragments through six distinct rAd5 vectors, only one rAd5 construct expressing full-length Gag was given to animals in the study in question. We found that vaccinees in both cases recognized a similar number of Gag pools covering the same regions as the minigenes (P = 0.77). Also, the breadth of Gag-directed responses was indistinguishable between group 4 and recipients of a homologous rAd5 prime-boost regimen aimed at recapitulating the immunization protocol employed in the Step trial (P = 0.95) (72). It should be noted that these comparisons still suffer from caveats, such as differences in the total number of immunogens chosen for each study and potential competition between epitopes located within rAd5-derived proteins and the SIV inserts. Notwithstanding these limitations, these data imply that the gene fragmentation strategy tested here did not significantly expand the breadth of vaccine-induced T-cell responses and that priming with EP rDNA was the decisive factor for the broad T-cell repertoire elicited in groups 1 and 2.
A related goal was to determine whether gene fragmentation was able to engender responses to epitopes that are subdominant during primary SIVmac239 infection. For this analysis, we monitored if Mamu-A*01+ vaccinees in groups 1 to 4 developed CD8+ T cells to five Mamu-A*01-restricted epitopes with previously defined immunodominance hierarchies (62, 63). In addition to the dominant Gag181–189CM9 determinant, only one subdominant epitope (Vif100–109VL10) was consistently recognized by the Mamu-A*01+ vaccinees. Despite some variability in their expansion kinetics, CD8+ T cells targeting these epitopes peaked at similar levels within each Mamu-A*01+ macaque in groups 2 to 4. The group 1 vaccinees were an exception, as they had barely detectable CD8+ T cells targeting Gag181–189CM9, while their contemporaneous Vif100–109VL10-specific responses reached modest to high frequencies. This analysis emphasizes the difficulty of overcoming immunodominance by vaccination. Likewise, a recent attempt by our group to induce epitope-specific subdominant CD8+ T cells by using both novel and traditional vector platforms also met with limited success (73). Thus, maneuvers other than gene fragmentation may be required to significantly alter immunodominance hierarchies of vaccine-induced CD8+ T-cell responses in primates.
The inability of rAd5 to boost Gag181–189CM9-specific CD8+ T cells in the two Mamu-A*01+ macaques in group 1 was intriguing, since these responses underwent robust expansion in MHC class I-matched vaccinees in groups 2 and 3. This unusual pattern may be due to the lymphopenia experienced by group 1 animals after their third EP rDNA immunization, which was associated with the large dose of the IL-12-expressing plasmid delivered at that occasion. Given that exogenous administration of recombinant IL-12 has been shown to abrogate immunodominance of CD8+ T-cell responses in mice (74), it is possible that the overdose of IL-12 produced shortly after the last EP rDNA immunization perturbed the immunodominant rank of the Gag181–189CM9-specific CD8+ T-cell response elicited in the Mamu-A*01+ vaccinees in group 1.
We determined vaccine efficacy by challenging all animals in groups 1 to 4, plus eight unvaccinated control macaques (group 5), with repeated, titered inoculations of SIVmac239 via the intrarectal route. This regimen aimed at simulating clinically relevant human exposures to HIV-1 through anal receptive intercourse (64). Our efficacy endpoints included reductions in peak and set point viral loads compared to those in group 5. Although our study was not adequately powered to detect an effect on SIV acquisition rates, we conducted this analysis anyway and found no difference between vaccinated and control groups. Unfortunately, none of the four vaccine regimens tested here affected peak viremia. It should be noted, however, that it is exceedingly difficult to contain acute-phase viral replication following rectal transmission of SIV, and presumably HIV-1. Even in experimentally controlled settings, in which SIV inoculations are performed atraumatically, viral particles can rapidly penetrate the rectal epithelia and disseminate to other organs. Indeed, Ribeiro dos Santos et al. recently reported that SIV can reach mucosal lymphoid aggregates and colic lymph nodes as early as 4 h after challenge (75). Alarmingly, PBMC-associated SIV DNA was detected 20 h later. Given this fast entry kinetics, AIDS vaccine approaches should elicit immune responses not only at mucosal sites of transmission but also in lymph nodes where virus amplification can occur.
Vaccinees in groups 1 and 2 significantly reduced set point viral loads by 1 log compared to group 5. This reduction was more pronounced in group 1. Of note, Winstone et al. recently reported robust protection in macaques vaccinated with an EP rDNA plus pIL-12 prime followed by a rAd5 boost encoding the entire SIVmac239 proteome (60). After repeated intrarectal challenges with the same SIVmac239 stock and dose used in the present study, vaccinees decreased peak and set point viral loads by 2.6 and 4.4 logs, respectively, compared to the mock-immunized group. Moreover, two immunized animals remained uninfected after 13 challenges. This level of control is substantially better than the outcome observed in groups 1 and 2 here. Variations in the dose of pIL-12 given to the group 1 macaques and the additional recombinant vectors used in group 2 may have contributed to these divergent outcomes. However, a more likely explanation for the positive results reported by Winstone et al. is that their vaccine encoded an envelope protein that was identical in amino acid sequence to that of the challenge virus. Consequently, in addition to mounting cellular responses directed against all viral proteins, those vaccinees were also equipped with Env-specific antibodies that probably provided an extra line of defense against the virus. While these data suggest that combining cellular and humoral immunity can improve the efficacy of HIV-1 vaccine regimens, engendering these responses will require novel vector platforms in light of the futility of all rAd5-based human vaccines tested so far (2, 4, 5).
The group 1 and group 2 vaccine regimens generated high frequencies of SIV-specific CD4+ T cells, which participate in key immunological processes, including antibody production and generation of CD8+ T-cell responses (76–78). Interestingly, we observed that vaccine-elicited CD4+ T cells in groups 1 and 2 demonstrated different functional profiles before and after SIV infection. While the majority of SIV-specific CD4+ T cells elaborated IL-2 following the rAd5 boost, this subset was virtually absent at day 14 postinfection. This change in phenotype was likely due to the elimination of IL-2-secreting CD4+ T cells after infection, since production of this cytokine has been associated with a higher susceptibility to HIV replication (66). We also noticed that a considerable fraction of the anamnestic SIV-specific CD4+ T-cell responses expanding after infection of group 1 and group 2 vaccinees comprised CD107a+ CD4+ T cells. Strikingly, CD4+ T cells displaying this cytolytic phenotype were not detected during the vaccine phase. Of note, similar phenotypic changes in CD4+ T-cell responses have been observed in one human subject who was vaccinated with a recombinant canarypox vector and later acquired HIV-1 infection (79). Notably, Soghoian et al. recently described cytolytic virus-specific CD4+ T cells during acute HIV-1 infection (69). Further characterization of these cells revealed that they were capable of inhibiting HIV replication in vitro and were associated with delayed progression to AIDS. However, in contrast to these findings, the magnitude of SIV-specific CD107a+ CD4+ T-cell responses in the present study did not correlate with improved outcome after infection (data not shown). Overall, these results have implications for the evaluation of CD4+ T-cell responses induced by HIV-1 vaccine strategies, as the phenotype of these responses might change early after infection.
The single rAd5 immunization delivered to group 4 macaques also afforded a modest (1.6-log) reduction in their median set point compared to that in group 5, but this difference was not significant after correcting for multiple comparisons. This outcome was rather surprising, given that group 4 vaccinees developed the lowest levels of SIV-specific cellular responses after vaccination. While homologous rAd5 vaccination has been largely ineffective in stringent monkey trials (18, 72, 80), including more antigens to this regimen, in addition to Gag, has been shown to result in more favorable outcomes (19, 20). Thus, the fact that group 4 animals were vaccinated not only with Gag but also with Vif and Nef sequences might explain their partial control of chronic-phase viremia. Importantly, since the rAd5 immunization was the only source of SIV-specific T-cell responses in these animals, group 4 can serve as a reference to evaluate the contribution of the additional immunizations used in groups 1 to 3 to their outcome after challenge. Notably, the median peak and set point viral loads in groups 1 to 3 were similar, if not superior, to those in group 4. The proximity of these outcomes indicates that the rBCG, EP rDNA plus pIL-12, and rYF17D vectors administered to vaccinees in groups 1 to 3, despite augmenting their overall SIV-specific T-cell responses, did not enhance their ability to control viral replication. Although larger sample sizes would be required to validate this hypothesis, our data suggest that increasing the magnitude, breadth, and functional heterogeneity of vaccine-induced T-cell responses does not necessarily translate into greater antiviral activity. Likewise, none of the nine immunological parameters included in our correlates analysis was associated with control of chronic-phase viremia, confirming that conventional T-cell measurements are poor predictors of vaccine efficacy against AIDS virus infection (21, 81). Indeed, the homologous rAd5 regimens employed in the STEP and Phambili trials and the rDNA prime/rAd5 boost assessed in the recent HVTN 505 study did not afford any protection against HIV-1 infection and yet induced acceptable levels of T-cell responses in the majority of vaccinees (2, 4, 5, 82). Collectively, these results stress the difficulty of harnessing the potential of cellular immunity in vaccine strategies without knowledge of the immunological correlates of T-cell-mediated control of AIDS virus replication.
In conclusion, here we tested whether vaccinating rhesus macaques with minigenes expressing fragments of Gag, Vif, and Nef would result in broadly targeted T-cell responses capable of controlling SIV replication. We delivered these minigenes through combinations of rBCG, EP rDNA plus pIL-12, rYF17D, and rAd5 vectors arranged in four distinct immunization protocols. Vaccinees in groups 1 and 2, who were primed with EP rDNA plus pIL-12, developed the highest levels of SIV-specific T-cell responses. A close examination of CD8+ T-cell responses elicited in Mamu-A*01+ animals and comparisons between the present experiment and previous SIV efficacy trials conducted in our laboratory suggested that the gene fragmentation strategy employed here had only a minor impact on immunodominance. Unfortunately, none of the four vaccine regimens decreased peak viremia after repeated mucosal challenges with SIVmac239, but a modest (1-log) reduction in set point viral loads was observed in groups 1, 2, and possibly 4. These findings are relevant for the development and testing of new HIV-1 vaccine modalities, since they demonstrate that broad, high-frequency, and polyfunctional T-cell responses elicited by conventional vector platforms may not be sufficient to substantially decrease AIDS virus replication.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded in part by Public Health Service grants R01 AI076114 and R01 AI049120 from the National Institute of Allergy and Infectious Diseases awarded to D.I.W., Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and FIOCRUZ. In addition, this work was supported by grant P51 RR000167 from the National Center for Research Resources to the Wisconsin National Primate Research Center at the University of Wisconsin—Madison and in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. Additional support was provided by the International AIDS Vaccine Initiative (IAVI) and its generous supporters, including the United States Agency for International Development (USAID); the Bill and Melinda Gates Foundation; the Ministry of Foreign Affairs of Denmark; Irish Aid; the Ministry of Finance of Japan; the Ministry of Foreign Affairs of the Netherlands; the Norwegian Agency for Development Cooperation (NORAD); and the United Kingdom Department for International Development (DFID). This research was conducted in part at a facility constructed with support from Research Facilities Improvement Program grants RR15459 and RR020141.
We thank Jerald C. Sadoff for providing the rBCG constructs used in this study. We are also thankful to Barbara Felber and George Pavlakis for providing the AG157 plasmid used in the electroporated rDNA vaccinations. We are also grateful to Chrystal Glidden, Gretta Borchardt, and Debra Fisk for MHC and TRIM5 typing of animals, and Saverio Capuano III, Eric Peterson, and Kristin Crosno for providing excellent care of the rhesus macaques used in this study. We thank the AIDS Research and Reference Reagent Program for providing the SIVmac239 peptides used in our immunological assays.
Footnotes
Published ahead of print 16 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00601-14.
REFERENCES
- 1.Joint United Nations Programme on HIV/AIDS. 2013. Global report: UNAIDS report on the global AIDS epidemic. UNAIDS Programme, Geneva, Switzerland: http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf [Google Scholar]
- 2.Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, Gilbert PB, Lama JR, Marmor M, Del Rio C, McElrath MJ, Casimiro DR, Gottesdiener KM, Chodakewitz JA, Corey L, Robertson MN. 2008. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 372:1881–1893. 10.1016/S0140-6736(08)61591-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flynn NM, Forthal DN, Harro CD, Judson FN, Mayer KH, Para MF. 2005. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 191:654–665. 10.1086/428404 [DOI] [PubMed] [Google Scholar]
- 4.Gray GE, Allen M, Moodie Z, Churchyard G, Bekker LG, Nchabeleng M, Mlisana K, Metch B, de Bruyn G, Latka MH, Roux S, Mathebula M, Naicker N, Ducar C, Carter DK, Puren A, Eaton N, McElrath MJ, Robertson M, Corey L, Kublin JG. 2011. Safety and efficacy of the HVTN 503/Phambili study of a clade-B-based HIV-1 vaccine in South Africa: a double-blind, randomised, placebo-controlled test-of-concept phase 2b study. Lancet Infect. Dis. 11:507–515. 10.1016/S1473-3099(11)70098-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammer SM, Sobieszczyk ME, Janes H, Karuna ST, Mulligan MJ, Grove D, Koblin BA, Buchbinder SP, Keefer MC, Tomaras GD, Frahm N, Hural J, Anude C, Graham BS, Enama ME, Adams E, DeJesus E, Novak RM, Frank I, Bentley C, Ramirez S, Fu R, Koup RA, Mascola JR, Nabel GJ, Montefiori DC, Kublin J, McElrath MJ, Corey L, Gilbert PB. 2013. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N. Engl. J. Med. 369:2083–2092. 10.1056/NEJMoa1310566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pitisuttithum P, Gilbert P, Gurwith M, Heyward W, Martin M, van Griensven F, Hu D, Tappero JW, Choopanya K. 2006. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J. Infect. Dis. 194:1661–1671. 10.1086/508748 [DOI] [PubMed] [Google Scholar]
- 7.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361:2209–2220. 10.1056/NEJMoa0908492 [DOI] [PubMed] [Google Scholar]
- 8.Burton DR, Ahmed R, Barouch DH, Butera ST, Crotty S, Godzik A, Kaufmann DE, McElrath MJ, Nussenzweig MC, Pulendran B, Scanlan CN, Schief WR, Silvestri G, Streeck H, Walker BD, Walker LM, Ward AB, Wilson IA, Wyatt R. 2012. A blueprint for HIV vaccine discovery. Cell Host Microbe 12:396–407. 10.1016/j.chom.2012.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burton DR, Desrosiers RC, Doms RW, Koff WC, Kwong PD, Moore JP, Nabel GJ, Sodroski J, Wilson IA, Wyatt RT. 2004. HIV vaccine design and the neutralizing antibody problem. Nat. Immunol. 5:233–236. 10.1038/ni0304-233 [DOI] [PubMed] [Google Scholar]
- 10.Allen TM, Altfeld M, Geer SC, Kalife ET, Moore C, O'sullivan KM, Desouza I, Feeney ME, Eldridge RL, Maier EL, Kaufmann DE, Lahaie MP, Reyor L, Tanzi G, Johnston MN, Brander C, Draenert R, Rockstroh JK, Jessen H, Rosenberg ES, Mallal SA, Walker BD. 2005. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J. Virol. 79:13239–13249. 10.1128/JVI.79.21.13239-13249.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goulder PJ, Watkins DI. 2004. HIV and SIV CTL escape: implications for vaccine design. Nat. Rev. Immunol. 4:630–640. 10.1038/nri1417 [DOI] [PubMed] [Google Scholar]
- 12.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53. 10.1038/nm1520 [DOI] [PubMed] [Google Scholar]
- 13.Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68:4650–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, Irwin CE, Safrit JT, Mittler J, Weinberger L, Kostrikis LG, Zhang L, Perelson AS, Ho DD. 1999. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189:991–998. 10.1084/jem.189.6.991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matano T, Shibata R, Siemon C, Connors M, Lane HC, Martin MA. 1998. Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J. Virol. 72:164–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak MJ, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. 10.1038/nature10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawada M, Tsukamoto T, Yamamoto H, Iwamoto N, Kurihara K, Takeda A, Moriya C, Takeuchi H, Akari H, Matano T. 2008. Gag-specific cytotoxic T-lymphocyte-based control of primary simian immunodeficiency virus replication in a vaccine trial. J. Virol. 82:10199–10206. 10.1128/JVI.01103-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu J, O'Brien KL, Lynch DM, Simmons NL, La Porte A, Riggs AM, Abbink P, Coffey RT, Grandpre LE, Seaman MS, Landucci G, Forthal DN, Montefiori DC, Carville A, Mansfield KG, Havenga MJ, Pau MG, Goudsmit J, Barouch DH. 2009. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature 457:87–91. 10.1038/nature07469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson NA, Reed J, Napoe GS, Piaskowski S, Szymanski A, Furlott J, Gonzalez EJ, Yant LJ, Maness NJ, May GE, Soma T, Reynolds MR, Rakasz E, Rudersdorf R, McDermott AB, O'Connor DH, Friedrich TC, Allison DB, Patki A, Picker LJ, Burton DR, Lin J, Huang L, Patel D, Heindecker G, Fan J, Citron M, Horton M, Wang F, Liang X, Shiver JW, Casimiro DR, Watkins DI. 2006. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J. Virol. 80:5875–5885. 10.1128/JVI.00171-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson NA, Keele BF, Reed JS, Piaskowski SM, MacNair CE, Bett AJ, Liang X, Wang F, Thoryk E, Heidecker GJ, Citron MP, Huang L, Lin J, Vitelli S, Ahn CD, Kaizu M, Maness NJ, Reynolds MR, Friedrich TC, Loffredo JT, Rakasz EG, Erickson S, Allison DB, Piatak MJ, Lifson JD, Shiver JW, Casimiro DR, Shaw GM, Hahn BH, Watkins DI. 2009. Vaccine-induced cellular responses control simian immunodeficiency virus replication after heterologous challenge. J. Virol. 83:6508–6521. 10.1128/JVI.00272-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koup RA, Graham BS, Douek DC. 2011. The quest for a T cell-based immune correlate of protection against HIV: a story of trials and errors. Nat. Rev. Immunol. 11:65–70. 10.1038/nri2890 [DOI] [PubMed] [Google Scholar]
- 22.Masopust D, Picker LJ. 2012. Hidden memories: frontline memory T cells and early pathogen interception. J. Immunol. 188:5811–5817. 10.4049/jimmunol.1102695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranasinghe S, Flanders M, Cutler S, Soghoian DZ, Ghebremichael M, Davis I, Lindqvist M, Pereyra F, Walker BD, Heckerman D, Streeck H. 2012. HIV-specific CD4 T cell responses to different viral proteins have discordant associations with viral load and clinical outcome. J. Virol. 86:277–283. 10.1128/JVI.05577-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adland E, Carlson JM, Paioni P, Kloverpris H, Shapiro R, Ogwu A, Riddell L, Luzzi G, Chen F, Balachandran T, Heckerman D, Stryhn A, Edwards A, Ndung'u T, Walker BD, Buus S, Goulder P, Matthews PC. 2013. Nef-specific CD8+ T cell responses contribute to HIV-1 immune control. PLoS One 8:e73117. 10.1371/journal.pone.0073117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwamoto N, Takahashi N, Seki S, Nomura T, Yamamoto H, Inoue M, Shu T, Naruse TK, Kimura A, Matano T. 2014. Control of simian immunodeficiency virus replication by vaccine-induced Gag- and Vif-specific CD8+ T cells. J. Virol. 88:425–433. 10.1128/JVI.02634-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martins MA, Wilson NA, Reed JS, Ahn CD, Klimentidis YC, Allison DB, Watkins DI. 2010. T-cell correlates of vaccine efficacy after a heterologous simian immunodeficiency virus challenge. J. Virol. 84:4352–4365. 10.1128/JVI.02365-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mudd PA, Martins MA, Ericsen AJ, Tully DC, Power KA, Bean AT, Piaskowski SM, Duan L, Seese A, Gladden AD, Weisgrau KL, Furlott JR, Kim YI, Veloso de Santana MG, Rakasz E, Capuano S, III, Wilson NA, Bonaldo MC, Galler R, Allison DB, Piatak MJ, Haase AT, Lifson JD, Allen TM, Watkins DI. 2012. Vaccine-induced CD8+ T cells control AIDS virus replication. Nature 491:129–133. 10.1038/nature11443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barouch DH. 2008. Challenges in the development of an HIV-1 vaccine. Nature 455:613–619. 10.1038/nature07352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McElrath MJ, Haynes BF. 2010. Induction of immunity to human immunodeficiency virus type-1 by vaccination. Immunity 33:542–554. 10.1016/j.immuni.2010.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watkins DI, Burton DR, Kallas EG, Moore JP, Koff WC. 2008. Nonhuman primate models and the failure of the Merck HIV-1 vaccine in humans. Nat. Med. 14:617–621. 10.1038/nm.f.1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yewdell JW. 2006. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity 25:533–543. 10.1016/j.immuni.2006.09.005 [DOI] [PubMed] [Google Scholar]
- 32.Im EJ, Hong JP, Roshorm Y, Bridgeman A, Letourneau S, Liljestrom P, Potash MJ, Volsky DJ, McMichael AJ, Hanke T. 2011. Protective efficacy of serially up-ranked subdominant CD8+ T cell epitopes against virus challenges. PLoS Pathog. 7:e1002041. 10.1371/journal.ppat.1002041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Ewald BA, Lynch DM, Nanda A, Sumida SM, Barouch DH. 2006. Modulation of DNA vaccine-elicited CD8+ T-lymphocyte epitope immunodominance hierarchies. J. Virol. 80:11991–11997. 10.1128/JVI.01348-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mok H, Lee S, Wright DW, Crowe JEJ. 2008. Enhancement of the CD8+ T cell response to a subdominant epitope of respiratory syncytial virus by deletion of an immunodominant epitope. Vaccine 26:4775–4782. 10.1016/j.vaccine.2008.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez F, Harkins S, Slifka MK, Whitton JL. 2002. Immunodominance in virus-induced CD8+ T-cell responses is dramatically modified by DNA immunization and is regulated by gamma interferon. J. Virol. 76:4251–4259. 10.1128/JVI.76.9.4251-4259.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Li F, Liu Y, Hong K, Meng X, Chen J, Zhang Z, Huo Z, Sun M, Self SG, Shao Y. 2011. HIV fragment gag vaccine induces broader T cell response in mice. Vaccine 29:2582–2589. 10.1016/j.vaccine.2011.01.049 [DOI] [PubMed] [Google Scholar]
- 37.Singh RA, Barry MA. 2004. Repertoire and immunofocusing of CD8 T cell responses generated by HIV-1 gag-pol and expression library immunization vaccines. J. Immunol. 173:4387–4393 http://www.jimmunol.org/cgi/content/abstract/173/7/4387 [DOI] [PubMed] [Google Scholar]
- 38.Kaizu M, Borchardt GJ, Glidden CE, Fisk DL, Loffredo JT, Watkins DI, Rehrauer WM. 2007. Molecular typing of major histocompatibility complex class I alleles in the Indian rhesus macaque which restrict SIV CD8+ T cell epitopes. Immunogenetics 59:693–703. 10.1007/s00251-007-0233-7 [DOI] [PubMed] [Google Scholar]
- 39.Mothe BR, Weinfurter J, Wang C, Rehrauer W, Wilson N, Allen TM, Allison DB, Watkins DI. 2003. Expression of the major histocompatibility complex class I molecule Mamu-A*01 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 77:2736–2740. 10.1128/JVI.77.4.2736-2740.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang ZQ, Fu TM, Casimiro DR, Davies ME, Liang X, Schleif WA, Handt L, Tussey L, Chen M, Tang A, Wilson KA, Trigona WL, Freed DC, Tan CY, Horton M, Emini EA, Shiver JW. 2002. Mamu-A*01 allele-mediated attenuation of disease progression in simian-human immunodeficiency virus infection. J. Virol. 76:12845–12854. 10.1128/JVI.76.24.12845-12854.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loffredo JT, Maxwell J, Qi Y, Glidden CE, Borchardt GJ, Soma T, Bean AT, Beal DR, Wilson NA, Rehrauer WM, Lifson JD, Carrington M, Watkins DI. 2007. Mamu-B*08-positive macaques control simian immunodeficiency virus replication. J. Virol. 81:8827–8832. 10.1128/JVI.00895-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yant LJ, Friedrich TC, Johnson RC, May GE, Maness NJ, Enz AM, Lifson JD, O'Connor DH, Carrington M, Watkins DI. 2006. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J. Virol. 80:5074–5077. 10.1128/JVI.80.10.5074-5077.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, Kaur A, Hirsch VM, Johnson WE. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 8:e1000462. 10.1371/journal.pbio.1000462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reynolds MR, Sacha JB, Weiler AM, Borchardt GJ, Glidden CE, Sheppard NC, Norante FA, Castrovinci PA, Harris JJ, Robertson HT, Friedrich TC, McDermott AB, Wilson NA, Allison DB, Koff WC, Johnson WE, Watkins DI. 2011. The TRIM5α genotype of rhesus macaques affects acquisition of simian immunodeficiency virus SIVsmE660 infection after repeated limiting-dose intrarectal challenge. J. Virol. 85:9637–9640. 10.1128/JVI.05074-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosati M, von Gegerfelt A, Roth P, Alicea C, Valentin A, Robert-Guroff M, Venzon D, Montefiori DC, Markham P, Felber BK, Pavlakis GN. 2005. DNA vaccines expressing different forms of simian immunodeficiency virus antigens decrease viremia upon SIVmac251 challenge. J. Virol. 79:8480–8492. 10.1128/JVI.79.13.8480-8492.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jalah R, Patel V, Kulkarni V, Rosati M, Alicea C, Ganneru B, von Gegerfelt A, Huang W, Guan Y, Broderick KE, Sardesai NY, LaBranche C, Montefiori DC, Pavlakis GN, Felber BK. 2012. IL-12 DNA as molecular vaccine adjuvant increases the cytotoxic T cell responses and breadth of humoral immune responses in SIV DNA vaccinated macaques. Hum. Vaccine Immunother. 8:1620–1629. 10.4161/hv.21407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson RD, Haskell RE, Xia H, Roessler BJ, Davidson BL. 2000. A simple method for the rapid generation of recombinant adenovirus vectors. Gene Ther. 7:1034–1038. 10.1038/sj.gt.3301197 [DOI] [PubMed] [Google Scholar]
- 48.Bonaldo MC, Mello SM, Trindade GF, Rangel AA, Duarte AS, Oliveira PJ, Freire MS, Kubelka CF, Galler R. 2007. Construction and characterization of recombinant flaviviruses bearing insertions between E and NS1 genes. Virol. J. 4:115. 10.1186/1743-422X-4-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martins MA, Bonaldo MC, Rudersdorf RA, Piaskowski SM, Rakasz EG, Weisgrau KL, Furlott JR, Eernisse CM, Veloso de Santana MG, Hidalgo B, Friedrich TC, Chiuchiolo MJ, Parks CL, Wilson NA, Allison DB, Galler R, Watkins DI. 2013. Immunogenicity of seven new recombinant yellow fever viruses 17D expressing fragments of SIVmac239 Gag, Nef, and Vif in Indian rhesus macaques. PLoS One 8:e54434. 10.1371/journal.pone.0054434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cline AN, Bess JW, Piatak MJ, Lifson JD. 2005. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J. Med. Primatol. 34:303–312. 10.1111/j.1600-0684.2005.00128.x [DOI] [PubMed] [Google Scholar]
- 51.Robertson MJ, Cameron C, Atkins MB, Gordon MS, Lotze MT, Sherman ML, Ritz J. 1999. Immunological effects of interleukin 12 administered by bolus intravenous injection to patients with cancer. Clin. Cancer Res. 5:9–16 [PubMed] [Google Scholar]
- 52.Shiver JW, Emini EA. 2004. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Annu. Rev. Med. 55:355–372. 10.1146/annurev.med.55.091902.104344 [DOI] [PubMed] [Google Scholar]
- 53.Giraldo-Vela JP, Bean AT, Rudersdorf R, Wallace LT, Loffredo JT, Erickson P, Wilson NA, Watkins DI. 2010. Simian immunodeficiency virus-specific CD4+ T cells from successful vaccinees target the SIV Gag capsid. Immunogenetics 62:701–707. 10.1007/s00251-010-0473-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loffredo JT, Valentine LE, Watkins DI. 2006. Beyond Mamu-A*01+ Indian rhesus macaques: continued discovery of new MHC class I molecules that bind epitopes from the simian AIDS viruses. HIV Mol. Immunol. 2007:29–51 http://www.hiv.lanl.gov/content/immunology/pdf/2006_07/siv_epitopes_article.pdf [Google Scholar]
- 55.Hirao LA, Wu L, Khan AS, Hokey DA, Yan J, Dai A, Betts MR, Draghia-Akli R, Weiner DB. 2008. Combined effects of IL-12 and electroporation enhances the potency of DNA vaccination in macaques. Vaccine 26:3112–3120. 10.1016/j.vaccine.2008.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hirao LA, Wu L, Satishchandran A, Khan AS, Draghia-Akli R, Finnefrock AC, Bett AJ, Betts MR, Casimiro DR, Sardesai NY, Kim JJ, Shiver JW, Weiner DB. 2010. Comparative analysis of immune responses induced by vaccination with SIV antigens by recombinant Ad5 vector or plasmid DNA in rhesus macaques. Mol. Ther. 18:1568–1576. 10.1038/mt.2010.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalams SA, Parker SD, Elizaga M, Metch B, Edupuganti S, Hural J, De Rosa S, Carter DK, Rybczyk K, Frank I, Fuchs J, Koblin B, Kim DH, Joseph P, Keefer MC, Baden LR, Eldridge J, Boyer J, Sherwat A, Cardinali M, Allen M, Pensiero M, Butler C, Khan AS, Yan J, Sardesai NY, Kublin JG, Weiner DB. 2013. Safety and comparative immunogenicity of an HIV-1 DNA vaccine in combination with plasmid interleukin 12 and impact of intramuscular electroporation for delivery. J. Infect. Dis. 208:818–829. 10.1093/infdis/jit236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saade F, Petrovsky N. 2012. Technologies for enhanced efficacy of DNA vaccines. Expert Rev. Vaccines 11:189–209. 10.1586/erv.11.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vasan S, Hurley A, Schlesinger SJ, Hannaman D, Gardiner DF, Dugin DP, Boente-Carrera M, Vittorino R, Caskey M, Andersen J, Huang Y, Cox JH, Tarragona-Fiol T, Gill DK, Cheeseman H, Clark L, Dally L, Smith C, Schmidt C, Park HH, Kopycinski JT, Gilmour J, Fast P, Bernard R, Ho DD. 2011. In vivo electroporation enhances the immunogenicity of an HIV-1 DNA vaccine candidate in healthy volunteers. PLoS One 6:e19252. 10.1371/journal.pone.0019252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winstone N, Wilson AJ, Morrow G, Boggiano C, Chiuchiolo MJ, Lopez M, Kemelman M, Ginsberg AA, Mullen K, Coleman JW, Wu CD, Narpala S, Ouellette I, Dean HJ, Lin F, Sardesai NY, Cassamasa H, McBride D, Felber BK, Pavlakis GN, Schultz A, Hudgens MG, King CR, Zamb TJ, Parks CL, McDermott AB. 2011. Enhanced control of pathogenic simian immunodeficiency virus SIVmac239 replication in macaques immunized with an interleukin-12 plasmid and a DNA prime-viral vector boost vaccine regimen. J. Virol. 85:9578–9587. 10.1128/JVI.05060-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sardesai NY, Weiner DB. 2011. Electroporation delivery of DNA vaccines: prospects for success. Curr. Opin. Immunol. 23:421–429. 10.1016/j.coi.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Allen TM, Mothe BR, Sidney J, Jing P, Dzuris JL, Liebl ME, Vogel TU, O'Connor DH, Wang X, Wussow MC, Thomson JA, Altman JD, Watkins DI, Sette A. 2001. CD8+ lymphocytes from simian immunodeficiency virus-infected rhesus macaques recognize 14 different epitopes bound by the major histocompatibility complex class I molecule Mamu-A*01: implications for vaccine design and testing. J. Virol. 75:738–749. 10.1128/JVI.75.2.738-749.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mothe BR, Horton H, Carter DK, Allen TM, Liebl ME, Skinner P, Vogel TU, Fuenger S, Vielhuber K, Rehrauer W, Wilson N, Franchini G, Altman JD, Haase A, Picker LJ, Allison DB, Watkins DI. 2002. Dominance of CD8 responses specific for epitopes bound by a single major histocompatibility complex class I molecule during the acute phase of viral infection. J. Virol. 76:875–884. 10.1128/JVI.76.2.875-884.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McDermott AB, Mitchen J, Piaskowski S, De Souza I, Yant LJ, Stephany J, Furlott J, Watkins DI. 2004. Repeated low-dose mucosal simian immunodeficiency virus SIVmac239 challenge results in the same viral and immunological kinetics as high-dose challenge: a model for the evaluation of vaccine efficacy in nonhuman primates. J. Virol. 78:3140–3144. 10.1128/JVI.78.6.3140-3144.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. U. S. A. 103:5514–5519. 10.1073/pnas.0509996103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geldmacher C, Ngwenyama N, Schuetz A, Petrovas C, Reither K, Heeregrave EJ, Casazza JP, Ambrozak DR, Louder M, Ampofo W, Pollakis G, Hill B, Sanga E, Saathoff E, Maboko L, Roederer M, Paxton WA, Hoelscher M, Koup RA. 2010. Preferential infection and depletion of Mycobacterium tuberculosis-specific CD4 T cells after HIV-1 infection. J. Exp. Med. 207:2869–2881. 10.1084/jem.20100090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nemes E, Bertoncelli L, Lugli E, Pinti M, Nasi M, Manzini L, Manzini S, Prati F, Borghi V, Cossarizza A, Mussini C. 2010. Cytotoxic granule release dominates gag-specific CD4+ T-cell response in different phases of HIV infection. AIDS 24:947–957. 10.1097/QAD.0b013e328337b144 [DOI] [PubMed] [Google Scholar]
- 68.Soghoian DZ, Streeck H. 2010. Cytolytic CD4+ T cells in viral immunity. Expert Rev. Vaccines 9:1453–1463. 10.1586/erv.10.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Soghoian DZ, Jessen H, Flanders M, Sierra-Davidson K, Cutler S, Pertel T, Ranasinghe S, Lindqvist M, Davis I, Lane K, Rychert J, Rosenberg ES, Piechocka-Trocha A, Brass AL, Brenchley JM, Walker BD, Streeck H. 2012. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci. Transl. Med. 4:123ra25. 10.1126/scitranslmed.3003165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kedl RM, Rees WA, Hildeman DA, Schaefer B, Mitchell T, Kappler J, Marrack P. 2000. T cells compete for access to antigen-bearing antigen-presenting cells. J. Exp. Med. 192:1105–1113. 10.1084/jem.192.8.1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Willis RA, Kappler JW, Marrack PC. 2006. CD8 T cell competition for dendritic cells in vivo is an early event in activation. Proc. Natl. Acad. Sci. U. S. A. 103:12063–12068. 10.1073/pnas.0605130103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reynolds MR, Weiler AM, Piaskowski SM, Piatak MJ, Robertson HT, Allison DB, Bett AJ, Casimiro DR, Shiver JW, Wilson NA, Lifson JD, Koff WC, Watkins DI. 2012. A trivalent recombinant Ad5 gag/pol/nef vaccine fails to protect rhesus macaques from infection or control virus replication after a limiting-dose heterologous SIV challenge. Vaccine 30:4465–4475. 10.1016/j.vaccine.2012.04.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vojnov L, Bean AT, Peterson EJ, Chiuchiolo MJ, Sacha JB, Denes FS, Sandor M, Fuller DH, Fuller JT, Parks CL, McDermott AB, Wilson NA, Watkins DI. 2011. DNA/Ad5 vaccination with SIV epitopes induced epitope-specific CD4+ T cells, but few subdominant epitope-specific CD8+ T cells. Vaccine 29:7483–7490. 10.1016/j.vaccine.2011.07.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eberl G, Kessler B, Eberl LP, Brunda MJ, Valmori D, Corradin G. 1996. Immunodominance of cytotoxic T lymphocyte epitopes co-injected in vivo and modulation by interleukin-12. Eur. J. Immunol. 26:2709–2716. 10.1002/eji.1830261124 [DOI] [PubMed] [Google Scholar]
- 75.Ribeiro Dos Santos P, Rancez M, Pretet JL, Michel-Salzat A, Messent V, Bogdanova A, Couedel-Courteille A, Souil E, Cheynier R, Butor C. 2011. Rapid dissemination of SIV follows multisite entry after rectal inoculation. PLoS One 6:e19493. 10.1371/journal.pone.0019493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jennings SR, Bonneau RH, Smith PM, Wolcott RM, Chervenak R. 1991. CD4-positive T lymphocytes are required for the generation of the primary but not the secondary CD8-positive cytolytic T lymphocyte response to herpes simplex virus in C57BL/6 mice. Cell. Immunol. 133:234–252. 10.1016/0008-8749(91)90194-G [DOI] [PubMed] [Google Scholar]
- 77.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. 1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393:480–483. 10.1038/31002 [DOI] [PubMed] [Google Scholar]
- 78.Williams MA, Bevan MJ. 2007. Effector and memory CTL differentiation. Annu. Rev. Immunol. 25:171–192. 10.1146/annurev.immunol.25.022106.141548 [DOI] [PubMed] [Google Scholar]
- 79.Betts MR, Exley B, Price DA, Bansal A, Camacho ZT, Teaberry V, West SM, Ambrozak DR, Tomaras G, Roederer M, Kilby JM, Tartaglia J, Belshe R, Gao F, Douek DC, Weinhold KJ, Koup RA, Goepfert P, Ferrari G. 2005. Characterization of functional and phenotypic changes in anti-Gag vaccine-induced T cell responses and their role in protection after HIV-1 infection. Proc. Natl. Acad. Sci. U. S. A. 102:4512–4517. 10.1073/pnas.0408773102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Casimiro DR, Wang F, Schleif WA, Liang X, Zhang ZQ, Tobery TW, Davies ME, McDermott AB, O'Connor DH, Fridman A, Bagchi A, Tussey LG, Bett AJ, Finnefrock AC, Fu TM, Tang A, Wilson KA, Chen M, Perry HC, Heidecker GJ, Freed DC, Carella A, Punt KS, Sykes KJ, Huang L, Ausensi VI, Bachinsky M, Sadasivan-Nair U, Watkins DI, Emini EA, Shiver JW. 2005. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with DNA and recombinant adenoviral vaccine vectors expressing Gag. J. Virol. 79:15547–15555. 10.1128/JVI.79.24.15547-15555.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Makedonas G, Betts MR. 2011. Living in a house of cards: re-evaluating CD8+ T-cell immune correlates against HIV. Immunol. Rev. 239:109–124. 10.1111/j.1600-065X.2010.00968.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McElrath MJ, De Rosa SC, Moodie Z, Dubey S, Kierstead L, Janes H, Defawe OD, Carter DK, Hural J, Akondy R, Buchbinder SP, Robertson MN, Mehrotra DV, Self SG, Corey L, Shiver JW, Casimiro DR. 2008. HIV-1 vaccine-induced immunity in the test-of-concept Step Study: a case-cohort analysis. Lancet 372:1894–1905. 10.1016/S0140-6736(08)61592-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.