

Abstract
BACKGROUND AND PURPOSE
Infection with Gram-negative bacteria has been recognized as an initiator of rheumatoid arthritis, which is characterized by chronic inflammation and infiltration of immune cells. Carbon monoxide (CO) exhibits anti-inflammatory properties. Here we have investigated the detailed mechanisms of vascular cell adhesion molecule-1 (VCAM-1) expression induced by LPS and if CO inhibited LPS-induced leukocyte adhesion to synovial fibroblasts by suppressing VCAM-1 expression.
EXPERIMENTAL APPROACH
Human rheumatoid arthritis synovial fibroblasts (RASFs) were incubated with LPS and/or the CO-releasing compound CORM-2. Effects of LPS on VCAM-1 levels were determined by analysing mRNA expression, promoter activity, protein expression, and immunohistochemical staining. The molecular mechanisms were investigated by determining the expression, activation, and binding activity of transcriptional factors using target signal antagonists.
KEY RESULTS
CORM-2 significantly inhibited inflammatory responses in LPS-treated RASFs by down-regulating the expression of adhesion molecule VCAM-1 and leukocyte infiltration. The down-regulation of LPS-induced VCAM-1 expression involved inhibition of the expression of phosphorylated-NF-κB p65 and AP-1 (p-c-Jun, c-Jun and c-Fos mRNA levels). These results were confirmed by chromatin immunoprecipitation assay to detect NF-κB and AP-1 DNA binding activity.
CONCLUSIONS AND IMPLICATIONS
LPS-mediated formation of the TLR4/MyD88/TRAF6/c-Src complex regulated NF-κB and MAPKs/AP-1 activation leading to VCAM-1 expression and leukocyte adhesion. CORM-2, which liberates CO to elicit direct biological activities, attenuated LPS-induced VCAM-1 expression by interfering with NF-κB and AP-1 activation, and significantly reduced LPS-induced immune cell infiltration of the synovium.
Keywords: carbon monoxide, LPS, VCAM-1, leukocyte adhesion
Introduction
Rheumatoid arthritis synovial fibroblasts (RASFs) are important cells in joint erosion and actively contribute to inflammation (Neumann et al., 2010). Bacterial infection has been shown to initiate inflammation in synovial fibroblasts mediated through Toll-like receptors (TLRs) (Rosenstein et al., 2004; Kobayashi et al., 2008; receptor nomenclature follows Alexander et al., 2013). TLR4 is activated by LPS leading to the activation of MAPKs and transcription factors via adaptor molecules and induces the expression of proinflammatory molecules (Kim et al., 2010; Rego et al., 2011). Up-regulation of adhesion molecules, including vascular cell adhesion molecule-1 (VCAM-1) enhanced endothelial adhesion and migration in rheumatoid arthritis (RA) (McMurray, 1996). Moreover, adhesion molecules present on the RASFs surface regulate the trafficking of leukocytes into and/or through the synovial tissues (Carter and Wicks, 2001). Accordingly, blocking the expression of adhesion molecules may exert beneficial effects in RA (Okamoto et al., 2008).
Carbon monoxide (CO) is highly toxic because it binds haemoglobin with a higher affinity than oxygen, thereby forming carboxyhaemoglobin and causing severe respiratory dysfunction. However, an increasing number of publications indicate that administration of exogenous CO has beneficial effects, including the inhibition of LPS-induced production of cytokines both in vivo and in vitro studies, and other cytoprotective and anti-inflammatory effects in acute inflammation (Ryter et al., 2006; Sun et al., 2008b; Lee et al., 2009). CO, as a gas, has often been used to investigate CO as an anti-inflammatory agent (Otterbein et al., 2000; Nakao et al., 2003), although this approach has limitations in terms of knowing the exact amount of CO produced at cellular levels.
CO covalently bound to a transition metal can be carried and released under appropriate conditions and this is the basis for the design of CO-releasing molecules (CORMs) as therapeutic agents aimed at delivering controlled amounts of CO to tissues and organs (Motterlini and Otterbein, 2010). CORMs have potent anti-inflammatory effects, decreasing the production of inflammatory mediators and attenuating levels of NO and TNF-α in macrophages stimulated with LPS (Otterbein et al., 2000; Sawle et al., 2005). The first lipid-soluble metal carbonyl complex, exhibiting CO-releasing properties, to be identified was tricarbonyl dichlororuthenium (II) dimer {[Ru(CO)3Cl2]2}, and is known as CORM-2. CORM-2 is able to transfer CO spontaneously to myoglobin and exerts typical CO-mediated pharmacological effects such as vasodilation and hypotension (Motterlini et al., 2002). Moreover, CORM-2 attenuated the sequestration of leukocytes in renal tissues by interfering with NF-κB activation, ICAM-1 expression and thus suppressing the pro-adhesive phenotype of endothelial cells (Sun et al., 2008a). These studies have shown a wide range of therapeutic benefits of CO, including cytoprotection and restoration of homeostasis in various cell types. However, the precise cellular and molecular targets that involve downstream gene regulation have not been investigated in human RASFs. Therefore, in this study, we used CORM-2 to assess the effects and potential mechanisms of CO released locally, in the modulation of VCAM-1 expression and leukocyte infiltration in LPS-challenged RASFs.
Methods
Cell culture
RASFs were obtained from 30 patients with RA who underwent knee or hip surgery. Informed consent was obtained from all patients, and the experimental protocol was approved by the Institutional Review Board, Chang Gung Memorial Hospital. Cells were isolated and grown as previously described (Luo et al., 2010). More than 95% of the cells were fibroblasts, which were characterized by an immunofluorescence staining using an antibody specific for the fibroblast protein marker vimentin (Luo et al., 2010). Experiments were performed using the cells from passages 3 to 6. Cells were pretreated with CORM-2 (25 μM) or iCORM-2 (25 μM) for 16 h followed by incubation with LPS (100 μg·mL−1). iCORM-2, a compound that does not release CO, served as a negative control.
Animals
All animal care and experimental procedures complied with the Guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care Committee of Chang Gung University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 18 mice were used in this study.
ICR mice (aged 4–6 weeks) were purchased from the National Laboratory Animal Centre (Taipei, Taiwan). Mice were randomly divided into six groups (n = 3 per group) and were intra-articularly injected with PBS (sham), PP1 (0.8 μg·kg−1 of body weight), tanshinone IIA (0.09 μg·kg−1 of body weight), helenalin (0.08 μg·kg−1 of body weight) for 2 h or CORM-2 (8 μg·kg−1 of body weight) for 16 h before treatment with LPS (100 μg·kg−1) and were killed after 24 h (Chi et al., 2011).
Immunohistochemistry
To examine the cellular expression and localization of VCAM-1 protein, immunohistochemical staining was performed on the serial sections of the ankle joints or RA synovial tissues as previously described (Chi et al., 2011). Briefly, the first section was incubated with an anti-VCAM-1 antibody at 37°C for 1 h and then with horseradish peroxidase-conjugated anti-rabbit IgG antibodies at room temperature for 1 h. The second section was incubated with an anti-vimentin antibody for the positive localization and identification of synovial fibroblasts and observed using an optical microscope. Images were obtained with light microscopy at a magnification ×200. Pictures of 3–5 random fields across three replicates were captured for quantification using Image J software (NIH, Bethesda, MD, USA). The brown DAB image was converted to binary image and grey value measured. In general, 50–100 counts per field of vimentin-positive cells (RASFs) were seen in the synovial layer, whereas 75–100 counts per field of VCAM-1-positive cells were seen in the presence of LPS. The quantitative data of immunohistochemical staining were calculated as the ratio (as %) of the number of VCAM-1-positive cells to the number of vimentin-positive cells, in the microscopic field.
Preparation of cell extracts and Western blot analysis
RASFs were incubated with LPS at 37°C for the indicated time intervals. The cells were washed, scraped, collected and centrifuged at 45 000× g at 4°C for 1 h to yield the whole cell extract, as previously described (Wu et al., 2004).
Plasmids and transfection
The shRNAs (p42, p44, JNK1, JNK2 and p38) were kindly provided by Dr. C.P. Tseng (Chang Gung University, Taiwan). For construction of the VCAM-1-luc plasmid, human VCAM-1 promoter, a region spanning −1716 to −119 bp (kindly provided by Dr. W.C. Aird, Beth Israel Deaconess Medical Center, Boston, MA, USA) was cloned into a pGL3-basic vector (Promega, Madison, WI, USA). The AP-1 luciferase reporter construct (AP-1-Luc) and NF-κB luciferase reporter construct (NF-κB-Luc) were purchased from Clontech (San Diego, CA, USA). SMARTpool RNA duplexes corresponding to human haem oxygenase (HO)-1, TLR4, MyD88, TRAF6, TRAF2, c-Src, c-Jun, c-Fos and scrambled #2 siRNAs were from Dharmacon (Lafayette, CO, USA). siRNA (100 nM) was formulated with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the protocol of the manufacturer, as previously described (Chi et al., 2011).
Total RNA extraction and real-time PCR analysis
Total RNA was isolated from RASFs treated with LPS in 10 cm culture dishes with Trizol (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the protocol of the manufacturer. The levels of mRNA expression were determined by real-time PCR, as previously described (Wu et al., 2010). The primers and probes used for real-time PCR of human VCAM-1and GAPDH were obtained from Invitrogen.
Electrophoretic mobility shift assays (EMSA)
DNA-protein binding assays were performed with nuclear extracts of RASFs, treated with LPS (100 μg·mL−1). Synthetic complementary oligonucleotides 3′-biotinylated were from MDBio (Taipei, Taiwan). The sequences of the oligonucleotides used are 5′-TCTTTTGATGGTTCATTTTTTAAAA-3′ and 5′-TGCCCTGGGTTTCCCCTTGAAGGGATTTCCCTCCGCCTCTGCA-3′ for wild-type AP-1 and NF-κB. Binding reactions were carried out for 20 min at room temperature in the presence of 50 ng·μL−1 poly(dI-dC), 0.05% Nonidet P-40, 5 mM MgCl2, 10 mM EDTA and 2.5% glycerol in 1X binding buffer [LightShift™ chemiluminescent EMSA kit, Pierce, Rockford, IL, USA] using 20 fmol of biotin-end-labelled target DNA and 9 μg of nuclear extract. Unlabelled target DNA (4 pmol) was added per 20 μL of binding reaction. To test the effect of antibodies on DNA protein binding, nuclear extracts were pre-incubated with antibodies of c-Fos and c-Jun for 30 min at room temperature. Assays were loaded onto native 4% polyacrylamide gels pre-electrophoresed for 60 min in 0.5X Tris borate/EDTA and electrophoresed at 100 V before being transferred onto a positively charged nylon membrane (Hybond™-N+) for 30 min. Transferred DNAs were cross-linked to the membrane at 120 mJ·cm−2 and detected using a HRP-conjugated streptavidin.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed as previously described (Chi et al., 2011). Soluble chromatin was immunoprecipitated using an anti-p65, anti-c-Jun, anti-c-Fos or anti-IgG antibody. The purified DNA was subjected to PCR amplification using primers specific for the VCAM-1 promoter: 5′-TGGGCTATGTGTGTGCAAG-3′ (sense) and 5′-TTTAAGTTGCTGTCGTGAT- 3′ (antisense). PCR fragments were analysed on 2% agarose in 1x Tris-acetate-EDTA gel containing ethidium bromide.
Isolation of cell fractions
RASFs were seeded in 10 cm dishes. When they reached 90% confluence, the cells were starved for 24 h in serum-free DMEM/F-12 medium. After incubation, the cells were washed once with ice-cold PBS. Homogenization buffer A (200μL; 20 mM Tris–HCl, pH 8.0, 10 mM EGTA, 2 mM EDTA, 2 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 25 μg·mL−1 aprotinin, 10 μg·mL−1 leupeptin) was added to each dish, and the cells were scraped into a 1.5 mL tube. Cells were centrifuged at 5000× g for 15 min at 4°C. The pellet was collected as the nuclear fraction. The supernatant was centrifuged at 15 000× g at 4°C for 60 min to yield the pellet (membrane fraction) and the supernatant (cytosolic fraction).
Immunofluorescence staining
RASFs were grown on 6-well culture plates with coverslips. Confluent cells were shifted to DMEM/F-12 containing 1% FBS for 24 h and incubated with 100 μg·mL−1 LPS. Cells were fixed, permeabilized and stained using an anti-NF-κB (p65) antibody as previously described (Wu et al., 2004).
Measurement of luciferase activity
VCAM-1-Luc, AP-1-Luc or NF-κB-Luc activity was determined as previously described (Wu et al., 2010) using a luciferase assay system (Promega). Firefly, luciferase activities were normalized to that of β-galactosidase activity.
Leukocyte adhesion assay
Leukocyte-RASF adhesion was measured as previously described (Lee et al., 2012). Confluent RASFs in 6-well plates were incubated with BCECF/AM-labelled THP-1 cells (2 × 106 cells per mL) at 37°C for 1 h. The numbers of adherent THP-1 cells were determined by counting four fields per 200X high-power field well using a fluorescence microscope (Axiovert 200M, Zeiss, Jena, Germany).
Data analysis
Data are reported as the mean ± SEM of at least three independent experiments. Comparisons of ≥3 populations were made using a GraphPad Prism Program, and analysed by one-way anova followed by Tukey's post hoc test. P-values less than 0.05 were considered significant.
Materials
Anti-VCAM-1, anti-TLR4, anti-MyD88, anti-TRAF2, anti-TNF receptor-associated factor 6 (TRAF6), anti-c-Src, anti-c-Jun, anti-c-Fos, anti-IκBα and anti-NF-κB (p65) antibodies were from Santa Cruz (Santa Cruz, CA, USA). Anti-GAPDH antibody was from Biogenesis (Boumemouth, UK). PhosphoPlus IKKα/β, IκBα, p42/p44 MAPK, p38 MAPK and JNK1/2 antibodies were from Cell Signaling (Danver, MA, USA). U0126, SB202190, SP600125, curcumin, tanshinone IIA and helenalin were from Biomol (Plymouth Meeting, PA, USA). Tricarbonyldichlororuthenium (II) dimer (CORM-2) and ruthenium (III) chloride (inactive CORM-2) were from Sigma (St. Louis, MO, USA). We obtained ultrapure preparations of LPS from InvivoGen (San Diego, CA, USA).
Results
LPS/TLR4 induces VCAM-1 expression through the MyD88-dependent pathway
LPS has been shown to induce VCAM-1 expression in various cell types (Lin et al., 2007; Fernandez-Pisonero et al., 2012; Lee et al., 2012). Thus, we first investigated whether LPS could induce VCAM-1 expression in RASFs, by measuring VCAM-1 protein in the cell lysates and on plasma membrane of RASFs, by Western blot. As shown in Figure 1A and B, exposure of RASFs to LPS induced the expression of VCAM-1 in both cell lysates and plasma membrane fraction, in a time-dependent manner. In addition, LPS enhanced VCAM-1 mRNA expression and promoter activity in a time-dependent manner with maximal responses within 6 h (Figure 1C). LPS-triggered TLR4 signalling has been shown to activate both MyD88-dependent and MyD88-independent signalling pathways in various cell types (Akira and Takeda, 2004; Sheedy and O'Neill, 2007; Carpenter and O'Neill, 2009), and leading to the expression of inflammatory genes (Funakoshi-Tago et al., 2003; Lee et al., 2008; Lin et al., 2010). Moreover, MyD88 recruits IL-1 receptor-associated kinase and then TRAF6, which activates the prototypic inflammatory transcription factor NF-κB (O'Neill, 2008), which encodes inflammatory genes such as VCAM-1 (Luo et al., 2010). In RASFs, transfection with siRNA of TLR4, MyD88 or TRAF6 down-regulated the expression of TLR4, MyD88 or TRAF6 protein and attenuated LPS-induced VCAM-1 protein and mRNA expression, and promoter activity (Figure 1D and E). In contrast, transfection with TRAF2 siRNA had no effect on LPS-induced responses.
Figure 1.

MyD88 and TRAF6 are necessary for LPS-induced VCAM-1 expression. (A) RASFs were treated with various concentrations of LPS for the indicated time intervals and collected for Western blot analysis of VCAM-1 protein expression. (B) Cells were treated with LPS (100 μg·mL−1) for the indicated time intervals. Membrane fraction was prepared and examined by Western blotting. Gαs was used as a marker protein for the membrane fraction. (C) The levels of mRNA and VCAM-1 expression were analysed by real-time PCR and promoter luciferase activity respectively. (D) RASFs were transfected with TLR4, MyD88, TRAF2 or TRAF6 siRNA, and then treated with 100 μg·mL−1 LPS for (D) 6 h or (E) 4 h. (D) The protein expression of TLR4, MyD88, TRAF2, TRAF6 and VCAM-1 were determined by Western blot. (E) The levels of VCAM-1 mRNA and promoter activity were determined by real-time PCR and promoter activity. All analyses were performed on samples from three RA patients. Results are representative of three independent experiments. Values are the mean ± SEM. In B, #P ≤ 0.01 versus vehicle alone. In D, #P ≤ 0.01 versus LPS alone.
LPS-induced VCAM-1 expression is mediated through the formation of a TLR4/MyD88/TRAF6/c-Src complex
VCAM-1 gene expression is mediated through c-Src kinase in various cell types (Morel et al., 2002; Lin et al., 2008a,b; Lee et al., 2012). As shown in Figure 2A, pretreatment of RASFs with PP1 (c-Src kinase inhibitor) concentration-dependently attenuated LPS-induced VCAM-1 expression. Also, transfection with c-Src siRNA down-regulated c-Src protein expression and attenuated LPS-induced VCAM-1 expression (Figure 2B). As shown in Figure 2C, LPS stimulated c-Src phosphorylation in a time-dependent manner, which was attenuated by PP1.
Figure 2.

LPS enhances VCAM-1 expression via TLR4/MyD88/TRAF6/c-Src. (A) RASFs were pre-incubated with PP1 for 1 h, and then stimulated with 100 μg·mL−1 LPS for 6 h. (B) RASFs were transfected with scrambled (scrb), or c-Src siRNA, and then treated with 100 μg·mL−1 LPS for 6 h. (C) Cells were pretreated with PP1 and then incubated with LPS for the indicated time intervals. (D, E) Cells were incubated with LPS for the indicated time intervals or transfected with scrb, MyD88, TRAF6 or c-Src siRNA and then stimulated with LPS for 5 min. Cell lysates were immunoprecipitated (IP) and determined by immunoblotting (IB) using the indicated antibodies. (F) Cells were transfected with scrb, TLR4, MyD88, TRAF6 or c-Src siRNA and then incubated with LPS for the indicated times or for 3 min. The levels of various proteins were analysed by Western blot using the antibodies indicated. All analyses were performed on samples from three RA patients. Results are representative of three independent experiments. Values are the mean ± SEM. In A, C, F, *P ≤ 0.05; #P ≤ 0.01 versus LPS alone.
We used an in vitro co-immunoprecipitation to investigate if TLR4, MyD88 and TRAF6 were associated with c-Src, following LPS stimulation. As shown in Figure 2D and E (left panel), the protein levels of MyD88, TRAF6 and c-Src were time-dependently increased in a TLR4-immunoprecipitated complex, which were attenuated by transfection with siRNA of MyD88, TRAF6 or c-Src. In addition, transfection with these siRNAs also decreased the protein levels of TLR4, MyD88 or TRAF6 in a c-Src-immunoprecipitated complex (Figure 2E, right panel). To confirm these findings, transfection with siRNA of TLR4, MyD88, TRAF6 or c-Src also attenuated LPS-stimulated c-Src phosphorylation (Figure 2F), indicating that c-Src was downstream of TLR4, MyD88 and TRAF6.
LPS induces VCAM-1 expression via c-Src-dependent MAPKs activation
Some studies have indicated that VCAM-1 expression is regulated by MAPK family members (p42/p44 MAPK, p38 MAPK and JNK1/2; Wu et al., 2004; Luo et al., 2010). In our experiments, we used pharmacological inhibitors selective for different members of the MAPK family: U0126 for MEK1/2, SB202190 for p38 MAPK and SP600125 for JNK1/2. As shown in Figure 3A, LPS-induced VCAM-1 protein expression was concentration-dependently inhibited by pretreatment with U0126, SB202190 or SP600125. We also used transfection with shRNA of p44, p42, p38, JNK1 or JNK2 to down-regulate the expression of their respective proteins and found they subsequently attenuated LPS-induced VCAM-1 expression (Figure 3B). We then showed that treatment of RASFs with LPS stimulated p42/p44 MAPK, p38 MAPK or JNK1/2 phosphorylation in a time-dependent manner, which was attenuated by U0126, SB202190 or SP600125 (Figure 3C). Others have shown that MAPKs activation is mediated through c-Src (Funakoshi-Tago et al., 2003; Cheng et al., 2010). We therefore used transfection with c-Src siRNA to down-regulate the expression of the protein and found this to inhibit the LPS-stimulated phosphorylation of p42/p44 MAPK, p38 MAPK and JNK1/2.
Figure 3.

LPS induces VCAM-1 expression via c-Src-dependent MAPKs activation. (A) RASFs were pretreated with U0126, SB202190 or SP600125 for 1 h and then incubated with LPS for 6 h. (B) Cells were transfected with empty vector (pTOPO-U6) or with the indicated shRNA and then incubated with LPS for 6 h. (C) RASFs were pretreated with U0126 (U0; 10 μM), SB202190 (SB; 10 μM) or SP600125 (SP; 10 μM) and then incubated with LPS for the indicated time intervals. (D) Cells were transfected with an empty vector or c-Src shRNA and then incubated with LPS for the indicated time intervals. The levels of various proteins were analysed by Western blot using the antibodies indicated. All analyses were performed on samples from three RA patients. Results are representative of three independent experiments. Values (A–D) are the mean ± SEM. *P ≤ 0.05; #P ≤ 0.01 versus LPS alone.
Involvement of AP-1 in LPS-induced VCAM-1 expression
The promoter region of VCAM-1 gene contains several potential regulatory elements including that for AP-1 (Iademarco et al., 1992; Sawa et al., 2008). As shown in Figure 4A, pretreatment of RASFs with the inhibitors of AP-1, curcumin or tanshinone IIA, concentration-dependently inhibited LPS-induced VCAM-1 expression. Within the AP-1 subfamily, c-Jun and c-Fos are important transcriptional activators, acting on VCAM-1 promoter AP-1 motifs (Lin et al., 2009). We used siRNAs for c-Jun or for c-Fos and found them to reduce expression of the respective proteins and to inhibit LPS-induced VCAM-1 expression (Figure 4B). Moreover, c-Jun and c-Fos are transcriptionally regulated by various stimuli (Reddy and Mossman, 2002; Lin et al., 2009). In our experiments, LPS stimulated c-Jun and c-Fos mRNA expression in a time-dependent manner (Figure 4C), with a maximal response within 30 min. Up-regulation of c-Fos or c-Jun protein is mediated through activation of p42/p44 MAPK, p38 MAPK and JNK1/2 (Lin et al., 2009). In human RASFs, LPS induced both c-Fos and c-Jun mRNA expression which were attenuated by pretreatment with U0126, SP600125 or PP1. However, pretreatment with SB202190 attenuated LPS-induced c-Jun mRNA, but not c-Fos mRNA expression, revealing the different regulatory roles of p38 MAPK in c-Fos and c-Jun expression.
Figure 4.

LPS induces VCAM-1 expression via c-Src/MAPKs/AP-1. (A) RASFs were pretreated with tanshinone IIA or curcumin for 1 h and then incubated with LPS for 6 h. (B) Cells were transfected with an empty vector (scrb), c-Jun or c-Fos siRNA and then incubated with LPS for 6 h. The levels of VCAM-1 expression were analysed by Western blot. (C) Cells were treated with LPS (100 μg·mL−1) for the indicated time intervals. (D) RASFs were pretreated with the inhibitors and then incubated with LPS for 30 min. The levels of c-Jun and c-Fos mRNA were determined by real-time PCR. (E) Cells were transfected with AP-1 reporter gene together with a β-galactosidase plasmid, pretreated with the inhibitors for 1 h, and then incubated with LPS for 1 h. AP-1 promoter activity was determined. (F) Nuclear extracts were prepared from RASFs pretreated with U0126 (U0; 10 μM), SB202190 (SB; 10 μM), SP600125 (SP; 10 μM) or curcumin (Cur; 1 μM) and then incubated with LPS for 1 h. The complexes were subjected to EMSA followed by ECL detection. All analyses were performed on samples from three RA patients. Results are representative of three independent experiments. In A, C, *P ≤ 0.05; #P ≤ 0.01 versus vehicle alone. Values in B, D and E are the mean ± SEM. *P ≤ 0.05; #P ≤ 0.01 versus LPS alone.
To investigate whether up-regulation of c-Jun and c-Fos mRNA in response to LPS was related to the transcriptional activity of AP-1, RASFs were transfected with AP-1-luci plasmids. As shown in Figure 4E, LPS-stimulated AP-1 luciferase activity was inhibited by pretreatment with PP1, U0126, SB202190 or SP600125. To confirm the involvement of the MAPKs in the activation of AP-1 by LPS, DNA binding activity of AP-1 was determined, by EMSA, in nuclear extracts of RASFs. As shown in Figure 4F, LPS enhanced the DNA binding activity of AP-1, which was inhibited by pretreatment with U0126, SB202190, SP600125 or curcumin.
LPS-induced VCAM-1 expression via c-Src-dependent NIK/IKK/NF-κB activation
In addition to AP-1, the promoter region of VCAM-1 contains a NF-κB binding site that is regulated by several external stimuli in different cell types (Iademarco et al., 1992; Collins et al., 1995; Basta et al., 2005). To investigate the involvement of NF-κB activation in VCAM-1 expression by LPS, we used helenalin, a sesquiterpene lactone known to be a specific inhibitor of NF-κB (Lyss et al., 1998) and showed that helenalin concentration-dependently attenuated LPS-induced VCAM-1 expression in RASFs (Figure 5A). The NF-κΒ-inducing kinase (NIK) directly interacts with IKKα and IKKβ, and the phosphorylation of IκBα by IKKα and IKKβ, is enhanced by NIK co-expression (Shambharkar et al., 2007). To confirm the involvement of these components (Figure 5B), we transfected RASFs with the dominant negative mutants of MyD88, NIK, IKKα or IKKβ and found them to attenuate LPS-induced VCAM-1 expression. Cells activated by cytokines lead to the degradation of IκBα, accompanied by NF-κB translocation into the nucleus. We therefore stimulated RASFs with LPS and measured IκBα degradation and NF-κB translocation in the cytosolic and nuclear fractions respectively. As shown in Figure 5C, the levels of p-IκBα were increased and those of IκBα were decreased in the cytosolic fraction and, in the nuclear fraction, the p65 subunit of NF-κB was increased in a time-dependent manner, indicating that p65 subunit was translocated from the cytoplasm into the nucleus. Further, as shown in Figure 5D, LPS-stimulated phosphorylation of IκBα and translocation of NF-κB was inhibited by helenalin and PP1, but not by U0126, SB202190 and SP600125. Moreover, immunofluorescence staining demonstrated that pretreatment with helenalin and PP1, but not U0126, SB202190 or SP600125, also attenuated LPS-induced NF-κB (p65) translocation (Figure 5E). Using EMSA, we found that the DNA binding activity of NF-κB was enhanced by LPS and inhibited by helenalin but not U0126, SB202190 and SP600125 (Figure 5F). The LPS-stimulated NF-κB promoter activity was inhibited by pretreatment with PP1 and helenalin, but not U0126, SB202190 and SP600125 (Figure 5G and H). On the other hand, transfection with c-Src siRNA significantly reduced c-Src protein expression and LPS-induced IKKα/β phosphorylation in RASFs (Figure 5I).
Figure 5.

LPS induces VCAM-1 expression via c-Src/NF-κB. (A) RASFs were pretreated with helenalin (HLN) and then incubated with LPS for 6 h. (B) Cells were transfected with a dominant negative mutant of MyD88, NIK, IKKα or IKKβ, and then treated with 100 μg·mL−1 LPS for 6 h. The protein expression of VCAM-1 was determined. (C) Cells were treated with LPS (100 μg·mL−1) for the indicated time intervals. (D) Cells were pretreated with the indicated inhibitors and then incubated with LPS for 1 h. Cytosol or nuclear extracts were examined by Western blotting. Lamin A and GAPDH were used as marker proteins for nuclear and cytosolic fractions respectively. (E) NF-κB nuclear translocation was identified by immunofluorescence staining. (F) RASFs were pretreated with the indicated inhibitors and then incubated with LPS for 1 h. Nuclear extracts were subjected to EMSA. (G) Cells were transfected with NF-κB reporter gene, treated with LPS (100 μg·mL−1) for the indicated time intervals. (H) Cells were pretreated with the inhibitors for 1 h, and then incubated with LPS for 1 h. NF-κB promoter activity was determined. (I) Cells were transfected with c-Src siRNA, treated with LPS (100 μg·mL−1) for the indicated time intervals. The phosphorylation of IKKα/β were determined by Western blot. All analyses were performed on samples from three RA patients. Results are representative of three independent experiments. In G, *P ≤ 0.05; #P ≤ 0.01 versus vehicle alone. Values in A and H are the mean ± SEM. *P ≤ 0.05; #P ≤ 0.01 versus LPS alone.
c-Src, MAPKs-dependent AP-1 and NF-κB are vital to LPS-induced VCAM-1 promoter activity, mRNA up-regulation and leukocyte adhesion
We further examined whether c-Src, MAPKs, AP-1 and NF-κB were involved in VCAM-1 expression occurring at the transcriptional level in these cells. The up-regulation of VCAM-1 gene transcription was confirmed by gene promoter luciferase activity assay and real-time PCR. As shown in Figure 6A, LPS-stimulated VCAM-1 promoter activity and mRNA expression was significantly attenuated by pretreatment with the inhibitor of c-Src (PP1), MEK1/2 (U0126), p38 (SB202190), JNK1/2 (SP600125), NF-κB (helenalin) or AP-1 (tanshinone IIA). Furthermore, as shown in Figure 6B, the increased number of leukocytes adhering to RASFs treated with LPS was attenuated by pretreatment with U0126, SB202190, SP600125, PP1, helenalin or tanshinone IIA. Pretreatment with U0126, SB202190, SP600125, helenalin or tanshinone IIA also reduced expression of VCAM-1 in the plasma membrane fraction (Figure 6C).
Figure 6.

Involvement of MAPKs, AP-1 and NF-κB in VCAM-1 promoter activity, mRNA expression, and leukocyte adhesion stimulated by LPS. (A) RASFs were transfected without or with VCAM-1 reporter gene, pretreated with 1 μM U0126 (U0), 10 μM SB202190 (SB), 1 μM SP600125 (SP), 1 μM helenalin (HLN) or 1 μM tanshinone IIA (TSIIA) for 1 h, and then incubated with LPS for 6 h (promoter) or 4 h (mRNA). Promoter activity and mRNA were determined. (B) Cells were pretreated with the indicated inhibitors for 1 h and then incubated with LPS for 8 h prior to addition of leukocytes. The leukocyte adherence was measured. Data are expressed as the mean and SEM of three separate experiments. In A and B, *P < 0.05; #P < 0.01, versus cells incubated with LPS alone. (C) Cells were pretreated with the indicated inhibitors for 1 h and then incubated with LPS for 16 h. Membrane fraction was prepared and analysed by Western blotting. (D) Immunohistochemical staining for VCAM-1 and vimentin in serial sections of ankle joints from PBS-treated (sham; panels a–c), LPS-injected (panels d–f), PP1-pretreated (panels g–i), tanshinone IIA-pretreated (panels j–l), and helenalin-pretreated mice (panels m–o) are shown. Arrowheads indicate positive staining. The quantities of immunohistochemical results are representative of three mice per experimental group.
To confirm these results in vivo, mice were injected intra-articularly with PP1, tanshinone IIA or helenalin and, 2 h later, injected with LPS. The images of immunostaining in the articular joints, taken 24 h after LPS, showed that the number of VCAM-1-expressing cells was higher in LPS-treated mice than in PBS-treated mice (Figure 6D, panels a, d). Moreover, LPS-induced expression of VCAM-1 on the synovial layer of the articular joints was significantly attenuated by treatment with PP1, tanshinone IIA or helenalin (Figure 6D, panels d, g, j, m).
CORM-2 inhibits leukocyte adhesion to RASFs challenged with LPS via suppressing AP-1 and NF-κB activation
Administration of exogenous CO inhibits LPS-induced production of cytokines and consequently exerts anti-inflammatory effects (Lee and Chau, 2002). We therefore pretreated RASFs with CORM-2 or inactive CORM-2 (iCORM-2, which does not liberate CO) before exposure to LPS. As shown in Figure 7A and B, LPS-induced VCAM-1 protein and mRNA expression, and promoter activity was attenuated by pretreatment with CORM-2, but not iCORM-2. CORM-2, but not iCORM-2, also decreased adhesion of leukocytes to RASFs (Figure 7C). Treatment with CORM-2 suppressed the increased NF-κB and AP-1 promoter activities, following LPS (Figure#x2009;7D). As a confirmation, chromatin was immunoprecipitated using an anti-c-Fos, anti-c-Jun or anti-p65 antibody, and the VCAM-1 promoter region was amplified by PCR. Pretreatment with CORM-2, but not iCORM-2 (Figure 7E), inhibited the LPS-induced c-Jun, c-Fos and p65 binding to the VCAM-1 promoter. The transcriptional activity of NF-κB is enhanced upon phosphorylation of its p65 subunit on serine residues (Zhong et al., 1998). In RASFs, Figure 7F, the LPS-stimulated phosphorylation of p65 and c-Jun was attenuated by CORM-2 but not iCORM-2. As shown in Figure 7G, pretreatment with CORM-2 also attenuated LPS-induced expression of mRNA for both c-Jun and c-Fos, in RASFs.
Figure 7.

Inhibition of AP-1 and NF-κB prevents LPS-induced leukocyte adhesion by CORM-2. (A–G) Cells were pretreated with CORM-2 or iCORM-2 for 16 h and then incubated with LPS for additional 6 h (A), 4 h (B), 8 h (C), 1 h (D, E) or 30 min (G). The levels of VCAM-1 protein, mRNA and promoter (A, B), the leukocyte adherence (C), the AP-1 and NF-κB promoter activities (D), ChIP assays (E), the levels of phospho-p65 and phospho-c-Jun (F), and the expression of mRNA for c-Jun and c-Fos (G) were determined. All analyses were performed on samples from three RA patients. Results are representative of three independent experiments. Values are the mean ± SEM. *P ≤ 0.05; #P ≤ 0.01 versus LPS alone. (H) Immunohistochemical staining for VCAM-1 or vimentin in sections of synovial tissues from RA patients or ankle joints from mice injected with the indicated inhibitors. Arrowheads indicate positive staining. The summary immunohistochemical data are from three mice per experimental group.
To confirm these results in vivo, mice were intra-articularly injected with CORM-2 for 16 h, and then with LPS. 24 h later, the articular joints were stained immunohistochemically. The increase in VCAM-1-expressing cells after LPS (Figure 7H, panels a, b) was attenuated by CORM-2 (Figure 7H, panels b, c). The levels of VCAM-1 expression normalized to vimentin are summarized in the bar graph (Figure 7H, left panel).
Discussion and conclusions
Infection of bacteria has been recognized to initiate the pathology of RA, which is characterized with chronic inflammation and immune cell infiltration (Higashiyama et al., 1995; Tak et al., 1995). LPS, released from Gram-negative bacteria, may promote the expression of adhesion molecules and thus contribute to the adhesiveness between RASFs and immune cells. VCAM-1 deficiency affords substantial protection against tissue inflammation in mice with collagen-induced arthritis (Carter et al., 2002). Endogenous CO is derived from HO-catalysed metabolism and plays an important role in the modulation of pathological processes (Ryter et al., 2002). For instance, CO suppressed LPS-induced production of proinflammatory cytokines in vascular endothelial cells (Otterbein et al., 2000). An effective and convenient means of generating CO in situ is to use CORMs, compounds containing a heavy metal such as ruthenium, cobalt or iron surrounded by carbonyl groups as coordinated ligands (Motterlini, 2007). For each mole of CORM-2, approximately 0.7 mole of CO is liberated and the effects of these CORMs are similar to those of endogenous, HO-1-derived, CO, exerting a variety of biological activities, including anti-inflammatory effects. To explore the potential mechanisms responsible for CORM-2 effects, we assessed the interference of different signal transduction pathways in LPS-induced VCAM-1 expression. Our results showed that LPS-induced VCAM-1 expression is mediated through TLR4/MyD88/TRAF6/c-Src-dependent NF-κB and MAPKs associated with AP-1 activation in RASFs.
TLR4 is a pattern recognition receptor that functions as a LPS sensor and whose activation results in the production of several proinflammatory, antiviral and antibacterial cytokines (Kim et al., 2010; Rego et al., 2011). In addition, two signalling pathways have been described following TLR4 activation, the MyD88-dependent and -independent pathways and, in this study, LPS induced VCAM-1 expression in RASFs via a MyD88-dependent pathway. Verstak et al. (2009) indicated that MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR4-mediated NF-κB proinflammatory responses. In RASFs, we established that LPS enhanced VCAM-1 expression via a TLR4/MyD88/TRAF6 pathway. c-Src, a protein frequently participating in the cross-talk between the cytoplasmic protein TKs and receptors, mediated LPS signalling in human renal mesangial cells (Lee et al., 2012). Here, we demonstrated that LPS not only induced c-Src activation but also induced formation of the TLR4/MyD88/TRAF6/c-Src complex in RASFs. In mammals, MAPKs consist of more than a dozen MAPK enzymes that coordinately regulate cell proliferation, differentiation, motility and survival. The best known are the conventional MAPKs, including p42/p44 MAPK, JNK1/2 and p38 MAPK. MAPKs were crucial in the modulation of VCAM-1 expression by LPS, in human tracheal smooth muscle cells (Lin et al., 2007) and, here, we also found that LPS induced inflammatory responses in RASFs via a c-Src-dependent MAPKs pathway.
Many biological effects attributed to CO have been linked to its ability to modulate MAPKs signalling pathways. In rat primary hepatocytes, CO blocked glucose deprivation-induced hepatocyte cytotoxicity by suppressing ERK1/2 activation, but not p38 MAPK (Choi et al., 2003) and against apoptosis initiated by oxidative stress, via inhibition of JNK1/2 activity (Conde de la Rosa et al., 2008). Effects of CO gas or CO-RMs on MAPKs activation are variable and depend on the cell types. In airway smooth muscle cells and T-cells, CO exerts its antiproliferative effects via inhibition of ERK1/2 phosphorylation, whereas in pancreatic stellate cells, CO acts through activation of the p38 MAPK pathway (Schwer et al., 2010). In chondrocytes from patients with osteoarthritis (OA), IL-1β-induced ERK1/2 and JNK1/2 phosphorylation was strongly reduced by CORM-2 at 100 and 200 μM (Garcia-Arnandis et al., 2011). In contrast, we observed that LPS-stimulated phosphorylation of c-Src and MAPKs was only slightly inhibited by CORM-2 at 2 μM (Supporting Information Fig. S1). This difference may be due to the different concentrations used. Moreover, we found that treatment of RASFs with 100 μM CORM-2 for 24 h caused an approximate 30–40% loss in cell viability (data not shown). In addition, it has been reported that overexpression of HO-1 is responsible for the catabolism of haem to produce CO, which inhibits the activation of c-Src in gastric epithelial cells infected with Helicobacter pylori (Gobert et al., 2013). In contrast, treatment of RASFs with CORM-2 had no effect on LPS-stimulated c-Src phosphorylation (Supporting Information Fig. S1). Hence, our data indicated that the c-Src-dependent MAPKs pathway may be not a target of the anti-inflammatory effects of CORM-2, which is involved in LPS-induced VCAM-1 expression in RASFs. Instead, these anti-inflammatory effects of CO might target the transcriptional control of VCAM-1 expression through the suppression of the activity of transcription factors, in particular AP-1 and NF-κB.
We demonstrated that LPS induced VCAM-1 expression via a MAPKs-dependent activation of AP-1 transcriptional activity. CO participates in HO-1 inhibition of TPA-induced tumour invasion of breast carcinoma cells via suppressing ERK/c-Jun/AP-1 activation, and CO interaction with the MMP-9 protein may contribute to its inhibition of MMP-9 enzyme activity in vitro (Lin et al., 2008a,b). In RAW264.7 macrophages, CO reduced IL-6 expression through interference with LPS-induced JNK1/2 activation and impairment of AP-1 signalling (Morse et al., 2003). Although, in OA synoviocytes, CORM-2 reduced production of MMP-1 and MMP-3 through reduced binding of AP-1 to DNA, the underlying mechanism was still unclear (Garcia-Arnandis et al., 2011). Increased AP-1 activity in response to stimuli is due to increased c-Jun and c-Fos expression and may also be due to enhanced c-Jun phosphorylation. Thus, one possible mechanism is modulation of c-Jun and c-Fos expression by CORM-2 which leads to inhibition of AP-1 activation. In this study, LPS-induced mRNA levels of c-Jun and c-Fos, c-Jun phosphorylation or AP-1 DNA binding ability and promoter activity were all blocked by CORM-2, suggesting that suppression of AP-1 is probably involved in CORM-2-mediated down-regulation of VCAM-1 expression by LPS.
We established that LPS stimulated NF-κB activation leading to VCAM-1 expression via a MAPK-independent cascade. Inhibition of NF-κB activation by CORM-2-released CO was associated with the reduced expression of ICAM-1 protein in LPS-stimulated HUVECs (Sun et al., 2008b). In contrast, CO has no effect on NF-κB in activated endothelial cells (Soares et al., 2004) and the protection against hepatic ischaemia-reperfusion injury by CO administration was not associated with inhibition of NF-κB signalling (Kaizu et al., 2005). In our study, CORM-2 suppresses the activity of NF-κB induced by LPS in RASFs. Furthermore, we identified the kinases relevant for p65 phosphorylation that was also inhibited by CORM-2 treatment. Similarly, inhibition of NF-κB activation by CORM-2 was dependent on the inhibition of p65 phosphorylation in cytokine-stimulated Caco-2 cells (Megias et al., 2007). We further confirmed that the down-regulation of adhesion molecules by CORM-2 was associated with decreased phosphorylation of the NF-κB-p65 subunit, which results in the prevention of NF-κB DNA binding to VCAM-1 promoter region. In contrast, CO released by CORM-3 induced rapid activation of NF-κB and Nrf2 in the myocardium (Stein et al., 2012). To our knowledge, CO immediately induces accumulation of Nrf2 in the nucleus, but phosphorylation of the p65 subunit is delayed, leading to the up-regulation of cytoprotective genes, such as HO-1. Further work would be necessary to identify transcription factors involved in the expression of cytoprotective genes that may be induced by CORM-2 treatment. Our present data indicate that CORM-2 alone has no effect on the phosphorylation of p65 and c-Jun, promoter activity of NF-κB and AP-1 in RASFs (Figure 7D and F).
In summary, as shown in Figure 8, CORM-2 effectively inhibited activation of NF-κΒ and AP-1, which was accompanied by a decrease of the expression of VCAM-1. In parallel, leukocyte adhesion to RASFs challenged with LPS was markedly decreased by CORM-2 administration. The results support the hypothesis that decreased expression or function of VCAM-1, induced by CORMs, could be of therapeutic value in RA.
Figure 8.
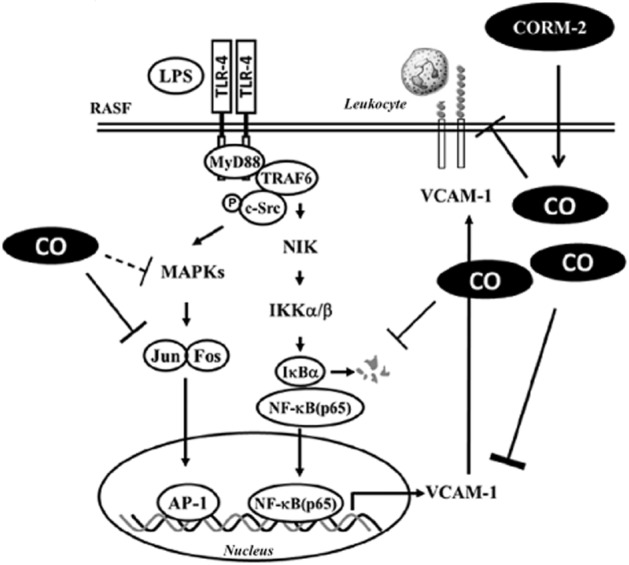
Schematic diagram illustrating the possible mechanisms responsible for CORM-2 effects on LPS-induced VCAM-1 expression in RASFs. LPS activates a TLR4/MyD88/TRAF6/c-Src pathway leading to enhance two distinct signalling cascades, including NIK/IKKα/β-dependent activation of NF-κB and MAPKs-dependent activation of AP-1 transcription factors in RASFs. CORM-released CO decreased inflammatory responses by interfering with activation of NF-κΒ and AP-1, and expression of VCAM-1 protein, thereby suppressing leukoyte adhesion to RASFs.
Acknowledgments
The authors appreciate Dr. C. P. Tseng (Department of Medical Biotechnology and Laboratory Science, Chang Gung University, Taiwan) for providing shRNAs. This work was supported by NSC101-2321-B-182-011, NSC101-2320-B-182-039-MY3 and NSC101-2314-B-182-182A-112 from National Science Council, Taiwan; EMRPD1C0261 and EMRPD1C0271 from Ministry of Education, Taiwan; and CMRPD1C0102, CMRPD1B0383 and CMRPG3B1091 from Chang Gung Medical Research Foundation, Taiwan.
Glossary
- ChIP
chromatin immunoprecipitation
- CORM
carbon monoxide-releasing molecule
- EMSA
electrophoretic mobility shift assay
- HO
haem oxygenase
- OA
osteoarthritis
- RA
rheumatoid arthritis
- RASFs
rheumatoid arthritis synovial fibroblast
- TLR
Toll-like receptor
- TRAF6
TNF receptor-associated factor 6
- VCAM-1
vascular cell adhesion molecule-1
Conflict of interests
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12680
Figure S1 Effects of CORM-2 on LPS-induced phosphorylation of Src and MAPKs. Cells were pretreated with CORM-2 for 16 h and then incubated with LPS for the indicated time intervals. The levels of phospho-Src, phospho-p38 MAPK, phospho-p44/42 MAPKand phospho-JNK1/2 were determined.
References
- Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basta G, Lazzerini G, Del TS, Ratto GM, Schmidt AM, De CR. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arterioscler Thromb Vasc Biol. 2005;25:1401–1407. doi: 10.1161/01.ATV.0000167522.48370.5e. [DOI] [PubMed] [Google Scholar]
- Carpenter S, O'Neill LA. Recent insights into the structure of Toll-like receptors and post-translational modifications of their associated signaling proteins. Biochem J. 2009;422:1–10. doi: 10.1042/BJ20090616. [DOI] [PubMed] [Google Scholar]
- Carter RA, Wicks IP. Vascular cell adhesion molecule 1 (CD106): a multifaceted regulator of joint inflammation. Arthritis Rheum. 2001;44:985–994. doi: 10.1002/1529-0131(200105)44:5<985::AID-ANR176>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Carter RA, Campbell KI, O'Donnel KL, Wicks IP. Vascular cell adhesion molecule-1 (VCAM-1) blockade in collagen-induced arthritis reduces joint involvement and alters B cell trafficking. Clin Exp Immunol. 2002;128:44–51. doi: 10.1046/j.1365-2249.2002.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SE, Lee IT, Lin CC, Kou YR, Yang CM. Cigarette smoke particle-phase extract induces HO-1 expression in human tracheal smooth muscle cells: role of the c-Src/NADPH oxidase/MAPK/Nrf2 signaling pathway. Free Radic Biol Med. 2010;48:1410–1422. doi: 10.1016/j.freeradbiomed.2010.02.026. [DOI] [PubMed] [Google Scholar]
- Chi PL, Luo SF, Hsieh HL, Lee IT, Hsiao LD, Chen YL, et al. cPLA2 induction by IL-1β via the MyD88-dependent and cooperation of p300, Akt, and NF-κB pathway in human rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2011;63:2905–2917. doi: 10.1002/art.30504. [DOI] [PubMed] [Google Scholar]
- Choi BM, Pae HO, Kim YM, Chung HT. Nitric oxide-mediated cytoprotection of hepatocytes from glucose deprivation-induced cytotoxicity: involvement of heme oxygenase-1. Hepatology. 2003;37:810–823. doi: 10.1053/jhep.2003.50114. [DOI] [PubMed] [Google Scholar]
- Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-κB and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- Conde de la Rosa L, Vrenken TE, Hannivoort RA, Buist-Homana M, Havingaa R, Slebosb DJ, et al. Carbon monoxide blocks oxidative stress-induced hepatocyte apoptosis via inhibition of the p54 JNK isoform. Free Radic Biol Med. 2008;44:1323–1333. doi: 10.1016/j.freeradbiomed.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Fernandez-Pisonero I, Duenas AI, Barreiro O, Montero O, Sanchez-Madrid F, Garcia-Rodriguez G. Lipopolysaccharide and sphingosine-1-phosphate cooperate to induce inflammatory molecules and leukocyte adhesion in endothelial cells. J Immunol. 2012;189:5402–5410. doi: 10.4049/jimmunol.1201309. [DOI] [PubMed] [Google Scholar]
- Funakoshi-Tago M, Tago K, Sonoda Y, Tominaga S, Kasahara T. TRAF6 and C-SRC induce synergistic AP-1 activation via PI3-kinase-AKT-JNK pathway. Eur J Biochem. 2003;270:1257–1268. doi: 10.1046/j.1432-1033.2003.03487.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Arnandis I, Guillen MI, Gomar F, Castejon MA, Alcaraz MJ. Control of cell migration and inflammatory mediators production by CORM-2 in osteoarthritic synoviocytes. PLoS ONE. 2011;6:e24591. doi: 10.1371/journal.pone.0024591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert AP, Verriere T, de Sablet T, Peek RM, Jr, Chaturvedi R, Wilson KT. Haem oxygenase-1 inhibits phosphorylation of the Helicobacter pylori oncoprotein CagA in gastric epithelial cells. Cell Microbiol. 2013;15:145–156. doi: 10.1111/cmi.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashiyama H, Saito I, Hayashi Y, Miyasaka N. In situ hybridization study of vascular cell adhesion molecule-1 messenger RNA expression in rheumatoid synovium. J Autoimmun. 1995;8:947–957. doi: 10.1016/s0896-8411(95)80028-x. [DOI] [PubMed] [Google Scholar]
- Iademarco MF, McQuillan JJ, Rosen GD, Dean DC. Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1) J Biol Chem. 1992;267:16323–16329. [PubMed] [Google Scholar]
- Kaizu T, Nakao A, Tsung A, Toyokawa H, Sahai R, Geller DA, et al. Carbon monoxide inhalation ameliorates cold ischemia/reperfusion injury after rat liver transplantation. Surgery. 2005;138:229–235. doi: 10.1016/j.surg.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Jung Y, Shin BS, Kim H, Song H, Bae SH, et al. Redox regulation of lipopolysaccharide-mediated sulfiredoxin induction, which depends on both AP-1 and Nrf2. J Biol Chem. 2010;285:34419–34428. doi: 10.1074/jbc.M110.126839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Momohara S, Kamatani N, Okamoto H. Molecular aspects of rheumatoid arthritis: role of environmental factors. FEBS J. 2008;275:4456–4462. doi: 10.1111/j.1742-4658.2008.06581.x. [DOI] [PubMed] [Google Scholar]
- Lee IT, Wang SW, Lee CW, Chang CC, Lin CC, Luo SF, et al. Lipoteichoic acid induces HO-1 expression via the TLR2/MyD88/c-Src/NADPH oxidase pathway and Nrf2 in human tracheal smooth muscle cells. J Immunol. 2008;181:5098–5110. doi: 10.4049/jimmunol.181.7.5098. [DOI] [PubMed] [Google Scholar]
- Lee IT, Luo SF, Lee CW, Wang SW, Lin CC, Chang CC, et al. Overexpression of HO-1 protects against TNF-α-mediated airway inflammation by down-regulation of TNFR1-dependent oxidative stress. Am J Pathol. 2009;175:519–532.s. doi: 10.2353/ajpath.2009.090016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IT, Shih RH, Lin CC, Chen JT, Yang CM. Role of TLR4/NADPH oxidase/ROS-activated p38 MAPK in VCAM-1 expression induced by lipopolysaccharide in human renal mesangial cells. Cell Commun Signal. 2012;10:33. doi: 10.1186/1478-811X-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- Lin CC, Lee IT, Yang YL, Lee CW, Kou YR, Yang CM. Induction of COX-2/PGE(2)/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: role of TLR4-dependent NADPH oxidase activation. Free Radic Biol Med. 2010;48:240–254. doi: 10.1016/j.freeradbiomed.2009.10.047. [DOI] [PubMed] [Google Scholar]
- Lin CW, Shen SC, Hou WC, Yang LY, Chen YC. Heme oxygenase-1 inhibits breast cancer invasion via suppressing the expression of matrix metalloproteinase-9. Mol Cancer Ther. 2008a;7:1195–1206. doi: 10.1158/1535-7163.MCT-07-2199. [DOI] [PubMed] [Google Scholar]
- Lin WN, Luo SF, Lee CW, Wang CC, Wang JS, Yang CM. Involvement of MAPKs and NF-κB in LPS-induced VCAM-1 expression in human tracheal smooth muscle cells. Cell Signal. 2007;19:1258–1267. doi: 10.1016/j.cellsig.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Lin WN, Luo SF, Wu CB, Lin CC, Yang CM. Lipopolysaccharide induces VCAM-1 expression and neutrophil adhesion to human tracheal smooth muscle cells: involvement of Src/EGFR/PI3-K/Akt pathway. Toxicol Appl Pharmacol. 2008b;228:256–268. doi: 10.1016/j.taap.2007.11.026. [DOI] [PubMed] [Google Scholar]
- Lin WN, Luo SF, Lin CC, Hsiao LD, Yang CM. Differential involvement of PKC-dependent MAPKs activation in lipopolysaccharide-induced AP-1 expression in human tracheal smooth muscle cells. Cell Signal. 2009;21:1385–1395. doi: 10.1016/j.cellsig.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Luo SF, Fang RY, Hsieh HL, Chi PL, Lin CC, Hsiao LD, et al. Involvement of MAPKs and NF-κB in tumor necrosis factor alpha-induced vascular cell adhesion molecule 1 expression in human rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2010;62:105–116. doi: 10.1002/art.25060. [DOI] [PubMed] [Google Scholar]
- Lyss G, Knorre A, Schmidt TJ, Pahl HL, Merfort I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-κB by directly targeting p65. J Biol Chem. 1998;273:33508–33516. doi: 10.1074/jbc.273.50.33508. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray RW. Adhesion molecules in autoimmune disease. Semin Arthritis Rheum. 1996;25:215–233. doi: 10.1016/s0049-0172(96)80034-5. [DOI] [PubMed] [Google Scholar]
- Megias J, Busserolles J, Alcaraz MJ. The carbon monoxide-releasing molecule CORM-2 inhibits the inflammatory response induced by cytokines in Caco-2 cells. Br J Pharmacol. 2007;150:977–986. doi: 10.1038/sj.bjp.0707184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel JC, Park CC, Zhu K, Kumar P, Ruth JH, Koch AE. Signal transduction pathways involved in rheumatoid arthritis synovial fibroblast interleukin-18-induced vascular cell adhesion molecule-1 expression. J Biol Chem. 2002;277:34679–34691. doi: 10.1074/jbc.M206337200. [DOI] [PubMed] [Google Scholar]
- Morse D, Pischke SE, Zhou Z, Davis RJ, Flavell RA, Loop T, et al. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J Biol Chem. 2003;278:36993–36998. doi: 10.1074/jbc.M302942200. [DOI] [PubMed] [Google Scholar]
- Motterlini R. Carbon monoxide-releasing molecules (CO-RMs): vasodilatory, anti-ischaemic and anti-inflammatory activities. Biochem Soc Trans. 2007;35:1142–1146. doi: 10.1042/BST0351142. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9:728–743. doi: 10.1038/nrd3228. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Clark JE, Foresti R, Sarathchandra P, Mann BE, Green CJ. Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ Res. 2002;90:E17–E24. doi: 10.1161/hh0202.104530. [DOI] [PubMed] [Google Scholar]
- Nakao A, Moore BA, Murase N, Liu F, Zuckerbraun BS, Bach FH, et al. Immunomodulatory effects of inhaled carbon monoxide on rat syngeneic small bowel graft motility. Gut. 2003;52:1278–1285. doi: 10.1136/gut.52.9.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann E, Lefevre S, Zimmermann B, Gay S, Muller-Ladner U. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol Med. 2010;16:458–468. doi: 10.1016/j.molmed.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Hoshi D, Kiire A, Yamanaka H, Kamatani N. Molecular targets of rheumatoid arthritis. Inflamm Allergy Drug Targets. 2008;7:53–66. doi: 10.2174/187152808784165199. [DOI] [PubMed] [Google Scholar]
- O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–18. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- Otterbein LE, Bach FH, Alam J, Soares M, Tao LH, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- Reddy SP, Mossman BT. Role and regulation of activator protein-1 in toxicant-induced responses of the lung. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1161–L1178. doi: 10.1152/ajplung.00140.2002. [DOI] [PubMed] [Google Scholar]
- Rego D, Kumar A, Nilchi L, Wright K, Huang S, Kozlowski M. IL-6 production is positively regulated by two distinct Src homology domain 2-containing tyrosine phosphatase-1 (SHP-1)-dependent CCAAT/enhancer-binding protein beta and NF-κB pathways and an SHP-1-independent NF-κB pathway in lipopolysaccharide-stimulated bone marrow-derived macrophages. J Immunol. 2011;186:5443–5456. doi: 10.4049/jimmunol.1003551. [DOI] [PubMed] [Google Scholar]
- Rosenstein ED, Greenwald RA, Kushner LJ, Weissmann G. Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation. 2004;28:311–318. doi: 10.1007/s10753-004-6641-z. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Otterbein LE, Morse D, Choi AM. Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem. 2002;234–235:249–263. doi: 10.1023/A:1015957026924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- Sawa Y, Ueki T, Hata M, Iwasawa K, Tsuruga E, Kojima H, et al. LPS-induced IL-6, IL-8, VCAM-1, and ICAM-1 expression in human lymphatic endothelium. J Histochem Cytochem. 2008;56:97–109. doi: 10.1369/jhc.7A7299.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW264.7 murine macrophages. Br J Pharmacol. 2005;145:800–810. doi: 10.1038/sj.bjp.0706241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer CI, Mutschler M, Stoll P, Goebel U, Humar M, Hoetzel A, et al. Carbon monoxide releasing molecule-2 inhibits pancreatic stellate cell proliferation by activating p38 mitogen-activated protein kinase/heme oxygenase-1 signaling. Mol Pharmacol. 2010;77:660–669. doi: 10.1124/mol.109.059519. [DOI] [PubMed] [Google Scholar]
- Shambharkar PB, Blonska M, Pappu BP, Li H, You Y, Sakurai H, et al. Phosphorylation and ubiquitination of the IkappaB kinase complex by two distinct signaling pathways. EMBO J. 2007;26:1794–1805. doi: 10.1038/sj.emboj.7601622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, O'Neill LA. The troll in toll: mal and tram as bridges for TLR2 and TLR4 signaling. J Leukoc Biol. 2007;82:196–203. doi: 10.1189/jlb.1206750. [DOI] [PubMed] [Google Scholar]
- Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, et al. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol. 2004;172:3553–3563. doi: 10.4049/jimmunol.172.6.3553. [DOI] [PubMed] [Google Scholar]
- Stein AB, Bolli R, Dawn B, Sanganalmath SK, Zhu Y, Wang OL, et al. Carbon monoxide induces a late preconditioning-mimetic cardioprotective and antiapoptotic milieu in the myocardium. J Mol Cell Cardiol. 2012;52:228–236. doi: 10.1016/j.yjmcc.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Sun Z, Jin Q, Chen X. CO-releasing molecules (CORM-2)-liberated CO attenuates leukocytes infiltration in the renal tissue of thermally injured mice. Int J Biol Sci. 2008a;4:176–183. doi: 10.7150/ijbs.4.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Zou X, Chen Y, Zhang P, Shi G. Preconditioning of carbon monoxide releasing molecule-derived CO attenuates LPS-induced activation of HUVEC. Int J Biol Sci. 2008b;4:270–278. doi: 10.7150/ijbs.4.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak PP, Thurkow EW, Daha MR, Kluin PM, Smeets TJ, Meinders AE, et al. Expression of adhesion molecules in early rheumatoid synovial tissue. Clin Immunol Immunopathol. 1995;77:236–242. doi: 10.1006/clin.1995.1149. [DOI] [PubMed] [Google Scholar]
- Verstak B, Nagpal K, Bottomley SP, Golenbock DT, Hertzog PJ, Mansell A. MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-κB proinflammatory responses. J Biol Chem. 2009;284:24192–24203. doi: 10.1074/jbc.M109.023044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Hsieh HL, Jou MJ, Yang CM. Involvement of p42/p44 MAPK, p38 MAPK, JNK and nuclear factor-κB in interleukin-1β-induced matrix metalloproteinase-9 expression in rat brain astrocytes. J Neurochem. 2004;90:1477–1488. doi: 10.1111/j.1471-4159.2004.02682.x. [DOI] [PubMed] [Google Scholar]
- Wu CY, Chi PL, Hsieh HL, Luo SF, Yang CM. TLR4-dependent induction of vascular adhesion molecule-1 in rheumatoid arthritis synovial fibroblasts: roles of cytosolic phospholipase A(2)alpha/cyclooxygenase-2. J Cell Physiol. 2010;223:480–491. doi: 10.1002/jcp.22059. [DOI] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.