

Abstract
Objectives
Nosocomial pathogens such as Acinetobacter baumannii are a growing public health threat, due in part to their increasing resistance to antibiotics. Since some strains are resistant to all available antibiotics, novel therapies are urgently needed. Plasmablasts are short-lived B cells found in the blood that can be collected and harnessed to produce therapeutic antibodies. We set out to determine whether plasmablasts are induced during infection with A. baumannii and other nosocomial pathogens.
Methods
We obtained blood samples from patients infected with antibiotic-resistant nosocomial pathogens, and analysed their plasmablast response by flow cytometry.
Results
We observed a strong induction of plasmablasts in patients with antibiotic-resistant A. baumannii infection. Furthermore, plasmablasts were also induced in response to other drug-resistant nosocomial pathogens.
Conclusions
These data suggest that plasmablasts may be broadly harnessed to develop therapeutic antibodies to combat otherwise untreatable antibiotic-resistant infections.
Keywords: Acinetobacter baumannii, nosocomial infections, antibody therapy
Introduction
Nosocomial infections are a growing problem in hospitals worldwide. In the USA alone there are over 2 million hospital-acquired infections, resulting in 100 000 deaths annually.1 Antibiotics are the primary treatment for such infections, but rates of resistance are rapidly increasing among common nosocomial pathogens. These include the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter species and Escherichia coli), which are often highly drug resistant and can cause infections that are very difficult to treat.2 In fact, some strains are now resistant to all antibiotics,3 making them essentially untreatable. Thus, there is a critical need to develop new therapies to combat these antibiotic-resistant bacteria.
Development of new classes of antibiotics is a long and costly endeavour that, despite the increased need, has slowed considerably.4 An alternative therapeutic approach is using antibodies to target bacteria. Human plasmablasts are short-lived B cells that transiently peak in the blood after viral infection (influenza and dengue fever).5,6 These cells can easily be collected, facilitating the subsequent isolation of antibodies that they produce. Although the plasmablast response has been well characterized in viral infections, it is not clear if there is a similar induction of these cells during nosocomial bacterial infections. We set out to determine whether plasmablasts are induced during acute nosocomial bacterial infections, which would raise the exciting possibility of harnessing these cells to generate novel therapeutic antibodies to combat antibiotic-resistant bacteria for which no treatment is currently available.
Materials and methods
Patient samples
Blood samples (in EDTA-coated tubes) were obtained from patients with A. baumannii, K. pneumoniae, Enterobacter aerogenes or E. coli infections at Grady Memorial Hospital (n = 17) or Emory University Hospital (n = 3). Blood samples from healthy controls (n = 5) were obtained from the Emory Vaccine Center blood donor programme. All samples were obtained in accordance with Emory Institutional Review Board regulations. As these were residual blood samples that had already been collected by hospital staff, patient consent was not needed or obtained for this study. Infection was confirmed by blood, urine or respiratory culture at the time of each blood collection, and patients were culture positive for only the indicated pathogen. Identification of bacteria and antibiotic resistance profiles was performed using automated microbial identification systems (VITEK and MicroScan), with confirmatory testing using Etest diffusion (bioMérieux) on Mueller–Hinton media. Complete blood counts were recorded for each sample.
Flow cytometry
For flow cytometric analysis, whole blood was split into four 140 μL samples, each of which was stained with a different antibody panel. Erythrocytes were then lysed (FACS lysis buffer, BD) and fixed with 1% paraformaldehyde. Antibodies were obtained from Beckman Coulter [CD19-FITC (J3-119), CD27-PE (1A4CD27), IgG1-FITC, IgG1-PE and IgG1κ-APC] or BD Pharmingen [Ki-67-FITC (MOPC-21), CD3-PerCP (SK7), CD20-PerCP (L27) and CD38-APC (HB7)]. Staining for intracellular Ki-67 was performed by permeabilizing the cells (FACS permeabilization buffer, BD Biosciences) after erythrocyte lysis and then adding anti-Ki-67 antibody. Samples were analysed with a BD FACSCalibur flow cytometer (BD Biosciences) using isotype controls to determine gating. Flow cytometry data were analysed using FlowJo software.
Results
Plasmablasts are induced in response to antibiotic-resistant A. baumannii infection
We obtained residual blood from daily blood draws of patients in the intensive care unit (patient data are summarized in Table S1, available as Supplementary data at JAC Online). A. baumannii infections were confirmed by blood, urine or respiratory culture (no other pathogens were cultured from these samples). Two strains were susceptible to all antibiotics tested, while one strain was resistant to fewer than three classes of antibiotics and three strains were resistant to at least three classes and were classified as multidrug resistant (Table S2, available as Supplementary data at JAC Online). Seven strains were resistant to drugs in at least seven antibiotic classes (one was resistant to all drugs tested), and were classified as extremely drug resistant (XDR).3 Consistent with bacterial infection, patients were leucocytotic (Table S1) and lymphopenic (data not shown).
Plasmablasts were defined as lymphocytes positive for the surface markers CD27 and CD38 and negative for CD3 and CD20 (CD27+ CD38+ CD3− CD20−),6 and were quantified in the blood of these patients by flow cytometry (Figure S1, available as Supplementary data at JAC Online). Backgating analysis showed that nearly all cells defined as plasmablasts using this strategy were additionally CD19+ (data not shown). Strikingly, plasmablast levels were elevated in samples from all but one patient, being at least 1% in the total population of CD3− C20− lymphocytes and ranging as high as 21% (Figure 1a). In contrast, healthy controls showed no induction of plasmablasts, with levels <0.5%, a baseline level established previously.6 These data clearly demonstrate that plasmablasts are robustly induced during A. baumannii infection.
Figure 1.

A. baumannii infection induces cycling human plasmablasts. (a) Peak plasmablast levels (percentage of CD27+ CD38+ plasmablasts of total CD3− CD20− lymphocytes) observed in healthy controls (n = 5) and patients with A. baumannii infection (n = 13). (b) Percentage of recently proliferated plasmablasts (Ki-67+) in observed peak plasmablast samples from healthy controls (n = 5) and patients with A. baumannii infection (n = 8). (c) Average percentage of plasmablasts across all samples from A. baumannii-infected patients 0–7, 8–16 or ≥17 days after culture positivity (n = 13). (d) Time course of plasmablast levels from an A. baumannii-infected patient (isolate 5). Flow cytometry plots were gated for lymphocytes by forward and side scatter, and then as CD3− CD20−. Graphs shown represent contour plots indicating areas of higher cell density. Square gates represent cells defined as plasmablasts, with the percentage of cells within the gate quantified. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Plasmablasts are generated through the proliferation and differentiation of memory B cells.7 To determine whether the observed plasmablasts were recently derived, we quantified the expression of Ki-67, a marker of recent proliferation. The majority of plasmablasts (∼60%) in infected patients exhibited high Ki-67 expression, indicating that these cells were newly derived and proliferating. In contrast, only 30% of the plasmablasts in healthy controls expressed high levels of Ki-67, indicating that the majority of these cells were not proliferating (Figure 1b). These data further confirm that A. baumannii infection induces the production of plasmablasts.
In order to study the kinetics of plasmablast induction, we sampled blood from patients several times over the course of infection. Patients from whom multiple samples were obtained exhibited increases in plasmablast levels at early timepoints or decreases in plasmablast levels at later timepoints (data not shown). Aggregation of the data from all timepoints and all A. baumannii-infected patients revealed a transient induction that peaked between 8 and 16 days after culture positivity [Figure 1c and Figure S2 (available as Supplementary data at JAC Online)]. To further study the kinetics of this response in a single patient, we were able to obtain several samples over a 17 day timecourse from one individual infected with XDR A. baumannii. Plasmablast levels in this patient were at baseline on day 5 (0.39%), increased on day 6 (2.21%), further increased to an observed peak at day 11 (2.63%) and returned to baseline at day 16 (0.25%) (Figure 1d). Taken together, these data demonstrate a clear induction and retraction phase in the plasmablast response to A. baumannii infection.
Plasmablast induction in response to diverse antibiotic-resistant bacteria
After observing robust plasmablast induction during A. baumannii infection, we set out to determine whether a similar response occurred during infection with other nosocomial bacteria. We quantified plasmablasts in samples from patients with K. pneumoniae, E. aerogenes or E. coli infection. All but one patient exhibited plasmablast levels above the baseline of healthy controls (Figure 2). Furthermore, one patient with E. coli infection had a peak plasmablast level of 40%, an incredibly robust response. While these are a small number of samples and analysis of many more would be required to confirm these data, the results reveal that plasmablasts are strongly induced following infection with diverse nosocomial bacterial pathogens.
Figure 2.
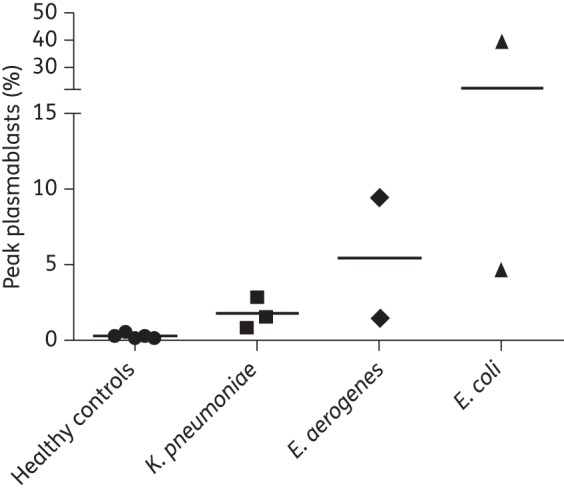
Plasmablast induction during infection with diverse antibiotic-resistant nosocomial bacteria. Peak plasmablast levels (percentage of CD27+ CD38+ plasmablasts of total CD3− CD20− lymphocytes) observed in healthy controls and patients with the indicated bacterial infections.
Discussion
We demonstrate a robust induction of plasmablasts in human patients with nosocomial bacterial infections. The magnitude of the response in some patients was comparable to the very high levels observed during viral infections,6 ranging up to 40% of CD3− CD20− lymphocytes in one patient infected with E. coli. Furthermore, the peak level of plasmablasts was probably higher in many patients than what we observed, since we quantified levels of these cells at only a few timepoints during the course of infection. The expansion of plasmablasts after nosocomial bacterial infection may indicate that these cells play an important role in the protective host response. The rapid production of antibodies during the acute phase of infection provided by these cells may be critical for limiting bacterial replication. In a mouse model, antibodies elicited from immunization with A. baumannii provide protection against infection.8 Further, these antibodies are effective therapeutically as a treatment for antibiotic-resistant A. baumannii.9,10
Given the aforementioned problem of antibiotic resistance among nosocomial bacterial pathogens, as well as the efficacy of antibody therapy, the robust expansion of plasmablasts in infected patients raises the potential of harnessing these cells therapeutically. As was shown by Wrammert et al.,5 plasmablasts may be used to develop monoclonal antibodies that can effectively treat the infections that induced their expansion. The specificity of the plasmablasts observed in this study remains to be determined. If these cells are in fact specific for the bacteria causing the infections, these cells would be especially useful in targeting such highly antibiotic-resistant pathogens. Furthermore, a wide range of antibodies against a single pathogen can be collected. A panel of antibodies against different targets could impede the ability of bacteria to develop resistance to such a treatment. In addition, these antibodies would be human derived and not have to be humanized in order to avoid the hyperinflammatory reactions that can occur with antibodies derived from other organisms.11 The results presented here provide insight into the host response to nosocomial infections, and lay the foundation for harnessing human plasmablasts to combat highly antibiotic-resistant pathogens.
Funding
This work was supported by The Emory University Center For AIDS Research Immunology Core (grant number P30-A01050409) and National Institutes of Health (grant number U54-AI057157) from the Southeastern Regional Center of Excellence for Emerging Infections and Biodefense to D. S. W., who is also supported by a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease award.
Transparency declarations
None to declare.
Supplementary data
Acknowledgements
We would like to thank Dr John Altman for helpful discussions and Dr Edward Mocarski and Dr William Shafer for critical reading of this manuscript prior to submission.
References
- 1.Reed D, Kemmerly SA. Infection control and prevention: a review of hospital-acquired infections and the economic implications. Ochsner J. 2009;9:27–31. [PMC free article] [PubMed] [Google Scholar]
- 2.Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis. 2008;197:1079–81. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 3.Magiorakos AP, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18:268–81. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 4.White AR. Effective antibacterials: at what cost? The economics of antibacterial resistance and its control. J Antimicrob Chemother. 2011;66:1948–53. doi: 10.1093/jac/dkr260. [DOI] [PubMed] [Google Scholar]
- 5.Wrammert J, Smith K, Miller J, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453:667–71. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wrammert J, Onlamoon N, Akondy RS, et al. Rapid and massive virus-specific plasmablast responses during acute dengue virus infection in humans. J Virol. 2012;86:2911–8. doi: 10.1128/JVI.06075-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radbruch A, Muehlinghaus G, Luger EO, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6:741–50. doi: 10.1038/nri1886. [DOI] [PubMed] [Google Scholar]
- 8.McConnell MJ, Dominguez-Herrera J, Smani Y, et al. Vaccination with outer membrane complexes elicits rapid protective immunity to multidrug-resistant Acinetobacter baumannii. Infect Immun. 2011;79:518–26. doi: 10.1128/IAI.00741-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo TA, Beanan JM, Olson R, et al. The K1 capsular polysaccharide from Acinetobacter baumannii is a potential therapeutic target via passive immunization. Infect Immun. 2013;81:915–22. doi: 10.1128/IAI.01184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo G, Lin L, Ibrahim AS, et al. Active and passive immunization protects against lethal, extreme drug resistant-Acinetobacter baumannii infection. PLoS One. 2012;7:e29446. doi: 10.1371/journal.pone.0029446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Presta LG. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv Drug Deliv Rev. 2006;58:640–56. doi: 10.1016/j.addr.2006.01.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.