

Abstract
BACKGROUND AND PURPOSE
Leukotrienes (LTs) are inflammatory mediators produced via the 5-lipoxygenase (5-LOX) pathway and are linked to diverse disorders, including asthma, allergic rhinitis and cardiovascular diseases. We recently identified the benzimidazole derivative BRP-7 as chemotype for anti-LT agents by virtual screening targeting 5-LOX-activating protein (FLAP). Here, we aimed to reveal the in vitro and in vivo pharmacology of BRP-7 as an inhibitor of LT biosynthesis.
EXPERIMENTAL APPROACH
We analysed LT formation and performed mechanistic studies in human neutrophils and monocytes, in human whole blood (HWB) and in cell-free assays. The effectiveness of BRP-7 in vivo was evaluated in rat carrageenan-induced pleurisy and mouse zymosan-induced peritonitis.
KEY RESULTS
BRP-7 potently suppressed LT formation in neutrophils and monocytes and this was accompanied by impaired 5-LOX co-localization with FLAP. Neither the cellular viability nor the activity of 5-LOX in cell-free assays was affected by BRP-7, indicating that a functional FLAP is needed for BRP-7 to inhibit LTs, and FLAP bound to BRP-7 linked to a solid matrix. Compared with the FLAP inhibitor MK-886, BRP-7 did not significantly inhibit COX-1 or microsomal prostaglandin E2 synthase-1, implying the selectivity of BRP-7 for FLAP. Finally, BRP-7 was effective in HWB and impaired inflammation in vivo, in rat pleurisy and mouse peritonitis, along with reducing LT levels.
CONCLUSIONS AND IMPLICATIONS
BRP-7 potently suppresses LT biosynthesis by interacting with FLAP and exhibits anti-inflammatory effectiveness in vivo, with promising potential for further development.
Keywords: 5-lipoxygenase, 5-lipoxygenase activating protein, leukotriene, benzimidazole, inflammation
Introduction
The leukotrienes (LTs) comprise two different classes of pro-inflammatory lipid mediators derived from arachidonic acid (AA) with distinct biological activities. Whereas LTB4 acts as a chemoattractant for leukocytes and promotes immunological responses, the cysteinyl-containing LTS (cysLTs) C4, D4 and E4 induce bronchoconstriction and mucus secretion, cause plasma extravasation and stimulate fibrocyte proliferation (Peters-Golden and Henderson, 2007). Consequently, intervention with LTs represents a pertinent pharmacological approach against inflammatory diseases, and anti-LT therapy has been validated in clinical trials of asthma and allergic rhinitis, with potential in other respiratory and allergic disorders (Peters-Golden and Henderson, 2007), as well as in cardiovascular diseases such as atherosclerosis, myocardial infarction, stroke and abdominal aortic aneurysm (Riccioni and Back, 2012).
The biosynthesis of LTs requires first the liberation of AA from membrane phospholipids that is conferred by the cytosolic PLA2 (cPLA2) (Uozumi et al., 1997). Then, 5-lipoxygenase (5-LOX) catalyses the incorporation of molecular oxygen into AA to generate 5-hydroperoxyeicosatetraenoic acid (5-HPETE) that is dehydrated by 5-LOX into LTA4 (for nomenclature see Alexander et al., 2013). This unstable epoxide intermediate is further metabolized to bioactive LTs by LTA4 hydrolase (yielding LTB4) or LTC4 synthase (yielding LTC4) (Radmark et al., 2007). Substantial experimental evidence indicates that the cellular production of LTs from endogenous AA requires the so-called 5-LOX-activating protein (FLAP), which may play an essential role in AA transfer to 5-LOX (Abramovitz et al., 1993; Ferguson et al., 2007). FLAP is a trimeric 18 kD protein localized at the nuclear membrane and belongs to the superfamily of membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG). However, neither an enzymatic function nor modulation by GSH has been revealed for FLAP thus far (Evans et al., 2008; Ferguson, 2012). Apparently, FLAP acts as a scaffold for 5-LOX at the nuclear envelope (Bair et al., 2012), where it facilitates access of AA to 5-LOX (Ferguson et al., 2007). Importantly, genetic ablation or pharmacological interference of FLAP fully abolishes the generation of 5-LOX-derived products (Byrum et al., 1997; Evans et al., 2008), implying its crucial role in LT biosynthesis.
In the past, two chemotypes of FLAP inhibitors, namely the indole series (e.g. MK886) and quinoline-based compounds (e.g. BAY X-1005/DG-031) or hybrids thereof (MK591), have been developed and evaluated in clinical trials, but these compounds were not further explored (Evans et al., 2008; Sampson, 2009; Ferguson, 2012). Recent re-assessment of FLAP inhibitors of the indole series provided GSK2190915 as a promising candidate that is currently undergoing phase II trials in patients with asthma (Stock et al., 2011; Bain et al., 2013; Follows et al., 2013; Kent et al., 2013; Snowise et al., 2013). Also, the anti-inflammatory compound licofelone, which was originally developed as a dual inhibitor of the COX and 5-LOX pathways (Laufer et al., 1994a,b), targets FLAP (Fischer et al., 2007) and has reached clinical phase III for osteoarthritis (Raynauld et al., 2009).
Inspired by the therapeutic potential of FLAP inhibitors, we attempted to discover novel chemotypes using a virtual screening approach targeting FLAP. By means of a combined ligand- and structure-based pharmacophore model, we recently identified BRP-7 [1-(2-chlorobenzyl)-2-(1-(4-isobutylphenyl)ethyl)-1H-benzimidazole] (Figure 1A) as a LT synthesis inhibitor in intact neutrophils, without direct effects on 5-LOX (Banoglu et al., 2012). Here, we show that (i) BRP-7 inhibits 5-LOX product synthesis with typical features of well-recognized FLAP inhibitors; (ii) is effective in human whole blood (HWB); and (iii) has anti-inflammatory effects in two LT-related functional in vivo models, suggesting promising potential for further development.
Figure 1.
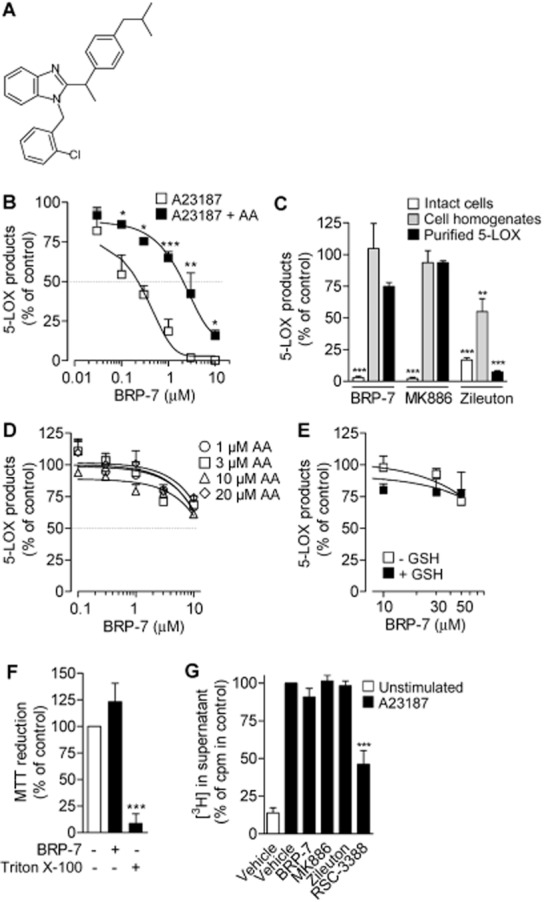
BRP-7 has features resembling other FLAP inhibitors. (A) BRP-7 structure. (B) 5-LOX product formation in human neutrophils stimulated with A23187 (2.5 μM), without or with AA (40 μM), after pre-incubation with BRP-7 or vehicle (0.1% DMSO). (C) Effects of 10 μM BRP-7, 100 nM MK886 or 10 μM zileuton on 5-LOX product formation in intact neutrophils and in neutrophil homogenates, and on the activity of human recombinant 5-LOX. Intact neutrophils were stimulated with A23187 (2.5 μM). For homogenates and purified 5-LOX, test compounds were added 5 min before the addition of 1 mM Ca2+ plus 40 μM AA. (D) Effects of BRP-7 on the activity of human recombinant 5-LOX at increasing AA substrate concentrations. (E) Reducing conditions do not restore 5-LOX inhibition by BRP-7. GSH (5 mM) or vehicle was added to homogenates of neutrophils, prior to BRP-7 addition. After 10 min on ice, 1 mM CaCl2 and 40 μM AA were added for 10 min at 37°C. The 100% controls correspond to 5-LOX products in (B, C, E) intact cells, 117.5 ± 17.5 and 831.3 ± 50.9 ng·mL−1, for A23187 or A23187 + AA, respectively; purified 5-LOX, 947.8 ± 125.2 ng·mL−1; homogenates, 484.7 ± 72 ng·mL−1 and 429.6 ± 111.8 ng·mL−1, without GSH and after addition of 5 mM GSH, respectively; (D) 37.9 ± 6.1, 153.1 ± 4.6, 610.3 ± 37.8 and 812.3 ± 22.6 ng·mL−1, in the presence of 1, 3, 10 and 20 μM AA respectively. (F) Cell viability of human neutrophils after 30 min of pre-incubation at 37°C with vehicle (0.1% DMSO), 10 μM BRP-7 or 1% Triton X-100. (G) Analysis of radioactivity in supernatants of [3H]-AA-labelled neutrophils after pre-incubation with 10 μM BRP-7, 100 nM MK886, 10 μM zileuton or 1 μM cPLA2 inhibitor (RSC-3388) and stimulation with 2.5 μM A23187 plus 50 μM thiomersal; 100% corresponds to 18 073 ± 1735 cpm. Data are means + SEM; n = 3–9; *P < 0.05; **P < 0.01; ***P < 0.001 (B) versus inhibition by the corresponding BRP-7 concentration without the addition of AA; (C–F) versus 100% control; anova plus Bonferroni.
Methods
Materials
BRP-7 was synthesized and characterized as reported previously (Banoglu et al., 2012). RSC-3388 (N-{(2S,4R)-4-(biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl}-3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl]acrylamide) was purchased from Calbiochem (Bad Soden, Germany), zileuton from Sequoia Research Products (Oxford, UK), MK886 from Cayman Chemical (Ann Arbor, MI, USA), and zymosan and λ-carrageenan type IV isolated from Gigartina aciculairis and Gigartina pistillata from Sigma (Milan, Italy). HPLC solvents were from VWR International GmbH (Darmstadt, Germany). AA, Ca2+ ionophore A23187, celecoxib, LPS, N-formyl-methionyl-leucyl-phenylalanine (fMLP), indomethacin and all other fine chemicals were from Sigma (Taufkirchen, Germany), unless stated otherwise. BRP-7 probes for fishing approach were synthesized as indicated in the Supporting Information Appendix S1.
Cells
Neutrophils and monocytes were isolated from buffy coats from adult healthy volunteers obtained at the Institute of Transfusion Medicine, University Hospital Jena. Neutrophils were immediately isolated by dextran sedimentation and centrifugation on Nycoprep cushions (PAA Laboratories, Linz, Austria) and hypotonic lysis of erythrocytes as described previously (Pergola et al., 2008). Monocytes were separated from peripheral blood mononuclear cells by adherence to culture flasks as described previously (Pergola et al., 2011). To exclude any cytotox effects of the test compounds, cell viability was analysed by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay as described previously (Tretiakova et al., 2008).
Determination of 5-LOX product formation
For assays in intact cells, neutrophils or monocytes [5 × 106 and 2 × 106 mL−1, respectively; in PBS (pH 7.4) containing 1 mg·mL−1 glucose and 1 mM CaCl2 (PGC buffer); incubation volume, 1 mL] were pre-incubated with the compounds or vehicle (0.1% DMSO) for 15 min at 37°C. Then, 2.5 μM A23187 plus AA was added. For neutrophils, 40 μM AA was used as a standard concentration, as in previous studies (Werz et al., 2002), whereas 10 μM AA was used for monocytes, because of substrate inhibition at higher AA concentrations. The reaction was stopped after 10 min with 1 mL methanol and 30 μL 1 N HCl, and 200 ng PGB1 and 500 μL of PBS were added.
For assays in cell homogenates, 1 mM EDTA was added to cells re-suspended in PBS. Samples were cooled on ice (5 min), sonicated (3 × 10 s) at 4°C, and 1 mM ATP and 1 mM DTT were added, as indicated. For assays with isolated 5-LOX, Escherichia coli BL21 was transformed with pT3-5LO plasmid, human recombinant 5-LOX protein was expressed at 30°C as described previously (Pergola et al., 2012), and partially purified 5-LOX was added to 1 mL of a 5-LOX reaction mix (PBS, pH 7.4, 1 mM EDTA, 1 mM ATP). Samples (0.5–2 μg of partially purified 5-LOX, resulting in about 1000 ng·mL−1 5-LOX products in an activity test performed with 20 μM AA; or cell homogenates, corresponding to 5 × 106 and 2 × 106 cells·mL−1 per sample, for neutrophils and monocytes, respectively) were incubated for 10 min at 4°C with vehicle or test compounds, pre-warmed for 30 s at 37°C, and 2 mM CaCl2 and the indicated concentrations of AA were added. The reaction was stopped as indicated for intact cells.
For assays in HWB, freshly withdrawn blood from healthy adult donors was obtained by venipuncture and collected in monovettes containing 16 IE heparin·mL−1. Aliquots of 2 mL were primed with 1 μg·mL−1 LPS for 15 min at 37°C, followed by incubation with vehicle (0.1% DMSO) or test compounds for 15 min at 37°C, and stimulation with 1 μM fMLP for 15 min at 37°C. Samples were prepared for extraction of metabolites and then extracted and analysed by HPLC as described previously (Pergola et al., 2012).
5-LOX products include LTB4, its all-trans isomers and 5-HPETE. The cysteinyl leukotrienes (cysLTs) C4, D4 and E4 were not detected. For the determination of cysLTs in supernatants of monocytes, a cysLT elisa kit from Enzo Life Sciences International Inc. (Lörrach, Germany) was used.
Determination of 3H-labelled AA release
Release of 3H-labelled AA from neutrophils and monocytes was analysed as previously described (Fischer et al., 2005). Briefly, freshly isolated cells were re-suspended at 1 × 107 in 1 mL of RPMI 1640 medium containing 5 nM [3H]-AA (corresponding to 0.5 μCi·mL−1, specific activity 200 Ci·mmol−1) and incubated for 120 min at 37°C in 5% CO2 atmosphere. Cells were then washed twice with PBS containing 1 mg·mL−1 glucose and 2 mg·mL−1 fatty acid-free bovine albumin to remove unincorporated [3H]-AA. Labelled cells (2 × 107 mL−1 PMNL and 5 × 106 mL−1 monocytes) were re-suspended in 1 mL of PGC containing 2 mg·mL−1 fatty acid-free bovine albumin and pre-incubated with 0.1% DMSO or test compounds (15 min, 37°C). Neutrophils were stimulated with 2.5 μM A23187 in the presence of 50 μM thiomersal to block the re-acylation of unconverted AA into membranes (Zarini et al., 2006), and monocytes were stimulated with 2.5 μM A23187. The samples were then placed on ice, centrifuged and aliquots (300 μL) of the supernatants were assayed for radioactivity by scintillation counting (Micro Beta Trilux; PerkinElmer, Waltham, MA, USA).
Analysis of subcellular redistribution of 5-LOX
For analysis of 5-LOX subcellular redistribution, neutrophils (3 × 107 mL−1 PGC buffer) were pre-incubated with 0.1% DMSO or BRP-7 for 15 min at 37°C and stimulated with 2.5 μM A23187. After 5 min, the reaction was stopped on ice and samples were centrifuged (200× g for 5 min at 4°C). Subcellular fractionation was performed by mild detergent (0.1% NP40) lysis, yielding a nuclear and a non-nuclear fraction as described previously (Werz et al., 2001). Aliquots (20 μL) of nuclear and non-nuclear fractions were analysed by SDS-PAGE and Western blotting as described previously (Pergola et al., 2012). 5-LOX antiserum (1551, AK7, kindly provided by Dr Olof Rådmark, Karolinska Institutet, Stockholm, Sweden) was used at 1:100 dilution; alkaline phosphatase-conjugated IgGs (Sigma-Aldrich) were used at 1:1000 dilution. Proteins were visualized with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolylphosphate in detection buffer [100 mM Tris/HCl (pH 9.5), 100 mM NaCl, 5 mM MgCl2].
Analysis of 5-LOX subcellular localization by indirect immunofluorescence microscopy (IFM) was performed as described previously (Pergola et al., 2008; 2011). In brief, neutrophils or monocytes were pre-incubated with 0.1% DMSO or test compounds (15 min at 37°C), then placed onto poly-l-lysine (MW 150 000–300 000; Sigma-Aldrich)-coated glass coverslips, and activated by the addition of 2.5 μM A23187 for 3 min at 37°C. Cells were fixed in methanol (−20°C, 30 min) and permeabilized with 0.1% Tween 20 in PBS (RT, 10 min), followed by three washing steps with PBS. Samples were blocked with 10% non-immune goat serum (Invitrogen, Darmstadt, Germany) for 10 min at RT, washed with PBS and incubated with mouse anti-5-LOX (kindly provided by Dr Dieter Steinhilber, University of Frankfurt, Germany) and rabbit anti-FLAP (1:200; Abcam, Cambridge, UK) for 1 h at RT. The coverslips were washed 10 times with PBS, incubated for 10 min at RT in the dark with Alexa Fluor 488 goat anti-rabbit (1:1500) and Alexa Fluor 555 anti-mouse IgG (1:1000) (Invitrogen) diluted in PBS, and washed 10 times with PBS. Where indicated, the DNA was stained with 0.1 μg·mL−1 DAPI in PBS for 3 min at RT in the dark. The coverslips were then mounted on glass slides with Mowiol (Calbiochem) containing 2.5% n-propyl gallate (Sigma-Aldrich). The fluorescence was visualized with a Zeiss Axio Observer.Z1 microscope (Carl Zeiss, Jena, Germany) and a Plan-Apochromat 100×/1.40 Oil DIC M27 objective. Images were taken with an AxioCam MR3 camera and were acquired, cut, linearly adjusted in the overall brightness and contrast, and exported to TIF by the AxioVision 4.8 software (Carl Zeiss).
Immobilization of BRP-7 derivatives and pull-down assays
Toyopearl AF-Amino-650 M resin (Tosoh Bioscience, Stuttgart, Germany) was washed five times with water and acetate buffer (0.1 M sodium acetate, 0.5 M NaCl; pH 4) and once with 80% MeOH (pH 5). Then, 100 μmol of 1a and 2a carboxylate analogues were solubilized in 10 mL of 80% MeOH by adding 1 M NaOH until the compounds were completely dissolved. Subsequently, pH was adjusted to 5 with 1 M HCl. Toyopearl resin (500 μL) and 40 mg 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide were added to form amides 1b and 2b (48 h, pH 5). Probes were washed with acetate buffer and stored in 20% MeOH until use.
For preparation of nuclear membranes, 1.5 × 109 neutrophils were lysed by a mild detergent (0.1% NP40) lysis with 1.6 mL of lysis buffer [10 mM Tris–HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 1 mM EDTA, 0.1% NP-40, 1 mM PMSF, 60 μg·mL−1 soybean trypsin inhibitor (STI) and 10 μg·mL−1 leupeptin], vortexed (3 × 5 s), kept on ice for 10 min and centrifuged (1000× g, 10 min, 4°C) (Werz et al., 2001). The resulting pellets (nuclear fractions) were re-suspended in 500 μL of TKM buffer [50 mM Tris–HCl (pH 7.4), 25 mM KCl, 5 mM MgCl2, 250 mM sucrose, 1 mM EDTA, 1 mM PMSF, 60 μg·mL−1 STI and 10 μg·mL−1 leupeptin]. Nuclei were disrupted by sonication (3 × 5 s), samples were cleared by centrifugation (5 min, 10 000× g, 4°C) and the resulting supernatants were centrifuged for 1 h at 100 000× g, for isolation of membranes. The pellet was then re-suspended in 100 μL of membrane buffer [100 mM Tris–HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA, 0.5 mM DTT, 5% glycerol, 0.05% Tween 20, 1 mM PMSF, 60 μg·mL−1 STI, 10 μg·mL−1 leupeptin], resulting in a protein concentration of about 6.5 mg·mL−1.
For pull-down assays, 50 μL of resin was added to 500 μL of binding buffer [50 mM HEPES (pH 7.4), 200 mM NaCl, 1 mM EDTA] and 8 μL of the neutrophil nuclear membranes (corresponding to about 50 μg of protein), incubated (2.5 h, 4°C) and washed three times with 500 μL of binding buffer. Proteins were eluted with 50 μL of 2× SDS-b (SDS-PAGE sample loading buffer) (5 min, 96°C). Equal amounts of protein were loaded onto 16% acrylamide gels and analysed for FLAP by Western blotting using rabbit anti-FLAP (Abcam), anti-rabbit IRDye 800CW (Li-Cor Biosciences, Lincoln, NE, USA), and detection with an Odyssey Infrared Imaging System (Li-Cor Bioscience), and analysis by the Odyssey application software (version 3.0.25).
Activity assays of COX-1 and -2, of microsomal PGE2 synthase-1 (mPGES-1)
Activities of the purified ovine COX-1 (50 units) or human recombinant COX-2 (20 units) (both from Cayman Chemical) were analysed as described previously (Koeberle et al., 2008). Briefly, the COX enzymes were pre-incubated with the test compound for 5 min at 4°C, samples were pre-warmed for 60 s at 37°C, AA (5 μM for COX-1, 2 μM for COX-2) was added, and after 5 min at 37°C, the COX product 12-hydroxy-5,8,10-heptadecatrienoic acid (12-HHT) was analysed by HPLC. Indomethacin (10 μM) was used as a reference inhibitor.
To analyse the effect of BRP-7 on cellular COX-1 activity, freshly isolated human platelets (108 mL−1 PGC buffer) were pre-incubated with test compounds for 15 min at 37°C and stimulated for 10 min at 37°C with 5 μM AA. The COX reaction was stopped and 12-HHT was analysed as for the isolated enzyme.
To analyse the effect of BRP-7 on cellular COX-2 activity, A549 cells were stimulated with 2 ng·mL−1 IL-1β for 72 h to induce COX-2 expression, washed twice with PBS, re-suspended in PGC buffer (2 × 106 cells·mL−1) and pre-incubated with the indicated compounds for 15 min at 37°C. After stimulation for 15 min at 37°C with 3 μM AA, the reaction was stopped on ice, the supernatants were recovered after centrifugation at 60× g for 10 min at 4°C, and 6-keto PGF1α formation was measured by elisa (6-keto PGF1α from Sapphire Bioscience, Waterloo, Australia).
Preparation of A549 cells and determination of mPGES-1 activity was performed as described previously (Koeberle et al., 2008). In brief, cells (2 × 106 cells in 20 mL of medium) were plated, incubated for 16 h (37°C, 5% CO2) and then the culture medium was replaced by fresh DMEM/high glucose (4.5 g·L−1) medium containing 2% (v v−1) FCS. mPGES-1 expression was induced by 2 ng·mL−1 IL-1β for 72 h. Cells were sonicated and the microsomal fraction was prepared by differential centrifugation at 10 000× g for 10 min and at 174 000× g for 1 h. The pellet was re-suspended in 1 mL of homogenization buffer [0.1 M potassium phosphate buffer (pH 7.4), 1 mM PMSF, 60 μg·mL−1 STI, 1 μg·mL−1 leupeptin, 2.5 mM glutathione and 250 mM sucrose] and then serially diluted in 0.1 M potassium phosphate buffer (pH 7.4) containing 2.5 mM glutathione for activity test. A dilution leading to about 10 μM PGE2 (using 20 μM PGH2 as substrate; incubation volume, 100 μL) was finally used for the experiments. The microsomal membranes (total volume, 100 μL) were pre-incubated with the test compounds or 0.1% DMSO. After 15 min, PGE2 formation was initiated by the addition of PGH2 (20 μM). After 1 min at 4°C, the reaction was terminated with 100 μL of stop solution (40 mM FeCl2, 80 mM citric acid and 10 μM of 11β-PGE2 as internal standard), and PGE2 was separated by solid-phase extraction (RP-18 material) and analysed by HPLC.
Evaluation of human ether-a-go-go gene (hERG) and CYP3A4 inhibition
The experiments for evaluation of hERG and cytochrome P450 (CYP3A4) inhibition were performed by Cyprotex Discovery Ltd (Macclesfield, UK), as indicated in the Supporting Information Appendix S1. Briefly, the hERG assay was performed with CHO cells stably transfected with hERG and electrophysiology measurements by an IonWorks™ HT instrument (Molecular Devices Corporation, Sunnyvale, CA, USA) and specialized multi-well plate (PatchPlate™). For CYP3A4 assay, the test compound was incubated with human liver microsomes and NADPH in the presence of the specific CYP3A4 substrate, midazolam. 1-Hydroxymidazolam was monitored by LC-MS/MS.
Animal studies
The animal studies are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Male Wistar Han rats (200–230 g; Harlan, San Pietro al Natisone, Italy, and Charles River, Calco, Italy) and male CD-1 mice (35–40 g; Charles River) (30 rats, 50 mice) were housed at the Department of Pharmacy (Naples, Italy) in a controlled environment (21 ± 2°C) and provided with standard rodent chow and water. Animals were allowed to acclimatize for 4 days before the experiments and were subjected to a 12 h light–12 h dark schedule. Experiments were conducted during the light phase. Animal care was in compliance with Italian regulations on protection of animals used for experimental and other scientific purpose (Ministerial Decree 116/92) as well as with the European Economic Community regulations (Official Journal of the European Community L 358/1 12/18/1986). The animal studies were approved by the local ethical committee of the University of Naples Federico II on 10 February 2011 (approval number 2011/0017635) and on 28 June 2011 (approval number 2011/0075376).
The carrageenan-induced pleurisy in rats was performed as described previously (Pergola et al., 2012). Thus, BRP-7 or MK886 at the indicated dose or vehicle (1.5 mL of 0.9% saline solution containing 4% DMSO) was given i.p. 30 min before λ-carrageenan type IV 1% (w v−1; 0.2 mL), which was injected into the pleural cavity. The animals were killed by inhalation of CO2 at 4 h, the pleural exudates were collected, the cells in the exudates were counted and the amounts of LTB4 were assayed by EIA kit (Cayman Chemical), according to manufacturer's instructions.
For zymosan-induced peritonitis in mice, BRP-7 or MK-886 at the indicated dose or vehicle (0.5 mL of 0.9% saline solution containing 2% DMSO) was given i.p. 30 min before zymosan i.p. injection (0.5 mL of suspension of 2 mg mL−1 in 0.9% w/v saline). Mice were killed by inhalation of CO2 at the indicated time, followed by a peritoneal lavage with 3 mL of cold PBS. Exudates were collected, the cells in the exudates were counted and LTC4 was measured by EIA (Cayman Chemical). Vascular permeability was assessed by the Evans blue method according to Kolaczkowska et al. (2002).
Statistics
Results are expressed as mean + SEM of n observations, where n represents the number of experiments performed on different days in duplicates or the number of animals, as indicated. The IC50 values were determined by interpolation on semi-logarithmic graphs and validated with GraphPad Prism software (GraphPad, San Diego, CA, ISA). Statistical evaluation of the data was performed by one-way anova) for independent or correlated samples, followed by Tukey's HSD (honestly significant difference) post hoc tests. Where appropriate, Student's t-test was applied. A P-value < 0.05 (*) was considered significant.
Results
Differential inhibition of 5-LOX product synthesis by BRP-7 in cell-based and cell-free assays
Firstly, we studied the ability of BRP-7 to interfere with 5-LOX product synthesis and we addressed whether BRP-7 shares typical mechanistic properties with FLAP inhibitors. As we observed no significant differences in the inhibitory potency between the individual enantiomers (Sardella et al., 2013), racemic BRP-7 was used. In agreement with our previous results (Banoglu et al., 2012), BRP-7 concentration-dependently inhibited the formation of 5-LOX-derived products (i.e. LTB4 and its trans isomers and 5-HPETE) in A23187-stimulated human neutrophils (IC50 = 0.15 ± 0.08 μM; Figure 1B). However, BRP-7 did not inhibit 5-LOX product formation in cell-free assays (i.e. human recombinant 5-LOX or neutrophil homogenates), which was similar to the FLAP inhibitor MK886, whereas the direct 5-LOX inhibitor zileuton was active in this respect (Figure 1C). Failure to inhibit 5-LOX in such cell-free assays was independent of the substrate concentration (Figure 1D), excluding a competitive mode of action, and may indicate the lack of a direct effect on 5-LOX, but it may also be related to a lack of effect under non-reducing conditions, as reported for non-redox-type 5-LOX inhibitors (Werz et al., 1998). In contrast to this class of compounds, however, inclusion of GSH in homogenates did not restore the inhibitory effect of BRP-7 on 5-LOX (Figure 1E). Also, BRP-7 was ineffective as a radical scavenger in a DPPH (1,1-diphenyl-2-picrylhydrazyl) assay (not shown), excluding interference by redox-related mechanisms. Together, direct interference of BRP-7 with 5-LOX can be excluded. Moreover, non-specific effects of BRP-7 in intact cells due to cell toxicity are unlikely because BRP-7 had no significant effects on cell viability (Figure 1F). Of interest, as described for MK886 (Fischer et al., 2007), the efficiency of BRP-7 in A23187-stimulated neutrophils was significantly reduced when excess (40 μM) exogenous AA was included (Figure 1A), despite it not inducing a significant inhibition of AA release from [3H]-AA-labelled neutrophils (Figure 1G).
BRP-7 blocks 5-LOX co-localization with FLAP at the nuclear envelope in neutrophils
FLAP inhibitors were shown to block 5-LOX translocation to the nucleus seemingly by preventing the association between 5-LOX/FLAP (Evans et al., 2008). As shown in Figure 2A, upon stimulation of neutrophils with A23187, cytosolic 5-LOX is translocated to the nucleus, and this was concentration-dependently inhibited by BRP-7, starting at 1 μM, although complete inhibition was not observed even at 10 μM. IFM allowed more detailed analysis of 5-LOX subcellular localization and also of FLAP (Figure 2B). In unstimulated cells, FLAP was localized to the nuclear envelope, whereas 5-LOX was distributed within the cytosol, and co-localization of 5-LOX and FLAP was not evident. Upon A23187 stimulation, 5-LOX clearly co-localized with FLAP at the nuclear envelope. Pre-incubation of neutrophils with BRP-7 or MK886 (10 μM and 100 nM, concentrations that completely inhibited 5-LOX product formation) prevented the 5-LOX association with FLAP, although only to a partial extent, indicating that inhibition of 5-LOX translocation by FLAP inhibitors is not directly related to LT suppression.
Figure 2.
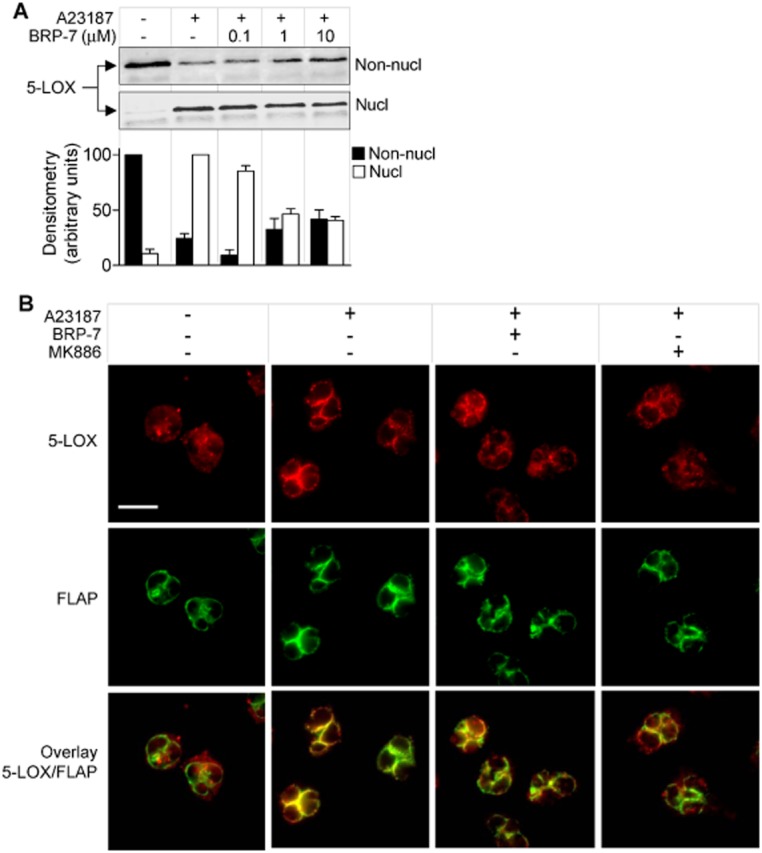
BRP-7 reduces the nuclear translocation of 5-LOX in human neutrophils. Analysis of 5-LOX localization by (A) subcellular fractionation and (B) IFM. (A) Neutrophils were pre-incubated for 15 min at 37°C with vehicle (0.1% DMSO) or BRP-7 and then stimulated with 2.5 μM A23187 for 5 min at 37°C and lysed by mild detergent (0.1% NP-40). Non-nuclear (non-nucl) and nuclear (nucl) fractions were analysed by Western blot and densitometric analysis was performed. Data are given as means + SEM; n = 3. (B) Neutrophils were pre-incubated with vehicle (0.1% DMSO), BRP-7 (10 μM), MK-886 (100 nM) and stimulated by A23187 (2.5 μM; 3 min). Cells were fixed, permeabilized and incubated with mouse anti-5-LOX and rabbit anti-FLAP, followed by Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 555 goat anti-mouse IgG. Red: 5-LOX; green: FLAP. Scale bar: 10 μm. The pictures shown are representative of three similar samples.
To analyse the interaction of BRP-7 with FLAP, we used an affinity chromatography method using an insoluble BRP-7 matrix and solubilized FLAP (from neutrophil membranes) as the protein source. Thus, the 2′-chlorine in BRP-7 was replaced by a methylene ether linked either via an amide function to a sepharose matrix (yielding matrix 1b) or to ethyl carboxylate (yielding 1a) (Figure 3A). Compound 1a inhibited 5-LOX product synthesis, although with a lower potency as compared to BRP-7 (IC50 for 1a = 2.6 ± 1.1 μM). The truncated analogues thereof, that is, matrix 2b and compound 2a, were used as negative, inactive controls (Figure 3A; IC50 for 2a was >10 μM). Interestingly, FLAP was enriched in pull-downs using matrix 1b that carries the bioactive probe, as compared to matrix 2b (Figure 3B). Excess BRP-7 (100 μM) prevented the FLAP enrichment in pull-downs with matrix 1b (Figure 3B), as expected. 5-LOX protein was not detected in the pull-downs (not shown), excluding possible indirect effects on FLAP.
Figure 3.
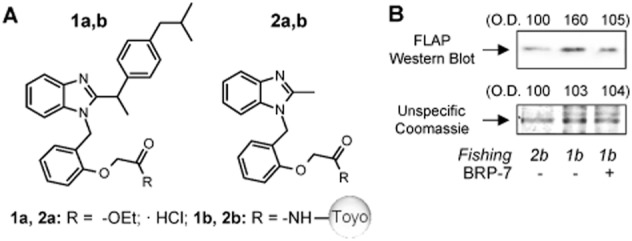
FLAP binds to an immobilized BRP-7 derivative. (A) Structures of BRP-7 derivatives used for the evaluation of IC50 values for 5-LOX products in A23187-stimulated human neutrophils (compounds 1a and 2a), and for protein fishing experiments after coupling on amino-activated Toyopearl AF-Amino-650 (compounds 1b and 2b). Compounds 2a and 2b were used as inactive controls. In (B), nuclear membrane proteins of human neutrophils were incubated with 1b or 2b functionalized beads. For 1b functionalized beads, competition was performed with 100 μM BRP-7, as indicated. Precipitated proteins were analysed by Western blot for FLAP or by Coomassie staining followed by densitometry. Similar results were obtained in two additional experiments.
Cellular pharmacology of BRP-7 in monocytes
Besides neutrophils, monocytes are a major source of LTs (Surette et al., 1993; Pergola et al., 2011). We thus determined the effects of BRP-7 on 5-LOX product formation in human monocytes stimulated with A23187 in the presence or absence of exogenous AA (10 μM). BRP-7 potently inhibited the formation of 5-LOX products (LTB4 and its trans isomers and 5-HPETE) in the absence of AA with an IC50 = 0.04 ± 0.01 μM (Figure 4A). In the presence of 10 μM AA, BRP-7 still inhibited 5-LOX activity, but, similar to that observed for neutrophils, with lower potency (IC50 = 0.25 ± 0.1 μM). In contrast to neutrophils, monocytes may convert LTA4 by LTC4 synthase to LTC4, and BRP-7 efficiently also suppressed the synthesis of cysLTs (IC50 = 0.03 ± 0.01 μM; Figure 4B). In monocyte homogenates, BRP-7 up to 0.3 μM failed to significantly inhibit 5-LOX activity and this was also found for MK886 but not for zileuton, as expected (Figure 4A). Analysis of cell viability revealed no compromising effect of BRP-7 (10 μM) as compared to staurosporine (1 μM), used as control (Figure 4C). In addition to 5-LOX products, monocytes also generate other related eicosanoids from AA including 12- and 15-HPETE, as well as the COX product 12-HHT (Pergola et al., 2011). However, neither the formation of 12- and 15-HPETE, nor 12-HHT, nor the amount of radioactivity in the supernatant (from [3H]-AA) after A23187 stimulation was affected by 10 μM BRP-7 (Figure 4D,E), implying it has a selective effect on 5-LOX product synthesis.
Figure 4.
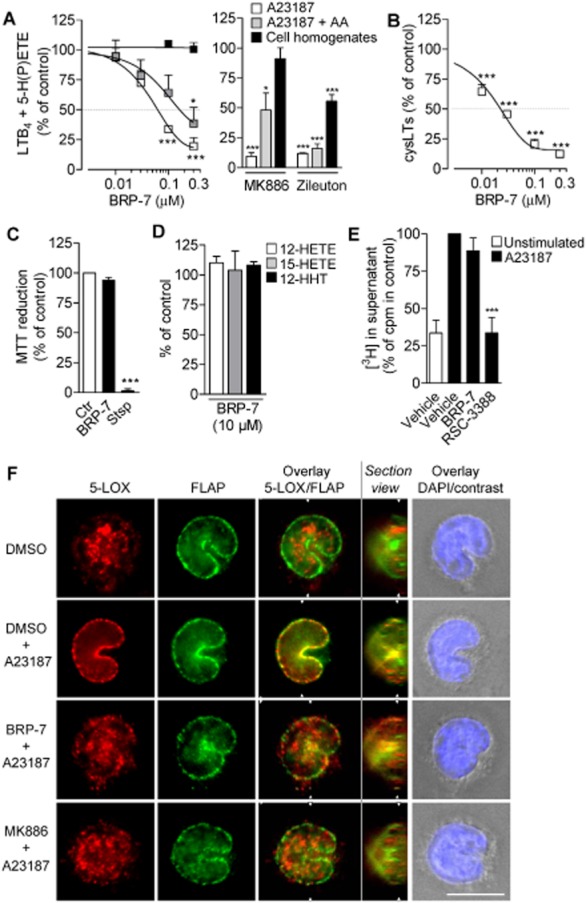
Cellular pharmacology of BRP-7 in human monocytes. (A, B) Effect of BRP-7, 100 nM MK886 or 10 μM zileuton on (A) LTB4 + 5-H(P)ETE and (B) cysLT formation in intact human monocytes stimulated with A23187 (2.5 μM) in the presence or absence of exogenous AA (10 μM) or in monocyte homogenates treated with 1 mM Ca2+ plus 10 μM AA. Pre-incubation with compounds or vehicle (0.1% DMSO) was performed for 15 min at 37°C for intact cells or 5 min at 4°C for homogenates. The 100% controls correspond to (A) A23187, 42.0 ± 4.1 ng·mL−1; A23187 + AA, 29.2 ± 1.3 ng·mL−1; homogenates, 49.7 ± 13.2 ng·mL−1; (B) 583 ± 85 pg·mL−1. (C) Cell viability of human monocytes after a 30 min pre-incubation at 37°C with vehicle (0.1% DMSO), 10 μM BRP-7 or 1 μM staurosporine (stsp). (D) Effect of 10 μM BRP-7 on the formation of 12-HETE, 15-HETE and 12-HHT in monocytes stimulated for 10 min at 37°C with A23187 (2.5 μM). The 100% controls correspond to 12-HETE, 127.5 ± 27.2 ng·mL−1; 15-HETE, 3.9 ± 1.2 ng·mL−1; 12-HHT, 19.4 ± 2.6 ng·mL−1. (E) Analysis of radioactivity in supernatants of [3H]-AA-labelled monocytes after pre-incubation with 10 μM BRP-7 or 1 μM cPLA2 inhibitor (RSC-3388) and stimulation with 2.5 μM A23187;100% corresponds to 25 437 ± 4226 cpm. (F) Analysis of 5-LOX localization by indirect IFM after pre-incubation of adherent monocytes for 15 min at 37°C with vehicle (0.1% DMSO), BRP-7 (10 μM) or MK-886 (100 nM) and stimulation with A23187 (2.5 μM, 3 min). Cells were fixed, permeabilized and incubated with mouse anti-5-LOX and rabbit anti-FLAP, followed by Alexa Fluor 488 goat anti-rabbit IgG, Alexa Fluor 555 goat anti-mouse IgG and DAPI. Red, 5-LOX; green, FLAP; blue, DNA. Scale bar: 10 μm. Sections were generated with the AxioVision 4.8 software from 0.2 μm step Z stacks of adherent monocytes (adhesion substrate on the right side of the view). The cutting plane is indicated in the overlays by white triangles. In (A)–(C), data are expressed as percentage of vehicle control (100%) and are means + SEM; n = 3; *P < 0.05; **P < 0.01; ***P < 0.001 (A) versus 100% control; anova plus Bonferroni.
As observed previously (Pergola et al., 2011), the bulk of the 5-LOX protein in resting monocytes was localized inside the nucleus, whereas FLAP was found at the nuclear membrane (Figure 4F). A23187 stimulation redistributed 5-LOX to the nuclear membrane, where it co-localized with FLAP. BRP-7 and MK886 efficiently prevented the co-localization of 5-LOX and FLAP, even though the compounds failed to restore the pattern to that of resting cells with clear intranuclear 5-LOX. These data indicate that FLAP inhibitors prevent the association of 5-LOX with FLAP in stimulated monocytes but do not generally interfere with the redistribution of 5-LOX in these stimulated cells.
Effects of BRP-7 on COX-1 and -2, mPGES-1, CYP3A4 and hERG
FLAP inhibitors such as MK886 tend also to inhibit other enzymes involved in the AA cascade, including COX-1 and mPGES-1 (Claveau et al., 2003; Koeberle et al., 2009), which let us to evaluate their selectivity. BRP-7 (at 10 μM) did not inhibit COX-1 or COX-2 in both cell-free (Figure 5A,B) and cellular (Figure 5C,D) assays, and also failed to significantly block mPGES-1 activity in a cell-free assay (Figure 5E). Reference compounds for COX enzymes (indomethacin) and for mPGES-1 (MK886) confirmed the functionality of the assays respectively. Finally, we tested the ability of BRP-7 to inhibit CYP3A4, the most common CYP-isoforms in drug metabolism for prediction of probable drug interactions. BRP-7 did not significantly inhibit CYP3A4 activity up to 25 μM as compared to the reference drug ketoconazole (which showed an IC50 of 0.04 μM; n = 7). In addition, BRP-7 as a lipophilic benzimidazole derivative could bind to the human ether-a-go-go gene (hERG) voltage-gated potassium channel thereby prolonging the drug-induced QT interval (Yao et al., 2008). However, there was no appreciable hERG inhibition up to 10 μM (0.97% inhibition; n = 19).
Figure 5.
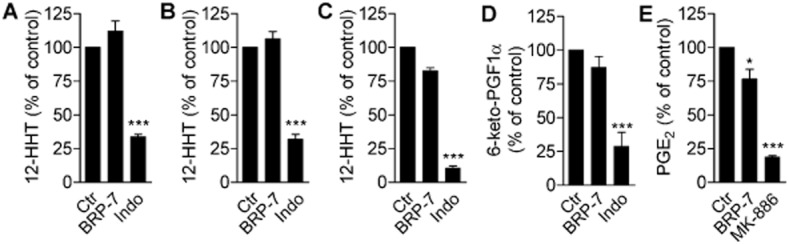
Effect of BRP-7 on COX-1, COX-2 and mPGES-1 activity. Effect of BRP-7 (10 μM) on the activity of (A) purified ovine COX-1 or (B) human recombinant COX-2 as measured by HPLC analysis of COX-derived 12-HHT after incubation with AA (5 and 2 μM, for COX-1 and COX-2 respectively). Effect of BRP-7 (10 μM) on the activity of (C) COX-1 by HPLC analysis of 12-HHT production in freshly isolated human platelets stimulated with 5 μM AA or of (D) COX-2 by EIA analysis of 6-keto PGF1α in IL-1β-stimulated A549 cells treated with 3 μM AA. (E) Effect of BRP-7 (10 μM) on mPGES-1 activity, measured by HPLC analysis of PGE2 produced in microsomes of IL-1β-stimulated A549 cells treated with 20 μM PGH2. (A–D) 10 μM indomethacin (indo) and (E) 10 μM MK886 were used as positive controls. The 100% controls correspond to (A) 132.0 ± 13.2 ng·mL−1 12-HHT; (B) 103.8 ± 13.4 ng·mL−1 12-HHT; (C) 138.5 ± 24.1 ng·mL−1 12-HHT; (D) 406.8 ± 145.9 pg·mL−1 6-keto PGF1α; (E) 1.1 ± 0.2 nmol PGE2 in 100 μL reaction buffer. Data are expressed as percentage of stimulated vehicle control (100%) and are means + SEM; n = 3–5; *P < 0.05; ***P < 0.001 versus 100% control; anova plus Bonferroni.
Efficiency of BRP-7 in HWB and efficacy in vivo
Given the favourable efficiency and selectivity of BRP-7, we next aimed to estimate its effectiveness and pharmacological relevance under more complex systems and in vivo. Thus, we analysed 5-LOX product synthesis in HWB stimulated with the pathophysiological relevant stimuli LPS and fMLP, which includes important variables such as plasma protein binding, presence of co-factors and cell–cell interactions. BRP-7 significantly inhibited 5-LOX product synthesis in blood with an IC50 of 4.8 ± 1 μM (Figure 6A), which is slightly higher, in the single-digit micromolar range, than that for MK886 (IC50 of 1.1 ± 0.4 μM; not shown).
Figure 6.
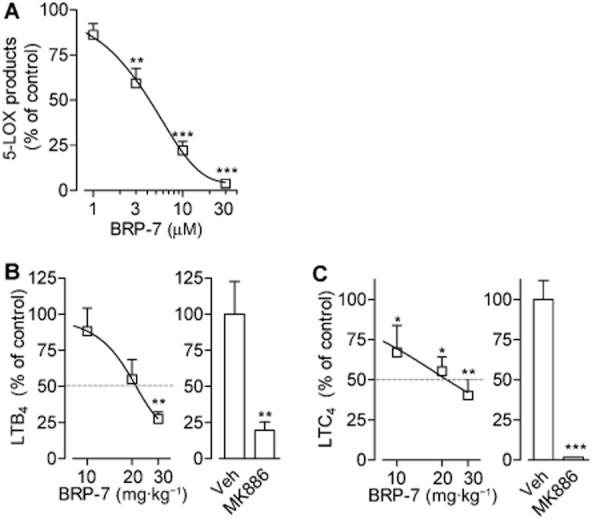
BRP-7 suppresses 5-LOX product formation in human whole blood. Effect of BRP-7 on (A) 5-LOX product formation in human whole blood stimulated with LPS (1 μg·mL−1; 30 min, 37°C) and fMLP (1 μM; 15 min, 37°C). BRP-7 or vehicle (0.1% DMSO) were added 10 min before fMLP. 5-LOX products were assessed by HPLC and are means + SEM of percentage of vehicle control (100%, corresponding to 41.3 ± 6.5 ng·mL−1, n = 6); (B) LTB4 levels in carrageenan-induced pleurisy in rats; (C) LTC4 levels in zymosan-induced peritonitis in mice. In (B) and (C), male rats or mice (n = 5–7, each group) were treated i.p. with 10, 20 or 30 mg·kg−1 BRP-7, 1 mg·kg−1 MK886 or vehicle (4 or 2% DMSO, for rat pleurisy and mouse peritonitis, respectively), 30 min before (B) intrapleural injection of carrageenan or (C) i.p. injection of zymosan. Analysis was performed (B) 4 h after carrageenan injection or (C) 30 min after zymosan injection. Data are means + SEM % of vehicle control; 100% corresponds to (B) 0.573 ± 0.130 ng·mL−1 LTB4 or (C) 142.3 ± 17.1 ng·mL−1 LTC4. **P < 0.01; ***P < 0.001 versus vehicle control; anova plus Bonferroni.
We next studied the effects of BRP-7 in vivo using the carrageenan-induced pleurisy in rats, an animal model of acute inflammation involving 5-LOX. The i.p. pretreatment (30 min before carrageenan administration) of rats with 10, 20 or 30 mg·kg−1 of BRP-7 significantly reduced the inflammatory reaction, measured as exudate volume, inflammatory cell numbers and LTB4 levels in the pleural exudates (Table 1, Figure 6B), with an ID50 value for LTB4 inhibition of about 20 mg·kg−1. For comparison, the reference FLAP inhibitor MK886 (at the maximally effective dose of 1 mg·kg−1, i.p.) also significantly reduced exudate volume, infiltrated cells and LTB4 (Table 1, Figure 6B). In zymosan-induced peritonitis in mice, another well-recognized model of acute inflammation, BRP-7 significantly impaired levels of LTC4 with an ID50 of about 20 mg·kg−1 (Figure 6C). At this dose, BRP-7 reduced vascular permeability by 38% and inhibited neutrophil infiltration by 30% (Table 2), compared to reductions of 76 and 37% by MK886 (1 mg·kg−1), respectively, implying that BRP-7-induced inhibition of LT biosynthesis in vivo is associated with its anti-inflammatory efficacy.
Table 1.
Effect of BRP-7 on carrageenan-induced pleurisy in rats
| Treatmenta | LTB4 (ng pro rat)b | Exudate volume (mL)b | Inflammatory cells (× 106)b |
|---|---|---|---|
| Vehicle | 0.573 ± 0.130 | 0.45 ± 0.13 | 50.0 ± 7.2 |
| 10 mg·kg−1 BRP-7 | 0.505 ± 0.092 | 0.14 ± 0.09* | 31.0 ± 1.6** |
| 88.2% | 31.1% | 62.0% | |
| 20 mg·kg−1 BRP-7 | 0. 314 ± 0.078 | 0.20 ± 0.08* | 26.7 ± 3.2*** |
| 54.8% | 44.4% | 53.4% | |
| 30 mg·kg−1 BRP-7 | 0.157 ± 0.028 | 0.03 ± 0.08** | 18.6 ± 3.9** |
| 27.4% | 6.6% | 37.2% | |
| 1 mg·kg−1 MK886 | 0.113 ± 0.032 | 0.08 ± 0.08** | 36.7 ± 2.7* |
| 19.7% | 17.7% | 73.4% |
Data are means ± SEM, n = 4–7. Values in italics are percentage of vehicle control.
Male rats (n = 5–7) were treated i.p. with 10, 20 or 30 mg·kg−1 BRP-7, vehicle (4% DMSO) or 1 mg·kg−1 MK886, as indicated, 30 min before intrapleural injection of carrageenan.
Analysis was performed 4 h after carrageenan injection.
P < 0.05;
P < 0.01;
P < 0.001 versus vehicle (anova + Bonferroni).
Table 2.
Effect of BRP-7 on zymosan-induced peritonitis in mice
| Treatmenta | LTC4 (ng·mL−1)b | Vascular permeability (A650)b | Inflammatory cells (× 106)c |
|---|---|---|---|
| Vehicle | 144.1 ± 20.0 | 1.35 ± 0.12 | 24.5 ± 1.9 |
| BRP-7 | 70.4 ± 16.0* | 0.84 ± 0.09* | 17.7 ± 1.0** |
| 48.8% | 62.2% | 69.7% | |
| MK886 | 7.5 ± 1.6*** | 0.32 ± 0.05*** | 16.1 ± 0.5** |
| 5.2% | 23.7% | 63.4% |
Data are means ± SEM, n = 6. Values in italics are percentage of vehicle control.
Male mice (n = 6, each group) were treated i.p. with 20 mg·kg−1 BRP-7, 1 mg·kg−1 MK886 or vehicle (2% DMSO), 30 min before i.p. injection of zymosan.
Analysis was performed 30 min after zymosan injection.
Analysis was performed 4 h after zymosan injection.
P < 0.05;
P < 0.01;
P < 0.001 versus vehicle (Student's t-test).
Discussion and conclusions
There is currently a strong interest in the development of FLAP inhibitors because of their promising potential as therapeutics against inflammatory and allergic disorders as well as against cardiovascular disease (Evans et al., 2008; Back, 2009; Sampson, 2009; Ferguson, 2012; Bain et al., 2013; Kent et al., 2013). However, recent development of drug candidates targeting FLAP (e.g. GSK2190915) concentrates on the classical FLAP inhibitor structure of MK886 and novel chemotypes are rare but urgently in demand. Here, we provide evidence that BRP-7, a recently identified hit from an in silico FLAP inhibitor discovery approach (Banoglu et al., 2012), acts on FLAP and thereby efficiently suppresses 5-LOX product synthesis in vitro and in vivo with anti-inflammatory effectiveness. Our results demonstrate that BRP-7 (i) efficiently inhibits cellular 5-LOX product synthesis and shares typical features of FLAP inhibitors; (ii) interferes with 5-LOX subcellular localization and co-localization with FLAP; (iii) does not inhibit other related or relevant enzymes involved in AA metabolism; (iv) does not interfere with CYP3A4 or hERG in vitro; and (iv) is effective in HWB and in animal models of acute inflammation and this is accompanied by reduced LT levels. Therefore, BRP-7 might represent a novel basic structure for the development of alternative FLAP inhibitors suitable for exploitation as tools and/or potential therapeutics.
Pharmacological intervention with the LT pathway encompasses essentially three different strategies, that is, inhibition of enzymes involved in LT biosynthesis (e.g. 5-LOX, LTA4 hydrolase or LTC4 synthase), antagonism of LT receptors (e.g. the cysLT1 antagonist montelukast) and inhibition of LT formation by interference with FLAP. As compared to LT receptor antagonism and to inhibition of LTA4 hydrolase or LTC4 synthase, targeting 5-LOX or FLAP appears advantageous because it leads to suppression of both classes of LTs (LTB4 and cysLT) and is appealing for its higher efficacy as an anti-inflammatory therapy. Although the possibility of direct inhibition of 5-LOX as a medical intervention has been extensively explored, the successful development of 5-LOX inhibitors has been strongly hampered due to their toxicity or lack of in vivo efficacy (Pergola and Werz, 2010), and until today, only one 5-LOX inhibitor (zileuton) could possibly enter the market.
In contrast, targeting FLAP has demonstrated promise in animal studies and in early clinical phases, but no FLAP inhibitor has yet received marketing authorization (Hofmann and Steinhilber, 2013). In fact, the discovery and evaluation of new FLAP-interfering chemotypes has been strongly limited by the fact that FLAP has no specific enzymatic functionality, which might be utilized as a measure for a simple and convenient compound screening approach. For this reason, experimental evidence for direct and functional FLAP targeting is limited and pharmacological evaluation of FLAP inhibitors has to rely on comparative analysis with previously recognized compounds (e.g. in terms of inhibition of 5-LOX product formation in cellular assays without direct 5-LOX inhibition), which has hampered the identification of novel clinically relevant compounds. In fact, research has mainly focused upon the structural optimization of the only two known chemotypes for FLAP inhibition, that is, the indole and quinoline cores (Sampson, 2009). Interestingly, the crystal structure of FLAP bound to two inhibitors has recently been elucidated and these studies have also provided functional insights into the role of FLAP in aiding 5-LOX for LTA4 synthesis, by revealing the location of the inhibitor-binding site (Ferguson et al., 2007; Ferguson, 2012). Although the resolution of the FLAP structure was too low for reliable structure-based design, we made use of the structural knowledge and combined it with ligand-based evidence to implement a rapid virtual screening strategy for the identification of new chemotypes for FLAP inhibitors (Banoglu et al., 2012). Screening of a pre-compiled collection of 2.8 mio vendor compounds (153 mio conformations) using this three-dimensional pharmacophore query led to 1792 compounds matching the desired pharmacophoric features, and after filtering according to forcefield refinement and rescoring function, 192 virtual hits remained. Then by use of protein–ligand interaction fingerprint application of MOE, BRP-7 was revealed as the overall hit, but its pharmacological profile remained unexplored.
BRP-7, a non-acidic benzimidazole derivative bearing the isobutylphenylethyl fingerprint of ibuprofen, is a novel structure that lacks typical pharmacophoric moieties of FLAP inhibitors, such as indole core, carboxylic acid or a quinoline residue (Evans et al., 2008; Sampson, 2009). Our present data show that BRP-7 displays the typical profile of FLAP inhibitors and exhibits pharmacological properties, which support its further development, that is, in vivo efficacy. Thus, similar to other FLAP inhibitors that typically fail to inhibit soluble 5-LOX in cell-free assays (Miller et al., 1990; Hatzelmann et al., 1993), BRP-7 failed to inhibit the activity of 5-LOX in cell homogenates (neutrophils and monocytes) or to inhibit isolated human recombinant 5-LOX at concentrations up to 100-fold higher than those needed to block cellular 5-LOX product synthesis by 50%. Similar features were observed for the FLAP inhibitor MK-866, whereas the direct 5-LOX inhibitor zileuton (Carter et al., 1991) blocked 5-LOX activity in the cell-free assay equally well as in intact neutrophils, as expected.
A strong loss of potency in cell-free assays also holds true for direct 5-LOX inhibitors of the non-redox type, because of the lack of a reducing environment (Werz et al., 1998; Fischer et al., 2004), and this may also apply to BRP-7. However, unlike the non-redox-type 5-LOX inhibitors (Werz et al., 1998; Fischer et al., 2004), supplementation of neutrophil homogenates with GSH did not confer 5-LOX inhibitory properties to BRP-7, implying that the failure of BRP-7 to inhibit 5-LOX in cell-free assays is not due to a lack of reducing environment, but rather suggesting that BRP-7 acts on FLAP.
As FLAP may confer high 5-LOX-mediated product synthesis by increasing the utilization of endogenously released AA by 5-LOX (Abramovitz et al., 1993; Ferguson, 2012), exogenous addition of AA as a substrate to some extent circumvents the requirement of FLAP. Accordingly, the potency of BRP-7 in the presence of exogenously added AA was reduced and no reduction of AA release was observed, again suggesting that BRP-7 interfers with FLAP. Finally, FLAP bound to the BRP-7-affinity matrix 1b more than the control matrix 2b carrying an inactive analogue, and this was reduced after competition with free BRP-7. Note that as compared to other FLAP inhibitors such as MK-866, BRP-7 showed a certain degree of selectivity with respect to other AA metabolizing enzymes (e.g. COX, 12/15-LOX) and MAPEG (e.g. mPGES-1). These data confirm the inhibition of AA transfer to 5-LOX as a primary effect of FLAP-interference, as suggested by previous studies (Ferguson et al., 2007). Interestingly, these effects on 5-LOX translocation and 5-LOX/FLAP localization were only observed at higher concentrations than those required for inhibition of LTs, suggesting that BRP-7's direct effects on the interaction between 5-LOX and FLAP may be a secondary effect. It is notable that BRP-7 showed a higher potency in monocytes than in neutrophils, which may relate to differences in the role of FLAP or variations in the accessibility of the compound to the nuclear membrane between the different cell types.
BRP-7 suppressed 5-LOX product formation in HWB and was effective in vivo in reducing LT levels and the inflammatory reaction, as observed in two well-recognized models of acute inflammation (i.e. carrageenan-induced pleurisy in rats and zymosan-induced peritonitis in mice). This is in contrast to several 5-LOX inhibitors, which are often found to be inactive under these conditions (Pergola and Werz, 2010). Although the actual potency of BRP-7 appears slightly lower as compared to optimized indole- or quinoline-containing derivatives, the chemical nature of BRP-7 and its synthetic accessibility offer several possibilities for lead optimization. Importantly, we did not observe significant inhibition of CYP3A4 by BRP-7, whereas CYP inhibition has been a critical issue for certain FLAP inhibitors (Hutchinson et al., 2009). Indeed, BRP-7 was also inactive on hERG and did not show significant cellular toxicity or obvious toxic effects in mice and rats. Together, we propose BRP-7 as novel chemotype that inhibits LT biosynthesis by targeting FLAP, without affecting related enzymes, such as 5-LOX, 12/15-LOX, COX-1, COX-2 and mPGES-1, but with pharmacological relevance in vivo and potential for further development as a lead compound.
Acknowledgments
We gratefully acknowledge the financial support by The Scientific and Technological Research Council of Turkey (TÜBİTAK Grant No. 112S596). We thank Julia Seegers, Katrin Fischer, Petra Wiecha and Monika Listing for expert technical assistance.
Glossary
- 12-HHT
12-hydroxy-5,8,10-heptadecatrienoic acid
- 5-HPETE
5-hydroperoxyeicosatetraenoic acid
- 5-LOX
5-lipoxygenase
- AA
arachidonic acid
- cPLA2
cytosolic PLA2
- cysLTs
cysteinyl LTs
- DPPH
1,1-diphenyl-2-picrylhydrazyl
- FLAP
5-lipoxygenase-activating protein
- fMLP
N-formyl-methionyl-leucyl-phenylalanine
- hERG
human ether-a-go-go gene
- HWB
human whole blood
- IFM
immunofluorescence microscopy
- mPGES-1
microsomal PGE2 synthase-1
- MTT
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
- PGC buffer
PBS pH 7.4 containing 1 mg·mL−1 glucose and 1 mM CaCl2
- SDS-b
SDS-PAGE sample loading buffer
- STI
soybean trypsin inhibitor
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12625
Appendix S1 Supporting methods.
References
- Abramovitz M, Wong E, Cox ME, Richardson CD, Li C, Vickers PJ. 5-Lipoxygenase-activating protein stimulates the utilization of arachidonic acid by 5-lipoxygenase. Eur J Biochem. 1993;215:105–111. doi: 10.1111/j.1432-1033.1993.tb18012.x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ, CGTP Collaborators The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back M. Inhibitors of the 5-lipoxygenase pathway in atherosclerosis. Curr Pharm Des. 2009;15:3116–3132. doi: 10.2174/138161209789058020. [DOI] [PubMed] [Google Scholar]
- Bain G, King CD, Schaab K, Rewolinski M, Norris V, Ambery C, et al. Pharmacodynamics, pharmacokinetics and safety of GSK2190915, a novel oral anti-inflammatory 5-lipoxygenase-activating protein inhibitor. Br J Clin Pharmacol. 2013;75:779–790. doi: 10.1111/j.1365-2125.2012.04386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bair AM, Turman MV, Vaine CA, Panettieri RA, Jr, Soberman RJ. The nuclear membrane leukotriene synthetic complex is a signal integrator and transducer. Mol Biol Cell. 2012;23:4456–4464. doi: 10.1091/mbc.E12-06-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banoglu E, Caliskan B, Luderer S, Eren G, Ozkan Y, Altenhofen W, et al. Identification of novel benzimidazole derivatives as inhibitors of leukotriene biosynthesis by virtual screening targeting 5-lipoxygenase-activating protein (FLAP) Bioorg Med Chem. 2012;20:3728–3741. doi: 10.1016/j.bmc.2012.04.048. [DOI] [PubMed] [Google Scholar]
- Byrum RS, Goulet JL, Griffiths RJ, Koller BH. Role of the 5-lipoxygenase-activating protein (FLAP) in murine acute inflammatory responses. J Exp Med. 1997;185:1065–1075. doi: 10.1084/jem.185.6.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GW, Young PR, Albert DH, Bouska J, Dyer R, Bell RL, et al. 5-Lipoxygenase inhibitory activity of zileuton. J Pharmacol Exp Ther. 1991;256:929–937. [PubMed] [Google Scholar]
- Claveau D, Sirinyan M, Guay J, Gordon R, Chan CC, Bureau Y, et al. Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J Immunol. 2003;170:4738–4744. doi: 10.4049/jimmunol.170.9.4738. [DOI] [PubMed] [Google Scholar]
- Evans JF, Ferguson AD, Mosley RT, Hutchinson JH. What's all the FLAP about? 5-Lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol Sci. 2008;29:72–78. doi: 10.1016/j.tips.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Ferguson AD. Structure-based drug design on membrane protein targets: human integral membrane protein 5-lipoxygenase-activating protein. Methods Mol Biol. 2012;841:267–290. doi: 10.1007/978-1-61779-520-6_12. [DOI] [PubMed] [Google Scholar]
- Ferguson AD, McKeever BM, Xu S, Wisniewski D, Miller DK, Yamin TT, et al. Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science. 2007;317(5837):510–512. doi: 10.1126/science.1144346. [DOI] [PubMed] [Google Scholar]
- Fischer L, Steinhilber D, Werz O. Molecular pharmacological profile of the nonredox-type 5-lipoxygenase inhibitor CJ-13,610. Br J Pharmacol. 2004;142:861–868. doi: 10.1038/sj.bjp.0705860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L, Poeckel D, Buerkert E, Steinhilber D, Werz O. Inhibitors of actin polymerisation stimulate arachidonic acid release and 5-lipoxygenase activation by upregulation of Ca2+ mobilisation in polymorphonuclear leukocytes involving Src family kinases. Biochim Biophys Acta. 2005;1736:109–119. doi: 10.1016/j.bbalip.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Fischer L, Hornig M, Pergola C, Meindl N, Franke L, Tanrikulu Y, et al. The molecular mechanism of the inhibition by licofelone of the biosynthesis of 5-lipoxygenase products. Br J Pharmacol. 2007;152:471–480. doi: 10.1038/sj.bjp.0707416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follows RM, Snowise NG, Ho SY, Ambery CL, Smart K, McQuade BA. Efficacy, safety and tolerability of GSK2190915, a 5-lipoxygenase activating protein inhibitor, in adults and adolescents with persistent asthma: a randomised dose-ranging study. Respir Res. 2013;14:54. doi: 10.1186/1465-9921-14-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzelmann A, Fruchtmann R, Mohrs KH, Raddatz S, Muller-Peddinghaus R. Mode of action of the new selective leukotriene synthesis inhibitor BAY X 1005 ((R)-2-[4-(quinolin-2-yl-methoxy)phenyl]-2-cyclopentyl acetic acid) and structurally related compounds. Biochem Pharmacol. 1993;45:101–111. doi: 10.1016/0006-2952(93)90382-7. [DOI] [PubMed] [Google Scholar]
- Hofmann B, Steinhilber D. 5-Lipoxygenase inhibitors: a review of recent patents (2010–2012) Expert Opin Ther Pat. 2013;23:895–909. doi: 10.1517/13543776.2013.791678. [DOI] [PubMed] [Google Scholar]
- Hutchinson JH, Li Y, Arruda JM, Baccei C, Bain G, Chapman C, et al. 5-Lipoxygenase-activating protein inhibitors: development of 3-[3-tert-butylsulfanyl-1-[4-(6-methoxy-pyridin-3-yl)-benzyl]-5-(pyridin-2-ylmethoxy)-1H-indol-2-yl]-2,2-dimethyl-propionic acid (AM103) J Med Chem. 2009;52:5803–5815. doi: 10.1021/jm900945d. [DOI] [PubMed] [Google Scholar]
- Kent SE, Boyce M, Diamant Z, Singh D, O'Connor BJ, Saggu PS, et al. The 5-lipoxygenase-activating protein inhibitor, GSK2190915, attenuates the early and late responses to inhaled allergen in mild asthma. Clin Exp Allergy. 2013;43:177–186. doi: 10.1111/cea.12002. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberle A, Siemoneit U, Buhring U, Northoff H, Laufer S, Albrecht W, et al. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2008;326:975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- Koeberle A, Siemoneit U, Northoff H, Hofmann B, Schneider G, Werz O. MK-886, an inhibitor of the 5-lipoxygenase-activating protein, inhibits cyclooxygenase-1 activity and suppresses platelet aggregation. Eur J Pharmacol. 2009;608:84–90. doi: 10.1016/j.ejphar.2009.02.023. [DOI] [PubMed] [Google Scholar]
- Kolaczkowska E, Shahzidi S, Seljelid R, van Rooijen N, Plytycz B. Early vascular permeability in murine experimental peritonitis is co-mediated by resident peritoneal macrophages and mast cells: crucial involvement of macrophage-derived cysteinyl-leukotrienes. Inflammation. 2002;26:61–71. doi: 10.1023/a:1014837110735. [DOI] [PubMed] [Google Scholar]
- Laufer S, Tries S, Augustin J, Dannhardt G. Pharmacological profile of a new pyrrolizine derivative inhibiting the enzymes cyclo-oxygenase and 5-lipoxygenase. Arzneimittelforschung. 1994a;44:629–636. [PubMed] [Google Scholar]
- Laufer SA, Augustin J, Dannhardt G, Kiefer W. (6,7-Diaryldihydropyrrolizin-5-yl)acetic acids, a novel class of potent dual inhibitors of both cyclooxygenase and 5-lipoxygenase. J Med Chem. 1994b;37:1894–1897. doi: 10.1021/jm00038a021. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DK, Gillard JW, Vickers PJ, Sadowski S, Leveille C, Mancini JA, et al. Identification and isolation of a membrane protein necessary for leukotriene production. Nature. 1990;343(6255):278–281. doi: 10.1038/343278a0. [DOI] [PubMed] [Google Scholar]
- Pergola C, Werz O. 5-Lipoxygenase inhibitors: a review of recent developments and patents. Expert Opin Ther Pat. 2010;20:355–375. doi: 10.1517/13543771003602012. [DOI] [PubMed] [Google Scholar]
- Pergola C, Dodt G, Rossi A, Neunhoeffer E, Lawrenz B, Northoff H, et al. ERK-mediated regulation of leukotriene biosynthesis by androgens: a molecular basis for gender differences in inflammation and asthma. Proc Natl Acad Sci U S A. 2008;105:19881–19886. doi: 10.1073/pnas.0809120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pergola C, Rogge A, Dodt G, Northoff H, Weinigel C, Barz D, et al. Testosterone suppresses phospholipase D, causing sex differences in leukotriene biosynthesis in human monocytes. FASEB J. 2011;25:3377–3387. doi: 10.1096/fj.11-182758. [DOI] [PubMed] [Google Scholar]
- Pergola C, Jazzar B, Rossi A, Northoff H, Hamburger M, Sautebin L, et al. On the inhibition of 5-lipoxygenase product formation by tryptanthrin: mechanistic studies and efficacy in vivo. Br J Pharmacol. 2012;165:765–776. doi: 10.1111/j.1476-5381.2011.01605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- Radmark O, Werz O, Steinhilber D, Samuelsson B. 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem Sci. 2007;32:332–341. doi: 10.1016/j.tibs.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Raynauld JP, Martel-Pelletier J, Bias P, Laufer S, Haraoui B, Choquette D, et al. Protective effects of licofelone, a 5-lipoxygenase and cyclo-oxygenase inhibitor, versus naproxen on cartilage loss in knee osteoarthritis: a first multicentre clinical trial using quantitative MRI. Ann Rheum Dis. 2009;68:938–947. doi: 10.1136/ard.2008.088732. [DOI] [PubMed] [Google Scholar]
- Riccioni G, Back M. Leukotrienes as modifiers of preclinical atherosclerosis? ScientificWorldJournal. 2012;2012:490968. doi: 10.1100/2012/490968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson AP. FLAP inhibitors for the treatment of inflammatory diseases. Curr Opin Investig Drugs. 2009;10:1163–1172. [PubMed] [Google Scholar]
- Sardella R, Levent S, Ianni F, Caliskan B, Gerstmeier J, Pergola C, et al. Chromatographic separation and biological evaluation of benzimidazole derivative enantiomers as inhibitors of leukotriene biosynthesis. J Pharm Biomed Anal. 2013;89C:88–92. doi: 10.1016/j.jpba.2013.10.039. [DOI] [PubMed] [Google Scholar]
- Snowise NG, Clements D, Ho SY, Follows RM. Addition of a 5-lipoxygenase-activating protein inhibitor to an inhaled corticosteroid (ICS) or an ICS/long-acting beta-2-agonist combination in subjects with asthma. Curr Med Res Opin. 2013;29:1663–1674. doi: 10.1185/03007995.2013.842163. [DOI] [PubMed] [Google Scholar]
- Stock NS, Bain G, Zunic J, Li Y, Ziff J, Roppe J, et al. 5-Lipoxygenase-activating protein (FLAP) inhibitors. Part 4: development of 3-[3-tert-butylsulfanyl-1-[4-(6-ethoxypyridin-3-yl)benzyl]-5-(5-methylpyridin-2-ylmethoxy)-1H-indol-2-yl]-2,2-dimethylpropionic acid (AM803), a potent, oral, once daily FLAP inhibitor. J Med Chem. 2011;54:8013–8029. doi: 10.1021/jm2008369. [DOI] [PubMed] [Google Scholar]
- Surette ME, Palmantier R, Gosselin J, Borgeat P. Lipopolysaccharides prime whole human blood and isolated neutrophils for the increased synthesis of 5-lipoxygenase products by enhancing arachidonic acid availability: involvement of the CD14 antigen. J Exp Med. 1993;178:1347–1355. doi: 10.1084/jem.178.4.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretiakova I, Blaesius D, Maxia L, Wesselborg S, Schulze-Osthoff K, Cinatl J, Jr, et al. Myrtucommulone from Myrtus communis induces apoptosis in cancer cells via the mitochondrial pathway involving caspase-9. Apoptosis. 2008;13:119–131. doi: 10.1007/s10495-007-0150-0. [DOI] [PubMed] [Google Scholar]
- Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390(6660):618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- Werz O, Szellas D, Henseler M, Steinhilber D. Nonredox 5-lipoxygenase inhibitors require glutathione peroxidase for efficient inhibition of 5-lipoxygenase activity. Mol Pharmacol. 1998;54:445–451. doi: 10.1124/mol.54.2.445. [DOI] [PubMed] [Google Scholar]
- Werz O, Klemm J, Samuelsson B, Radmark O. Phorbol ester up-regulates capacities for nuclear translocation and phosphorylation of 5-lipoxygenase in Mono Mac 6 cells and human polymorphonuclear leukocytes. Blood. 2001;97:2487–2495. doi: 10.1182/blood.v97.8.2487. [DOI] [PubMed] [Google Scholar]
- Werz O, Burkert E, Samuelsson B, Radmark O, Steinhilber D. Activation of 5-lipoxygenase by cell stress is calcium independent in human polymorphonuclear leukocytes. Blood. 2002;99:1044–1052. doi: 10.1182/blood.v99.3.1044. [DOI] [PubMed] [Google Scholar]
- Yao X, Anderson DL, Ross SA, Lang DG, Desai BZ, Cooper DC, et al. Predicting QT prolongation in humans during early drug development using hERG inhibition and an anaesthetized guinea-pig model. Br J Pharmacol. 2008;154:1446–1456. doi: 10.1038/bjp.2008.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarini S, Gijon MA, Folco G, Murphy RC. Effect of arachidonic acid reacylation on leukotriene biosynthesis in human neutrophils stimulated with granulocyte-macrophage colony-stimulating factor and formyl-methionyl-leucyl-phenylalanine. J Biol Chem. 2006;281:10134–10142. doi: 10.1074/jbc.M510783200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.