

Introduction
Statins reduce risk of cardiovascular disease (CVD) by decreasing plasma LDL concentrations, as well as reducing inflammation and improving endothelial function. Despite their documented efficacy, there is considerable interindividual variation in effects of statins on CVD biomarkers. In the studies summarized here we used complementary metabolomics platforms to define global effects of a statin (simvastatin) on metabolism and to identify markers indicative of mechanisms that contribute to variation in plasma LDL response to statin treatment.
Statins are the largest class of drugs prescribed worldwide for reducing risk of CVD. They act by competitively inhibiting HMG CoA reductase, resulting in reduced cellular cholesterol synthesis and upregulation of LDL receptors, consequently lowering plasma LDL levels. Other effects of HMG CoA reductase inhibition include reduced synthesis of multiple isoprenoids, as well as intermediates that prenylate proteins involved in a number of cellular processes including inflammation. Despite the well-proven efficacy of statins, clinical trials have demonstrated a residual CVD risk of ~50-80% in statin-treated patients. In this regard, there is considerable variability in LDL response to treatment, due in part to genetic differences, as well as age and environmental factors such as smoking [1]. In addition statins have a variety of pleiotropic effects, and treatment has been associated with a small but significant incidence of adverse events. While severe statin-related myopathy is relatively rare (~ 5 cases/100,000 patient years), milder symptoms are more prevalent and may lead to cessation of treatment. In addition, statin use has recently been associated with an increase in risk for incident type 2 diabetes[2].
Metabolomics offers tools to map effects of statin on metabolism and to [2]. identify statin-influenced pathways that may contribute to variability in clinical efficacy, as well as to risk of adverse events. We here describe application of three different metabolomic and lipidomics platforms to analysis of plasma samples derived from the Cholesterol and Pharmacogenetic (CAP) study, a 6 week trial of simvastatin 40 mg/d in a group of 944 men and women of European and African-American ancestry selected on the basis of baseline plasma cholesterol levels of 160-400 mg/dL . Efforts to ensure compliance in CAP resulted in a median compliance score [1]of 98% based on pill count and consistent with plasma activated simvastatin concentrations measured on the morning following bedtime dosing. Statin response was assessed as percent change in LDL-cholesterol (LDL-C), using the averages of two independent measurements at baseline and two values on treatment (4 wk and 6 wk). The changes varied across a normal distribution with a median of 41% and a range from approximately −80% to +24%, consistent with the range of LDL-C responses observed in other placebo-controlled statin intervention trials. Metabolomic analyses were performed using stored frozen plasma aliquots (−70°C) from 48 non-smoking individuals, half drawn from the upper and half from the lower 10% of the LDL-C response distribution (good and poor responders, respectively) and matched for sex (2/3 women), race (2/3 Caucasian), and age (within 10 years). Additional analyses were performed in samples from 100 individuals selected randomly across the full distribution of LDL-C response.
Targeted gas chromatography (GC)-based lipidomics platform
Using a targeted lipidomics platform we measured over 300 lipid species within eight lipid [3] classes and determined that metabolic changes in responders were more comprehensive than those seen in non-responders. Baseline cholesterol ester and phospholipid metabolites correlated with LDL-C response to treatment. Among the most consistent changes in both “good” and “poor” responders were increases in the mole percentages of arachidonic acid (20:4n6 ) and decreases in mole percentages of linoleic acid (18:2n6) within multiple lipid classes, primarily phosphatidylcholine and cholesteryl esters. However only “good” responders had a significant increase in the ratio of 20:4n6 to its precursor 20:3n6, which serves as a measure of the activity of delta-5 desaturase.
Previous studies have documented that statin-induced increases in arachidonic acid and reductions in linoleic acid are mediated by upregulation of expression of the genes encoding delta-5 and delta-6 desaturases (FADS1 and FADS2, respectively) via the sterol-responsive transcription factor SREBP-1c. However, the earlier work did not establish the widespread enrichment of arachidonic acid among multiple lipid classes. The metabolic or clinical impact of statin-induced upregulation of the fatty acid desaturases has not been established. However, since arachidonic acid is a precursor of multiple eicosanoids with both pro-and antiinflammatory properties, it is possible that this statin effect contributes to both clinical efficacy as well as adverse events.
Statin-induced changes in C-reactive protein (CRP), a marker of inflammation, were significantly positively correlated with baseline concentrations of phosphatidylethanolamine plasmalogens and inversely with levels of phosphatidylcholine plasmalogens. Given the involvement of plasmalogens in inflammatory processes, these findings suggest a role for inter-individual variation in plasmalogen metabolism in modifying the documented anti-inflammatory effects of statins. Notably, while there were significant correlations between changes in CRP and a number of polyunsaturated fatty acid metabolites, there was no overlap with metabolites whose changes correlated with LDL-C response to simvastatin, suggesting that distinct metabolic pathways mediate statin effects on these two biomarkers of statin efficacy.
Targeted GC-MS sterol and bile acid metabolomics platform
Using this platform, baseline levels of three secondary (bacterial-derived [4].) bile acids were found to predict, wih nominal statistical significance, the magnitude of statin-induced LDL-C lowering in “good” vs. “poor” responders (lithocholic acid, taurolithocholic acid, and glycolithocholic acid), as did coprostanol, which is produced in the intestine by enteric bacterial reduction of endogenous cholesterol (Table). Moreover, levels of several secondary bile acids were correlated with plasma levels of simvastatin, likely due to the fact that bile acids share transporters in the liver and intestine, notably, the organic anion transporter SLC01B1. A single nucleotide polymorphism (SNP), rs4149056, in the gene encoding this transporter has been associated with both statin LDL-C lowering efficacy and risk of statin-associated myopathy, In the CAP study, this SNP was associated with both activated simvastatin levels and plasma concentrations of seven bile acids, further reinforcing the potential role of intestinal bile acid metabolism in modulating simvastatin efficacy through effects on tissue transport. These findings, along with recent evidence for the role of the intestinal microbiome in generating metabolites that increase CVD risk, suggest that both genetic and environmental influences on gut flora can play an important role in modifying the therapeutic impact of statin therapy.
Table 1.
Associations of metabolites at baseline with good vs.poor response categories
| Metabolic Pathway |
Metabolite | Association | P value | Q value |
|---|---|---|---|---|
|
Sterol
Synthesis |
Lanosterol | negative | 0.90 | 0.96 |
| Lathosterol | positive | 0.44 | 0.79 | |
| 7 Dehydroxycholesterol | positive | 0.34 | 0.79 | |
| Desmosterol | positive | 0.15 | 0.47 | |
| Cholesterol | positive | 0.12 | 0.42 | |
| Cholestanol | negative | 0.85 | 0.96 | |
| 7 α-hydroxycholesterol | positive | 0.83 | 0.96 | |
|
Dietary
Sterols |
β sitosterol | positive | 0.92 | 0.96 |
| Campesterol | negative | 0.70 | 0.96 | |
| Coprostanol | positive | 0.02 | 0.20 | |
| Stigmasterol | negative | 0.39 | 0.79 | |
|
Primary Bile
Acids |
Cholic acid | negative | 0.41 | 0.79 |
| Chenodeoxycholic acid | negative | 0.10 | 0.41 | |
| Taurocholic acid | positive | 0.46 | 0.79 | |
| Glycocholic acid | positive | 0.84 | 0.96 | |
| Taurochenodeoxycholic acid | positive | 0.76 | 0.96 | |
| Glycochenodeoxycholic acid | positive | 0.77 | 0.96 | |
|
Secondary
Bile Acids |
Deoxycholic acid | positive | 0.69 | 0.96 |
| Ursodeoxycholic acid | positive | 0.99 | 0.99 | |
| Lithocholic acid | positive | 0.04 | 0.28 | |
| Taurodeoxycholic acid | positive | 0.06 | 0.28 | |
| Glycodeoxycholic acid | positive | 0.24 | 0.66 | |
| Glycoursodeoxycholic acid | positive | 0.48 | 0.79 | |
| Taurolithocholic acid | positive | 0.02 | 0.20 | |
| Glycolithocholic acid | positive | 0.02 | 0.20 |
Metabolites with nominal significance for prediction of good vs. poor responders are shown in bold. Postive associations indicate that higher levels are associated with good response.
Gas chromatography-time-of-flight mass-spectrometry-based platform.[5].
Using this unbiased platform we measured over 300 metabolites of intermediary metabolism, of which 160 were known chemical identities. The pre- and post-treatment values for the 100 individuals randomly selected across the full range of LDL-C response are shown in Supplementary Table 1. Pathway enrichment analysis indicated that these biomarkers of drug exposure were enriched for the pathway class amino acid degradation, suggesting previously unrecognized effects of statins on amino acid metabolism. These effects are consistent with the observation that among the metabolites whose change correlated with LDL-C lowering response to simvastatin were cystine, glutamine, urea cycle intermediates, and the dibasic amino acids ornithine, citrulline and lysine (Figure). The changes in ornithine and citrulline suggest an increased flux through the urea cycle that could be linked to amino acid degradation. Moreover, the dibasic amino acids share plasma membrane transporters with arginine, the rate-limiting substrate for nitric oxide synthase (NOS). Upregulation of NO by statin treatment, and the resultant increase in nitric oxide production, is an important mediator of statin’s benefit on endothelial function, and hence on CVD risk. Moreover, several studies have shown that manipulation of NOS enzymes in mouse models produces substantial changes in lipoprotein metabolism, with increased expression leading to improved levels of atherogenic lipoproteins.
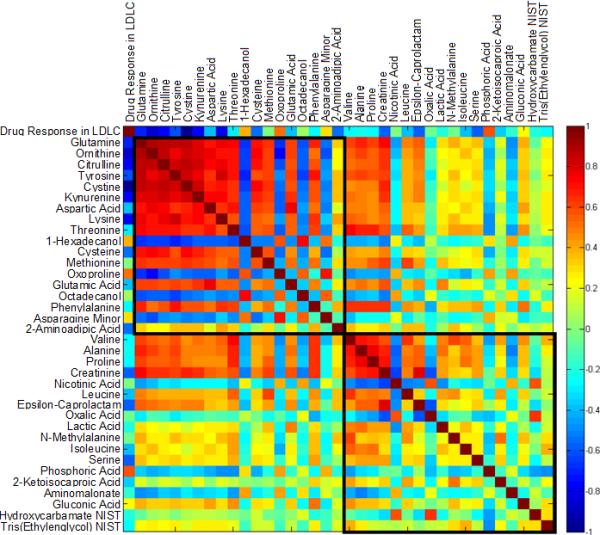
Correlation matrix illustrating two clusters of compounds correlated with LDL-C response to simvastatin 40 mg/day in 100 individuals. The two clusters were identified in a clustering analysis for the change of all metabolites according to their pairwise correlations as described previously [5]. The color scheme corresponds to correlation strength as shown by the color bar. Red: Better response, more reduction of the metabolite. Blue: Better response, less reduction or increase of the metabolite. NIST: National Institute of Standards and Technology.
The metabolite most significantly different between good and poor simvastatin responders at baseline was the purine metabolite xanthine, the substrate of xanthine oxidase, which produces hydrogen peroxide and hence is implicated in mechanisms of oxidative stress. Since free radicals are known to decouple NOS enzymatic activity, the lower basal level of xanthine in good responders might be expected to yield more robust NOS signaling, further supporting a link between the benefits of statins on lipids and endothelial function.
Baseline levels of 2-hydroxyvaleric acid (2-hydroxypentanoic acid) also strongly discriminated “good” and “poor” statin responders, with lower levels associated with greater response. Moreover, a reduction in its levels was a significant component of the metabolomic signature of simvastatin response. Studies of the effects of incubating simvastatin with intestinal bacteria ex vivo have demonstrated the production of a set of metabolites including and similar to 2-hydroxyvaleric acid. Hence it is possible that lower endogenous production of 2-hydroxyvaleric acid, and further reduction with simvastatin treatment, might represent reduced activity of one or more enzymes produced by intestinal bacteria that would result in lower rates of simvastatin degradation and hence enhanced efficacy. This observation reinforces the hypothesis that variation in the gut microbiome can contribute to interindividual differences in statin efficacy, In this regard, it was found that in “good” responders, simvastatin increased plasma levels of shikimic acid, an indole precursor of phenylalanine, tyrosine and tryptophan that is produced by plants and bacteria, but not animals, suggesting another potential interaction between simvastatin and intestinal microflora.
Taken together, the studies summarized here demonstrate that simvastatin has a wide range of metabolic effects beyond those directly involved in cholesterol metabolism that may contribute to its efficacy in reducing risk of CVD, as well as to its pleiotropic effects and the risk of adverse events. Further, the findings point to important interactions involving the metabolome, the genome, and the microbiome that may underlie interindividual differences in response to statin, as well as other drug therapies.
Supplementary Material
Acknowledgments
We thank the following individuals for their contributions as co-investigators on the studies reviewed in this paper: Rebecca A. Baillie , Oliver Fiehn, Peter D. Karp, Uyen Thao Nguyen, Miles Trupp, Steven M. Watkins, Michelle M. Wiest, William R. Wikoff, Katie Wojnoonski, and Zhao-Bang Zeng.
Funding:
This work was supported by National Institute of General Medical Sciences grants R24, GM078233, “The Metabolomics Research Network for Drug Response Phenotype” (RKD, RMK); and RC2GM092729 as part of the American Recovery and Reinvestment Act (ARRA) and U01 HL069757, “Pharmacogenomics and Risk of Cardiovascular Disease” (RMK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Competing Interests: All authors are inventors on one or more patents in the metabolomics field.
References
- [1].Simon JA, Lin F, Hulley SB, et al. Phenotypic predictors of response to simvastatin therapy among African-Americans and Caucasians: the Cholesterol and Pharmacogenetics (CAP) Study. The American Journal of Cardiology. 2006;97:843–850. doi: 10.1016/j.amjcard.2005.09.134. [DOI] [PubMed] [Google Scholar]
- [2].Naci H, Brugts J, Ades T. Comparative tolerability and harms of individual statins: A study-level network meta-analysis of 246, 955 participants from 135 randomized controlled trials. Circulation. Cardiovascular Quality and Outcomes. 2013 doi: 10.1161/CIRCOUTCOMES.111.000071. [DOI] [PubMed] [Google Scholar]
- [3].Kaddurah-Daouk R, Baillie RA, Zhu H, et al. Lipidomic analysis of variation in response to simvastatin in the Cholesterol and Pharmacogenetics Study. Metabolomics. 2010;6:191–201. doi: 10.1007/s11306-010-0207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kaddurah-Daouk R, Baillie RA, Zhu H, et al. Enteric microbiome metabolites correlate with response to simvastatin treatment. PLoS One. 2011;6:e25482. doi: 10.1371/journal.pone.0025482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Trupp M, Zhu H, Wikoff WR, et al. Metabolomics reveals amino acids contribute to variation in response to simvastatin treatment. PLoS One. 2012;7:e38386. doi: 10.1371/journal.pone.0038386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.