

ABSTRACT
Genogroup II, genotype 4 (GII.4) noroviruses are known to rapidly evolve, with the emergence of a new primary strain every 2 to 4 years as herd immunity to the previously circulating strain is overcome. Because viral genetic diversity is higher in chronic than in acute infection, chronically infected immunocompromised people have been hypothesized to be a potential source for new epidemic GII.4 strains. However, while some capsid protein residues are under positive selection and undergo patterned changes in sequence variation over time, the relationships between genetic variation and antigenic variation remains unknown. Based on previously published GII.4 strains from a chronically infected individual, we synthetically reconstructed virus-like particles (VLPs) representing early and late isolates from a small-bowel transplant patient chronically infected with norovirus, as well as the parental GII.4-2006b strain. We demonstrate that intrahost GII.4 evolution results in the emergence of antigenically distinct strains over time, comparable to the variation noted between the chronologically predominant GII.4 strains GII.4-2006b and GII.4-2009. Our data suggest that in some individuals the evolution that occurs during a chronic norovirus infection overlaps with changing antigenic epitopes that are associated with successive outbreak strains and may select for isolates that are potentially able to escape herd immunity from earlier isolates.
IMPORTANCE Noroviruses are agents of gastrointestinal illness, infecting an estimated 21 million people per year in the United States alone. In healthy individuals, symptomatic infection typically resolves within 24 to 48 h. However, symptoms may persist for years in immunocompromised individuals, and development of successful treatments for these patients is a continuing challenge. This work is relevant to the design of successful norovirus therapeutics for chronically infected patients; provides support for previous assertions that chronically infected individuals may serve as reservoirs for new, antigenically unique emergent strains; and furthers our understanding of genogroup II, genotype 4 (GII.4) norovirus immune-driven molecular evolution.
INTRODUCTION
Noroviruses are the leading cause of gastrointestinal illness worldwide. While typically an acute disease, norovirus infections can be serious in the young, old, and immunocompromised, as these groups are at risk for more severe disease and death (1–3). Norovirus is spread rapidly in environments where people are found in close proximity, including schools and daycare centers, nursing homes, cruise ships, and hospitals. Importantly, hospital outbreaks can result in significant economic damage, with direct and indirect costs from a single outbreak reaching $650,000 (4).
Noroviruses are members of the family Caliciviridae and contain an ∼7.5-kb single-stranded, positive-polarity RNA genome. They are divided into 5 genogroups; genogroups I and II are responsible for the majority of human disease and are further subdivided into at least 9 and 22 genotypes, respectively (5). The human norovirus genome contains three open reading frames (ORFs) encoding the nonstructural proteins, the ORF2 major capsid protein (VP1), and the ORF3 minor capsid protein (VP2) (6). VP1 is further divided into the shell (S) and protruding (P) domains, with the P domain comprised of the P1 and P2 subdomains (6). Phylogenetic studies indicate that the P2 subdomain is the most variable region of the norovirus genome (7, 8). The P2 subdomain is also the most surface-exposed region of the norovirus capsid, interacting with antibodies and histo-blood group antigens (HBGAs), which serve as binding ligands and putative receptors for human norovirus docking and entry.
Genogroup II, genotype 4 (GII.4) strains cause over 70% of all norovirus outbreaks (9), and epidemic outbreaks occur every 2 to 4 years, involving new antigenically distinct strains (7, 10). Studies of antigenic variation in GII.4 norovirus have shown that the P2 region is involved in strain-specific antibody recognition (7, 11, 12) and contains at least three blockade (potential neutralization) epitopes (13–15). In epidemic strains, genetic variation in P2 is linked to antigenic changes over time, indicating that molecular evolution in the P2 subdomain is likely driven by escape from human herd immunity (12–17).
Noroviruses typically cause acute infection in healthy individuals, resulting in symptomatic infection for 24 to 48 h, followed by virus shedding for 2 to 4 weeks (18, 19). However, some immunocompromised individuals, such as transplant patients on immunosuppressive drugs, those with primary immunodeficiencies, cancer patients undergoing chemotherapy, and those with HIV infections, may develop chronic norovirus infection. Symptomatic infection and virus shedding in these patients can persist for weeks to years (20–25) and can result in medical issues, such as dehydration and nutrient deficiencies (26), making development of treatment options for these patients an important priority. Unfortunately, there are no approved therapeutics or vaccines for controlling norovirus infections. Attempted methods to control chronic infection have included treatment with drugs effective against other diarrheal diseases (27), adjustment of the immunosuppressive drug type or dosage (28), and oral or enteral administration of human IgG (29–32). Although reduction in immunosuppression coupled with IgG administration has shown promise for some transplant patients, IgG therapy has failed in other studies, and reduction of immunosuppression is not always possible.
Existing studies provide a basis to investigate important questions about chronic norovirus infection. Although unconfirmed, one recent hypothesis is that chronically infected norovirus patients may be important sources of infection, both in health care settings (33) and as potential reservoirs for new, emergent GII.4 norovirus strains (20, 23, 25). Although the fitness and the infectivity of chronically shed virus are currently unknown, potential accounts of chronic norovirus shedders involved in hospital outbreaks and transmission of virus to both immunocompromised and immunocompetent individuals have been documented (21, 33, 34).
The virus capsid sequence and phylogenetic data from chronically infected patients have shown substantial genetic variation over the course of infection in many, but not all, patients (22, 23, 35). Siebenga et al. found that the capsid mutation rate was linked to immune impairment, suggesting that immune-driven selection drives evolution in the capsid during chronic infection (35) and explains differences in evolution, depending on the level of immunosuppression. Additional studies have corroborated a role for intrahost immune-driven selection by demonstrating that virus isolated from chronically infected patients undergoes positive selection and exhibits higher genetic diversity in the capsid protein than virus from acutely infected individuals (23, 25). In some chronically infected patients with GII.4 strain infections, many of the changes occur in blockade epitopes, areas of known or predicted antigenic importance, but antigenic comparisons have not been performed (13–15, 22, 35).
Here, we compare and contrast the antigenic differences using a panel of polyclonal and monoclonal antibodies and time-ordered virus-like particles (VLPs) derived from early (day 1 [P.D1]) and late (day 302 [P.D302]) capsid protein amino acid sequences from a chronically infected immunocompromised patient (23). Our data demonstrate significant antigenic differences between intrahost variants that mirror the degree of variation seen in major successive norovirus strains, suggesting that chronic norovirus infections can evolve antigenically unique variants with the potential to seed future norovirus outbreaks.
MATERIALS AND METHODS
Sequences and structural-homology models.
The GenBank (NCBI sequence database) sequences used in this study were JQ478409.1 (GII.4-2006b) (15), JQ417309 (P_04.2009, or P.D1) (23), JQ417327 (P_02.2010, or P.D302) (23), JN595867.1 (GII.4-2009) (15), and JX459908.1 (GII.4-2012) (36), and the VA387 crystal structure is available from the RCSB Protein Data Bank, identifier 2OBT (37). We refer to the sequences originally named P_04.2009 and P_02.2010 as P.D1 and P.D302, respectively, for simplicity here. Homology models of these sequences were constructed using Modeler (Max-Planck Institute for Developmental Biology) and modeled in PyMOL.
Production of VRPs.
Virus replicon particles (VRPs) carrying the norovirus major capsid gene were produced as previously described (38). Briefly, expression vector pVR21 carries the Venezuelan equine encephalomyelitis (VEE) genome with the VEE structural genes replaced with a commercially synthesized norovirus ORF2 gene (BioBasic) behind the 26S promoter. The VEE norovirus ORF2 construct and two separate plasmids expressing either the VEE 3526 E1 and E2 glycoproteins or the VEE 3526 capsid protein were used to make RNA. RNA from all three constructs was electroporated into BHK cells, and 48 h later, VRPs were harvested and purified by high-speed centrifugation. VRP titers were determined by counting fluorescent cells detected with fluorescein isothiocyanate (FITC)-labeled antibody. Virus-like particle (VLP) production from VRPs and structural integrity were confirmed by electron microscopy (EM).
Production of VLPs.
VLPs were produced as previously described (13, 15). Briefly, commercially synthesized norovirus ORF2 (BioBasic) from a chronically infected patient sequence or outbreak strain sequence was cloned into expression vector pVR21 behind the 26S promoter, and genome length RNAs were synthesized in vitro using T7 RNA polymerase. RNA from the VEE ORF2 construct and helper RNAs was electroporated into BHK cells, and 24 h later, VLPs were harvested and purified by high-speed centrifugation. The VLP concentration was determined by bicinchoninic acid (BCA) protein assay (Pierce), and structural integrity was confirmed for all VLPs by EM.
HBGA binding assay.
HBGA assays were performed as previously described (7). Briefly, avidin-coated plates (Pierce) were coated with 10 μg/ml synthetic biotinylated HBGAs (GlycoTech), followed by addition of 2 μg/ml VLPs. HBGA binding was detected with strain-specific mouse polyclonal sera, followed by anti-mouse IgG-horseradish peroxidase (HRP) (GE Healthcare) and then One-Step Ultra TMB HRP substrate. Positive reactivity for each HBGA was defined as an optical density at 450 nm (OD450) signal above or equal to 3 times the background binding (background range, 0.049 to 0.066) after background subtraction.
EIAs.
Reactivity with mouse and human monoclonal antibodies (MAbs) was determined by enzyme-linked immunoassay (EIA). Plates were coated with 0.5 μg/ml VLPs in phosphate-buffered saline (PBS), and then 2-fold serial dilutions of the MAb starting at 1 μg/ml MAb were added. Anti-mouse or -human IgG-HRP (GE Healthcare), followed by One-Step Ultra TMB EIA HRP substrate solution, was used for detection. Positive reactivity was defined as a mean OD450 of ≥0.2 after background subtraction. The data represent a combination of three independent trials, with each VLP run in duplicate in each trial. Sigmoidal dose-response analysis was performed as previously described (14) using the reactivity at 1 μg/ml as 100% binding. Fifty percent effective concentrations (EC50s) among VLPs were compared using one-way analysis of variance (ANOVA) with Dunnett's posttest. A P value of <0.05 was considered significant. VLPs with maximum reactivity below a mean OD450 of 0.2 were assigned a value of zero for graphical representations.
VLP-carbohydrate ligand-binding antibody blockade assays.
Blockade assays using pig gastric mucin (PGM) type III (Sigma Chemicals) were performed as previously described (14). PGM-bound VLPs were detected by rabbit anti-GII.4 norovirus polyclonal sera. The percent control binding was defined as the VLP ligand-binding level in the presence of test antibody or sera compared to the binding level in the absence of antibody multiplied by 100. All MAbs and sera were tested for blockade potential at 2-fold serial dilutions ranging from 0.0039 to 2 μg/ml (mouse MAbs), 0.0039 to 16 μg/ml (human MAbs), and 0.0098 to 5% (mouse sera). The data from blockade experiments using monoclonal antibodies represent a combination of three independent trials, with each VLP run in duplicate in each trial. The data from blockade experiments using polyclonal mouse sera represent a combination of two independent trials in which sera from five individual mice were tested for each VLP. Sigmoidal dose-response analysis was performed as previously described, and EC50s among VLPs were compared using one-way ANOVA with a Bonferroni posttest. A P value of <0.05 was considered significant. Blockade assays utilize VLP concentrations in the low nanomolar range; therefore, the assay does not discriminate between antibodies with subnanomolar affinities.
Monoclonal antibodies and mouse polyclonal sera.
Mouse (12) and human (14) monoclonal antibodies were isolated as previously described. BALB/c mice (five per group) were immunized by footpad injection with 5 × 104 VRPs expressing the norovirus capsid gene (GII.4-1987, GII.4-2002, GII.4-2006b, GII.4-2009, P.D1, or P.D302). Mice were boosted on day 21 and euthanized 7 days postboost, and sera were harvested. All animal studies were approved and conducted under the guidelines of UNC Chapel Hill's Institutional Animal Care and Use Committee (web ID 23433).
Antigenic cartography.
We utilized multidimensional-scaling (MDS) approaches as described and implemented within the AntigenMap 3D software (39, 40). The EC50 blockade titers of various sera against a panel of VLPs were normalized to the maximum blockade titer of each serum, as well as to the maximum overall blockade titer across sera (normalization method 1 in AntigenMap 3D). The normalized values were used to calculate Euclidean distances (D) between each pair of VLPs. For greater analytical, visualization, and graphical purposes, we then utilized the Matlab8.1 (MathWorks Inc., Natick, MA) cmdscale function to determine the XYZ coordinates so that the data could be displayed in 3 dimensions while maintaining the underlying Euclidean distances directly calculated from the data. We utilized R (www.r-project.org), with the package rgl for three-dimensional (3D) visualization of these data. We confirmed the output of our pipeline with that produced by AntigenMap 3D.
RESULTS
Comparison of sequence changes among chronic-infection isolates and GII.4-2006b.
Previous work had shown that changes identified in a few key surface-exposed epitopes correlate with shifts in GII.4 norovirus antigenicity (11, 13–15), including residues in epitope A (294, 296 to 298, 368, and 372) (13), epitope D (393 to 395) (14), and epitope E (407, 412, and 413) (15). Changes in these residues likely alter the ability of preexisting immunity to neutralize the virus, selecting for the emergence of new epidemic strains.
To study the within-host antigenic evolution of noroviruses during a chronic human infection, we aligned the sequences of the capsid P2 domains of GII.4-2006b, P.D1, and P.D302 to examine sequential amino acid changes from GII.4-2006b through P.D302 after at least 10 months of within-host evolution (23). Note that day 1 and day 302 refer to the days of sample collection and not from the beginning of infection, as the time between the beginning of infection and the collection of the day 1 sample is unknown. Between VP1 amino acid positions 248 and 434, there are 9 differences between GII.4-2006b and P.D1. After 10 months, there were 15 additional differences between P.D1 and P.D302 and 20 differences between GII.4-2006b and P.D302 located between these amino acid positions (Fig. 1A). Similarly, there are 16 differences spanning this domain between GII.4-2006b and subsequent epidemic strains, GII.4-2009 and GII.4-2012. Two of the differences between GII.4-2006b and P.D1 (S368A and S393G) and four of the differences between GII.4-2006b and P.D302 (A294G, S296T, S368A, and N412D) are located within blockade epitope sites (Fig. 1B). Four differences in blockade epitope residues also exist between P.D1 and P.D302 (A294G, S296T, G393S, and N412D) (Fig. 1B). We synthesized GII.4-2006b, P.D1, and P.D302 genes; expressed VLPs representing these strains; and measured differences in antigenicity and HBGA binding among the chronic-infection isolates and GII.4-2006b using biological assays. In addition, several amino acid substitutions present in the chronic-infection strains that are conserved in past epidemic strains may also influence the antigenic and HBGA binding characteristics of epitope sites A (292, 295, and 373), D (391), and E (414) (Fig. 1B) based on their positions relative to these epitopes in GII.4 homology models (Fig. 1C).
FIG 1.

Sequence changes in chronically infected patient strains compared to GII.4-2006b. (A) Available capsid amino acid sequences for GII.4-2006b, P.D1, and P.D302 were aligned using Clustal Omega, and sequence differences among GII.4-2006, P.D1, and P.D302 are shown. GII.4-2006b residues are shown in purple. P.D1 and P.D302 differences from GII.4-2006b are indicated in light blue, while orange indicates reversion to the GII.4-2006b residues. (B) Alignment of GII.4-2006b, P.D1, and P.D302 amino acid sequences in and around epitopes A, D, and E. Green indicates a position within a defined epitope, while white indicates nearby residues that may impact antigenicity in these epitopes. Yellow indicates an amino acid position newly defined as part of epitope A. (C) Structural homology models of GII.4-2006b, P.D1, and P.D302 capsid P2 dimers shown from a top view. Purple shows the locations of epitopes A, D, and E on the capsid P2 dimer, while green shows changed amino acid residues in P.D1 and P.D302 compared to GII.4-2006b.
Comparison of HBGA binding in chronic-infection isolates to that in GII.4-2006b.
To evaluate differences in HBGA binding preferences among GII.4-2006b, P.D1, and P.D302, we measured VLP binding to synthetic biotinylated carbohydrates (A, B, Lea, Leb, Lex, Ley, H type 1, and H type 3). As previously reported, GII.4-2006b bound A, B, Leb, Ley, and H type 3 (41). In contrast, chronic-infection strain VLPs exhibited HBGA binding profiles different from those of GII.4-2006b and from each other (Table 1). P.D1 was able to bind A, B, and H type 3, while P.D302 bound only B and H type 3 synthetic biotinylated HBGAs. This indicates that HBGA binding preferences may be altered over time during chronic infection, perhaps influenced by individual within-host HBGA expression phenotypes.
TABLE 1.
Chronic-infection strain HBGA binding preferences
| Strain | Bindinga |
|||||||
|---|---|---|---|---|---|---|---|---|
| A | B | Lea | Leb | Lex | Ley | H type 1 | H type 3 | |
| GII.4–2006 | + | + | − | + | − | + | − | + |
| P.D1 | + | + | − | − | − | − | − | + |
| P.D302 | − | + | − | − | − | − | − | + |
VLPs representing GII.4-2006b, P.D1, and P.D302 were assayed for the ability to bind synthetic biotinylated HBGAs A, B, Lea, Leb, Lex, Ley, H type 1, and H type 3 by carbohydrate binding assay. Positive reactivity (+) was defined as a value greater than or equal to 3 times the background binding value.
Reactivity with GII.4 mouse and human MAbs.
To measure antigenic differences among VLPs representing GII.4-2006b and chronic strains P.D1 and P.D302, we performed EIAs using mouse and human MAbs. We tested five GII.4-2006b mouse MAbs (G2, G3, G4, G6, and G7) and two GII.4 human MAbs (NVB111 and NVB43.9), all of which target epitope site A residues (294, 296 to 298, 368, and 372), for EIA binding with GII.4-2006b, P.D1, and P.D302 VLPs. GII.4-2006b and P.D1 differ in one epitope site A position, where P.D1 contains S368A compared to GII.4-2006b. P.D302 is different from GII.4-2006b at 3 of 6 epitope site A residues—A294G, S296T, and S368A—while P.D1 and P.D302 are different at 2 of 6 epitope site A residues—A294G and S296T. We also tested the reactivities of these VLPs with one human MAb (NVB97), which targets epitope site D residues (393 to 395). While GII.4-2006b and P.D302 share identical epitope site D residues, P.D1 has an S393G change compared to GII.4-2006b. In addition, we tested one human MAb (NVB71.4) that targets an unmapped conserved GII.4 epitope (14). Consistent with previously reported results, all the MAbs reacted strongly with GII.4-2006b VLPs (12, 14) (Table 2). In contrast, EC50s for P.D1 VLPs were significantly different (P < 0.05) from those for GII.4-2006b VLPs for mouse MAbs G2, G4, and G6 and human MAbs NVB43.9 and NVB111 (Table 2). Moreover, EC50s for P.D302 VLPs were significantly different from those for GII.4-2006b VLPs for all MAbs except NVB71.4 and different from those for P.D1 VLPs for all but NVB71.4 and NVB111 (Table 2). This indicates that epitope sites A and D are antigenically distinct among GII.4-2006b, P.D1, and P.D302, demonstrating antigenic variation over the course of chronic infection in important blockade epitopes.
TABLE 2.
GII.4 mouse and human MAb EIA reactivities with chronic-infection strains
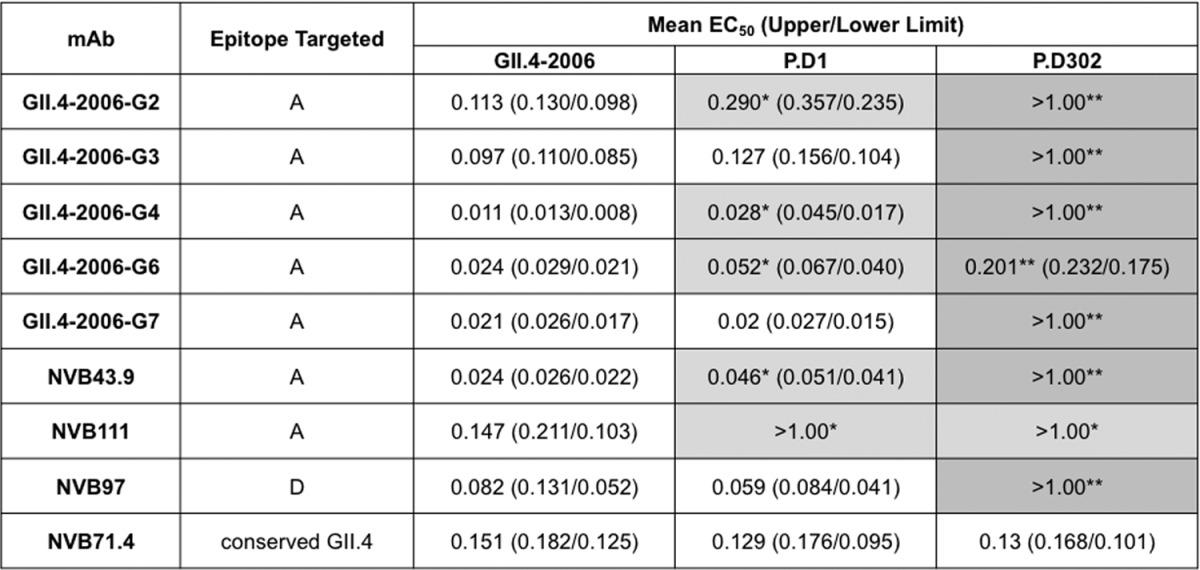
Mouse and human GII.4 monoclonal antibodies were assayed for reactivity with GII.4-2006b, P.D1, and P.D302 VLPs by multiple-dilution EIA. The mean percent binding (the percentage of the VLP bound to antibody in the dilution course compared to the amount of VLP bound with antibody at 1 μg/ml) of each VLP was fitted with a sigmoidal curve, and the mean EC50 (μg/ml) EIA reactivity titers for GII.4-2006b, P.D1, and P.D302 were calculated. Mean EC50 EIA reactivity titers for the test VLP that were significantly different from the mean EC50 for GII.4-2006b (light gray) (*) or were significantly different from both GII.4-2006b and P.D1 (P < 0.05) (dark gray) (**) are indicated. Monoclonal antibodies that did not demonstrate EIA reactivity at or above an OD450 of 0.2 at 1 μg/ml with a particular VLP are denoted by an EC50 of >1 μg/ml. The statistics were calculated by one-way ANOVA with a Bonferroni posttest.
Blockade activities for GII.4 mouse and human MAbs.
Compared to EIA, neutralization is a more relevant measure of functional antigenic change. To test the potential neutralization activities of MAbs (GII.4-2006b G2, G3, G4, G6, and G7 and NVB43.9, NVB71.4, NVB97, and NVB111) against GII.4-2006b, P.D1, and P.D302 VLPs, we performed blockade assays, a correlate of protective immunity (42) and a neutralization surrogate. Consistent with previous findings, all the MAbs were able to block ligand-VLP interactions for GII.4-2006b (12, 14) (Fig. 2). Likewise, P.D1 was blocked by all the MAbs (Fig. 2). However, EC50 blockade titers for two out of five GII.4-2006b mouse MAbs, G2 (Fig. 2A) and G7 (Fig. 2E), and two of four GII.4 human MAbs, NVB111 (Fig. 2G) and NVB71.4 (Fig. 2I), were significantly different, requiring 7.1 times, 2 times, 2 times, and 3.2 times more antibody, respectively, for blockade than GII.4-2006b VLPs. P.D302 VLP-ligand binding was blocked by GII.4-2006b mouse MAbs G2 (Fig. 2A), G6 (Fig. 2D), and G7 (Fig. 2E), but not by G3 (Fig. 2B) or G4 (Fig. 2C), and blocked by GII.4 human MAb NVB71.4 (Fig. 2I), but not by NVB43.9 (Fig. 2F), NVB111 (Fig. 2G), or NVB97 (Fig. 2H). EC50 blockade titers were significantly different between GII.4-2006b and P.D302 for G2, G6, G7, and NVB71.4, requiring 12.6 times, 15.9 times, 12 times, and 6.8 times more MAb than for GII.4-2006b, respectively. Overall, EC50 blockade titers were significantly higher for P.D302 than for both GII.4-2006 and P.D1 for all tested MAbs, demonstrating major antigenic changes in epitope sites A and D over the course of chronic norovirus infection.
FIG 2.

GII.4 mouse and human MAb blockade responses against chronic-infection strains. (A to I) Mouse and human GII.4 monoclonal antibodies were assayed for the ability to block GII.4-2006b, P.D1, and P.D302 VLP interaction with carbohydrate ligand. The mean percent control binding (the percentage of the VLP bound to carbohydrate ligand in the presence of an antibody compared to the amount of VLP bound with no antibody present) of each VLP was fitted with a sigmoidal curve, and the mean EC50 (μg/ml) blockade titers for GII.4-2006b, P.D1, and P.D302 were calculated. The error bars represent 95% confidence intervals. The asterisks indicate that the mean EC50 blockade titer for the test VLP was significantly different from the mean EC50 for GII.4-2006b (P < 0.05) (*) or was significantly different from those of both GII.4-2006b and P.D1 (P < 0.05) (**). Monoclonal antibodies that did not block a particular VLP were assigned an EC50 of 2 times the upper limit of detection for statistical analysis and are shown on the graph as data points above the upper limit of detection (dashed lines). Statistics were calculated by one-way ANOVA with a Bonferroni posttest.
Blockade responses of strain-specific mouse polyclonal sera.
While monoclonal antibodies are informative about the changes in a single epitope, polyclonal sera are needed to evaluate global antigenic changes. To measure differences in the total antibody response, we immunized mice with VRPs expressing the capsid genes from GII.4-2006b, P.D1, and P.D302 or GII.4-2009, the consecutive outbreak strain following GII.4-2006, and measured the induced serum blockade responses (Fig. 3). Mice immunized with GII.4-2006b VRPs mounted a robust blockade response against homotypic GII.4-2006b VLPs, while significantly more serum was needed to block GII.4-2009, P.D1, and P.D302 VLPs (16 times, 9.4 times, and 12.7 times, respectively) (Fig. 3A). Sera from mice immunized with GII.4-2009 VRPs induced a strong blockade response against GII.4-2009 VLPs; however, significantly more serum was needed to block GII.4-2006b and P.D302 VLPs, with 39 times more serum needed to block P.D302 VLPs than GII.4-2009 (Fig. 3B). Sera from mice immunized with P.D1 VRPs most efficiently blocked homotypic P.D1 VLPs. The EC50s indicated that more serum is required to block GII.4-2009 (3 times) and P.D302 (25.8 times) than P.D1, while GII.4-2006b and P.D1 EC50 titers were not significantly different (Fig. 3C). Sera from mice immunized with P.D302 VRPs efficiently blocked P.D302 VLP-ligand interactions and weakly blocked GII.4-2006b and P.D1, requiring 92 times and 61 times more serum, respectively. P.D302 serum was unable to block GII.4-2009 VLPs (Fig. 3D). These data show that chronic-isolate VLPs induce antibody responses that are different from those of the parental strain and each other, demonstrating major changes in total antibody response over the course of chronic infection.
FIG 3.

Blockade activity of mouse polyclonal sera against homotypic and heterotypic VLPs. Mice were immunized with VRPs expressing the capsid genes of GII.4-2006b, GII.4-2009, P.D1, and P.D302, and sera collected from the mice were tested for blockade activity against GII.4-2006b, GII.4-2009, P.D1, and P.D302 VLPs. Shown is the blockade activity of sera from mice immunized against GII.4-2006b (A), GII.4-2009 (B), P.D1 (C), and P.D302 (D) with homotypic and heterotypic VLPs. The mean percent control binding (the percentage of the VLP bound to carbohydrate ligand in the presence of sera compared to the amount of VLP bound with no serum present) of each VLP was fitted with a sigmoidal curve, and the mean EC50 (% sera) blockade titers for GII.4-2006b, GII.4-2009, P.D1, and P.D302 were calculated. The error bars represent 95% confidence intervals. *, the mean EC50 blockade titer for the test VLP was significantly different from the mean EC50 for the homotypic strain (P < 0.05). Sera that did not block a particular VLP were assigned an EC50 of 10% sera for statistical analysis and are shown on the graph as data points above the upper limit of detection (dashed lines). Statistics were calculated by one-way ANOVA with a Bonferroni posttest.
Antigenic cartography.
In order to further describe and visualize the differences between the antigenic properties of virus strains, we utilized the MDS approach known as antigenic cartography (39, 40). Specifically, we used the pipeline described in AntigenMap 3D (39) to measure and visualize the antigenic relationships among outbreak strains GII.4-1987, GII.4-1997, GII.4-2002, GII.4-2006b, GII.4-2009, and GII.4-2012, as well as chronic isolates P.D1 and P.D302, explicitly contrasting antigenic relationships between naturally occurring epidemic strains, as well as intrahost variants. The antigenic distances between strains were measured using GII.4-1987, GII.4-2002, GII.4-2006b, GII.4-2009, P.D1, and P.D302 mouse serum EC50 blocking titers against VLPs representative of the specified GII.4 strains, and Euclidean distance values were calculated based on these titers (Fig. 4A). Consistent with earlier findings (12), early (GII.4-1987, GII.4-1997, and GII.4-2002) and late (GII.4-2006b, GII.4-2009, and GII.4-2012) strains formed distinct clusters (Fig. 4B and C). Not surprisingly, the early within-host variant, P.D1, grouped closely with late strains (Fig. 4B and C), reflecting its origins from the GII.4-2006b lineage. In contrast, P.D302 did not group with any other strain and was antigenically distant from both the early and contemporary isolates. In order to confirm the visual analysis of these antigenic similarities, we compared Euclidean distances (D) between each pair of VLPs across all sera utilized for antigenic cartography (the Euclidean distance measures the straight-line distance between two points in a multidimensional space). We first examined the groupings of early and late GII.4 outbreak strains. The average distance within a group was 3.79 (range, 2.11 to 6.39), while the average distance between early and contemporary clusters was 10.7 (range, 8.49 to 13.32), with each distance unit corresponding to a roughly 1.25-fold difference in blockade response between viruses (Fig. 4A). As shown in Fig. 4B and C, P.D1 grouped closely with late outbreak strain VLPs, with an average D value of 3.46 (range, 2.26 to 5.09) (Fig. 4A). In contrast, P.D302 was quite distinct from both early and late outbreak strain viruses, as well as from P.D1, with an average D value of 9.92 (range, 8.73 to 11.62) (Fig. 4A). During an ∼10-month chronic infection in this individual, our data demonstrate that intrahost evolution can generate novel variants with unique HBGA binding patterns and encoding unique antigenic differences that are as dramatically distinct as those of time-ordered, epidemic outbreak strains that emerge in human populations.
FIG 4.

Antigenic cartography for GII.4 noroviruses. MDS was used to identify the antigenic relationships between different norovirus strains. (A) Euclidean antigenic distances between virus strains were calculated based on the EC50 efficacies of antisera raised against GII.4-1987, GII.4-2002, GII.4-2006b, GII.4-2009, P.D1, and P.D302 VLPs. The green squares represent distances within either the early (1987, 1998, and 2002) or late (2006, 2009, and 2012) virus groups. The purple squares show the distances between early and late virus groups. (B and C) We determined XYZ coordinates that maintain the underlying Euclidean distances between viruses while illustrating the relationships between GII.4 norovirus strains, with each map distance roughly corresponding to an ∼1.25-fold change in blockade response. (B) Early strains GII.4-1987 (yellow), GII.4-1997 (red), and GII.4-2002 (light blue) grouped together (lower right), and the late strains GII.4-2006b (light purple), GII.4-2009 (dark blue), and GII.4-2012 (dark purple) grouped together (lower left). P.D1 grouped with late strains, closest to GII.4-2006b, while P.D302 was separate from either late or early strains (top). (C) Side view of the same 3D graph as in panel B, showing the antigenic differences between strains.
Expansion of epitope site A.
We next determined whether novel sites of within-host evolution can refine existing epitope maps and identify potential immunogenic changes in epidemic strains of the future. Amino acid position 373 exhibited an N373H change between P.D1 and P.D302 but was conserved in major GII.4 epidemic strains until an N373R substitution emerged in GII.4-2012 Sydney. Although not supported with empirical data, recent work by Allen et al. (43) suggests that this change in the Sydney strain may have impacted its emergence. Since changes to residue 373 have never been shown to influence immunogenicity, and it is not included as a diagnostic A epitope site residue, this potentially hampers new epidemic strain identification. To determine whether position 373 contributes to antigenic differences in epitope A, we used the blockade assay to test potential neutralization of VLPs representing the parental strains GII.4-2009 New Orleans and GII.4-2012 Sydney and chimeric sequences GII.4-2012.09A, GII.4-2012.09A.R373N, and GII.4-2012.R373N (Fig. 5) by epitope A targeting human MAb 43.9. GII.4-2009 was efficiently blocked by MAb 43.9, while GII.4-2012 required significantly more (55.3 times) MAb for blockade. Blockade response was partially restored in chimeras GII.4-2012.09A and GII.4-2012.R373N but required 1.5 times and 4.8 times more MAb, respectively, for blockade than GII.4-2009. EC50 blockade titers were not statistically different between GII.4-2012.09A.R373N and GII.4-2009 VLPs. Similar trends were seen using mouse MAbs targeting epitope A residues (data not shown).
FIG 5.
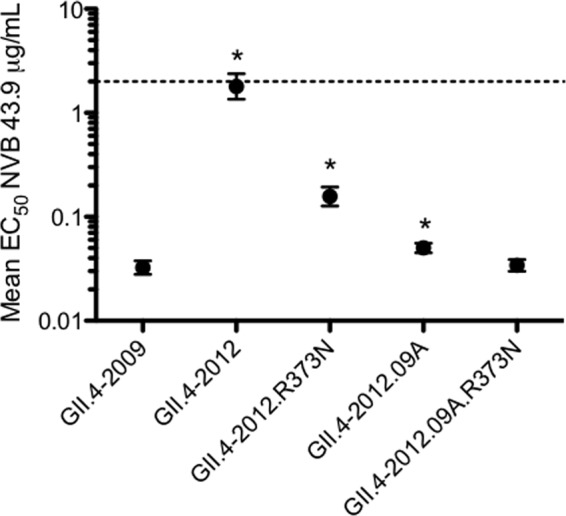
Expansion of epitope site A. Epitope A targeting human GII.4 MAb 43.9 was assayed for its ability to block GII.4-2009 New Orleans, GII.4-2012 Sydney, GII.4-2012.09A, GII.4-2012.R373N, and GII.4-2012.09A.R373N VLP interaction with carbohydrate ligand. The mean percent control binding (the percentage of the VLP bound to carbohydrate ligand in the presence of an antibody compared to the amount of VLP bound with no antibody present) of each VLP was fitted with a sigmoidal curve, and the mean EC50 (μg/ml) blockade titers for all VLPs were calculated. The error bars represent 95% confidence intervals. Statistics were calculated by one-way ANOVA with Dunnett's posttest. *, the mean EC50 blockade titer was significantly different from that of GII.4-2009.
DISCUSSION
Noroviruses are an important cause of gastroenteritis in immunocompromised individuals (44, 45), who are at increased risk for severe disease outcomes (1, 44). Recent vaccine trials utilizing a VLP-based vaccine approach support the idea that efficacious vaccines that elicit short-term protection in some healthy individuals can be generated, but vaccines may not protect immunocompromised populations, making development of therapeutics that effectively treat or prevent norovirus infections a top health priority.
In immunocompetent people, norovirus infection results in acute disease outcomes (46). In contrast, immunocompromised individuals can develop symptomatic disease and high-titer virus shedding for up to years. Unfortunately, the literature on specific chronically infected norovirus patient populations is sparse, and the duration and severity of chronic norovirus infections are likely influenced by several factors, including the underlying condition, drug treatment regime, degree of immunosuppression, and rate of within-host virus evolution, making it difficult to define the characteristics of a typical chronic norovirus case. From these limited studies, it is difficult to discern whether there are characteristics of chronic norovirus infection that are broadly applicable to all populations and characteristics that are true to specific populations, or whether characteristics vary by each individual case. Previous work has shown that during the course of chronic infection, virus genetic diversity can expand quickly (22, 23, 25, 35); however, it was previously unknown whether this genetic variation translated into antigenic variation or the emergence of antigenically unique isolates that differ significantly from contemporary epidemic strains. For the first time, our work clearly demonstrates the potential for significant antigenic variation over the course of chronic infection within an individual, which is important in terms of therapeutic treatment considerations and for studying the potential role of chronic shedders as reservoirs for evolving new outbreak strains.
Since there is no known animal reservoir for human noroviruses (47), the available data indicate that new GII.4 strains likely arise naturally within the human population by epochal evolution, immune-driven selection, and interhost transmission over time (12–14, 16, 17). The occurrence of frequent long-term chronic infections in immunosuppressed patients also represents a possible source of new GII.4 norovirus strains with epidemic potential (23, 25, 35), as these patients may provide an appropriate environment for sustained immune-directed molecular evolution by targeting previously identified surface-exposed blockade epitopes for mutation-driven escape. Evidence supporting this hypothesis includes sequence data from chronically infected patients that demonstrate the emergence of genetic changes in GII.4 blockade epitopes that modulate interhost antigenicity (22, 35). This diverse pool may contain variants antigenically distinct from the predominant circulating strain, allowing the emergence of a new strain under the right conditions (25). However, host and environmental factors, coupled with the type and degree of immunosuppression, may affect the rate and complexity of intrahost evolution that occurs over time (23), and future work that evaluates the roles of different immunosuppressive conditions in intrahost norovirus evolution is needed.
Our work demonstrated intrahost antigenic changes within epitope site A (amino acids 294, 296 to 298, 368, and 372). Interestingly, P.D302 contained residue substitutions in amino acid positions 292, 295, and 373, which are conserved in major GII.4 outbreak strains, except for 373, which was altered in the most recent predominant strain, GII.4-2012 Sydney. Changes in these residues likely impact epitope A antibody binding and blockade response, either by altering the conformational landscape of the epitope or by directly inhibiting the interaction of the antibody with the capsid. Using GII.4-2009/GII.4-2012 chimeric VLPs, we demonstrated that residues at position 373 impact the blockade response of human MAb NVB43.9, an antibody that targets epitope A. This demonstrates that position 373 is part of epitope A, expanding the epitope to 7 positions. Furthermore, we suggest that monitoring intrahost-evolved strains may provide a novel diagnostic strategy to map key residues capable of mediating antigenic changes in future outbreaks. While positions 292 and 295 have been conserved in previous predominant circulating GII.4 strains, their ability to change in this patient and their proximity to known epitope A residues suggest that these residues could potentially impact antigenic change in epitope A in future epidemics, as residue 373 did in GII.4-2012 Sydney.
Reactivity and blockade response data for antibody NVB97 demonstrate antigenic evolution in epitope site D during chronic infection. Epitope site D minimally includes residues 393 to 395 and is in close proximity to the carbohydrate binding pocket (37), and previous work demonstrated that modulation of residues within the epitope modulates HBGA specificity (7). Evolution in this epitope site is likely driven both by antibody selective pressure and pressure to maintain binding to one or more HBGAs. Despite conservation of residues 393 to 395 between GII.4-2006b and P.D302, antigenic phenotypes differ significantly, demonstrating that NVB97 recognition is modulated by amino acid positions outside the previously defined epitope site D residues. Position 391, which is close to the carbohydrate binding pocket, is conserved in major outbreak strains and between GII.4-2006b and P.D1, and previous work demonstrated that an alanine substitution at this residue had little impact on HBGA binding (48). Neither the antigenic consequences of residue changes nor the impacts of other residue substitutions on HBGA binding at this position have been rigorously evaluated, meaning that the D391N change in P.D302 may contribute to both the HBGA reactivity and antibody blockade differences observed for P.D302. To explore this possibility, we created homology models of these P2 domains and compared the predicted polar interactions present in residues 390 to 395 (Fig. 6) among GII.4-2006b, P.D1, and P.D302.
FIG 6.

Comparison of epitope site D polar interactions among GII.4-2006 and chronic-infection strains. PyMOL was used to model the polar interactions within residues 390 to 395 (A to C) and interactions between these residues and surrounding residues (D to F). GII.4-2006b is shown in purple (A and D), P.D1 is shown in teal (B and E), and P.D302 is shown in pink (C and F). Residues 390 to 395 are shown in orange for GII.4-2006b, yellow for P.D1, and aqua for P.D302. The dotted lines represent structure-based predicted polar interactions. (D to F) The dark-purple residues represent positions that interact with HBGAs.
Conformational comparisons between GII.4-2006b and P.D1 show general similarities in the shapes created by residues 390 to 395, with exceptions being the loss of a side chain in 393 of P.D1 and slight shifts in position for side chains in residues 394 and 395 (Fig. 6A and B). These conformational changes appear to impact the polar interactions within these residues, as the loss of the side chain in residue 393 ablates the hydrogen bond present in GII.4-2006b. In addition, the positional shifts in residues 394 and 395 in P.D1 appear to prevent the formation of another hydrogen bond present in GII.4-2006b. Conformational comparisons between GII.4-2006b and P.D302 demonstrate that the residue change at 391 has a significant impact on the shape and hydrogen-bonding networks for residues 390 to 395 (Fig. 6A and C). In P.D302, position 391 is bent downward, which differs from the position of the amino acid in GII.4-2006b and P.D1. The result of this change is the formation of a hydrogen bond between the side chain and the main chain of 391. In addition, though residue 393 is conserved between GII.4-2006b and P.D302, the side chain is shifted downward in P.D302 compared to GII.4-2006b, shifting the position of the hydrogen bond found at this residue. The formation of two additional novel hydrogen bonds between 390 and 393 and between 390 and 395 suggests that the 391 residue change and resulting conformational changes allowed these increased polar interactions. A slight conformational shift in residue 395 in P.D302 appears to ablate a polar interaction found in GII.4-2006b at this position. We also compared polar interactions of GII.4-2006, P.D1, and P.D302 to residues outside 390 to 395 (Fig. 6D to F). GII.4-2006b and P.D1 displayed five conserved polar interactions with surrounding amino acids (Fig. 6D and E), while P.D302 lost the polar interaction at residue 391 and gained an additional bond at residue 394 (Fig. 6F).
In addition to epitope site D being an antibody blockade epitope, these residues modulate HBGA binding, so evolution in this region is likely driven both by antibody selective pressure and by pressure to maintain binding to one or more HBGAs. Interestingly, all three structures maintained the two hydrogen bonds to positions 443 and 444. Residue 443 is in the HBGA binding site (37), and maintaining interaction with the residue may have been selected for in this individual in order to retain HBGA binding. The altered HBGA binding profile and reduced NBV97 binding and blockade for P.D302 may be explained by these polar differences, although this cannot be confirmed without a crystal structure of these P2 domains bound to NVB97 and HBGAs.
Our demonstration of intrahost changes in HBGA binding profiles in a chronically infected immunocompromised patient suggests that selection may favor variants that bind patient-specific HBGAs. While speculative, the potential emergence of intrahost variants that target patient-specific HBGA expression profiles could select for the emergence of novel strains that recognize unique or broad combinations of HBGA patterns, allowing altered pathogenicity and transmission efficiencies in an individual or across select human populations. We could not evaluate this possibility in our study because the HBGA expression profile of this chronically infected patient is unknown. Future research could evaluate these HBGA phenotypic and FUT 2/3 genotypic relationships using saliva and cells from chronically infected patients.
How much intrahost and interhost antigenic variation is necessary to give rise to a new strain that could escape herd immunity in the general population? Using blockade EC50 data from mouse sera against GII.4-2006b, GII.4-2009 (representative of a successive outbreak strain), P.D1, and P.D302, we demonstrated that the antigenic variation between P.D1 and P.D302 is 1.5 times greater than that seen between GII.4-2006b and GII.4-2009. To further address this question, we used antigenic cartography, which provides easily interpretable measures and visualization of multidimensional antigenic relationships and has previously been used to study antigenic differences in influenza virus strains (40, 49). This analysis provided further support for the idea that within-host changes in the virus can equal or exceed those differences seen across successive outbreak strains, with the antigenic space between P.D302 and both GII.4-2006b (D = 9.91) and P.D1 (D = 9.15) being greater than the average between the consecutive outbreak strains used in this study (average D = 4.98; range, 2.11 to 12.11), and mirrors the global difference between early GII.4 isolates (1987, 1997, and 2002) and contemporary strains (2006b, 2009, and 2012).
Antigenic cartography is a relatively new and powerful method with which to simply describe the multidimensional antigenic differences between virus strains. As such, there is room for improvement within these methods. Indeed, more complex statistical models underlying antigenic-cartography approaches are being developed to better account for uncertainty within these data sets (49), and more comprehensive surveys of both antisera and natural GII.4 isolates over a 30-year time span will better allow the characterization of antigenic change within noroviruses. In this study, the use of mouse sera permitted us to use an immunologically clean background with no preexposure history and provides a clearer starting point to evaluate specific relationships among outbreak strains and the intrahost isolates. Future work will require well-defined, time-ordered human sera from natural epidemic outbreaks; time-ordered sera from intrahost chronic infections; and synthetic reconstruction of capsids representing both outbreak and unique panels of interhost variants over time. Unfortunately, to date, we have been unable to obtain the samples necessary to pursue this comprehensive investigation. Our data suggest that intrahost evolution over a 10-month period can yield sufficient antigenic change to escape existing herd immunity. Clearly, additional work examining norovirus infectivity after prolonged shedding is needed in order to clarify whether chronically infected patients are a probable source of novel epidemic strains.
Therapeutics are needed to alleviate clinical disease during long-term norovirus infection and to prevent the potential emergence of novel antigenic variants with epidemic potential in the general population. Some success using IgG to treat chronic norovirus infection (32), coupled with our data demonstrating that P.D1 is relatively antigenically similar to GII.4-2006b while P.D302 is antigenically divergent, suggests that treatment early during chronic infection may be important for virus clearance and also supports the possibility that similarly administered, broadly neutralizing antibodies may be viable treatment options for patients suffering from long-term norovirus infection. Our work demonstrates that the GII.4 broadly neutralizing MAb NVB71.4 retains blockade response against P.D1 and P.D302, even though both strains are antigenically distinct from GII.4-2006b, GII.4-2009, and presumably other major GII.4 strains. This suggests that NVB71.4 or other antibodies with broad cross-blockade activity could be isolated and successfully used as norovirus therapeutics. Importantly, different monoclonal antibodies that target other GI and GII strain chronic infections will be needed. Furthermore, increased surveillance of norovirus isolates from chronically infected patients, as well as deep sequencing of patient isolates, should be considered in order to better understand the transmission dynamics and genetic potential of norovirus isolates from these patients, since they are likely different from what is seen in the general population. Overall, our work supports the idea that chronically infected individuals are potential reservoirs for antigenically novel norovirus strains, and further work to characterize their roles in transmission and emergent norovirus outbreaks and development of therapeutics to combat chronic infections should be a top priority.
ACKNOWLEDGMENTS
This work was supported by grant AI056351 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, and by institutional training grant T32-AI007419 from the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank Victoria Madden and C. Robert Bagnell, Jr., of the Microscopy Services Laboratory, Department of Pathology and Laboratory Medicine, University of North Carolina—Chapel Hill (UNC-CH), for expert technical support. We also acknowledge the UNC-CH Genome Analysis Facility.
Footnotes
Published ahead of print 19 March 2014
REFERENCES
- 1.Mattner F, Sohr D, Heim A, Gastmeier P, Vennema H, Koopmans M. 2006. Risk groups for clinical complications of norovirus infections: an outbreak investigation. Clin. Microbiol. Infect. 12:69–74. 10.1111/j.1469-0691.2005.01299.x [DOI] [PubMed] [Google Scholar]
- 2.Murata T, Katsushima N, Mizuta K, Muraki Y, Hongo S, Matsuzaki Y. 2007. Prolonged norovirus shedding in infants ≤6 months of age with gastroenteritis. Pediatr. Infect. Dis. J. 26:46–49. 10.1097/01.inf.0000247102.04997.e0 [DOI] [PubMed] [Google Scholar]
- 3.Okada M, Tanaka T, Oseto M, Takeda N, Shinozaki K. 2006. Genetic analysis of noroviruses associated with fatalities in healthcare facilities. Arch. Virol. 151:1635–1641. 10.1007/s00705-006-0739-6 [DOI] [PubMed] [Google Scholar]
- 4.Johnston CP, Qiu H, Ticehurst JR, Dickson C, Rosenbaum P, Lawson P, Stokes AB, Lowenstein CJ, Kaminsky M, Cosgrove SE, Green KY, Perl TM. 2007. Outbreak management and implications of a nosocomial norovirus outbreak. Clin. Infect. Dis. 45:534–540. 10.1086/520666 [DOI] [PubMed] [Google Scholar]
- 5.Kroneman A, Vega E, Vennema H, Vinje J, White PA, Hansman G, Green K, Martella V, Katayama K, Koopmans M. 2013. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 158:2059–2068. 10.1007/s00705-013-1708-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290. 10.1126/science.286.5438.287 [DOI] [PubMed] [Google Scholar]
- 7.Lindesmith LC, Donaldson EF, Lobue AD, Cannon JL, Zheng DP, Vinje J, Baric RS. 2008. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 5:e31. 10.1371/journal.pmed.0050031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donaldson EF, Lindesmith LC, Lobue AD, Baric RS. 2008. Norovirus pathogenesis: mechanisms of persistence and immune evasion in human populations. Immunol. Rev. 225:190–211. 10.1111/j.1600-065X.2008.00680.x [DOI] [PubMed] [Google Scholar]
- 9.Fankhauser RL, Monroe SS, Noel JS, Humphrey CD, Bresee JS, Parashar UD, Ando T, Glass RI. 2002. Epidemiologic and molecular trends of “Norwalk-like viruses” associated with outbreaks of gastroenteritis in the United States. J. Infect. Dis. 186:1–7. 10.1086/341085 [DOI] [PubMed] [Google Scholar]
- 10.Siebenga JJ, Vennema H, Renckens B, de Bruin E, van der Veer B, Siezen RJ, Koopmans M. 2007. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 81:9932–9941. 10.1128/JVI.00674-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen DJ, Noad R, Samuel D, Gray JJ, Roy P, Iturriza-Gomara M. 2009. Characterisation of a GII-4 norovirus variant-specific surface-exposed site involved in antibody binding. Virol. J. 6:150. 10.1186/1743-422X-6-150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindesmith LC, Donaldson EF, Baric RS. 2011. Norovirus GII.4 strain antigenic variation. J. Virol. 85:231–242. 10.1128/JVI.01364-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Debbink K, Donaldson EF, Lindesmith LC, Baric RS. 2012. Genetic mapping of a highly variable norovirus GII.4 blockade epitope: potential role in escape from human herd immunity. J. Virol. 86:1214–1226. 10.1128/JVI.06189-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindesmith LC, Beltramello M, Donaldson EF, Corti D, Swanstrom J, Debbink K, Lanzavecchia A, Baric RS. 2012. Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLoS Pathog. 8:e1002705. 10.1371/journal.ppat.1002705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindesmith LC, Debbink K, Swanstrom J, Vinje J, Costantini V, Baric RS, Donaldson EF. 2012. Monoclonal antibody-based antigenic mapping of norovirus GII.4-2002. J. Virol. 86:873–883. 10.1128/JVI.06200-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindesmith LC, Costantini V, Swanstrom J, Debbink K, Donaldson EF, Vinje J, Baric RS. 2013. Emergence of a norovirus GII.4 strain correlates with changes in evolving blockade epitopes. J. Virol. 87:2803–2813. 10.1128/JVI.03106-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Debbink K, Lindesmith LC, Donaldson EF, Costantini V, Beltramello M, Corti D, Swanstrom J, Lanzavecchia A, Vinje J, Baric RS. 2013. Emergence of new pandemic GII.4 Sydney norovirus strain correlates with escape from herd immunity. J. Infect. Dis. 208:1877–1887. 10.1093/infdis/jit370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rockx B, De Wit M, Vennema H, Vinje J, De Bruin E, Van Duynhoven Y, Koopmans M. 2002. Natural history of human calicivirus infection: a prospective cohort study. Clin. Infect. Dis. 35:246–253. 10.1086/341408 [DOI] [PubMed] [Google Scholar]
- 19.Obara M, Hasegawa S, Iwai M, Horimoto E, Nakamura K, Kurata T, Saito N, Oe H, Takizawa T. 2008. Single base substitutions in the capsid region of the norovirus genome during viral shedding in cases of infection in areas where norovirus infection is endemic. J. Clin. Microbiol. 46:3397–3403. 10.1128/JCM.01932-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siebenga J, Kroneman A, Vennema H, Duizer E, Koopmans M. 2008. Food-borne viruses in Europe network report: the norovirus GII.4 2006b (for US named Minerva-like, for Japan Kobe034-like, for UK V6) variant now dominant in early seasonal surveillance. Euro Surveill. 13(2):pii=8009 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=8009 [PubMed] [Google Scholar]
- 21.Nilsson M, Hedlund KO, Thorhagen M, Larson G, Johansen K, Ekspong A, Svensson L. 2003. Evolution of human calicivirus RNA in vivo: accumulation of mutations in the protruding P2 domain of the capsid leads to structural changes and possibly a new phenotype. J. Virol. 77:13117–13124. 10.1128/JVI.77.24.13117-13124.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schorn R, Hohne M, Meerbach A, Bossart W, Wuthrich RP, Schreier E, Muller NJ, Fehr T. 2010. Chronic norovirus infection after kidney transplantation: molecular evidence for immune-driven viral evolution. Clin. Infect. Dis. 51:307–314. 10.1086/653939 [DOI] [PubMed] [Google Scholar]
- 23.Hoffmann D, Hutzenthaler M, Seebach J, Panning M, Umgelter A, Menzel H, Protzer U, Metzler D. 2012. Norovirus GII.4 and GII.7 capsid sequences undergo positive selection in chronically infected patients. Infect. Genet. Evol. 12:461–466. 10.1016/j.meegid.2012.01.020 [DOI] [PubMed] [Google Scholar]
- 24.Ludwig A, Adams O, Laws HJ, Schroten H, Tenenbaum T. 2008. Quantitative detection of norovirus excretion in pediatric patients with cancer and prolonged gastroenteritis and shedding of norovirus. J. Med. Virol. 80:1461–1467. 10.1002/jmv.21217 [DOI] [PubMed] [Google Scholar]
- 25.Bull RA, Eden JS, Luciani F, McElroy K, Rawlinson WD, White PA. 2012. Contribution of intra- and interhost dynamics to norovirus evolution. J. Virol. 86:3219–3229. 10.1128/JVI.06712-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koo HL, DuPont HL. 2009. Noroviruses as a potential cause of protracted and lethal disease in immunocompromised patients. Clin. Infect. Dis. 49:1069–1071. 10.1086/605558 [DOI] [PubMed] [Google Scholar]
- 27.Capizzi T, Makari-Judson G, Steingart R, Mertens WC. 2011. Chronic diarrhea associated with persistent norovirus excretion in patients with chronic lymphocytic leukemia: report of two cases. BMC Infect. Dis. 11:131. 10.1186/1471-2334-11-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boillat Blanco N, Kuonen R, Bellini C, Manuel O, Estrade C, Mazza-Stalder J, Aubert JD, Sahli R, Meylan P. 2011. Chronic norovirus gastroenteritis in a double hematopoietic stem cell and lung transplant recipient. Transpl Infect. Dis. 13:213–215. 10.1111/j.1399-3062.2010.00565.x [DOI] [PubMed] [Google Scholar]
- 29.Florescu DF, Hill LA, McCartan MA, Grant W. 2008. Two cases of Norwalk virus enteritis following small bowel transplantation treated with oral human serum immunoglobulin. Pediatr. Transplant. 12:372–375. 10.1111/j.1399-3046.2007.00875.x [DOI] [PubMed] [Google Scholar]
- 30.Florescu DF, Hermsen ED, Kwon JY, Gumeel D, Grant WJ, Mercer DF, Kalil AC. 2011. Is there a role for oral human immunoglobulin in the treatment for norovirus enteritis in immunocompromised patients? Pediatr. Transplant. 15:718–721. 10.1111/j.1399-3046.2011.01556.x [DOI] [PubMed] [Google Scholar]
- 31.Ebdrup L, Bottiger B, Molgaard H, Laursen AL. 2011. Devastating diarrhoea in a heart-transplanted patient. J. Clin. Virol. 50:263–265. 10.1016/j.jcv.2010.11.007 [DOI] [PubMed] [Google Scholar]
- 32.Chagla Z, Quirt J, Woodward K, Neary J, Rutherford C. 2013. Chronic norovirus infection in a transplant patient successfully treated with enterally administered immune globulin. J. Clin. Virol. 58:306–308. 10.1016/j.jcv.2013.06.009 [DOI] [PubMed] [Google Scholar]
- 33.Sukhrie FH, Siebenga JJ, Beersma MF, Koopmans M. 2010. Chronic shedders as reservoir for nosocomial transmission of norovirus. J. Clin. Microbiol. 48:4303–4305. 10.1128/JCM.01308-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaufman SS, Chatterjee NK, Fuschino ME, Morse DL, Morotti RA, Magid MS, Gondolesi GE, Florman SS, Fishbein TM. 2005. Characteristics of human calicivirus enteritis in intestinal transplant recipients. J. Pediatr. Gastroenterol. Nutr. 40:328–333. 10.1097/01.MPG.0000155182.54001.48 [DOI] [PubMed] [Google Scholar]
- 35.Siebenga JJ, Beersma MF, Vennema H, van Biezen P, Hartwig NJ, Koopmans M. 2008. High prevalence of prolonged norovirus shedding and illness among hospitalized patients: a model for in vivo molecular evolution. J. Infect. Dis. 198:994–1001. 10.1086/591627 [DOI] [PubMed] [Google Scholar]
- 36.Eden JS, Tanaka MM, Boni MF, Rawlinson WD, White PA. 2013. Recombination within the pandemic norovirus GII.4 lineage. J. Virol. 87:6270–6282. 10.1128/JVI.03464-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao S, Lou Z, Tan M, Chen Y, Liu Y, Zhang Z, Zhang XC, Jiang X, Li X, Rao Z. 2007. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 81:5949–5957. 10.1128/JVI.00219-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baric RS, Yount B, Lindesmith L, Harrington PR, Greene SR, Tseng FC, Davis N, Johnston RE, Klapper DG, Moe CL. 2002. Expression and self-assembly of Norwalk virus capsid protein from Venezuelan equine encephalitis virus replicons. J. Virol. 76:3023–3030. 10.1128/JVI.76.6.3023-3030.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnett JL, Yang J, Cai Z, Zhang T, Wan XF. 2012. AntigenMap 3D: an online antigenic cartography resource. Bioinformatics 28:1292–1293. 10.1093/bioinformatics/bts105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai Z, Zhang T, Wan XF. 2010. A computational framework for influenza antigenic cartography. PLoS Comput. Biol. 6:e1000949. 10.1371/journal.pcbi.1000949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cannon JL, Lindesmith LC, Donaldson EF, Saxe L, Baric RS, Vinje J. 2009. Herd immunity to GII.4 noroviruses is supported by outbreak patient sera. J. Virol. 83:5363–5374. 10.1128/JVI.02518-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reeck A, Kavanagh O, Estes MK, Opekun AR, Gilger MA, Graham DY, Atmar RL. 2010. Serological correlate of protection against norovirus-induced gastroenteritis. J. Infect. Dis. 202:1212–1218. 10.1086/656364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen DJ, Adams NL, Aladin F, Harris JP, Brown DW. 2014. Emergence of the GII-4 norovirus Sydney2012 strain in England, winter 2012–2013. PLoS One 9:e88978. 10.1371/journal.pone.0088978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roddie C, Paul JP, Benjamin R, Gallimore CI, Xerry J, Gray JJ, Peggs KS, Morris EC, Thomson KJ, Ward KN. 2009. Allogeneic hematopoietic stem cell transplantation and norovirus gastroenteritis: a previously unrecognized cause of morbidity. Clin. Infect. Dis. 49:1061–1068. 10.1086/605557 [DOI] [PubMed] [Google Scholar]
- 45.Glass RI, Parashar UD, Estes MK. 2009. Norovirus gastroenteritis. N. Engl. J. Med. 361:1776–1785. 10.1056/NEJMra0804575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atmar RL, Opekun AR, Gilger MA, Estes MK, Crawford SE, Neill FH, Graham DY. 2008. Norwalk virus shedding after experimental human infection. Emerg. Infect. Dis. 14:1553–1557. 10.3201/eid1410.080117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Etherington GJ, Ring SM, Charleston MA, Dicks J, Rayward-Smith VJ, Roberts IN. 2006. Tracing the origin and co-phylogeny of the caliciviruses. J. Gen. Virol. 87:1229–1235. 10.1099/vir.0.81635-0 [DOI] [PubMed] [Google Scholar]
- 48.Tan M, Xia M, Cao S, Huang P, Farkas T, Meller J, Hegde RS, Li X, Rao Z, Jiang X. 2008. Elucidation of strain-specific interaction of a GII-4 norovirus with HBGA receptors by site-directed mutagenesis study. Virology 379:324–334. 10.1016/j.virol.2008.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bedford T, Suchard MA, Lemey P, Dudas G, Gregory V, Hay AJ, McCauley JW, Russell CA, Smith DJ, Rambaut A. 2014. Integrating influenza antigenic dynamics with molecular evolution. Elife 3:e01914. 10.7554/eLife.01914 [DOI] [PMC free article] [PubMed] [Google Scholar]