

Abstract
Diabetes mellitus belongs to the most rapidly increasing diseases worldwide. Approximately 90–95% of these patients suffer from type 2 diabetes mellitus, which is characterized by peripheral insulin resistance and the progressive loss of beta-cell function and mass. Considering the complications of this chronic disease, a reliable anti-diabetic treatment is indispensable. An ideal oral anti-diabetic drug should not only correct glucose homeostasis but also preserve or even augment beta-cell function and mass, ameliorate the subclinical inflammation present under insulin-resistant conditions and prevent the macro- and microvascular consequences of diabetes in order to reduce the mortality. Despite the many anti-diabetic drugs already in use, there is an ongoing research for additional drugs, guided by different concepts of the pathogenesis of type 2 diabetes. This review will briefly summarize current oral anti-diabetic drugs. In addition, emerging strategies for the treatment of diabetes will be described, among them the inhibition of glucagon action and anti-inflammatory drugs. Their suitability as ‘ideal anti-diabetic drugs’ will be discussed.
Keywords: type 2 diabetes mellitus, anti-diabetic drugs, insulin resistance, inflammation, beta-cell function
Pathogenesis of type 2 diabetes mellitus
Diabetes mellitus belongs to the most rapidly increasing diseases worldwide. Among the consequences of diabetes are micro- and macrovascular complications such as retinopathy and nephropathy leading to blindness and renal insufficiency, respectively, and cardiovascular and cerebrovascular diseases (Alberti and Zimmet, 1998; Stratton et al., 2000). Indeed, more than 60% of type 2 diabetics die of myocardial infarction or stroke (Giorgino et al., 2013). In general, two forms of diabetes mellitus are distinguished: type 1 diabetes is caused by the autoimmune destruction of the insulin-producing beta-cells in early childhood and resulting in an absolute lack of insulin (American Diabetes Association, 2004). For the development of type 2 diabetes, obesity caused by the chronic imbalance between calorie intake and energy expenditure is the major risk factor (American Diabetes Association, 2004; Lazar, 2005). The excess of nutrients is stored mainly in the white adipose tissue (WAT), the liver and the skeletal muscle. However, under conditions of chronic over-nutrition, their storage capacity is eventually exceeded and mitochondrial dysfunction, oxidative stress, endothelial reticulum stress and abnormal post-translational modification of intracellular proteins ensue. The cellular stress activates diverse signalling pathways, including the JNKs and the IκB kinase β (IKKβ), which, in turn, inhibit insulin signalling pathways and trigger inflammation within the WAT and other tissues (Donath and Shoelson, 2011; Lumeng and Saltiel, 2011; Odegaard and Chawla, 2013). This subacute inflammation within the metabolic tissues leads to increased secretion of pro-inflammatory cytokines, which reinforces inflammatory signals and decreases the secretion of protective factors such as adiponectin (Hotamisligil et al., 1995; Hotta et al., 2000; Donath and Shoelson, 2011; Lumeng and Saltiel, 2011; Odegaard and Chawla, 2013). Furthermore, mainly via inhibitory serine/threonine phosphorylation of the insulin receptor substrate 1, some pro-inflammatory cytokines inhibit insulin signalling, thereby escalating insulin resistance (Tamemoto et al., 1994; Aguirre et al., 2000; Hirosumi et al., 2002; Ueki et al., 2004; Werner et al., 2004). In this scenario, insulin resistance might be considered protective as it prevents the further excess uptake of nutrients and the deterioration of the cells within the metabolic tissues (Lumeng and Saltiel, 2011; Saltiel, 2012; Odegaard and Chawla, 2013). Deregulated nutrient uptake itself can activate inflammation by various mechanisms (Pal et al., 2012; Tremaroli and Backhed, 2012; Odegaard and Chawla, 2013). Thus, obesity-induced insulin resistance contributing to the low-grade inflammation of metabolic tissues and the low-grade inflammation contributing to insulin resistance perpetuate each other. To compensate for the increased insulin demand under conditions of insulin resistance, beta-cell hypertrophy and hyperplasia develops, leading to hyperinsulinaemia (Butler et al., 2003; Rhodes, 2005). It should be noted that most of the obese, insulin-resistant humans do not become diabetic, implying additional mechanisms for the pathogenesis of type 2 diabetes mellitus. Indeed, dysfunction of the beta-cells with regard to insulin secretion and biosynthesis and a reduction of beta-cell mass were demonstrated in patients suffering from type 2 diabetes or impaired glucose tolerance (Butler et al., 2003; Kahn et al., 2009; Marchetti et al., 2012; Weir and Bonner-Weir, 2013) (Figure 1). Hence, the targets of an optimal anti-diabetic therapy are the attenuation of the inflammation of metabolic tissues and insulin resistance and the restoration or at least the amelioration of beta-cell function and mass to ultimately prevent the development of micro- and macrovascular complications.
Figure 1.
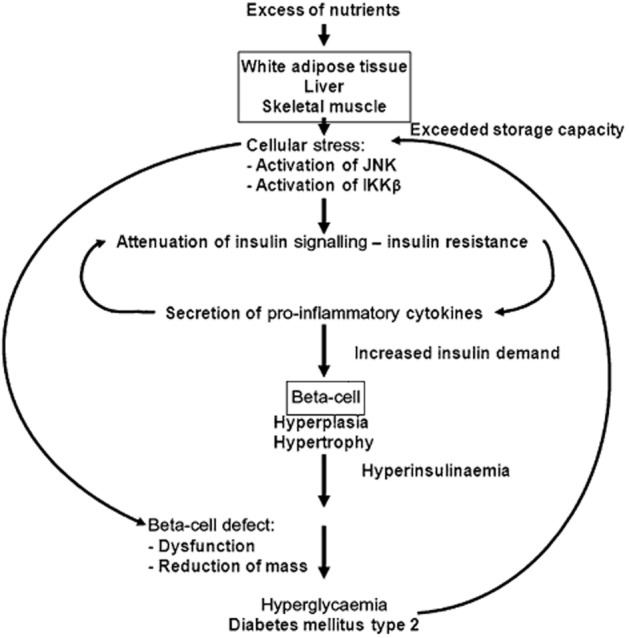
Pathogenesis of type 2 diabetes mellitus. Excess of nutrients is stored in the WAT, liver and skeletal muscle. When their storage capacity is exceeded, cellular stress leads to the activation of JNK and IKKβ, which, in turn, attenuates insulin signalling. Peripheral insulin resistance leads to secretion of pro-inflammatory cytokines mainly from macrophages migrated into the WAT. Pro-inflammatory cytokine signalling further aggravates insulin resistance. To compensate for the increased insulin demand, beta-cells hypertrophy and become hyperplastic, leading to hyperinsulinaemia. In addition, beta-cells themselves are exposed to cellular stress promoting beta-cell dysfunction and loss of beta-cells, and finally resulting in hyperglycaemia. The elevated blood glucose levels reinforce cellular stress.
Something old: current drugs for treating type 2 diabetes mellitus
The current drugs for treating type 2 diabetes mellitus can be roughly distinguished into those acting directly on beta-cells and those that do not. Sulfonylureas such as glibenclamide, tolbutamide and glimepiride have been in use since the 1950s. By inhibition of the ATP-dependent potassium channel (KATP, KIR6.2; channel nomenclature follows Alexander et al., 2013a) favouring membrane depolarization and subsequent calcium influx through the voltage-dependent L-type calcium channel (Cav1.x), they directly stimulate insulin secretion from the beta-cells. Increased insulin levels reduce blood glucose concentration but lead to weight gain, a most undesired effect in the typical obese type 2 diabetic. Another important adverse effect is hypoglycaemia. The United Kingdom Prospective Diabetes Study (UKPDS) demonstrated a significant reduction of microvascular complications after long-term treatment with glibenclamide (UKPDS 33) (UKPDS Group, 1998). In two observational studies, a higher risk for myocardial infarction and for mortality was found in patients treated with glibenclamide in comparison to treatment with glimepiride and gliclazide (Monami et al., 2006; Thisted et al., 2006). Another observational study found a higher incidence of cardiac events under glibenclamide in comparison to gliclazide only in patients with previously known ischaemic heart disease (Monami et al., 2007).
In addition to glucose, glucagon-like peptide-1 (GLP-1) secreted from the intestinal L-cells in response to meal ingestion is another important insulin secretagogue (Doyle and Egan, 2001). The short plasma half-life of GLP-1 of 2 min due to cleavage by dipeptidylpeptidase 4 (DPP 4; nomenclature follows Alexander et al., 2013b) makes the substitution of GLP-1 itself unsuitable. Therefore, s.c. injectable GLP-1 analogues with a mutated (i.e. exenatide) or a masked DPP 4 cleavage site (i.e. liraglutide) or orally available DPP 4 inhibitors (the gliptins sitagliptin, vildagliptin, saxagliptin) raising the endogenous GLP-1 levels are used. GLP-1 analogues and the gliptins have attracted much attention in the past years. As GLP-1 potentiates insulin secretion only in the presence of elevated glucose levels, the possibility of hypoglycaemic events is rather low. In rodents and in humans, GLP-1 has been shown to improve beta-cell function and increase beta-cell mass (Xu et al., 1999; Skoglund et al., 2000; Stoffers et al., 2000; Sonoda et al., 2008; Butler et al., 2013a). In addition, GLP-1 promotes satiety, slows down gastric emptying, inhibits the secretion of the glucogenic hormone glucagon from α-cells and results in weight loss (Thornberry and Gallwitz, 2009; Pabreja et al., 2014). However, recent reports have raised concerns about the safety profile of the GLP-1 receptor agonists and the DPP 4 inhibitors. In rodents, treatment with the long-acting GLP-1 receptor agonist exendin resulted in acinar cell death and inflammation and in accelerated metaplasia and lesion formation of pancreatic intraepithelial neoplasia (Nachnani et al., 2010; Gier et al., 2012). In humans, GLP-1-based therapy leads to increased proliferation and dysplasia within the exocrine pancreas (Butler et al., 2013a) and a meta-analysis revealed an association between GLP-1 therapy and increased risk for hospitalization for acute pancreatitis (Singh et al., 2013). In contrast, two recent studies evaluating the cardiovascular safety of saxagliptin and alogliptin, respectively, reported no increase in the incidence of pancreatitis; a slightly increased risk of heart failure was observed in the saxagliptin group (Scirica et al., 2013; White et al., 2013). It remains to be seen whether the undoubted benefits of GLP-1-based therapy outweigh its potential risks (Gallwitz et al., 2012; Butler et al., 2013b; Nauck, 2013).
As a result of the UKPDS (UKPDS Group, 1998), the biguanide metformin experienced a revival of its use and is now the first-choice anti-diabetic drug. A rare but potentially lethal effect is lactic acidosis, with an incidence of 4.3 cases in 100 000 patient-years (Salpeter et al., 2010). Still, there are several contraindications for metformin use, including cardiovascular, renal, hepatic and pulmonary diseases. In the case of metformin, the beneficial effects clearly outweigh its potential risks: metformin was shown to prevent cardiovascular mortality and disease (UKPDS 34, 1998) and might reduce cancer incidence (Evans et al., 2005; Libby et al., 2009; Noto et al., 2012; van Staa et al., 2012). In male mice, long-term treatment with metformin extended their lifespan (Martin-Montalvo et al., 2013). In pre-diabetic humans, both lifestyle intervention and metformin reduced the incidence of diabetes, but lifestyle intervention was more effective (Knowler et al., 2002). Metformin lowers blood glucose levels mainly through inhibition of hepatic gluconeogenesis; enhanced glucose uptake into the skeletal muscle has also been described. Metformin is weight neutral (Viollet et al., 2012; Rena et al., 2013). Although metformin has been in use since the 1950s, its molecular mechanisms are still not completely understood: inhibition of complex I in the mitochondrial electron transport chain, resulting in energy depletion with increased ADP/ATP and AMP/ATP ratios and activation of the AMP-dependent kinase (AMPK), a central cellular energy sensor and regulator of energy homeostasis have been proposed (Hardie et al., 2012; Logie et al., 2012; Hardie, 2013; Rena et al., 2013). Consistent with this, infusion of the direct activator of AMPK, 5-aminoimidazole-4-carboxamide riboside (AICAR) decreased hepatic glucose output, thus lowering blood glucose levels in type 2 diabetic patients (Boon et al., 2008). Shaw et al. (2005) suggested that AMPK-induced inhibition of the cAMP-regulated transcriptional co-activator (CRTC) 2 prevents the expression of gluconeogenic genes in hepatocytes, consistent with the findings that CRTC2 plays a pivotal role in hepatic glucose output under fasting conditions (Koo et al., 2005). However, metformin still exerted hypoglycaemic effects in mice lacking AMPK in the liver, suggesting that AMPK – and transcription-independent mechanisms – confer metformin-caused reduction of hepatic gluconeogenesis (Foretz et al., 2010). Another AMPK-independent mechanism of metformin action was proposed by Miller et al. (2013), showing that metformin attenuated glucagon-induced hepatic gluconeogenesis, by indirectly inhibiting the adenylate cyclase. Metformin is a hydrophilic base and is transported via organic cationic transporters (OCT) 1 and 2 into the liver, the gut and the kidney (Graham et al., 2011; Viollet and Foretz, 2013; transporter nomenclature follows Alexander et al., 2013c). In OCT1-deficient mice, hepatic metformin concentration was decreased and the drug no longer reduced fasting blood glucose levels, suggesting that OCT1 is important for hepatic metformin action (Shu et al., 2007).
From a pathophysiological point of view, the thiazolidinediones have a very favourable pattern of action: they enhance insulin sensitivity of skeletal muscle and liver, inhibit hepatic gluconeogenesis and are anti-inflammatory in various organs. However, fluid retention with associated peripheral oedema due to altered renal sodium and water reabsorption, the higher rate of fractures due to decreased bone formation and increased bone resorption, and the weight gain, in part, due to increased food intake and to increased adipogenesis greatly diminished the widespread use of rosiglitazone and pioglitazone (Ahmadian et al., 2013). Whereas pioglitazone was suggested to exert modest protective effects on the CVS (Dormandy et al., 2005; Lincoff et al., 2007), rosiglitazone has been associated with an increased risk of myocardial infarction (Nissen and Wolski, 2007; 2010), resulting in the withdrawal of this drug. However, rosiglitazone's increased risk of myocardial infarction remains a matter of debate, whereas an increased risk for heart failure is well documented for the thiazolidinediones (Home et al., 2009; Kaul and Diamond, 2011; Winterstein, 2011). In addition, increased incidence of bladder cancer has been reported for pioglitazone (Zhu et al., 2012). Thiazolidinediones are agonists of the PPARγ (NR1C; nomenclature follows Alexander et al., 2013d), a nuclear receptor that forms permissive heterodimers with retinoid X receptors (RXR, NR2B; Ahmadian et al., 2013). Specific endogenous PPARγ ligands are still elusive, but some fatty acids and their derivatives can bind and activate this nuclear receptor (Ahmadian et al., 2013). In addition to ligand binding, PPARγ activity is regulated by post-translational modifications, among them phosphorylation by distinct kinases, acetylation, sumoylation and ubiquitination (Ahmadian et al., 2013), thereby expanding the cell- or tissue-specific modulation of this nuclear receptor. To prevent the adverse effects of thiazolidinediones, but retaining the desired effects, in analogy to the selective oestrogen receptor modulators, selective PPAR modulators might be promising new anti-diabetic drugs (Bhalla et al., 2011; Choi et al., 2011; Weidner et al., 2012; 2013; Cheon, 2013). Dual PPARγ/α agonists represent an approach to combine the glucose-lowering effects of the PPARγ agonists with the lipid-lowering effects of the PPARα agonists (like the fibrates) to effectively manage glycaemic control and improve cardiovascular outcome in type 2 diabetic patients. Several dual agonists, called glitazars, have been developed with promising effects on lowering HbA1c and plasma lipid levels (Henry et al., 2009; Rosenson et al., 2012; Wilding, 2012). However, due to diverse safety concerns, the further development and in the case of aliglitazar phase 3 clinical trials were stopped (Rosenson et al., 2012; Wilding, 2012) (media release from Roche, http://www.roche.com/de/media/media.release/med-cor-2013-07-10.htm). Whereas the glucose-lowering effect of thiazolidinediones is due to many actions, dapagliflozin exerts its effect through inhibition of the sodium-glucose transporter 2 (SGLT2) in the proximal tubule of the kidney, thus preventing glucose reabsorption. The SGLT2 is a low-affinity, high-capacity transporter, reabsorbing most of the glucose in the urine. Its inhibition cannot result in hypoglycaemia because a fraction of the remaining glucose is reabsorbed by the SGLT1, a high-affinity, low-capacity transporter that is expressed in the more distal part of the renal tubular system (Tahrani et al., 2011).
Something new: potential drug targets and drugs
Given the epidemic dimensions of obesity and diabetes worldwide, it is not surprising that many more potential drug targets are currently under investigation. TAK-875, an agonist of the G-protein coupled receptor/free fatty acid receptor 1 (GPR40/FFAR1) highly expressed on beta-cells, is one example for a novel anti-diabetic drug (Yashiro et al., 2012). In isolated rat and human islets, TAK-875, by binding to its receptor, increased the intracellular calcium concentration and activated PKC, thereby potentiating glucose-stimulated insulin secretion (Yashiro et al., 2012). A phase 2 trial revealed that the glucose-lowering effects of TAK-875 and the sulfonylurea glimepiride are comparable, but less hypoglycaemic events occurred in the group treated with TAK-875. However, weight gain was similar in both patient groups treated either with glimepiride or with TAK-875 (Burant et al., 2012). The long-term effects of this novel drug-like protection or maintenance of beta-cell mass or the prevention of cardiovascular complications remain to be seen.
Another potential drug target within the beta-cell, but not exclusively there, is the activation of the glucokinase. In beta-cells, glucokinase acts as a glucose sensor and by phosphorylation of glucose triggers glucose oxidation, insulin biosynthesis and insulin secretion. In hepatocytes, this enzyme enhances glycolysis, glycogen synthesis and lipogenesis (Matschinsky et al., 2011; Matschinsky, 2013). Thus, activators of glucokinase effectively lower blood glucose levels by increased beta-cell insulin release and decreased hepatic glucose output. Heterozygous inactivating mutations of glucokinase cause maturity-onset diabetes of the young characterized by mild fasting hyperglycaemia; homozygous inactivating mutations result in permanent neonatal diabetes mellitus, demonstrating the importance of this enzyme for glucose homeostasis (Osbak et al., 2009). However, in a recent phase 2 trial, type 2 diabetic patients treated with the glucokinase activator MK0941 developed hyperlipidaemia and vascular hypertension, besides hypoglycaemic events. Furthermore, after 3–4 months of treatment, the drug failed (Meininger et al., 2011; Matschinsky, 2013). Two structurally distinct glucokinase activators induced hepatic steatosis in normoglycaemic and diabetic rodents (De Ceuninck et al., 2013). Hence, the long-term activation of glucokinase might not be beneficial at all (Rees and Gloyn, 2013).
Glucagon is another quite intriguing target for the therapy of diabetes. This peptide hormone is secreted from the pancreatic α-cells in response to mixed meal nutrients, amino acids and hypoglycaemia. Glucagon secretion and biosynthesis is inhibited by insulin and probably other factors, secreted by the neighbouring beta-cells (Philippe, 1989; Grzeskowiak et al., 2000; Gromada et al., 2007; Kawamori et al., 2009; D'Alessio, 2011; Unger and Cherrington, 2012). Glucagon binds to its Gs-protein coupled receptor (see Alexander et al., 2013e) on hepatocytes, thus stimulating gluconeogenesis and enhancing glucose output. Hence, glucagon, elevating fasting glucose levels, can be considered as a functional antagonist of insulin, decreasing postprandial glucose levels. The relevance for targeting glucagon receptors or α-cells to interfere with the pathogenesis of diabetes has long been neglected (D'Alessio, 2011; Unger and Cherrington, 2012). Glucagon levels are enhanced in poorly controlled type 1 and type 2 diabetes, and some type 2 diabetic patients with at least moderately controlled glucose levels show fasting hyperglucagonaemia (Reaven et al., 1987; D'Alessio, 2011). Dysfunction of the α-cells might contribute to the pathogenesis of type 2 diabetes: in diabetic patients, α-cell response to hyperglycaemia is blunted and glucagon secretion is enhanced by physiological stimuli to a greater extent than in non-diabetics (Unger, 1985). In addition, the α-cell itself might become insulin resistant, failing to reduce glucagon biosynthesis and secretion in response to insulin (Gonzalez et al., 2008; Kawamori et al., 2009). Hence, blocking glucagon action is a suitable target for treating type 2 diabetes mellitus. Glucagon-receptor knockout mice and treatment of several animal models with antibodies against glucagon or antisense oligonucleotides against the glucagon receptor support this notion (Gelling et al., 2003; Liang et al., 2004; Sorensen et al., 2006a,b; Conarello et al., 2007; D'Alessio, 2011; Tahrani et al., 2011). However, α-cell hyperplasia with elevated glucagon and GLP-1 levels, and hepatic steatosis have been observed in animal models (Gelling et al., 2003; Conarello et al., 2007; D'Alessio, 2011; Tahrani et al., 2011). It should be noted that already drugs exist that interfere with glucagon action: GLP-1 analogues and DPP 4 inhibitors reduce α-cell glucagon secretion (Thornberry and Gallwitz, 2009; Pabreja et al., 2014); metformin seems to inhibit hepatic glucagon action by indirectly inhibiting the adenylate cyclase of the Gs-coupled glucagon receptor (Miller et al., 2013).
Guided by the observation that an excess of glucocorticoids (Cushing's syndrome) shows similarities to the metabolic syndrome with obesity, insulin resistance and diabetes, decreasing local concentrations of hydrocortisone (cortisol) has become another alternative for the treatment of type 2 diabetes. Cortisol, produced and secreted from the adrenal glands, induces hyperglycaemia by promoting hepatic gluconeogenesis and glycogenolysis. In the presence of NADPH, 11β-hydroxysteroid dehydrogenase 1 (11β-HSD 1) converts the inactive cortisone to the active cortisol. This enzyme is mainly expressed in liver and the adipose tissue. The 11β-HSD 2 is mainly expressed in tissues that also express the mineralocorticoid receptor such as the kidney and oxidizes cortisol to cortisone, thus allowing aldosterone to bind to its receptor (Tahrani et al., 2011; Anagnostis et al., 2013). Mice deficient in 11β-HSD 1 or treated with specific 11β-HSD 1 inhibitors show improved glucose tolerance, reduced insulin resistance and decreases in body weight (Morgan et al., 2009; Tahrani et al., 2011; Anagnostis et al., 2013). The effects seem to be mainly due to the inhibition of the adipose tissue enzyme (Lavery et al., 2012). In type 2 diabetic patients treated for 12 weeks with INCB13739 in addition to metformin therapy, reduced HbA1c levels (by 0.6%) and fasting glucose levels were observed (Tahrani et al., 2011; Anagnostis et al., 2013). Other 11β-HSD 1 inhibitors tested in diabetic patients had negligible effects on glucose metabolism (Anagnostis et al., 2013).
Taking into account the inflammatory nature of diabetes, immune-modulating therapies may be another option for the treatment of type 2 diabetes mellitus (Donath and Shoelson, 2011; Lumeng and Saltiel, 2011). The pro-inflammatory cytokine IL-1β seems to initiate the migration of macrophages to the inflamed adipose tissue and islets (Brooks-Worrell et al., 2012). Furthermore, IL-1β is secreted from the beta-cells themselves in hyperglycaemia, inhibits insulin gene transcription and induces beta-cell apoptosis (Maedler et al., 2002; Oetjen et al., 2007). Hence, attenuating this cytokine's effect represents a promising target. In a double-blind trial, type 2 diabetic patients were randomized to placebo or to treatment with the recombinant human IL-1-receptor antagonist anakinra administered once daily s.c. for 13 weeks. Treatment with anakinra lowered HbA1c by 0.46% and reduced the markers of systemic inflammation (Larsen et al., 2007). In a 39 week follow-up study, the anti-inflammatory effect of anakinra was still present, whereas the improvement in HbA1c levels was no longer detectable (Larsen et al., 2009). Other IL-1β neutralizing antibodies are currently investigated in clinical trials (Brooks-Worrell et al., 2012). TNF-α is secreted from the adipose tissue in the pre-diabetic state and is elevated in obesity, insulin resistance and type 2 diabetes (Hotamisligil et al., 1993; Plomgaard et al., 2007). A recent study demonstrated that treatment of obese, insulin-resistant patients with the recombinant TNF-α receptor 2 etanercept for 6 months improved fasting glucose levels (Stanley et al., 2011). These findings suggest that targeting the chronic, low-grade inflammation might provide a useful drug target. In fact, the glucose-lowering effect of a well-known anti-inflammatory drug was described already more than 100 years ago (Ebstein, 1876).
Something very old: salicylic acid for the treatment of type 2 diabetes
In 1876, a report by Ebstein was published, describing the attenuation of classical diabetic symptoms (polyuria, polydipsia and elevated glucose concentration of the urine) in two middle-aged patients receiving salicylic acid natron. Without knowing the pathogenesis of diabetes or the pharmacodynamics of salicylic acid, Ebstein proposed that this drug might be used to treat diabetes (Ebstein, 1876). This report was long forgotten. However, with the realization of insulin resistance and diabetes as a chronic inflammatory process, salicylate as an anti-inflammatory drug was re-evaluated. Indeed, in type 2 diabetic patients, treatment with high-dose acetylsalicylic acid (approximately 7 g·day−1) reduced fasting glucose levels and hepatic glucose output, and improved insulin-stimulated peripheral glucose uptake (Hundal et al., 2002). Salicylates inhibit COX and therefore prostanoid synthesis (Higgs et al., 1984). In addition, they inhibit IKKβ, crucial for the activation of the pro-inflammatory transcription factor complex NF-κB (Yuan et al., 2001). As salicylates still confer anti-inflammatory actions in mice deficient in COX-2 or in a component of the NF-κB complex, other mechanisms must apply (Cronstein et al., 1999; Hawley et al., 2012). The finding that in AMPK-deficient mice the effects of salicylate, such as increased fat utilization and the decrease of plasma fatty acids, were lost indicates that AMPK might be a target of salicylate action. Indeed, salicylate was shown to directly bind and activate this kinase (Hawley et al., 2012). In addition, down-regulation of 11β-HSD 1 in adipose tissue by salicylate, thereby reducing intra-adipose glucocorticoid levels and improving insulin sensitivity, was reported (Nixon et al., 2012). Irrespective of its mechanism of action, salicylates were used in clinical trials. To reduce the adverse effects of acetylsalicylic acid (aspirin), such as its anti-thrombotic action and irritation of the gastrointestinal tract, the dimer of salicylic acid, salsalate, was used. Salsalate is only marketed in the US. In patients with newly diagnosed type 2 diabetes, 12 week salsalate treatment (3 g day−1) reduced fasting glucose and HbA1c (Faghihimani et al., 2013). In a randomized double-blind trial, patients with type 2 diabetes were treated for 48 weeks either with salsalate or placebo as ‘add on’ to their usual anti-diabetic therapy. In the salsalate treatment group, glycaemia improved concomitantly, with a reduction in the already existing diabetes medication. In addition, a reduction of inflammation estimated by lower circulating leukocyte, neutrophil and lymphocyte counts was observed (Goldfine et al., 2013b). The findings that in the same group body weight, plasma low-density lipoprotein cholesterol levels and urinary albumin levels increased warrants, however, further evaluation of salsalate (Goldfine et al., 2010; 2013b). In patients at risk for diabetes (insulin resistance, impaired fasting glucose or impaired glucose tolerance), salsalate lowered fasting glucose levels and inhibited adipose tissue NF-κB activity without changing peripheral insulin resistance (Goldfine et al., 2013a). The explanation for this unexpected finding might be that salsalate through activation of AMPK reduced hepatic glucose output (Hawley et al., 2012).
Conclusion and outlook
Considering the new concepts of the pathogenesis of type 2 diabetes, attenuation of cellular stress in the metabolic tissues thereby preventing insulin resistance and low-grade inflammation and protection of beta-cell function and mass are desirable goals in the treatment of this chronic progressive disease (Figure 1). Clearly, lifestyle intervention to balance nutrient intake and consumption, thus avoiding obesity is the best way to reach these goals (Figure 2), but the nearly pandemic dimensions of obesity argue against the realization of this approach. There is a broad range of oral anti-diabetic drugs, but none of them can cover all the needs in diabetes therapy, and for the new drugs (and the very old drug salsalate), long-term data on the prevention of macrovascular complications and drug safety are lacking. As it is the decompensation of beta-cell function and the loss of beta-cell mass that result in diabetes, protecting beta-cells against the beta-cell toxic signals present under insulin-resistant and subacute inflammatory conditions is supposed to prevent or at least slow the onset of clinically apparent diabetes. To date, this has only been shown for the GLP-1 analogues and the DPP 4 inhibitors, whose safety profile remains a matter of debate. Hence, to achieve the maintenance of beta-cell function and mass, the identification of new drug targets within the beta-cell and the subsequent development of drugs provide an additional and much needed strategy to treat diabetes mellitus.
Figure 2.

Action of old and new drugs against type 2 diabetes.
Acknowledgments
The authors thank H.J. Steinfelder (Göttingen) and R. Böger and T. Christ (Hamburg) for critically reading the manuscript. The work in our lab was supported by a grant from the Deutsche Forschungsgemeinschaft to E. O. (OE 181/3-1).
Glossary
- 11β-HSD
11β-hydroxysteroid dehydrogenase
- AICAR
5-aminoimidazole-4-carboxamide riboside
- AMPK
adenosine monophosphate (AMP)-dependent kinase
- CRTC
cAMP-regulated transcriptional co-activator
- DPP 4
dipeptidylpeptidase 4
- GLP-1
glucagon-like peptide-1
- GPR40/FFAR1
G-protein coupled receptor/free fatty acid receptor 1
- IKKβ
IκB kinase β
- OCT
organic cationic transporter
- SGLT
sodium-glucose transporter
- UKPDS
United Kingdom Prospective Diabetes Study
- WAT
white adipose tissue
Conflict of interest
The authors declare to have no conflict of interest.
References
- Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013a;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013c;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL,, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear Hormone Receptors. Br J Pharmacol. 2013d;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013e;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2004;27(Suppl. 1):S5–S10. doi: 10.2337/diacare.27.2007.s5. [DOI] [PubMed] [Google Scholar]
- Anagnostis P, Katsiki N, Adamidou F, Athyros VG, Karagiannis A, Kita M, et al. 11beta-Hydroxysteroid dehydrogenase type 1 inhibitors: novel agents for the treatment of metabolic syndrome and obesity-related disorders? Metabolism. 2013;62:21–33. doi: 10.1016/j.metabol.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Bhalla K, Hwang BJ, Choi JH, Dewi R, Ou L, McLenithan J, et al. N-Acetylfarnesylcysteine is a novel class of peroxisome proliferator-activated receptor gamma ligand with partial and full agonist activity in vitro and in vivo. J Biol Chem. 2011;286:41626–41635. doi: 10.1074/jbc.M111.257915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon H, Bosselaar M, Praet SFE, Blaak EE, Saris WHM, Wagenmakers AJM, et al. Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia. 2008;51:1893–1900. doi: 10.1007/s00125-008-1108-7. [DOI] [PubMed] [Google Scholar]
- Brooks-Worrell B, Narla R, Palmer JP. Biomarkers and immune-modulating therapies for type 2 diabetes. Trends Immunol. 2012;33:546–553. doi: 10.1016/j.it.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Burant CF, Viswanathan P, Marcinak J, Cao C, Vakilynejad M, Xie B, et al. TAK-875 versus placebo or glimepiride in type 2 diabetes mellitus: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2012;379(9824):1403–1411. doi: 10.1016/S0140-6736(11)61879-5. [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes. 2013a;62:2595–2604. doi: 10.2337/db12-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler PC, Elashoff M, Elashoff R, Gale EA. A critical analysis of the clinical use of incretin-based therapies: are the GLP-1 therapies safe? Diabetes Care. 2013b;36:2118–2125. doi: 10.2337/dc12-2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon HG. Latest research and development trends in non-insulin anti-diabetics. Arch Pharm Res. 2013;36:145–153. doi: 10.1007/s12272-013-0016-7. [DOI] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477(7365):477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conarello SL, Jiang G, Mu J, Li Z, Woods J, Zycband E, et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia. 2007;50:142–150. doi: 10.1007/s00125-006-0481-3. [DOI] [PubMed] [Google Scholar]
- Cronstein BN, Montesinos MC, Weissmann G. Salicylates and sulfasalazine, but not glucocorticoids, inhibit leukocyte accumulation by an adenosine-dependent mechanism that is independent of inhibition of prostaglandin synthesis and p105 of NFkappaB. Proc Natl Acad Sci U S A. 1999;96:6377–6381. doi: 10.1073/pnas.96.11.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessio D. The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes Obes Metab. 2011;13(Suppl. 1):126–132. doi: 10.1111/j.1463-1326.2011.01449.x. [DOI] [PubMed] [Google Scholar]
- De Ceuninck F, Kargar C, Charton Y, Goldstein S, Perron-Sierra F, Ilic C, et al. S 50131 and S 51434, two novel small molecule glucokinase activators, lack chronic efficacy despite potent acute antihyperglycaemic activity in diabetic mice. Br J Pharmacol. 2013;169:999–1010. doi: 10.1111/bph.12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- Doyle ME, Egan JM. Glucagon-like peptide-1. Recent Prog Horm Res. 2001;56:377–399. doi: 10.1210/rp.56.1.377. [DOI] [PubMed] [Google Scholar]
- Ebstein W. Zur therapie des diabetes mellitus, insbesondere uber die anwendung des salicylsauren natron bei demselben. Berl Klin Wochenschr. 1876;13:337–340. [Google Scholar]
- Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihimani E, Aminorroaya A, Rezvanian H, Adibi P, Ismail-Beigi F, Amini M. Salsalate improves glycemic control in patients with newly diagnosed type 2 diabetes. Acta Diabetol. 2013;50:537–543. doi: 10.1007/s00592-011-0329-2. [DOI] [PubMed] [Google Scholar]
- Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallwitz B, Rosenstock J, Rauch T, Bhattacharya S, Patel S, von Eynatten M, et al. 2-year efficacy and safety of linagliptin compared with glimepiride in patients with type 2 diabetes inadequately controlled on metformin: a randomised, double-blind, non-inferiority trial. Lancet. 2012;380:475–483. doi: 10.1016/S0140-6736(12)60691-6. [DOI] [PubMed] [Google Scholar]
- Gelling RW, Du XQ, Dichmann DS, Romer J, Huang H, Cui L, et al. Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A. 2003;100:1438–1443. doi: 10.1073/pnas.0237106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gier B, Matveyenko AV, Kirakossian D, Dawson D, Dry SM, Butler PC. Chronic GLP-1 receptor activation by exendin-4 induces expansion of pancreatic duct glands in rats and accelerates formation of dysplastic lesions and chronic pancreatitis in the Kras(G12D) mouse model. Diabetes. 2012;61:1250–1262. doi: 10.2337/db11-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgino F, Leonardini A, Laviola L. Cardiovascular disease and glycemic control in type 2 diabetes: now that the dust is settling from large clinical trials. Ann N Y Acad Sci. 2013;1281:36–50. doi: 10.1111/nyas.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med. 2010;152:346–357. doi: 10.1059/0003-4819-152-6-201003160-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Conlin PR, Halperin F, Koska J, Permana P, Schwenke D, et al. A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia. 2013a;56:714–723. doi: 10.1007/s00125-012-2819-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Fonseca V, Jablonski KA, Chen YD, Tipton L, Staten MA, et al. Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Ann Intern Med. 2013b;159:1–12. doi: 10.7326/0003-4819-159-1-201307020-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Boer U, Dickel C, Quentin T, Cierny I, Oetjen E, et al. Loss of insulin-induced inhibition of glucagon gene transcription in hamster pancreatic islet alpha cells by long-term insulin exposure. Diabetologia. 2008;51:2012–2021. doi: 10.1007/s00125-008-1134-5. [DOI] [PubMed] [Google Scholar]
- Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, et al. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011;50:81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Gromada J, Franklin I, Wollheim CB. α-Cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev. 2007;28:84–116. doi: 10.1210/er.2006-0007. [DOI] [PubMed] [Google Scholar]
- UKPDS Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- Grzeskowiak R, Amin J, Oetjen E, Knepel W. Insulin responsiveness of the glucagon gene conferred by interactions between proximal promoter and more distal enhancer-like elements involving the paired-domain transcription factor Pax6. J Biol Chem. 2000;275:30037–30045. doi: 10.1074/jbc.M000984200. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes. 2013;62:2164–2172. doi: 10.2337/db13-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. doi: 10.1126/science.1215327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RR, Lincoff AM, Mudaliar S, Rabbia M, Chognot C, Herz M. Effect of the dual peroxisome proliferator-activated receptor-α/γ agonist aleglitazar on risk of cardiovascular disease in patients with type 2 diabetes (SYNCHRONY): a phase II, randomised, dose-ranging study. Lancet. 2009;374:126–135. doi: 10.1016/S0140-6736(09)60870-9. [DOI] [PubMed] [Google Scholar]
- Higgs GA, Moncada S, Vane JR. Eicosanoids in inflammation. Ann Clin Res. 1984;16:287–299. [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Home PD, Pocock SJ, Beck-Nielsen H, Curtis PS, Gomis R, Hanefeld M, et al. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet. 2009;373(9681):2125–2135. doi: 10.1016/S0140-6736(09)60953-3. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20:1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, et al. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. 2002;109:1321–1326. doi: 10.1172/JCI14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SE, Zraika S, Utzschneider KM, Hull RL. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia. 2009;52:1003–1012. doi: 10.1007/s00125-009-1321-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul S, Diamond GA. Is there clear and convincing evidence of cardiovascular risk with rosiglitazone? Clin Pharmacol Ther. 2011;89:773–776. doi: 10.1038/clpt.2011.65. [DOI] [PubMed] [Google Scholar]
- Kawamori D, Kurpad AJ, Hu J, Liew CW, Shih JL, Ford EL, et al. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab. 2009;9:350–361. doi: 10.1016/j.cmet.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437(7062):1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care. 2009;32:1663–1668. doi: 10.2337/dc09-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavery GG, Zielinska AE, Gathercole LL, Hughes B, Semjonous N, Guest P, et al. Lack of significant metabolic abnormalities in mice with liver-specific disruption of 11β-hydroxysteroid dehydrogenase type 1. Endocrinology. 2012;153:3236–3248. doi: 10.1210/en.2012-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar MA. How obesity causes diabetes: not a tall tale. Science. 2005;307:373–375. doi: 10.1126/science.1104342. [DOI] [PubMed] [Google Scholar]
- Liang Y, Osborne MC, Monia BP, Bhanot S, Gaarde WA, Reed C, et al. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes. 2004;53:410–417. doi: 10.2337/diabetes.53.2.410. [DOI] [PubMed] [Google Scholar]
- Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–1188. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- Logie L, Harthill J, Patel K, Bacon S, Hamilton DL, Macrae K, et al. Cellular responses to the metal-binding properties of metformin. Diabetes. 2012;61:1423–1433. doi: 10.2337/db11-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–860. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti P, Bugliani M, Boggi U, Masini M, Marselli L. The pancreatic beta cells in human type 2 diabetes. Adv Exp Med Biol. 2012;771:288–309. doi: 10.1007/978-1-4614-5441-0_22. [DOI] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matschinsky FM. GKAs for diabetes therapy: why no clinically useful drug after two decades of trying? Trends Pharmacol Sci. 2013;34:90–99. doi: 10.1016/j.tips.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM, Zelent B, Doliba NM, Kaestner KH, Vanderkooi JM, Grimsby J, et al. Research and development of glucokinase activators for diabetes therapy: theoretical and practical aspects. Handb Exp Pharmacol. 2011;203:357–401. doi: 10.1007/978-3-642-17214-4_15. [DOI] [PubMed] [Google Scholar]
- Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2560–2566. doi: 10.2337/dc11-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monami M, Luzzi C, Lamanna C, Chiasserini V, Addante F, Desideri CM, et al. Three-year mortality in diabetic patients treated with different combinations of insulin secretagogues and metformin. Diabetes Metab Res Rev. 2006;22:477–482. doi: 10.1002/dmrr.642. [DOI] [PubMed] [Google Scholar]
- Monami M, Balzi D, Lamanna C, Barchielli A, Masotti G, Buiatti E, et al. Are sulphonylureas all the same? A cohort study on cardiovascular and cancer-related mortality. Diabetes Metab Res Rev. 2007;23:479–484. doi: 10.1002/dmrr.736. [DOI] [PubMed] [Google Scholar]
- Morgan SA, Sherlock M, Gathercole LL, Lavery GG, Lenaghan C, Bujalska IJ, et al. 11beta-Hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes. 2009;58:2506–2515. doi: 10.2337/db09-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachnani JS, Bulchandani DG, Nookala A, Herndon B, Molteni A, Pandya P, et al. Biochemical and histological effects of exendin-4 (exenatide) on the rat pancreas. Diabetologia. 2010;53:153–159. doi: 10.1007/s00125-009-1515-4. [DOI] [PubMed] [Google Scholar]
- Nauck MA. A critical analysis of the clinical use of incretin-based therapies: the benefits by far outweigh the potential risks. Diabetes Care. 2013;36:2126–2132. doi: 10.2337/dc12-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Rosiglitazone revisited: an updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch Intern Med. 2010;170:1191–1201. doi: 10.1001/archinternmed.2010.207. [DOI] [PubMed] [Google Scholar]
- Nixon M, Wake DJ, Livingstone DE, Stimson RH, Esteves CL, Seckl JR, et al. Salicylate downregulates 11β-HSD1 expression in adipose tissue in obese mice and in humans, mediating insulin sensitization. Diabetes. 2012;61:790–796. doi: 10.2337/db11-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS ONE. 2012;7:e33411. doi: 10.1371/journal.pone.0033411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339(6116):172–177. doi: 10.1126/science.1230721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oetjen E, Blume R, Cierny I, Schlag C, Kutschenko A, Kratzner R, et al. Inhibition of MafA transcriptional activity and human insulin gene transcription by interleukin-1beta and mitogen-activated protein kinase kinase kinase in pancreatic islet beta cells. Diabetologia. 2007;50:1678–1687. doi: 10.1007/s00125-007-0712-2. [DOI] [PubMed] [Google Scholar]
- Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanne-Chantelot C, Ellard S, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512–1526. doi: 10.1002/humu.21110. [DOI] [PubMed] [Google Scholar]
- Pabreja K, Mohd MA, Koole C, Wootten D, Furness SG. Molecular mechanisms underlying physiological and receptor pleiotropic effects mediated by GLP-1R activation. Br J Pharmacol. 2014;171:1114–1128. doi: 10.1111/bph.12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18 doi: 10.1038/nm.2851. [DOI] [PubMed] [Google Scholar]
- Philippe J. Glucagon gene transcription is negatively regulated by insulin in a hamster islet cell line. J Clin Invest. 1989;84:672–677. doi: 10.1172/JCI114214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomgaard P, Nielsen AR, Fischer CP, Mortensen OH, Broholm C, Penkowa M, et al. Associations between insulin resistance and TNF-alpha in plasma, skeletal muscle and adipose tissue in humans with and without type 2 diabetes. Diabetologia. 2007;50:2562–2571. doi: 10.1007/s00125-007-0834-6. [DOI] [PubMed] [Google Scholar]
- Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1987;64:106–110. doi: 10.1210/jcem-64-1-106. [DOI] [PubMed] [Google Scholar]
- Rees MG, Gloyn AL. Small molecular glucokinase activators: has another new anti-diabetic therapeutic lost favour? Br J Pharmacol. 2013;168:335–338. doi: 10.1111/j.1476-5381.2012.02201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia. 2013;56:1898–1906. doi: 10.1007/s00125-013-2991-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes CJ. Type 2 diabetes – a matter of beta-cell life and death? Science. 2005;307(5708):380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- Rosenson RS, Wright RS, Farkouh M, Plutzky J. Modulating peroxisome proliferator–activated receptors for therapeutic benefit? Biology, clinical experience, and future prospects. Am Heart J. 2012;164:672–680. doi: 10.1016/j.ahj.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salpeter SR, Greyber E, Pasternak GA, Salpeter EE. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus. Cochrane Database Syst Rev. 2010;(4) doi: 10.1002/14651858.CD002967.pub4. CD002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel AR. Insulin resistance in the defense against obesity. Cell Metab. 2012;15:798–804. doi: 10.1016/j.cmet.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–1326. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117:1422–1431. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Chang HY, Richards TM, Weiner JP, Clark JM, Segal JB. Glucagonlike peptide 1-based therapies and risk of hospitalization for acute pancreatitis in type 2 diabetes mellitus: a population-based matched case-control study. JAMA Intern Med. 2013;173:534–539. doi: 10.1001/jamainternmed.2013.2720. [DOI] [PubMed] [Google Scholar]
- Skoglund G, Hussain MA, Holz GG. Glucagon-like peptide 1 stimulates insulin gene promoter activity by protein kinase A-independent activation of the rat insulin I gene cAMP response element. Diabetes. 2000;49:1156–1164. doi: 10.2337/diabetes.49.7.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. Beta-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proc Natl Acad Sci U S A. 2008;105:6614–6619. doi: 10.1073/pnas.0710402105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen H, Brand CL, Neschen S, Holst JJ, Fosgerau K, Nishimura E, et al. Immunoneutralization of endogenous glucagon reduces hepatic glucose output and improves long-term glycemic control in diabetic ob/ob mice. Diabetes. 2006a;55:2843–2848. doi: 10.2337/db06-0222. [DOI] [PubMed] [Google Scholar]
- Sorensen H, Winzell MS, Brand CL, Fosgerau K, Gelling RW, Nishimura E, et al. Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes. 2006b;55:3463–3469. doi: 10.2337/db06-0307. [DOI] [PubMed] [Google Scholar]
- van Staa TP, Patel D, Gallagher AM, de Bruin ML. Glucose-lowering agents and the patterns of risk for cancer: a study with the General Practice Research Database and secondary care data. Diabetologia. 2012;55:654–665. doi: 10.1007/s00125-011-2390-3. [DOI] [PubMed] [Google Scholar]
- Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, et al. TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab. 2011;96:E146–E150. doi: 10.1210/jc.2010-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers DA, Kieffer TJ, Hussain MA, Drucker DJ, Bonner-Weir S, Habener JF, et al. Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein IDX-1 and increase islet size in mouse pancreas. Diabetes. 2000;49:741–748. doi: 10.2337/diabetes.49.5.741. [DOI] [PubMed] [Google Scholar]
- Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321(7258):405–412. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahrani AA, Bailey CJ, Del Prato S, Barnett AH. Management of type 2 diabetes: new and future developments in treatment. Lancet. 2011;378(9786):182–197. doi: 10.1016/S0140-6736(11)60207-9. [DOI] [PubMed] [Google Scholar]
- Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994;372(6502):182–186. doi: 10.1038/372182a0. [DOI] [PubMed] [Google Scholar]
- Thisted H, Johnsen SP, Rungby J. Sulfonylureas and the risk of myocardial infarction. Metabolism. 2006;55(Suppl. 1):S16–S19. doi: 10.1016/j.metabol.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Gallwitz B. Mechanism of action of inhibitors of dipeptidyl-peptidase-4 (DPP-4) Best Pract Res Clin Endocrinol Metab. 2009;23:479–486. doi: 10.1016/j.beem.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- Ueki K, Kondo T, Kahn CR. Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phosphorylation of insulin receptor substrate proteins by discrete mechanisms. Mol Cell Biol. 2004;24:5434–5446. doi: 10.1128/MCB.24.12.5434-5446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH. Glucagon physiology and pathophysiology in the light of new advances. Diabetologia. 1985;28:574–578. doi: 10.1007/BF00281991. [DOI] [PubMed] [Google Scholar]
- Unger RH, Cherrington AD. Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest. 2012;122:4–12. doi: 10.1172/JCI60016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Foretz M. Revisiting the mechanisms of metformin action in the liver. Ann Endocrinol (Paris) 2013;74:123–129. doi: 10.1016/j.ando.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122:253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner C, de Groot JC, Prasad A, Freiwald A, Quedenau C, Kliem M, et al. Amorfrutins are potent antidiabetic dietary natural products. Proc Natl Acad Sci U S A. 2012;109:7257–7262. doi: 10.1073/pnas.1116971109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner C, Wowro SJ, Freiwald A, Kawamoto K, Witzke A, Kliem M, et al. Amorfrutin B is an efficient natural peroxisome proliferator-activated receptor gamma (PPARγ) agonist with potent glucose-lowering properties. Diabetologia. 2013;56:1802–1812. doi: 10.1007/s00125-013-2920-2. [DOI] [PubMed] [Google Scholar]
- Weir GC, Bonner-Weir S. Islet beta cell mass in diabetes and how it relates to function, birth, and death. Ann N Y Acad Sci. 2013;1281:92–105. doi: 10.1111/nyas.12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner ED, Lee J, Hansen L, Yuan M, Shoelson SE. Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J Biol Chem. 2004;279:35298–35305. doi: 10.1074/jbc.M405203200. [DOI] [PubMed] [Google Scholar]
- White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327–1335. doi: 10.1056/NEJMoa1305889. [DOI] [PubMed] [Google Scholar]
- Wilding JPH. PPAR agonists for the treatment of cardiovascular disease in patients with diabetes. Diabetes Obes Metab. 2012;14:973–982. doi: 10.1111/j.1463-1326.2012.01601.x. [DOI] [PubMed] [Google Scholar]
- Winterstein AG. Rosiglitazone and the risk of adverse cardiovascular outcomes. Clin Pharmacol Ther. 2011;89:776–778. doi: 10.1038/clpt.2011.43. [DOI] [PubMed] [Google Scholar]
- Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48:2270–2276. doi: 10.2337/diabetes.48.12.2270. [DOI] [PubMed] [Google Scholar]
- Yashiro H, Tsujihata Y, Takeuchi K, Hazama M, Johnson PR, Rorsman P. The effects of TAK-875, a selective G protein-coupled receptor 40/free fatty acid 1 agonist, on insulin and glucagon secretion in isolated rat and human islets. J Pharmacol Exp Ther. 2012;340:483–489. doi: 10.1124/jpet.111.187708. [DOI] [PubMed] [Google Scholar]
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293(5535):1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Shen Z, Lu Y, Zhong S, Xu C. Increased risk of bladder cancer with pioglitazone therapy in patients with diabetes: a meta-analysis. Diabetes Res Clin Pract. 2012;98:159–163. doi: 10.1016/j.diabres.2012.05.006. [DOI] [PubMed] [Google Scholar]