

Abstract
Not surprisingly, the death of a cell is a complex and well controlled process. For several decades, apoptosis, the first genetically programmed death process to be identified has taken centre stage as the principal mechanism of programmed cell death (type I cell death) in mammalian tissues. Apoptosis has been extensively studied and its contribution to the pathogenesis of disease well documented. However, apoptosis does not function alone in determining the fate of a cell. More recently, autophagy, a process in which de novo formed membrane enclosed vesicles engulf and consume cellular components, has been shown to engage in complex interplay with apoptosis. As a result, cell death has been subdivided into the categories apoptosis (Type I), autophagic cell death (Type II), and necrosis (Type III). The boundary between Type I and II cell death is not completely clear and as we will discuss in this review and perhaps a discrete difference does not exist, due to intrinsic factors among different cell types and crosstalk among organelles within each cell type. Apoptosis may begin with autophagy and autophagy can often end with apoptosis, inhibition or a blockade of caspase activity may lead a cell to default into Type II cell death from Type I.
Keywords: Apoptosis, Autophagy, Sorafenib, Caspase, Autophagosome
1. Introduction
Autophagy is often observed in dying cells. In some aspects this may be an attempt by the cell to mitigate a given stress before resorting to apoptosis. Alternatively, activation of autophagy might reflect a crosstalk between the processes of autophagy and apoptosis. Autophagy is induced in response to many stresses that ultimately lead to apoptosis, such as, organelle dysfunction, metabolic stress, atrophy [1] and chemotherapeutic intervention [2], pathogen infection and starvation. If the pertinent stress is resolved, cells will typically restore homeostasis and return to their initial state. However, if the stress persists and autophagy is no longer able to support cell survival, cells can respond by activating the processes of apoptosis in order to ensure their controlled and efficient elimination, without triggering local inflammation. Therefore, regulatory ties between autophagy and apoptosis (and vice versa) may represent an evolutionary advantage to cells, since it allows for a more controlled response to a given stress signal. Thus, although autophagy and apoptosis clearly represent distinct cellular processes with fundamentally different biochemical and morphological features, the protein networks that control their regulation and execution can be highly inter connected. The cross talk between apoptosis and autophagy is complex and may often appear contradictory, but critical to the overall fate of the cell. Determining which interactions are important in the regulation of cell death by autophagy is of vital importance as we try to either protect the cells we do not want to die (as in neurodegenerative diseases), or cause diseased cells to die (as in cancer treatment).
Clearly, cell death cannot be neatly and discretely categorised and the different types of cell death overlap [3]. Multiple direct and indirect interactions between apoptosis and autophagy have been described, indicating a mechanistic overlap and interaction between the apoptosis machinery and autophagy proteins [4–6]. The majority of these interactions have currently been shown to be apoptotic evens altering autophagy; less is known at the mechanistic level about how autophagic events control apoptosis. Several laboratories have reported that molecules previously defined as intermediaries in the activation of apoptosis also function as intermediaries in the activation of autophagy, further calling into question the specific roles of both apoptosis and autophagic cell death, in addition to our ability to distinguish these processes. The anti cancer drug sorafenib, a potent multiple tyrosine kinase inhibitor currently approved for the treatment of refractory renal cell and hepatocellular carcinoma., serves to highlight this issue. Sorafenib is recognised as the standard systemic treatment for patients with advanced hepatocellular carcinoma. However, the direct functional mechanism of tumour lethality mediated by sorafenib remain to be fully characterised and the precise mechanisms of drug resistance are largely unknown. Studies have suggested that sorafenib can induce both autophagy and apoptosis [7]. We have shown in our laboratory that sorafenib can enhance the cytotoxicity of the anti folate pemetrexed through an autophagy dependent mechanism, the treatment increased autophagy and cell death in vitro and suppressed tumour growth in vivo. [8]. Similarly, other work has indicated that sorafenib induced autophagy in hepatocellular carcinoma mediated through Beclin 1 [9]. Conversely, recent data has suggested that the anti cancer effects of sorafenib are mediated by the induction of apoptotic cell death and that the simultaneous activation of autophagy via IRE1, led to an inhibition of apoptosis and thus limited the efficacy of the drug [10]. In agreement with this work, inhibition of autophagy has been demonstrated to potentiate the anti tumour effect of sorafenib in hepatocellular carcinoma [11]. We will show throughout this review, where pertinent, that sorafenib is intimately involved in both autophagic and/or apoptotic mediated cell death.
2. Regulation of apoptosis by autophagy proteins
The regulation of apoptosis by autophagy can currently be divided into a number of different mechanisms. We will first discuss the potential regulation of apoptosis by specific autophagy proteins. In this process specific autophagy proteins can regulate apoptosis via mechanisms which may not be related to their canonical role in autophagy signalling. The apoptotic function of these proteins does not need activation of the entire autophagic process. Specific autophagy related proteins may have evolved to regulate the apoptotic pathway through direct interaction with components of the apoptotic machinery. Inhibition or genetic alteration of these specific autophagy genes may affect apoptosis, since usually neither autophagosome formation, or autolysosomal function are required for their apoptotic function. Autophagosome formation requires the covalent conjugation of the autophagy proteins ATG12 and ATG5 in a ubiquitylation like process that involves ATG7 and ATG10. ATG5 and ATG12 are, therefore, integral parts of the autophagic machinery and are required for the induction of autophagy. ATG5 and ATG12 have been found to have key roles in the initiation of apoptosis in response to diverse stress signals (12). Non conjugated forms of ATG12 and ATG5 contribute to induction of apoptosis, implying that their role in apoptosis may be independent of their role in autophagy. In apoptotic cells, ATG5 is cleaved by calpains resulting the translocation of the N terminal fragment of ATG5 to the mitochondrion, where it mediates the release of cytochrome c by interacting with the pro survival BCL2 family member B cell lymphoma extra large (BCLXL) [12]. Similarly, sorafenib can affect a change in the pro survival activity of BCLXL, leading to the induction of apoptosis. Sorafenib mediated BIM activation has been shown to interfere with BCL2, BCLXL, and MCL1 activation. BIM was shown to bind to BCL2 and BCLX, possibly in response to sorafenib mediated release of BIM from MCL1 after down regulation of the latter [13]. ATG12 has been shown to be necessary for caspase activation in response to a range of apoptotic stress inducers. Knockdown of several other essential autophagic genes did not show a significant effect upon apoptosis, supporting a specific role for ATG12 in apoptosis, that does not require autophagosome formation [14]. Further, a BH3 like domain within ATG12 is able to bind to and inhibit BCL2 and MCL1, two anti apoptotic members of the BCL2 family [14]. Recently, a conjugate comprised form of ATG12 and ATG3 has been shown to be involved in regulation of mitochondrial homeostasis and cell death via apoptosis mediated by mitochondrial pathways, but has no effect on death receptor mediated apoptosis [15]. In addition, disrupting ATG12 conjugation to ATG3 renders cells resistant to mitochondrial cell death [16].
The regulation of apoptosis by other proteins essential for recruitment of autophagy proteins to sites of autophagosome formation include the class III phosphatidylinositol 3 kinase (PIK3C3)–Beclin 1 complex, which serves as a central regulator of autophagy downstream of mammalian target of rapamycin (mTOR), Fig. 1. Beclin 1, the mammalian orthologue of yeast Atg6, has a pivotal role in autophagy, a process of programmed cell survival, which is increased during periods of cell stress and extinguished during the cell cycle. Beclin 1 was identified as a new member of the BH3 only family of proteins [17]. Binding of BCL2 to the BH3 domain of Beclin 1 has been shown to inhibit autophagy [18]. The interaction between BCL2 and Beclin 1 may function as a rheostat that maintains autophagy at levels that are compatible with cell survival rather than cell death [2]. Mutations of either the BH3 only domain within Beclin 1, or the BH3 receptor domain within BCL2 or BCLXL, disrupted the Beclin 1–BCL2 complex, resulting in the stimulation of autophagy. Sorafenib may not play a role in altering Beclin1 BCL2 binding, since over expression of BIM does not affect the interaction between BCL2 and Beclin 1. However, sorafenib mediated BIM activation does interfere with BCL2, BCLXL, and MCL1 activation. BIM has been shown to functions as an inhibitor of autophagy, [19], but has been shown to lead to induction of apoptosis in a number of studies [13,20], possibly mediated via release of BIM from MCL1 after down regulation of the latter [13].
Fig. 1.

BCL2 and BCLXL can inhibit apoptosis by blocking activation of BAX and BAK by preventing subsequent release of cytochrome c and other apoptogenic proteins from the mitochondria.
It would be expected that binding of Beclin 1 to BCL2 would induce apoptosis through neutralising the anti apoptotic function of BCL2. However, apoptosis was not shown to be induced [1]. Further, BCL2 retained full anti apoptotic activity. [21]. The interaction between Beclin 1 and BCL2 is unidirectional, with inhibition of autophagy without a reciprocal effect on apoptotic events. In contrast, Beclin 1 can have an anti apoptotic role in several settings including TRAIL, chemotherapy, irradiation, immunotherapy, nutrient deprivation, angiogenesis inhibitors and hypoxia. With respect to TRAIL, the anti apoptotic effects of Beclin 1 can be overcome using sorafenib. Suppression of STAT3 by sorafenib has recently been shown to down regulate MCL1 and BCLXL and to sensitise cells to TRAIL mediated apoptosis [22]. In addition, Sorafenib has been shown to induce apoptosis of both endometrial cancer cell lines and human primary cultures and sensitises these cells to TRAIL induced apoptosis [23]. BID knockdown protects cancer cells against apoptosis and leads to induction of autophagy, by increased expression of Beclin 1. The precise mechanism of Beclin 1 mediated inhibition of apoptosis is not yet clear, but may be related to unregulated autophagy as an adaptive or anti injury mechanism, leading to the clearing of apoptotic cells [24]. In addition, ER localised BCL2, but not mitochondria localised BCL2 was shown to inhibit autophagy [25], consistent with the initial ideas that ER associated class III PI3K activity was crucial in the nucleation of autophagosome formation. Beclin 1 can co localise with BCLXL within mitochondria via its BH3 domain [26] suggesting a differential role of BCLXL in Beclin 1 complex when compared with BCL2.
A number of groups have observed caspase induced site specific cleavage of Beclin 1 following induction of apoptosis [27,28]. These results led to a hypothesis that Beclin 1cleavage may inhibit protective autophagy in cells committed to cell death. However an in vitro assay suggested that the C terminal fragment of Beclin 1 was shown to translocate to mitochondria and sensitise cells to induce release of cytochrome c in response to apoptotic signals, possibly indicating its pro apoptotic function in vivo.
[29,24]. This process may represent an amplifying loop for inducing massive apoptotic cell death. So, with respect to Beclin 1 and ATG5, activation of apoptosis can induce site specific cleavage of these autophagic proteins, which may serve to suppress protective autophagy and at the same time, generate mitochondria targeted cleavage products which may function as positive feedback loops leading to an enhanced apoptotic response, Fig. 2.
Fig. 2.

The autophagy regulator ATG5 induces caspase activation following cleavage of Atg 5 by calpain and translocation of the truncated N terminal Atg 5 protein fragment to the outermitichondrial membrane, leading to the induction mitochondrial cytochrome c release and caspase activation. Similarly, translocation of C terminal BECN1 fragment to the outer mitochondrial membrane can induce cytochrome-c release and apoptosis.
3. Regulation of autophagy by apoptotic proteins
Similar to apoptotic regulation by autophagic proteins, components of the apoptotic machinery can regulate autophagy via molecular interactions with autophagy proteins. The most extensively studied example for this type of regulation is the role played by BCL2 and the potential to inhibit both pathways. As previously mentioned, in resting cells, BCL2 is constitutively bound to Beclin 1, this allows for a low levels of autophagy. Under autophagic inducing cell stress, BCL2 can dissociate from Beclin 1, resulting in an increase in autophagy. Indeed, a combination of BCL2 dependent regulation and feedback loops between Beclin1 and caspases robustly enforces a sequential activation of cellular responses depending upon the intensity and duration of stress levels [30]. Mutations of either the BH3 only domain within Beclin 1, or the BH3 receptor domain within BCl2 or BCLXL, disrupted the Beclin 1–BCL2 complex, resulting in the stimulation of autophagy. A number of mechanisms that control the dissociation of BCL2 from Beclin 1 in cells under autophagic stress have been identified. Regulation of the BCL2–Beclin 1 has been demonstrated through competitive displacement of the Beclin 1 BH3 domain by other BCL2 family proteins, including BAD (1), tBID, BAD and BNIP3, but not by BAX and BAK [31]. Moreover, the pro apoptotic BH3 only proteins such as BNIP3, BAD, NOXA, PUMA, BIMEL and BIK are all able to induce autophagy. However, over expression of BIM does not affect the interaction between BCL2 and Beclin 1. BIM has been shown to functions as an inhibitor of autophagy, by anchoring Beclin 1 to microtubules [19]. Several studies have shown BIM to play an important functional role in sorafenib mediated apoptosis [13,20]. Sorafenib mediated BIM activation has been shown to interfere with BCL2, BCLXL, and MCL1 activation. BIM was shown to bind to BCL2 and BCLX, presumably in response to sorafenib mediated release of BIM from MCL1 after down regulation of the latter [13]. In a number of hepatic carcinoma cell lines, the sensitivity to sorafenib induced cytotoxicity was determined by BAD expression. Stable cell lines derived from Huh7 cells over expressing BAD were found to exhibit a striking five fold increase in their sensitivity to sorafenib induced cell death [32]. Recent work has shown that PUMA can be activated by sorafenib. Sorafenib treatment was shown to induce PUMA in a variety of cancer cells irrespective of their p53 status. In these studies, deficiency in PUMA abrogated sorafenib induced apoptosis and caspase activation and rendered sorafenib resistance in colony formation and xenograft tumour assays [33]. Conversely, another pro apoptotic protein, NOXA, has been shown to sharply down regulated by sorafenib [34].
Phosphorylation of Beclin 1 and BCL2 constitutes another mechanism controlling dissociation of BCL2 from Beclin 1. Phosphorylation of BCL2 at multiple sites by c Jun N terminal protein kinase 1 (JNK1) and extracellular signal related kinase (Erk) have been shown to reduce binding of BCL2 to Beclin 1, leading to the activation of autophagy [35–37] and Fig. 3. Further, phosphorylation of Beclin 1 within its BH3 domain by death associated protein kinase (DAPK) has been shown to induce autophagy by promoting its dissociation from BCLXL [38]. Sorafenib has been shown to activate autophagy in a dose and time dependent manner in a range of hepatic cell via release of beclin1 from MCL1. Sorafenib was shown to down regulated phospho STAT3 (P STAT3) and subsequently reduced the expression of MCL1, resulting in disruption of the beclin 1 MCL1 complex [9].
Fig. 3.

Phosphorylation of BCL2 at multiple sites by c Jun N terminal protein kinase 1 (JNK1) and extracellular signal related kinase (ERK) have been shown to reduce binding of BCL2 to Beclin 1, leading to the activation of autophagy However, once released from BCL2, as shown in figure 2, translocation of C terminal fragment of BECN1 to the outer mitochondrial membrane can induce cytochrome-c release and apoptosis.
Ceramide is associated with cell growth arrest and the induction of cell death, it is not only a potent inducer of apoptosis, as it can also trigger autophagic cell death in malignant glioma cells via activation of the pro apoptotic member of the BCL2 family, BNIP3 [39]. Further apoptotic regulation of autophagy can be seen in human leukaemic cells, down regulation of BCL2 has been shown to induce autophagy, leading to cell death [40]. In neural precursor cells, deprivation of growth factors leads to an autophagic cell death, which can be blocked by the anti apoptotic BCL2 [41], an involvement that has also been recognised in autophagic cell death induced by HSPin1, a molecule first identified as interacting with BCL/BCLXL [42].
Spatial separation of proteins to cellular compartments may represent an additional way to achieve the independent regulation of autophagy and apoptosis. Two cellular sources of BCL2 exist, one at the endoplasmic reticulum (ER) and the other at the mitochondria, both appear to regulate autophagy and apoptosis, respectively [20]. A group of ER proteins ensures the partitioning of BCL2 to the autophagy pathway. The regulation of autophagy by binding of BCL2 to Beclin 1 at the ER is facilitated by the ER protein, nutrient deprivation autophagy factor 1 (NAF1), see Fig. 4 and inhibited by the ER localised BH3 only protein (B cell interacting killer) BIK [43].
Fig. 4.
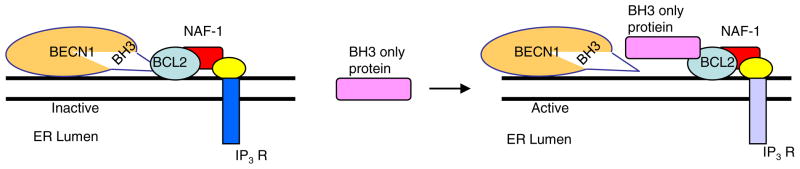
Beclin1/BCL2/NAF1 interaction and autophagy regulation at the endo[plasmic reticulum. IP3R—inositol triphosphate receptor.
Moreover, JNK1 specifically phosphorylates the ER pool of BCL2 during starvation to induce autophagy [35]. Conversely, interactions between BCL2 and BAX or BAK at the mitochondrion can control the activation of apoptosis [44,45]. Therefore, compartmentalisation of BCL2 together with organelle specific sets of associated interacting proteins, may allow for dynamic and independent regulation of autophagy and apoptosis.
The anti apoptotic protein MCL1 has also been shown to regulate autophagy. MCL1 degradation is an early event not only following induction of apoptosis, but also under nutrient deprivation conditions where MCL1 levels regulate activation of autophagy. Furthermore, deletion of MCL1 in cortical neurons of transgenic mice activates a strong autophagic response. This response has been demonstrated to be converted to apoptosis by either reducing the levels of the autophagy regulator Beclin 1, or by a concomitant activation of BAX [46], Fig. 4.
As indicated previously, the pivotal role played by MCL1 in induction of autophagy can be exploited in the relatively rapid (within three hours), sorafenib mediated dephosphorylated and/or down regulated in expression of MCL1, which has been shown to lead to caspase independent cell death [47]. In agreement with these results, sorafenib has been shown to induce apoptotic cell death in chronic lymphocytic leukemia. Sorafenib induces cell death was preceded by a rapid down regulation of MCL1 through the inhibition of protein translation. Subsequently, the cell intrinsic apoptotic pathway was activated, indicated by destabilisation of the mitochondrial membrane potential and activation of caspase 3 and 9 [48].
FLICE like inhibitor protein (FLIP) is an anti apoptotic protein that is implicated in suppression of death receptor mediated apoptosis [49]. FLIP has been shown recently, to inhibit autophagy by interfering with the function of ATG3 in the LC3 conjugation system, by competing with LC3 for binding of ATG3, attenuating LC3 lipidation [50]. In addition to sorafenib mediated alterations in MCL1 expression, FLIP is also rapidly dephosphorylated and/or down regulated in response cell exposure to sorafenib, resulting in caspase independent cell death. [46]. When autophagy is induced, the interaction between FLIP and ATG3 is substantially decreased. Notably, separate regions within the FLIP protein have been shown to control its anti autophagic and anti apoptotic activities, [50]. Similar to BCL2, FLIP can inhibit both autophagy and apoptosis. Since inhibition of apoptosis by FLIP takes place at the plasma membrane, and inhibition of autophagy probably occurs at sites of autophagosome formation, compartmentalisation of FLIP to these two sub cellular localisations could provide a way to achieve independent regulation of FLIP mediated autophagy and apoptosis.
p53, is a potent inducer of apoptosis, but multiple p53 target genes can also stimulate autophagy. This effect is often seen following inhibition of the signalling axis converging on the negative regulator of autophagy the mammalian target of rapamycin (mTOR). AMP activated protein kinase (AMPK), promotes autophagy by phosphorylating the tuberous sclerosis (TSC) complex proteins TSC1 and TSC2 which in turn down regulate mTOR activity [51]. Other studies have shown autophagy to be induced through increased expression of a direct p53 target gene called damage regulated autophagy modulator (DRAM), a lysosomal protein that functions at the crossroad between p53 induced autophagy and cell death [52]. Indeed, in HCT116 human colorectal cancer cells exposed to a prolonged nutrient deprivation, endogenous wt p53 was show, post transcriptionally, to down regulate LC3, a pivotal component of the autophagic machinery. This effect was shown to lead to a reduced, yet sustainable autophagic flux. Loss of p53 impaired this autophagic flux and causes excessive LC3 accumulation upon cellular starvation, culminating in apoptosis. [53]. However, LC3 accumulation may not always result in cell death. Sorafenib has been shown to inhibit the mammalian mTOR and induce accumulation of LC3 II, indicating enhanced autophagic activity, which conferred a survival advantage to the hepatoma cells used in this study. Concomitant pharmacological inhibition of the autophagic flux was shown to increase apoptotic cell death and decreased cell viability [11].
Contrasting with the fact that nuclear p53 functions as a pro autophagic transcription factor, the cytoplasmic pool of p53 has been shown to suppress autophagy in a number of experimental settings [55]. In human, mouse as well as nematode cells, knockdown of p53 by gene knockout, RNA interference or chemical agents was seen to induce autophagy that relied upon the canonical activation pathway converging on mTOR inhibition [54]. Human p53−/− colon cancer cells are characterized by increased baseline levels of autophagy that can be reduced by re introduction of wild type p53. Interestingly, these studies demonstrated that p53 inhibition caused autophagy mostly in the G0/G1 phase, to a lesser extent in the S phase, and practically spares G2/M phase of the cell cycle [55]. The suppression of autophagy by p53 correlates with its nuclear to cytosolic redistribution [56]. Inhibition of autophagy by p53 is maximal when the p53 nuclear localisation sequence is deleted, while it is entirely abolished by the deletion of the nuclear export signal [56,57] These observations prove firstly that the cytoplasmic pool of p53 inhibits autophagy, and that autophagy inhibition by p53 can be uncoupled from the pro apoptotic role of cytoplasmic p53. Sorafenib has been shown to be involved in p53 mediated cellular events. Glycogen synthase kinase 3β (GSK 3β) phosphorylates p53 on Ser33 [58], sorafenib increased the GSK 3β induced phosphorylation of p53 at Ser33. Inhibition of GSK 3β suggested that the GSK 3β activation induced by sorafenib did not contribute to the apoptosis, but exerted a protective effect when used alone [59].
4. Autophagosomes can play a role in apoptosis
Caspases can be activated via recruitment to the autophagosomes. At the moment, this mechanism of activation has been described for caspase 8. This caspase can be activated by a death inducing signalling complex (DISC) like complex, which has been shown to assemble on autophagosomal membranes [60]. A mechanism of sorafenib induced cyto toxicity may involve DISC formation. Sorafenib has been shown to cause CD95 tyrosine phosphorylation; DISC formation and the induction of cell death and autophagy, events which are abolished via knock down/knock out of Src family kinases [61]. Autophagy mediated activation of caspase 8, seen following treatment with proteasome inhibitors did not require death ligands, suggesting that autophagy links caspase 8 to apoptosis that is induced by intrinsic stress signals [62]. Two different routes for caspase 8 recruitment to the autophagosomes have currently been described. Ubiquitinated caspase 8 can bind to p62 through the ubiquitin binding domain of p62, and is subsequently recruited to the autophagosome through a direct interaction between p62 and LC3. Additionally, p62 is also required for activation of caspase 8, by facilitating self oligomerisation [60,63]. The second route of caspase 8 recruitment to the autophagosomes has been shown to take place through an interaction between FADD and ATG5 [64] Our lab has shown that sorafenib and vorinostat interacted in vitro in a synergistic fashion to kill hepatic, renal, and pancreatic adenocarcinoma cells and that cell killing was suppressed by inhibition of caspase 8. Indeed, sorafenib and vorinostat treatment increased surface levels of CD95 and CD95 association with caspase8. Knockdown of CD95 or FADD expression significantly was shown to reduce sorafenib/vorinostat mediated lethality [65].
The relative contribution of each route to caspase activation has not currently been determined, nor whether other caspases are activated in the same way. However, results to date have suggested that autophagosome mediated caspase activation requires the formation of autophagosomes (or at least autophagosomal membranes), but not autolysosomal activity. As a result, it was found that depletion of autophagy genes can lead to inhibition of caspase 8 activation, the pharmacological inhibition of lysosomal fusion was shown to enhance caspase 8 activation, this is probably due to the decreased turnover of autophagosomes [21]. Further, upon the inhibition of protein degradation pathways, cells upregulate the autophagic response to facilitate autophagolysosomal protein turnover, which is mediated by the lipidation and membrane localisation of LC3 This leads to the enrichment of LC3 on intracellular membranes that can serve as a molecular hub to recruit the ubiquitin binding protein SQSTM1/p62 and one of its binding partners, caspase 8. The induced oligomerisation and activation of caspase 8 subsequently initiate the downstream apoptosis cascade, protein turnover, which is mediated by the lipidation and membrane localisation of LC3 [66]. In agreement with these results, more recent studies have shown that over expression of wild type p62/SQSTM1 in U87MG human glioma cells was able to activate caspase 8 and then promote HAMLET (a complex of oleic acids and decalcified α lactalbumin that was discovered to selectively kill tumour cells both in vitro and in vivo), induced apoptosis, whereas knockdown of p62/SQSTM1 manifested the opposite effect [67]. This is another cellular system whereby sorafenib may potentiate cyto protective mechanisms is certain cells. Sorafenib has been shown to induce LC3 accumulation in autophagosomes, increase LC3 lipidation and p62 degradation. Further, the chemical inhibition of early and late autophagy with 3 methyladenine or chloroquine did not reduce, but rather potentiated sorafenib induced cell death, suggesting that, the cells used in this study, PC3 (a metastatic prostate cancer cell line), activate a cyto protective autophagic response following treatment with sorafenib. Conversely, sorafenib has been shown to induce caspase 8 cleavage and apoptosis in multiple myeloma cells in a time dependent manner, determined by flow cytometry [68].
5. Caspase regulation of autophagy
A number of autophagic proteins have been identified as targets of caspase mediated cleavage, which will ultimately lead to an inactivation of their autophagic function as shown in Fig. 5. Proteins include p62, Beclin 1, VPS34, ATG3, ATG4D and AMBRA1. Studies have shown that caspase 6 and 8 cleavage of p62 can inhibit autophagy, subtle effects of caspase inhibition on GFP LC3 lipidation were seen in these studies, but more marked effects on the formation of GFP LC3 puncta (a marker of autophagosome formation) and p62 degradation were evident, indicating that caspase cleavage of autophagy related proteins can affect the autophagic process, specifically p62 [69]. Other studies, as previously indicated, have shown that apoptosis blocks Beclin 1 dependent autophagosome synthesis. After cleavage, both N and C terminal Beclin 1 fragments change their localisations, these fragments do not interact normally with VPS34, which is required for autophagy. As a result, the cleavage of Beclin 1 has been shown to be a critical event whereby caspases inhibit autophagy [60,70].
Fig. 5.
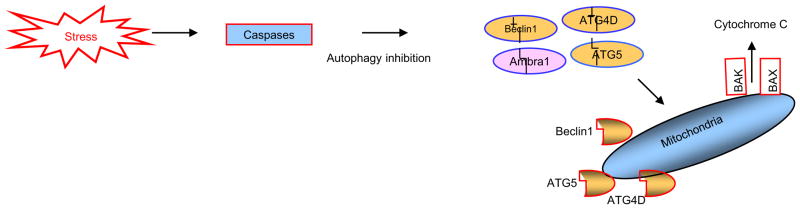
Apoptotic proteins can dismantle the autophagic machinery by degrading Ambra1, Beclin1, ATG4D and ATG5, through the actions of caspases and calpains. Of note is the fact that the proteolytic products of Beclin1 ATG5 and ATG4D may then translocate to the outer mitochondrial membrane and exhibit a pro-apoptotic activity.
A non cleavable Beclin 1 mutant was shown to restore autophagy [17]. ATG3 cleavage has been shown to be accompanied by both tumour necrosis factor α and tumour necrosis factor related apoptosis inducing ligand induced cell death, this event was inhibited by zVAD or a caspase 8 specific inhibitor zIETD. Indeed, caspase 8 over expression led to Atg3 degradation and this event depended upon caspase 8 enzymatic activity. Mutation of the caspase 8 cleavage site on Atg3 abolished its cleavage both in vitro and in vivo, demonstrating that Atg3 was a direct target of caspase 8. In these experiments, autophagy was seen to be inactive during apoptosis and blockage of caspases or over expression of a non cleavable Atg3 protein re established autophagic activity [71]. Atg4D over expression induces apoptosis, which is preceded by the caspase independent recruitment of Atg4D to mitochondria and is facilitated by a putative C terminal Bcl2 homology 3 (BH3) domain. Atg4D also acquires affinity for damaged mitochondria in cells treated with hydrogen peroxide. These data suggest that Atg4D is an autophagy regulator that links mitochondrial dysfunction with apoptosis [72].
Recently, Pagliarini et al [73], have demonstrated that apoptotic stimuli induce a rapid decrease in the level of the autophagic factor. Activating Molecule in Beclin1 Regulated Autophagy (AMBRA1) degradation was shown to be prevented by concomitant inhibition of caspases and calpains. Further, caspases were demonstrated to be responsible for Ambra1 cleavage at the D482 site, whereas calpains are involved in complete Ambra1 degradation. Finally, these workers showed that Ambra1 levels are critical for the rate of apoptosis induction. RNA interference mediated Ambra1 down regulation further sensitises cells to apoptotic stimuli, while Ambra1 over expression and more efficiently, a caspase non cleavable mutant counteract cell death by prolonging autophagy induction. This work suggests that Ambra1 is an important target of apoptotic proteases resulting in the dismantling of the autophagic machinery and the accomplishment of the cell death programme. This phenomenon may be explained in that inhibition of autophagy, which can often precedes apoptosis, serves to prevent the simultaneous activation of contradicting pro survival and pro death processes in the cell, enabling a transition in the direction of cell death. In agreement with this, expression of a mutant form of the autophagic protein AMBRA1 that cannot be cleaved by caspases, has been shown to confer partial protection from apoptotic cell death and a corresponding increase in autophagy [73]. As mentioned above, in some examples of caspase cleavage of autophagic proteins, a twist exists, in that the cleaved product of Beclin 1 acquires a pro apoptotic function, conferring an additional benefit for cells that are destined for demise [29]. Surprisingly, starvation induced autophagy in Drosophila nurse cells was shown to require the caspase Dcp1 and was inhibited by Bruce [74,75]. However, it is not known whether the autophagic function of Dcp1 requires proteolytic activity and if so, what the downstream autophagic substrates are.
6. Conclusion
The multiple layers of interaction between the processes of autophagy and apoptosis present themselves as a seamless balance between life and death in response to a given cellular stress. Any perturbation of this balance might be associated with a number of pathologies, such as cancer and neuro degeneration. Recently, emphasis has been placed upon identification and interpretation of the effects of direct protein–protein interactions between autophagic and apoptotic proteins. Such interactions will usually show a clear functional outcome for given interacting proteins. In this respect, future research appears warranted to assess a potential advantage to the cell in using an autophagy protein to regulate apoptosis (and vice versa). The autophagy proteins p62 and Beclin1 are likely to prove vital in the regulation of a number of pro and anti apoptotic molecules. Understanding of the balance between cell survival and death will prove crucial in determining the role of autophagy and apoptosis in the present and future treatment of cancer and other diseases.
References
- 1.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 2.Levine B, Yuan J. J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lockshin RA, Zahra Z. Int J Biochem Cell Biol. 2004;36:2405–2419. doi: 10.1016/j.biocel.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Eisenberg Lerner A, Bialik S, Simon HU, Kimchi A. Cell Death Differ. 2009;16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 5.White E, DiPaola RS. Clin Cancer Res. 2009;15:5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorburn A. Apoptosis. 2007;13:1–9. doi: 10.1007/s10495-007-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ullén A, Farnebo M, Thyrell L, Mahmoudi S, Kharaziha P, Lennartsson L, Grandér D, Panaretakis T, Nilsson S. Int J Oncol. 2010;37:15–20. doi: 10.3892/ijo_00000648. [DOI] [PubMed] [Google Scholar]
- 8.Bareford MD, Park MA, Yacoub A, Hamed HA, Tang Y, Cruickshanks N, Eulitt P, Hubbard N, Tye1 G, Burow ME, Fisher PB, Moran RG, Nephew KP, Grant S, Dent P. J Biol Chem. 2012;287:32113–32123. [Google Scholar]
- 9.Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS, Cheng AL, Chen PJ, Chen KF. Cell Death and Disease. 2013;4:e485. doi: 10.1038/cddis.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, Wang XY, Dai Z, Peng YF, Gu CY, Qiu SJ, Fan J. Autophagy. 2011;10:1159–1172. doi: 10.4161/auto.7.10.16818. [DOI] [PubMed] [Google Scholar]
- 11.Shimizu S, Takehara T, Hikita H, Kodama T, Tsunematsu H, Miyagi T, Ishida A, Tatsumi H, Kanto T, Hiramatsu T, Fujita K, Yoshimori N, Hayashi T. Int J Cancer. 2012;131(3):548–557. doi: 10.1002/ijc.26374. [DOI] [PubMed] [Google Scholar]
- 12.Perozzo YS, Schmid R, Ziemiecki I, Schaffner A, Scapozza T, Brunner L, Simon T. Nat Cell Biol. 2006;10:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 13.Rahmani M, Nguyen TK, Dent P, Grant S. Mol Pharmacol. 2007;72 (3):788–795. doi: 10.1124/mol.106.033308. [DOI] [PubMed] [Google Scholar]
- 14.Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. Mol Cell. 2011;44 (5):698–709. doi: 10.1016/j.molcel.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 15.Radoshevich L, Debnath J. Autophagy. 2011;1:109–111. doi: 10.4161/auto.7.1.13998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, Debnath J. Cell. 2010;142 (4):590–600. doi: 10.1016/j.cell.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oberstein A, Jeffrey PD, Shi Y. J Biol Chem. 2007;282:13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 18.Pattingre S, Levine B. Cancer Res. 2006;15 66(6):2885–2888. doi: 10.1158/0008-5472.CAN-05-4412. [DOI] [PubMed] [Google Scholar]
- 19.Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O, Rubinsztein DC. Mol Cell. 2012;10 47(3):359–370. doi: 10.1016/j.molcel.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang W, Konopleva M, Ruvolo VR, McQueen T, Evans RL, Bornmann WG, McCubrey J, Cortes J, Andreeff M. Leukemia. 2004;22 (4):808–818. doi: 10.1038/sj.leu.2405098. [DOI] [PubMed] [Google Scholar]
- 21.Ciechomska IA, Goemans GC, Skepper JN, Tolkovsky AM. Oncogene. 2009;28 (21):2128–2141. doi: 10.1038/onc.2009.60. [DOI] [PubMed] [Google Scholar]
- 22.Huang S, Sinicrope FA. Mol Cancer Ther. 2010;9(3):742–750. doi: 10.1158/1535-7163.MCT-09-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llobet D, Eritja N, Yeramian A, Pallares J, Sorolla A, Domingo M, Santacana M, Gonzalez-Tallada FJ, Matias-Guiu X, Dolcet X. Eur J Cancer. 2010;46 (4):836–850. doi: 10.1016/j.ejca.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 24.Kang RHJ, Zeh MT. Cell Death Differ. 2011;18 (4):571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Cell. 2005;122 (6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Noble CG, Dong JM, Manser E, Song H. J Biol Chem. 2008;283:26274–26282. doi: 10.1074/jbc.M804723200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Wang P, Yu J, Zhang L. Autophagy. 2011;7 (10):1239–1241. doi: 10.4161/auto.7.10.16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo S, Rubinsztein DC. Cell Death Differ. 2010;17 (2):268–277. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R, Verspurten J, Declercq W, Agostinis P, Vanden Berghe T, Lippens S, Vandenabeele P. Cell Death and Disease. 2010;1038:1–10. doi: 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapuy O, Vinod PK, Mandl J, Bánhegyi G. Mol Biosyst. 2013;2:296–306. doi: 10.1039/c2mb25261a. [DOI] [PubMed] [Google Scholar]
- 31.Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3 only protein. Oncogene. 2008;27(Suppl 1):S137–S148. doi: 10.1038/onc.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galmiche A, Ezzoukhry Z, François C, Louandre C, Sabbagh C, Nguyen-Khac E, Descamps V, Trouille N, Godin tC, Regimbeau JM, Joly JP, Barbare JC, Duverlie G, Mazière JC, Chatelain D. Mol Cancer Res. 2010;8 (8):1116–1125. doi: 10.1158/1541-7786.MCR-10-0029. [DOI] [PubMed] [Google Scholar]
- 33.Dudgeon C, Peng R, Wang P, Sebastiani A, Yu J, Zhang L. Oncogene. 2012;31 (46):4848–4858. doi: 10.1038/onc.2011.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panka DJ, Cho DC, Atkins MB, Mier JW. J Biol Chem. 2008;283 (2):726–732. doi: 10.1074/jbc.M705343200. [DOI] [PubMed] [Google Scholar]
- 35.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. Mol Cell. 2008;30 (6):678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. J Biol Chem. 2009;5:2719–2728. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, et al. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas Kramarski R, Kimchi A. EMBO Rep. 2009;10 (3):285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 40.Saeki K, You A, Okuma E, Yazaki Y, Susin SA, Kroemer G, Takaku F. Cell Death Differ. 2000;7:1263–1269. doi: 10.1038/sj.cdd.4400759. [DOI] [PubMed] [Google Scholar]
- 41.Cardenas-Aguayo Mdel C, Santa-Olalla J, Baizabal JM, Salgado LM, Covarrubias L. J Hematother Stem Cell Res. 2003;12:735–748. doi: 10.1089/15258160360732759. [DOI] [PubMed] [Google Scholar]
- 42.Yanagisawa H, Miyashita T, Nakano Y, Yamamoto D. Cell Death Differ. 2003;10:798–807. doi: 10.1038/sj.cdd.4401246. [DOI] [PubMed] [Google Scholar]
- 43.Chang NC, Nguyen M, Germain M, Shore GC. EMBO J. 2010;3:606–618. doi: 10.1038/emboj.2009.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams JM, Cory S. Curr Opin Immunol. 2007;19 (5):488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinou JC, Youle RJ. Dev Cell. 2011;21 (1):92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, Opferman JT, Slack RS. EMBO J. 2011;30:395–407. doi: 10.1038/emboj.2010.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katz SI, Zhou L, Chao G, Smith CD, Ferrara T, Wang W, Dicker DT, El-Deiry WD. Cancer Biol Ther. 2009;8 (24):2406–2416. doi: 10.4161/cbt.8.24.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huber S, Oelsner M, Decker TC, zum Büschenfelde M, Wagner M, Lutzny G, Kuhnt BT, Schmidt, Oostendorp RA, Peschel C, Ringshausen I. Leukemia. 2011;5:838–847. doi: 10.1038/leu.2011.2. [DOI] [PubMed] [Google Scholar]
- 49.Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, BodmerSchröter JLM, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Nature. 1997;388 (6638):190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 50.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. Nat Cell Biol. 2009;11 (11):1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng Z, Zhang H, Levine AJ, Jin S. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crighton D, Wilkinson S, O’Prey J, et al. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 53.Scherz-Shouval RH, Weidberg, Gonen C, Wilder S, Elazar Z, Oren M. Proc Natl Acad Sci U S A. 2010;107 (43):18511–18516. doi: 10.1073/pnas.1006124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, Amelio M, Criollo AD, Morselli E, Zhu C, Harper F, et al. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tasdemir E, Maiuri M, COrhon I, Kepp O, Morselli E, Criollo A, Kroemer G. Cell Cycle. 2008;7:3006–3011. doi: 10.4161/cc.7.19.6702. [DOI] [PubMed] [Google Scholar]
- 56.Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, Vicencio JM, Soussi T, Kroemer G. Cell Cycle. 2008;7:3056–3061. doi: 10.4161/cc.7.19.6751. [DOI] [PubMed] [Google Scholar]
- 57.Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D’Amelio M, et al. A dual role of p53 in the control of autophagy. Autophagy. 2008;4:810–814. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- 58.Turenne GA, Price BD. BMC Cell Biol. 2001;2:12–20. doi: 10.1186/1471-2121-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Panka DJ, Cho DC, Atkins MB, Mier JW. J Biol Chem. 2008;283 (2):726–732. doi: 10.1074/jbc.M705343200. [DOI] [PubMed] [Google Scholar]
- 60.Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, Kester M, Wang HG. J Biol Chem. 2012;15:12455–12468. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park MA, Reinehr R, Häussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A, Grant S, Dent P. Mol Cancer Ther. 2010;9 (8):2220–2231. doi: 10.1158/1535-7163.MCT-10-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laussmann MA, Passante E, Düssmann H, Rauen JA, Würstle ML, Delgado ME, Devocelle M, Prehn JH, Rehm M. Cell Death Differ. 2011;10:1584–1597. doi: 10.1038/cdd.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cell. 2009;137 (4):721–735. doi: 10.1016/j.cell.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 64.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y, Jung YK. J Biol Chem. 2005;280 (21):20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 65.Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, Curiel DT, Yacoub A, Graf M, Lee JD, Roberts PB, Fisher S, Grant P. Clin Cancer Res. 2008;14 (17):5385–5399. doi: 10.1158/1078-0432.CCR-08-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pan JA, Ullman E, Dou Z, Zong WX. Mol Cell Biol. 2011;31 (15):3158–3170. doi: 10.1128/MCB.05460-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang YB, Gong JL, Xing TY, Zheng SP, Ding W. Cell Death and Disease. 2013;4:1–10. doi: 10.1038/cddis.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramakrishnan V, Timm M, Haug JL, Kimlinger TK, Wellik LE, itzig TE, Rajkumar SV, Adjei AA, Kumar S. Oncogene. 2012;29:1190–1202. doi: 10.1038/onc.2009.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Norman JM. Autophagy. 2010;6 (8):1042–1056. doi: 10.4161/auto.6.8.13337. [DOI] [PubMed] [Google Scholar]
- 70.Cho DH, Jo YK, Hwang JJ, Lee YM, Roh SA, Kim JC. Cancer Lett. 2009;274 (1):95–100. doi: 10.1016/j.canlet.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 71.Oral O, Oz-Arslan D, Itah Z, Naghavi A, Deveci R, Karacali S, Gozuacik D. Apoptosis. 2012;8:810–820. doi: 10.1007/s10495-012-0735-0. [DOI] [PubMed] [Google Scholar]
- 72.Betin VM, Lane JD. Autophagy. 2009;5 (7):1057–1059. doi: 10.4161/auto.5.7.9684. [DOI] [PubMed] [Google Scholar]
- 73.Pagliarini V, Wirawan E, Romagnoli A, Ciccosanti F, Lisi G, Lippens S, Cecconi F, Fimia GM, Vandenabeele P, Corazzari M, Piacentini M. Cell Death Differ. 2012;19 (9):1495–1504. doi: 10.1038/cdd.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hou YC, Chittaranjan S, Barbosa SG, McCall K, Gorski SM. J Cell Biol. 2008;182 (6):1127–1139. doi: 10.1083/jcb.200712091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim YI, Ryu T, Lee J, Heo YS, Ahnn J, Lee SJ, Yoo O. BMC Cell Biol. 2010;25:11, 9. doi: 10.1186/1471-2121-11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]