

Abstract
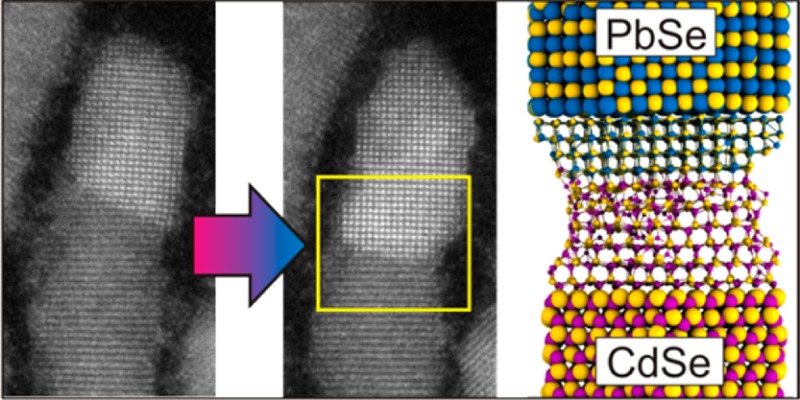
Here, we show a novel solid–solid–vapor (SSV) growth mechanism whereby epitaxial growth of heterogeneous semiconductor nanowires takes place by evaporation-induced cation exchange. During heating of PbSe-CdSe nanodumbbells inside a transmission electron microscope (TEM), we observed that PbSe nanocrystals grew epitaxially at the expense of CdSe nanodomains driven by evaporation of Cd. Analysis of atomic-resolution TEM observations and detailed atomistic simulations reveals that the growth process is mediated by vacancies.
Keywords: Colloidal Nanocrystals, Cation Exchange, Molecular Dynamics, Density Functional Theory, In Situ Transmission Electron Microscopy
Both the synthesis and design of heteronanocrystals (HNCs) have undergone a rapid development, whereby PbSe and CdSe NCs are key materials acting as functional building blocks within a wide variety of heterogeneous nanostructures.1−8 PbSe-CdSe HNCs are of particular interest as they can exhibit properties different from individual PbSe and CdSe dots. The presence of two semiconductor quantum dots connected via a well-defined interface opens new possibilities for tailoring the optoelectronic properties.1,4−7,9 Heat treatment of HNCs can induce new interface designs,5,10−13 exemplified by the transformation of PbSe/CdSe core/shell systems into PbSe-CdSe bihemispheres.5 Here, we report an in situ heating-induced epitaxial PbSe NC domain growth at the solid–solid PbSe-CdSe nanointerface through cation exchange. We show that Pb replaces Cd at the PbSe/CdSe interface, resulting in growth of the PbSe phase at the expense of the CdSe phase. The incorporated Pb is originating from Pb-oleate present as excess stabilizer at the surface of the mature PbSe/CdSe HNCs.
Vapor–liquid–solid (VLS)14−16 and vapor–solid–solid (VSS)17,18 growth mechanisms are commonly applied nowadays in nanochemistry to epitaxially grow semiconductor nanowires from the elements dissolved in a liquid (VLS) or solid (VSS) domain. In analogy with these growth mechanisms, the currently observed process could be called solid–solid–vapor (SSV) growth as the Cd evaporates, either as neutrally charged Cd atoms or in a molecular complex such as Cd-oleate.
Figure 1a shows a HAADF-STEM image (high angle annular dark field scanning transmission electron microscopy) of CdSe-PbSe dumbbell HNCs, consisting of CdSe nanorods with PbSe tips at both ends. In this imaging mode, the intensity scales with Z2, where Z is the atomic number. As Pb has a higher Z than Cd, PbSe NCs exhibit brighter contrast than the CdSe nanorods. When the HNCs were heated to 160 °C with a heating rate of 10 degrees/min and annealed at this temperature for 5 min, the bright contrast corresponding to PbSe was observed not only at the tips but also extended gradually inside the nanorod domain (solid arrows in Figure 1b), showing that the PbSe phase grows at the expense of the CdSe phase. When the HNCs were heated to 200 °C with the same heating rate and annealed at this temperature for 5 min, the bright contrast was observed over the entire nanorod in some nanorods (solid arrows in Figure 1c). The evolution of this growth was seen to initiate mostly from one PbSe tip domain (Supporting Information Movies S1 and S2), though it can also proceed from both PbSe tip domains (dashed arrows in Figure 1b and c).
Figure 1.

HAADF-STEM images and chemical mapping of the nanodumbbells before and after heating. (a) HAADF-STEM image of CdSe-PbSe nanodumbbells. The PbSe tips exhibit brighter contrast than the CdSe nanorods due to Z-contrast. (b,c) Dumbbell HNCs at 160 °C (b) and at 200 °C (c), showing gradual extension of PbSe domains at the expense of CdSe. A heating rate of 10 degrees/min was used in the in situ studies and the HNCs were annealed at the indicated temperatures for 5 min before imaging. Dumbbell HNCs with solid arrows transformed totally to brighter contrast with heating. This phenomenon occurred mostly from one side, though it can proceed from both PbSe domains as well (dumbbell with dashed arrows in panel c). (d–o) HAADF-STEM images and corresponding STEM-EDX elemental maps of dumbbell heteronanostructures annealed for 5 min at temperatures of (d–g) 100 °C, (h–k) 170 °C, and (l–o) 200 °C. In panels d–g, HNCs are in original dumbbell state with PbSe tips and CdSe nanorod. In panels h–k, a partially transformed nanorod is present. In panels l–o, two PbSe-CdSe HNCs became full PbSe domains. The Se remains in place during the transformation. Note that the contrast is maximized in each individual image; hence, intensities of different mappings cannot be directly compared. Quantitative analyses are provided in the Supporting Information.
Chemical mapping by means of energy-dispersive X-ray spectrometry (EDX) using a Chemi-STEM detector (see Methods and Supporting Information) was performed to provide further evidence of the chemical transition. Figure 1d–g shows the initial state of the HNCs at 100 °C with CdSe nanorods and PbSe tips. Figure 1f shows that Pb is also present at the lateral surfaces of the CdSe nanorods, pointing to adsorbed Pb-oleate molecules. The dumbbell depicted with an arrow in Figure 1h underwent a transformation, after which half the nanorod exhibited a bright contrast. With annealing at 170 °C for 5 min, the elemental maps of this dumbbell in Figure 1i–k shows that Pb is indeed present in the bright contrast regions and that Cd is absent. We, therefore, conclude that Cd started to sublimate (as neutral Cd atoms or in a molecular form) and that at the same time, PbSe was formed by Pb incorporation. Upon further heating to 200 °C and 5 min annealing at this temperature, two nanorods (indicated with arrows in Figure 1l) exhibited a bright contrast over their entire length. Elemental maps (Figure 1m–o) showed that Cd is no longer present and the nanorod completely transformed into PbSe. Disappearance of Cd from a nanostructure was also reported by De Trizio et al.12 during a heating of sandwich-morphology CdSe/Cu3P/CdSe HNCs. Note that a complete transformation occurred very rarely (in about one percent of the cases). Further heating of partially cation-exchanged nanodumbbells led to dissociation of the domains (Supporting Information Movie S3). The transformations took place everywhere on the substrate, not only in areas that were previously examined with the electron beam. The field of view was changed frequently in order to avoid beam effects when monitoring the evolution of the HNCs.
As a result of the cation exchange from CdSe to PbSe, the crystal structure transformed epitaxially from hexagonal wurtzite (WZ) to cubic rock-salt (RS). Figure 2 and Supporting Information Movie S4 show this transformation at atomic resolution. When the HNC was heated from 160 °C (Figure 2a) to 180 °C (Figure 2b) with a heating rate of 10 degrees/min, the brighter intensity corresponding to PbSe advanced into the CdSe region. The PbSe RS (200) lattice spacings started to appear along the nanorod domain instead of the CdSe WZ (0002) lattice spacings, as confirmed by the Fourier transformation (FT) patterns shown in the insets. It is clear that the cation exchange takes place at the PbSe/CdSe interface and propagates epitaxially (layer by layer) along the WZ ⟨0001⟩ direction. Two types of interfaces were observed: {100}PbSe/{0001}CdSe and {111}PbSe/{0001}CdSe, similar to the interfaces previously reported in the literature for PbSe/CdSe and PbS/CdS HNCs.7,19 Sometimes both types of interfaces were observed within one single dumbbell NC. Figure 2c shows a HNC with the interfaces of {111}PbSe/{0001}CdSe on the left (Figure 2d) and {100}PbSe/{0001}CdSe on the right (Figure 2f). It is clear from Figure 2 that epitaxial PbSe growth inside CdSe domain via cation exchange can advance from both PbSe/CdSe interfaces.
Figure 2.

Atomic-resolution HAADF-STEM images of CdSe-PbSe HNCs. PbSe has cubic rock salt (RS) crystal structure with a lattice constant20 of 6.13 Å, whereas CdSe has a hexagonal wurtzite (WZ) crystal structure with lattice parameters21a = 4.29 Å and c = 7.01 Å. The CdSe WZ (0002) spacing is 3.5 Å and PbSe RS (200) spacing is 3.1 Å. With heating from 160 °C (a) to 180 °C (b) with a heating rate of 10 degrees/min, WZ CdSe nanorods started to transform to RS PbSe. The insets are Fourier transforms (FTs) taken from the white squares in each image. The spot depicted with an arrow in the inset FT of panel a corresponds to WZ CdSe(0002) spacing. It disappeared in the inset FT of panel b, confirming the WZ to RS transformation. Supporting Information Movie S4 shows the transformation with atomic resolution. (c) HAADF-STEM image of a PbSe-CdSe dumbbell HNC. Stacking faults and a dislocation are present in the CdSe nanorod domain. The interface at the left-hand side is {111}PbSe/{0001}CdSe (panel d), whereas the interface at the right-hand side is {100}PbSe/{0001}CdSe (panel f).
Considering the source of Pb that is required for the epitaxial PbSe growth in CdSe via cation exchange, we note that PbSe NCs with excess Pb surface atoms (off-stoichiometric) have been reported in the literature.22−24 Pb atoms (possibly Pb-oleate molecules) are also present along the CdSe nanorods (Figure 1f). From the quantification of the elemental maps (see Methods and Supporting Information Table S1), it was found that the PbSe tips contained an excess of Pb, having a cation/anion ratio of 1.3 ± 0.2. After the transformation, the cation/anion ratio at these PbSe tips reduced to 1.02 ± 0.14. These findings indicate Pb diffusion from PbSe tips toward the PbSe/CdSe interface. Supporting Information Movie S4 verifies this, whereby the bright Pb contrast propagates into the initially CdSe nanorod, indicating the epitaxial growth of PbSe, whereas the (PbSe) tip domain starts to lose some of its brightness, indicating that excess Pb is consumed.
In the nanorod domains attached to the PbSe tips where cation exchange took place, the cation/anion ratio in the rod was reduced to 0.93 ± 0.11 due to Cd sublimation. That most nanodumbbells were not completely transformed must, hence, be due to the depletion of the source of Pb. The excess Pb atoms at the surfaces of the heteronanointerface diffuse toward the interface to form new layers of PbSe, but this process stops when all excess Pb has been depleted. As mentioned above, a complete transformation of the nanorods occurred only rarely. From an estimate of the number of Pb-oleate molecules that could cover the surface of the nanodumbbells (assuming a high surface density of 5 Pb-oleate molecules per square nanometer), it was found that for the typical dimensions of the nanodumbbells in this study, the number of surface Pb atoms is not sufficient to replace all the Cd atoms in the CdSe domain (the number of Cd sites is at least two times larger). Therefore, when a complete transformation did occur, likely also Pb atoms from neighboring HNCs contributed to the growth of the PbSe domain. This is in agreement with the observation that when the nanodumbbells were lying isolated on the SiN support membrane, the growth process did take place but always resulted in only a partial transformation of the HNCs as shown in Supporting Information Figure S21.
In order to better understand the nanoscopic growth mechanism at the PbSe/CdSe interface, force-field-based MD simulations were performed on HNC models, taking into account various possibilities for the PbSe/CdSe interfacial arrangements (details in Methods and Supporting Information). Surfactant molecules are not included in the simulation models, and therefore, the MD simulations serve only to study the structure of, and atomic mobility at the PbSe/CdSe interfaces. The isolated nanodumbbell models were equilibrated at 300 and 500 K for 5 ns sequentially. Figure 3a shows the final configuration of a nanodumbbell model after 5 ns at 500 K. This model has both types of the interfaces ({100}PbSe/{0001}CdSe and {111}PbSe/{0001}CdSe) in one HNC.
Figure 3.
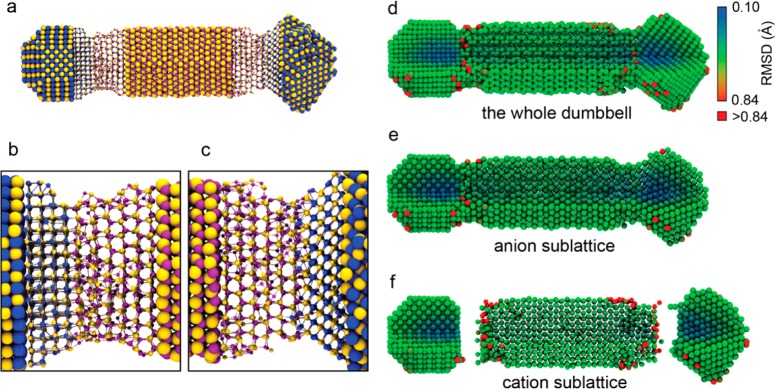
Force-field MD simulations of the PbSe-CdSe nanodumbbells. (a) Overview image showing the final configuration of a dumbbell obtained after MD simulation at a temperature of 500 K for 5 ns. The ball–stick presentation was used to show the structure of the interfaces. The yellow, purple, and blue spheres are Se, Cd, and Pb atoms, respectively. (b) Magnified image of the {100}PbSe/{0001}CdSe interface at the left-hand side of the dumbbell, and (c) magnified image of the {0001}CdSe/{111}PbSe interface at the right-hand side of the dumbbell. (d,e,f) The map of the root mean square displacement (RMSD) for each atom for the same PbSe-CdSe dumbbell model at 500 K. (d) The whole PbSe-CdSe dumbbell, (e) the anion sublattice, and (f) the cation sublattice. The dumbbell was cut so that both of the surface and inner atoms can be seen. The pure red atoms correspond to those having a RMSD larger than 0.84 Å.
The nanodumbbell model shown in Figure 3 is structurally and morphologically stable at temperatures up to 500 K. The middle part of the CdSe rod and both PbSe tips retain their initial WZ and RS structures, respectively. Structural disorder was mainly found in the CdSe domains near the interfaces. Compared to the {100}PbSe/{0001}CdSe interface, the CdSe domain near the {111}PbSe/{0001}CdSe interface is structurally more ordered. In the latter case, most of the Cd and Se atoms remain at the WZ lattice sites, which is likely due to the fact that the cation-terminated {0001}CdSe surface and the anion-terminated {111}PbSe surface form a continuous polar/polar interface, whereas the lattice mismatch is small. In contrast, the {100}PbSe/{0001}CdSe interface is a nonpolar/polar interface, which leads to stronger distortions in the atomic lattice due to Coulombic interactions. The simulations therefore suggest that the transformation at the {100}PbSe/{0001}CdSe interface will be more efficient than at the {111}PbSe/{0001}CdSe interface, although this could not be confirmed by the experiments as the orientation of the two crystals could be determined only in a limited number of cases. Not only is the atomic structure more disordered in the CdSe domains near the {100}PbSe/{0001}CdSe interfaces (see Supporting Information Figure S24 for a planar view of the atomic bilayers parallel to the interface), the simulations also show an unusually high mobility of the Cd atoms in the few first atomic layers from the PbSe/CdSe interface, as evidenced by the map of the root mean square displacement (RMSD) for each atom (Figure 3d–f). Those atoms with the highest mobility (red atoms) are mostly Cd atoms near the interfaces or on the surface, indicating that the cation exchange occurs only very close to the interface.
The experimental observations and the MD simulations suggest that the transformation is mediated by vacancies in the Cd and Pb sublattices; evaporation of Cd results in Cd vacancies at the CdSe surface. After migration of these Cd vacancies to the PbSe/CdSe interface, Pb atoms can jump into the vacant sites, thereby leaving behind vacancies on the Pb sublattice, which will eventually recombine with excess Pb absorbed at the surface of the PbSe domain. Density functional theory (DFT) calculations of defect energies (see Section F in Supporting Information) confirm that upon evaporation of Cd, both in CdSe and PbSe the defect energetics are ruled by vacancies. The DFT calculations also show (Supporting Information Table S10) that the Se-Frenkel defect energy (Se vacancy + Se interstitial) is considerably higher (6.00 for CdSe and 3.80 eV for PbSe) than the Cd-Frenkel and Pb-Frenkel defect energies (3.16 and 3.30 eV, respectively). It is, thus, energetically much more expensive to create defects on the Se sublattice. Because the Se sublattice is not much affected by the cation exchange that takes place on the (Pb,Cd) sublattice, the crystallographic orientation relation between the CdSe and PbSe nanodomains is retained during the transformation. This is the reason that the growth process is epitaxial in nature.
The most important driving force for the growth process is the evaporation of Cd. It is well known that a chemical reaction can be efficiently driven into one direction by bringing one reaction product in the gas phase. Assuming that the excess Pb originates from Pb-oleate coverage of the HNC and that the Cd evaporates in a molecular form, the chemical reaction can be summarized as follows:
| 1 |
In the CdSe lattice, the Cd and Se atoms can be modeled as ions. Bader charge analysis (details in Section E of the Supporting Information) performed on the electronic charge density obtained from DFT calculations shows that the effective charge of the Cd cation in CdSe bulk is approximately +0.8e. However, the Cd will evaporate only as a neutral species. Because the transition from a charged Cd+0.8 ion to a neutral Cd0 atom would require the nanocrystal to donate electrons, we consider it more likely that Cd at the surface of the nanocrystal binds to the surfactants (e.g., oleate), followed by evaporation. We mention here that heating in vacuum is an efficient method to detach surfactants from nanocrystals.20,25
From the available experimental and simulation data, a mechanism can now be deduced to describe the cation exchange. All processes take place close to the interfaces in a fast and volatile manner as demonstrated by Supporting Information Movie S4. The growth mechanism is shown schematically in Figure S1 of the Supporting Information and can be summarized as follows. (i) Cd sublimates from the surface of the CdSe nanodomains, whereby Cd vacancies are formed. (ii) The Cd vacancies occupy positions at the CdSe side of the PbSe/CdSe interface (Figure 3 and Supporting Information Figure S24). (iii) Cation replacement takes place as Pb atoms jump into vacant Cd sites in a layer by layer fashion, resulting in epitaxial growth of RS PbSe at the expense of WZ CdSe. (iv) The jumping Pb atoms leave behind vacancies, which migrate to the PbSe surface. (v) The Pb vacancies at the surface recombine with Pb ions from adsorbed Pb-oleate molecules. The oleate molecule remains adsorbed at the PbSe surface and possibly migrates to the CdSe domain where it combines with Cd and evaporates as Cd-oleate. (vi) The process is halted when the excess Pb (in the form of Pb-oleate molecules) in the system is depleted.
The atomistic mechanism described here most likely also takes place when HNCs undergo cation exchange in colloidal solutions, whereby instead of evaporating, the metal-molecule complex is dissolved in the solution. In the current solid–solid–vapor (SSV) growth mechanism, one solid phase grows epitaxially at the expense of another solid phase, efficiently driven by evaporation of one element (here, Cd) with simultaneous supply of another element (here, Pb coordinated with a molecule). Our results show that SSV growth can provide an alternative path for growing heterogeneous semiconductor nanowires, especially when the lattices have a partly ionic character, and therefore holds promise for generating new families of heterogeneous nanostructures.
Methods
The synthesis of PbSe/CdSe dumbbell nanostructures is detailed in the Supporting Information. TEM specimens were prepared by dropcasting 8 μL of the NC colloidal solution onto a MEMS microhot plate with electron-transparent SiN membranes, which was mounted onto a DENSsolutions low drift TEM heating holder.25 After dropcasting, the sample was plasma cleaned for 10 s in order to remove deposits from the solution that prevent high-resolution imaging in the TEM. The in situ experiments were performed in a 80–300 FEI Titan microscope equipped with a Chemi-STEM EDX detection system. During HAADF-STEM imaging, the microscope was operated at 300 kV. The camera length used in the experiments equals 91 mm in order to avoid diffraction effects and to guarantee Z-contrast imaging. In HAADF-STEM imaging, the intensity approximately scales with Z2. As Pb has a higher Z number than Cd, the PbSe domains appear with higher intensity in HAADF-STEM images in comparison to the CdSe domains.
The Chemi-STEM EDX experiments were performed using the same holder and in the same 80–300 FEI Titan microscope but operated at a lower acceleration voltage of 200 kV to reduce beam damage during mapping. A beam current of approximately 250 pA was used during acquisition. A representative spectrum is shown in Supporting Information Figure S20. In the quantification of the elemental maps, 18 PbSe NC maps were used to determine the cation/anion ratio at the PbSe tips at the initial state. For the PbSe tips from where cation exchange proceeded, the elemental composition of 10 different PbSe tips was quantified. For the nanorod domains attached to the PbSe tips where cation exchange took place, the elemental composition of 10 different nanorod (transformed)-domains was quantified. Additional TEM images, chemical maps, and quantitative analysis are provided in Figures S2–S19 and Tables S1–S6 of the Supporting Information.
For the force field MD simulations, a new interaction potential model for the Pb–Cd–Se system was developed. The potential was found to accurately describe physical parameters such as lattice parameters, elastic constants, and the relative stability of phases. Details of the potential model (Supporting Information Table S7) and a description of the nanodumbbell models are given in Section E of the Supporting Information. For simulations of the nanodumbbells, Coulomb and short-range interactions were calculated by taking into account all atom pairs. The equations of motion were integrated using the velocity Verlet algorithm with a time step of 1 fs. Periodic boundary conditions were not used and the nanodumbbell models were isolated in vacuum. Simulations of 5 ns were carried out in the NVT ensemble and 1 ns was used for equilibration.
All density functional theory (DFT) calculations on defect energies and energies of mixed PbSe-CdSe phases were carried out using the first-principles’ Vienna Ab Initio Simulation Program (VASP)26 using the projector augmented wave (PAW) method.27 The generalized gradient approximation (GGA) formulated by Perdew, Burke, and Ernzerhof (PBE) was employed for the exchange and correlation energy terms.28 The cutoff energy of the wave functions was 350.0 eV. The cutoff energy of the augmentation functions was about 500.0 eV. The electronic wave functions were sampled on a 4 × 4 × 2 grid using the Monkhorst and Pack method with 8 to 20 k-points depending on different symmetries of supercells (108 atoms). Structural optimizations were performed for both lattice parameters and coordinates of atoms. Different k-meshes and cutoff energies for waves were tested to have a good convergence (<2 meV/atom). Details are given in the Supporting Information.
Acknowledgments
This work is part of the research programme of the Foundation for Fundamental Research on Matter (FOM), which is part of The Netherlands Organization for Scientific Research (NWO). A.O.Y. and F.D.T. acknowledge European Soft Matter Infrastructure (ESMI) for Transnational Access Grant. DENSsolutions is acknowledged for providing MEMS based microhot plates and the low-drift TEM heating holder. Computational resources were supported by NWO Exacte Wetenschappen (Physical Sciences). This work was also sponsored by the Stichting Nationale Computerfaciliteiten (National Computing Facilities Foundation, NCF) for the use of supercomputing facilities, with financial support from NWO. M.A.v.H. acknowledges a VIDI grant from NWO. The authors acknowledge financial support from European Research Council (ERC Advanced Grants No. 24691-COUNTATOMS and No. 291667-HIERARSACOL, ERC Starting Grant No. 335078-COLOURATOMS). This work was supported by the Flemish Fund for Scientific Research (FWO Vlaanderen) through a Ph.D. research grant to B.G.
Supporting Information Available
Includes four movies (Movies S1, S2, S3, and S4), a description of the synthesis of the PbSe/CdSe HNCs, additional HAADF-STEM images, STEM-EDX chemical maps and quantification, force field MD simulations, and DFT calculations of the defect energies and mixed PbSe-CdSe phases. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
¶ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Son D. H.; Hughes S. M.; Yin Y.; Alivisatos A. P. Science 2004, 306, 1009–1012. [DOI] [PubMed] [Google Scholar]
- Robinson R. D.; Sadtler B.; Demchenko D. O.; Erdonmez C. K.; Wang L.-W.; Alivisatos A. P. Science 2007, 317, 355–358. [DOI] [PubMed] [Google Scholar]
- Bals S.; Casavola M.; van Huis M. A.; Van Aert S.; Batenburg K. J.; Van Tendeloo G.; Vanmaekelbergh D. Nano Lett. 2011, 11, 3420–3424. [DOI] [PubMed] [Google Scholar]
- Casavola M.; van Huis M. A.; Bals S.; Lambert K.; Hens Z.; Vanmaekelbergh D. Chem. Mater. 2012, 24, 294–302. [Google Scholar]
- Grodzińska D.; Pietra F.; van Huis M. A.; Vanmaekelbergh D.; de Mello Donegá C. J. Mater. Chem. 2011, 21, 11556–11565. [Google Scholar]
- Zhang Y.; Dai Q.; Li X.; Cui Q.; Gu Z.; Zou B.; Wang Y.; Yu W. W. Nanoscale Res. Lett. 2010, 5, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudera S.; Carbone L.; Casula M. F.; Cingolani R.; Falqui A.; Snoeck E.; Parak W. J.; Manna L. Nano Lett. 2005, 5, 445–449. [DOI] [PubMed] [Google Scholar]
- Overgaag K.; Evers W.; de Nijs B.; Koole R.; Meeldijk J.; Vanmaekelbergh D. J. Am. Chem. Soc. 2008, 130, 7833–7835. [DOI] [PubMed] [Google Scholar]
- Grodzińska D.; Evers W. H.; Dorland R.; van Rijssel J.; van Huis M. A.; Meijerink A.; de Mello Donegá C.; Vanmaekelbergh D. Small 2011, 7, 3493–3501. [DOI] [PubMed] [Google Scholar]
- van Huis M. A.; Figuerola A.; Fang C.; Béché A.; Zandbergen H. W.; Manna L. Nano Lett. 2011, 11, 4555–4561. [DOI] [PubMed] [Google Scholar]
- Figuerola A.; van Huis M. A.; Zanella M.; Genovese A.; Marras S.; Falqui A.; Zandbergen H. W.; Cingolani R.; Manna L. Nano Lett. 2010, 10, 3028–3036. [DOI] [PubMed] [Google Scholar]
- De Trizio L.; De Donato F.; Casu A.; Genovese A.; Falqui A.; Povia M.; Manna L. ACS Nano 2013, 7, 3997–4005. [DOI] [PubMed] [Google Scholar]
- Yalcin A. O.; de Nijs B.; Fan Z.; Tichelaar F. D.; Vanmaekelbergh D.; van Blaaderen A.; Vlugt T. J. H.; van Huis M. A.; Zandbergen H. W. Nanotechnology 2014, 25, 055601. [DOI] [PubMed] [Google Scholar]
- Wagner R. S.; Ellis W. C. Appl. Phys. Lett. 1964, 4, 89–90. [Google Scholar]
- Gudiksen M. S.; Lauhon L. J.; Wang J.; Smith D. C.; Lieber C. M. Nature 2002, 415, 617–620. [DOI] [PubMed] [Google Scholar]
- Wang H.; Zepeda-Ruiz L. A.; Gilmer G. H.; Upmanyu M. Nat. Commun. 2013, 4, 1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson A. I.; Larsson M. W.; Stenström S.; Ohlsson B. J.; Samuelson L.; Wallenberg L. R. Nat. Mater. 2004, 3, 677–681. [DOI] [PubMed] [Google Scholar]
- Campos L. C.; Tonezzer M.; Ferlauto A. S.; Grillo V.; Magalhães-Paniago R.; Oliveira S.; Ladeira L. O.; Lacerda R. G. Adv. Mater. 2008, 20, 1499–1504. [Google Scholar]
- Luther J. M.; Zheng H.; Sadtler B.; Alivisatos A. P. J. Am. Chem. Soc. 2009, 131, 16851–16857. [DOI] [PubMed] [Google Scholar]
- van Huis M. A.; Kunneman L. T.; Overgaag K.; Xu Q.; Pandraud G.; Zandbergen H. W.; Vanmaekelbergh D. Nano Lett. 2008, 8, 3959–3963. [DOI] [PubMed] [Google Scholar]
- Xu Y.-N.; Ching W. Y. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 48, 4335–4351. [DOI] [PubMed] [Google Scholar]
- Moreels I.; Lambert K.; De Muynck D.; Vanhaecke F.; Poelman D.; Martins J. C.; Allan G.; Hens Z. Chem. Mater. 2007, 19, 6101–6106. [Google Scholar]
- Moreels I.; Fritzinger B.; Martins J. C.; Hens Z. J. Am. Chem. Soc. 2008, 130, 15081–15086. [DOI] [PubMed] [Google Scholar]
- Petkov V.; Moreels I.; Hens Z.; Ren Y. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 81, 241304. [Google Scholar]
- van Huis M. A.; Young N. P.; Pandraud G.; Creemer J. F.; Vanmaekelbergh D.; Kirkland A. I.; Zandbergen H. W. Adv. Mater. 2009, 21, 4992–4995. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 47, 558–561. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Phys. Rev. Lett. 1996, 77, 3865–3868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.