

SNARE proteins can have functions unrelated to membrane fusion. The unassembled form of the SNARE protein syntaxin-8 interacts with the K+ channel TASK-1; both proteins are internalized via clathrin-mediated endocytosis in a cooperative manner. This is a novel mechanism for the control of endocytosis by cargo proteins.
Abstract
The endosomal SNARE protein syntaxin-8 interacts with the acid-sensitive potassium channel TASK-1. The functional relevance of this interaction was studied by heterologous expression of these proteins (and mutants thereof) in Xenopus oocytes and in mammalian cell lines. Coexpression of syntaxin-8 caused a fourfold reduction in TASK-1 current, a corresponding reduction in the expression of TASK-1 at the cell surface, and a marked increase in the rate of endocytosis of the channel. TASK-1 and syntaxin-8 colocalized in the early endosomal compartment, as indicated by the endosomal markers 2xFYVE and rab5. The stimulatory effect of the SNARE protein on the endocytosis of the channel was abolished when both an endocytosis signal in TASK-1 and an endocytosis signal in syntaxin-8 were mutated. A syntaxin-8 mutant that cannot assemble with other SNARE proteins had virtually the same effect as wild-type syntaxin-8. Total internal reflection fluorescence microscopy showed formation and endocytosis of vesicles containing fluorescence-tagged clathrin, TASK-1, and/or syntaxin-8. Our results suggest that the unassembled form of syntaxin-8 and the potassium channel TASK-1 are internalized via clathrin-mediated endocytosis in a cooperative manner. This implies that syntaxin-8 regulates the endocytosis of TASK-1. Our study supports the idea that endosomal SNARE proteins can have functions unrelated to membrane fusion.
INTRODUCTION
Membrane proteins are shuttled between subcellular compartments by carrier vesicles that bud from donor membranes and fuse with acceptor membranes. The “identity” of these vesicles is mainly determined by 1) specific phosphoinositides (Behnia and Munro, 2005; Di Paolo and De Camilli, 2006; Lemmon, 2008), 2) specific small GTPases and their associated regulatory factors (Conner and Schmid, 2003; Lee et al., 2009; Pucadyil and Schmid, 2009), and 3) specific soluble N-ethylmaleimide–factor attachment protein receptor (SNARE) proteins (McNew et al., 2000; Malsam et al., 2008). All of these components are capable of interacting with membrane proteins transported by the vesicles (the cargo; Di Paolo and De Camilli, 2006; Falkenburger et al., 2010). There has been growing support for the idea that there is a large variety of vesicle subtypes and that the identity and the itinerary of the carrier vesicles is at least partially determined by the cargo molecules (Lakadamyali et al., 2006; Doherty and McMahon, 2009; Loerke et al., 2009; Mettlen et al., 2010). One mechanism by which membrane proteins control their intracellular transport is the exposure of cytosolic sorting signals: short peptide motifs that interact with cytosolic proteins involved in the formation and budding of transport vesicles (Bonifacino and Glick, 2004; Traub, 2009). However, on their route between different compartments, cargo proteins also interact with many other proteins that influence their sorting decisions and trafficking kinetics (Bonifacino and Hurley, 2008; Mathie et al., 2010; McMahon and Boucrot, 2011; Smith et al., 2011). Thus the binding sites of such interacting proteins may also be regarded as trafficking signals in the wider sense, and they may provide for distinct itineraries for distinct cargoes.
The TREK-related acid-sensitive K+ channel 1 (TASK-1) is a two-pore-domain potassium channel (K2P channel) that is strongly inhibited by a decrease in extracellular pH (Duprat et al., 1997). It plays an important role in the regulation of the electrical activity of neurons (Meuth et al., 2003; Bayliss and Barrett, 2008) and cardiac muscle cells (Putzke et al., 2007; Decher et al., 2011; Limberg et al., 2011), in the release of steroid hormones by the adrenal gland (Heitzmann et al., 2008; Bandulik et al., 2010), and in the immune system (Bittner et al., 2009). In the present study we obtained evidence that the K+ channel TASK-1 can interact with the endosomal SNARE protein syntaxin-8, and we analyzed the mechanisms by which this SNARE protein influences the intracellular traffic of the channel.
In the human genome there are 36 different SNARE proteins, which are localized to distinct subcellular compartments. SNARE proteins are essential for the fusion of transport vesicles with their acceptor compartments (Malsam et al., 2008; Südhof and Rothman, 2009). There are four subclasses of SNARE proteins, Qa-, Qb-, Qc-, and R-SNAREs, which all possess a characteristic 16-turn SNARE helix (Fasshauer et al., 1998). Four SNARE helices, one from each subclass, assemble as a tetrameric coiled-coil complex that brings two membranes into close apposition and provides the energy for membrane fusion. The neuronal SNARE complex, consisting of syntaxin-1A (stx1A), synaptosome-associated protein 25 (SNAP-25), and vesicle-associated membrane protein 2 (VAMP2), is essential for fusion of synaptic vesicles with the presynaptic membrane and subsequent release of neurotransmitters into the synaptic cleft. After disassembly of the neuronal SNARE complex in the presynaptic membrane, R-SNAREs are retrieved by clathrin-mediated endocytosis using cargo-specific sorting adaptors such as adaptor protein 180 (AP180; Maritzen et al., 2012). The Qa-SNARE stx1A interacts with voltage-activated Ca2+ channels, and this interaction appears to be involved in the coupling between the electrical activity of nerve terminals and release of neurotransmitters (Hagalili et al., 2008; Atlas, 2013; Bachnoff et al., 2013). In addition, the neuronal SNARE proteins stx1A and VAMP2 were reported to interact with K+ channels present in neurons, insulin-secreting cells, and cardiomyocytes (Chao et al., 2011a, b; Dai et al., 2012) and to modulate their open probability.
The endosomal SNARE complex, consisting of the R-SNARE VAMP8 and the Q-SNAREs stx7, vesicle transport through interaction with target-SNARE analogue 1b (vti1b), and stx8, plays a role in fusion of early endosomes with later endosomal compartments. The entire endosomal SNARE complex has been reported to interact with the CFTR chloride channel and to reduce its surface expression (Bilan et al., 2004, 2008; Tang et al., 2011), but the underlying molecular mechanisms have not been studied. No information is available on the interaction of endosomal SNARE proteins with potassium or calcium channels, and very little is known about molecular mechanisms by which endosomal SNARE proteins are sorted to their cognate compartments.
In the present study we address the following questions: 1) Which part of the endosomal SNARE protein stx8 interacts with which part of the potassium channel TASK‑1? 2) Does the interaction with stx8 affect the gating or the intracellular traffic of TASK-1? 3) Which step of the intracellular transport of TASK-1 is modulated by stx8, and which sorting signals are involved? 4) Is it the unassembled SNARE protein stx8 or the endosomal SNARE complex that interacts with the channels? 5) How does stx8 reach its intracellular destination? 6) What is the functional role of the interaction between stx8 and TASK-1?
RESULTS
Syntaxin-8 interacts with the K+ channel TASK-1 and changes its surface expression
TASK-1 is a two-pore-domain potassium channel (K2P channel) with four transmembrane domains (M1–M4), two pore domains (P1 and P1), and a long cytosolic C-terminus (amino acids 243–394; Figure 1A). To identify proteins interacting with TASK‑1, we performed a yeast two-hybrid screen with a human brain cDNA library. We used the split-ubiquitin variant of the yeast two-hybrid system since it enables the use of full-length integral membrane proteins as baits and screens for interactions with integral membrane proteins and membrane-associated proteins. The C-terminal half of ubiquitin, along with an artificial transcription factor, was fused to the C-terminus of TASK-1. Screening with a brain cDNA library coding for proteins fused to the N-terminal half of ubiquitin (NubG-x; Molecular Biotechnology) yielded 63 putative interacting proteins. One of these proteins was the endosomal SNARE syntaxin-8 (stx8). The topology of stx8 is illustrated in Figure 1A. The protein has three N-terminal helices (Ha, Hb, and Hc), a SNARE domain (amino acids 145–207), and a C-terminal transmembrane domain (amino acids 216–233). In a specific yeast-two-hybrid assay using TASK-1 as bait and stx8 as prey, the robust interaction of TASK-1 with stx8 was confirmed (Figure 1B). With the closely related channel TASK-3 (Rajan et al., 2000) as bait, no interaction with stx8 was found. A stx8 mutant that cannot form SNARE complexes (stx8Q179A; because it lacks the critical glutamine residue in the “0” layer; Fasshauer et al., 1998) also showed robust interaction with TASK-1 (Figure 1B). In contrast, a mutant of stx8 in which the linker between the Hc helix and the SNARE domain was removed (stx8Δ100-140) showed no interaction (Figure 1B).
FIGURE 1:
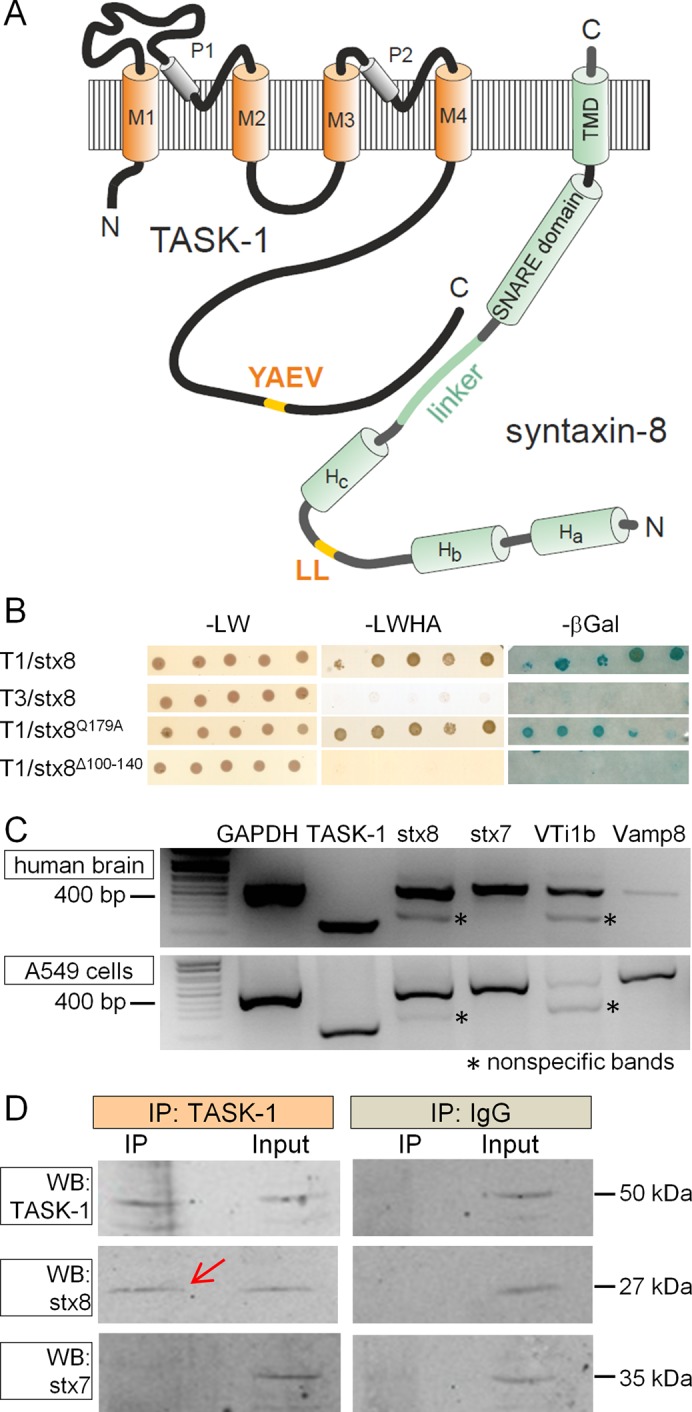
The K2P channel TASK-1 interacts with the SNARE protein syntaxin-8. (A) The topology of TASK-1 and stx8. (B) Membrane yeast two-hybrid screen with TASK-1 or TASK-3 as bait and stx8 or mutants thereof as prey. The Q179A mutant of stx8 cannot assemble with other SNARE proteins; in the Δ100–140 mutant the linker between the Hc domain and the SNARE domain was excised. (C) RT-PCR analysis of TASK-1 and endosomal SNARE proteins in human brain and in A549 cells. Asterisks represent nonspecific PCR products. (D) Coimmunoprecipitation of stx8 and TASK-1 endogenously expressed in A549 cells. The complex containing TASK-1 was precipitated from cell lysate with a TASK-1–specific antibody from Alomone (APC-024), and a Western blot of the precipitate was probed with TASK-1, stx8, and stx7 antibodies (left); the cell lysate (input) was used as positive control. Coimmunoprecipitation with an unrelated immunoglobulin G antibody (Santa Cruz Biotechnology) was used as a negative control (right).
In all of our membrane yeast-two hybrid experiments we used the positive and negative controls provided by the manufacturer (Supplemental Figure S1A). The positive controls clearly indicate that the bait proteins are expressed in the yeast strain. However, the negative result obtained with the prey protein stx8Δ100-140 (Figure 1B) might also be attributable to the lack of expression of this protein in yeast. Therefore we tested the expression of stx8 and its mutants in the yeast strain using Western blot analysis. We found that stx8, stx8Q179A, and stx8Δ100-140 were robustly expressed (Supplemental Figure S1B).
The interaction between TASK-1 and stx8 was confirmed by coimmunoprecipitation experiments. Myelocytomatosis oncogene–derived dekapeptide (myc)–tagged stx8 (mycstx8) or stx7 (mycstx7) was coexpressed with green fluorescent protein (GFP)–tagged human TASK‑1 (GFPTASK-1). Stx8 and stx7 were precipitated with anti-myc antibodies, and the precipitate was probed with anti-myc and anti-GFP antibodies. We found that GFPTASK-1 was coimmunoprecipitated with mycstx8 but not with mycstx7 (Supplemental Figure S2). We then performed coimmunoprecipitation experiments with cells that endogenously express TASK-1 and stx8. We analyzed the expression of the channel and all four endosomal SNARE proteins by reverse transcriptase (RT) PCR in several cell lines; the functionality of the primers was tested with human brain cDNA (Figure 1C). We found that A549 cells (a human alveolar adenocarcinoma cell line) express TASK-1, stx8, stx7, vti1b (weakly), and VAMP8 (Figure 1C). We precipitated the protein complex containing TASK-1 from the lysate of these cells using an anti–TASK-1 antibody from Alomone (APC-024). The Western blot of the precipitate showed a band of the predicted size of stx8 (red arrow, Figure 1D), which was also found in the lysate. No band corresponding to stx7 was detected in the precipitate (Figure 1D). Thus TASK‑1 and stx8 could be coprecipitated from nontransfected mammalian cells, which supports the idea that this interaction may be functionally relevant.
To study the functional consequences of the interaction between TASK-1 and stx8, we expressed the two proteins in another mammalian cell line (CHO cells, which do not express TASK‑1), and measured the acid-sensitive TASK-1 current (Duprat et al., 1997) using the patch-clamp technique. When the cells were transfected with rat TASK-1, a typical outwardly rectifying potassium current was recorded that could be blocked by extracellular acidification (Figure 2A). When the cells were cotransfected with rat TASK-1 and mycstx8, the shape of the current–voltage relation was unchanged, but the amplitude of the currents was reduced to ∼50% of control (Figure 2, B and C). Coexpression of mycstx7 had no effect on TASK-1 currents (Figure 2C). Cotransfection of human TASK-1 with mycstx8 or mycstx7 gave similar results as rat TASK-1 (Supplemental Figure S3). Western blot and immunohistochemistry showed that the expression of mycstx8 and mycstx7 was approximately equal (Supplemental Figure S4).
FIGURE 2:

Coexpression of TASK-1 with stx8 or stx7 in CHO cells and Xenopus oocytes. (A) Current–voltage relation of rTASK-1 expressed in CHO cells. The currents were measured using voltage ramps from −120 to +40 mV at pH 7.4 (black curve) and 6.0 (red curve). (B) TASK-1 current–voltage relation measured in the same batches of CHO cells 48 h after transfection of TASK-1 alone (black curve) and after cotransfection of TASK-1 with stx8 (green curve); mean values ± SEM of n = 28 cells. (C) Mean outward currents ± SEM measured in CHO cells at 0 mV after transfection with rat TASK‑1 alone (black) and after cotransfection of TASK-1 with stx8 (green) or stx7 (orange). (D) Typical current–voltage relation measured 48 h after injection of human TASK-1 cRNA (black curve) and after coinjection of TASK-1 and stx8 cRNA. For experiments with human TASK-1 we used the NQTASK-1 mutant, which displays a higher current amplitude (Zuzarte et al., 2009; Materials and Methods). (E) Mean outward currents ± SEM measured in Xenopus oocytes at 0 mV after injection of hTASK‑1 or hTASK-3 cRNA alone (black) or together with 1.5, 3, or 6 ng stx8 cRNA per oocyte as indicated. (F) Mean outward currents ± SEM measured in Xenopus oocytes at 0 mV measured after injection of hTASK‑1 cRNA alone (black) or together with 6 ng cRNA encoding stx8, VAMP8, vti1b, or stx7. (G) Mean surface expression of HA-tagged hTASK-1 channels (measured in relative light units [RLUs]) in Xenopus oocytes after injection of TASK-1 cRNA alone or together with 6 ng of stx8 or stx8Q179A. (H) Mean hTASK-1 current measured in Xenopus oocytes after injection of hTASK-1 cRNA alone or together with stx8 or stx8Q179A. In all bar graphs the number of oocytes or CHO cells from which the data were obtained is indicated in brackets. Note that in the series of experiments shown in E, F, and H, coinjection of 6 ng of stx8 cRNA caused a reduction of TASK-1 current to values between 13 and 23% of control, illustrating that there was a certain degree of variability among different batches of oocytes. For this reason, TASK-1 (and other) currents with and without coinjection of a second cRNA were always compared in the same batch of oocytes (measured on the same day); normalized current amplitudes of at least three different batches are combined in the bar graphs.
We then studied the effects of stx8 on hTASK-1 currents expressed in Xenopus oocytes. Coexpression of stx8 reduced current amplitude but did not change the shape of the current–voltage relation (Figure 2D). The extent of the reduction of TASK‑1 current depended on the amount of complementary RNA (cRNA) injected into the oocytes (Figure 2E). In contrast, the amplitude of TASK‑3 currents was not reduced by coexpression of stx8 (Figure 2E). We then asked whether the three other components of the endosomal SNARE complex can also interact with TASK-1. We found that coexpression of vti1b, VAMP8, or stx7 had no effect on the amplitude of TASK-1 currents (Figure 2F).
The current (I) produced by ion channels is given by I = γNPo, where γ is the single-channel conductance, N is the number of ion channels at the surface membrane, and Po is the open-state probability. To find out which of these parameters was changed by coexpression of stx8, we attached an extracellular hemagglutinin (HA) tag to TASK-1 and measured its surface expression with an enzyme-linked luminometric assay (Zuzarte et al., 2009). Coexpression of stx8 reduced the surface expression of TASK-1 in Xenopus oocytes to ∼24% (Figure 2G), which is similar to the relative reduction in current amplitude produced by stx8 in Xenopus oocytes (Figure 2, E and F). This finding suggests that the reduction in TASK-1 current observed in the presence of stx8 was due to a reduction of the copy number (N) of TASK‑1 channels at the surface membrane.
Dissection of the interacting regions of stx8 and TASK-1
We then asked whether the formation of a SNARE complex was necessary for the interaction between TASK-1 and stx8. To clarify this, we used a mutant of syntaxin-8, stx8Q179A, that is unable to form SNARE complexes (Fasshauer et al., 1998; Jahn and Scheller, 2006). We found that stx8Q197A caused a similar reduction of TASK-1 surface expression and TASK-1 current as wild-type stx8 (Figure 2, G and H). These findings suggest that it is the unassembled form of stx8 that interacts with TASK-1.
To identify the region of stx8 that interacts with TASK-1 channels, we constructed chimeras between stx8 and stx7 (which does not interact with the channel) and coexpressed them with TASK-1. We found that chimera 1 (Figure 3A), in which the SNARE motif of stx8 was replaced by that of stx7, had the same effect on current amplitude as wild-type stx8 (Figure 3B), which suggests that this construct was fully capable of interacting with TASK-1. Similar results were obtained with chimera 2, in which both the SNARE motif and the C-terminus of stx8 were replaced by the corresponding domains of stx7 (Figure 3, A and B). However, chimera 3, in which, in addition, the linker between the Hc helix and the SNARE domain of stx8 was replaced, had no significant effect on current amplitude (Figure 3, A and B). We then repeated these experiments with myc-tagged chimeras and obtained very similar results (Supplemental Figure S5, A and B). Western blotting with anti-myc antibodies showed that wild-type stx8 and the three stx8/stx7 chimeras were all strongly (and about equally) expressed in Xenopus oocytes (Supplemental Figure S5C). These results are consistent with the idea that the linker region (amino acids 100–140) proximal to the SNARE motif of stx8 interacts with the channel.
FIGURE 3:

Dissection of the interacting regions of stx8 and TASK-1. (A) Topology of stx8, stx7, and the stx8/stx7 chimeras. (B) TASK-1 currents measured in Xenopus oocytes expressing TASK-1 and stx8 or stx8/stx7 chimeras. (C) Normalized hTASK-1 currents measured in Xenopus oocytes expressing hTASK-1 and stx8 or deletion mutants of stx8. (D) Normalized currents measured in Xenopus oocytes expressing TASK-3/TASK-1 or TASK-1/TASK-3 chimeras alone or together with stx8 or stx8Q179A. (E) Schematic drawing of TASK-3/TASK-1 and TASK-1/TASK-3 chimeras.
To confirm the data obtained with the chimeras, we excised amino acids 100–140 of stx8 and measured the effect of this deletion mutant on TASK-1 current in Xenopus oocytes. Coexpression of stx8Δ100-140 had no effect on TASK-1 current (Figure 3C). This result is in agreement with our yeast two-hybrid analysis, which also showed no interaction with the stx8Δ100-140 mutant (Figure 1B). By attaching a myc tag to the deletion mutant, we verified that the protein expression of stx8Δ100-140 in Xenopus oocytes was comparable to that of wild-type stx8 (Supplemental Figure S5, B and C). Coexpression of stx8Δ100-119 or stx8Δ120-139 had a smaller effect on TASK-1 current than wild-type stx8 (Figure 3C). Interestingly, a mutant of stx8 in which the transmembrane region was removed (stxΔ216‑233) had no significant effect in TASK-1 current (Figure 3C, right), which suggests that stx8 needs to be anchored in the membrane for the interaction with TASK‑1 to occur. Taken together, the results of our yeast two-hybrid analysis (Figure 1B), our stx8/stx7 chimera experiments (Figure 3, A and B), and our deletion mutants (Figure 3C) suggest that the linker region between the Hc helix and the SNARE domain of stx8 is necessary for the interaction with TASK-1 channels.
To learn which part of TASK-1 may be responsible for the interaction with stx8, we used another set of chimeras (Figure 3, D and E) in which the C-terminus of TASK-3 was attached to TASK-1 (T1/T3 chimeras) or vice versa (T3/T1 chimeras; Renigunta et al., 2006). In the T1/T3 chimeras, stx8 had only a relatively small effect; in the T3/T1 chimeras, the reduction in current amplitude produced by coexpression of stx8 was as large as in wild-type TASK-1 channels (Figure 3D). These experiments suggest that it is (mainly) the C-terminus of TASK-1 that interacts with stx8. The current produced by the T3/T1 chimera was reduced by stx8 and by stx8Q179A to a similar extent (Figure 3D), in agreement with the results obtained with TASK-1 (Figure 2, G and H).
Cooperative endocytosis of TASK-1 and syntaxin-8
In searching for the possible mechanisms underlying the decrease in the surface expression of TASK-1 caused by stx8, we noted that both proteins harbor putative endocytosis signals (Bonifacino and Traub, 2003): TASK-1 has the sequence motif YAEV300-303, and stx8 has the sequence motif DRRQNLL77-83 (Figure 1A). We tested the hypothesis that stx8 may stimulate clathrin-mediated endocytosis of TASK-1 by coexpressing the channel and the SNARE protein with the C-terminus of the clathrin adaptor AP180 (AP180C), a construct that blocks clathrin-mediated endocytosis (Doherty and McMahon, 2009). Coexpression of TASK-1 with AP180C in oocytes caused a significant increase in TASK-1 current (Figure 4A), consistent with the idea that there may be constitutive endocytosis of TASK-1 channels (Mant et al., 2013). Furthermore, the effect of stx8 on TASK-1 current was substantially reduced by coexpression of AP180C (Figure 4A). These findings support the idea that the effects of stx8 on TASK-1 surface expression were at least partially due to stimulation of clathrin-mediated endocytosis. When we mutated the dileucine motif of stx8 to two alanines (stx8LL82,83AA), the effect of stx8 on wild-type TASK-1 current amplitude was diminished (Figure 4B). Similarly, when we replaced the tyrosine residue at position 300 in TASK-1 by alanine (TASK‑1Y300A), the effect of wild-type stx8 on TASK-1 current amplitude was diminished (Figure 4B). When we mutated both motifs, the effect of stx8 on TASK-1 currents was completely abolished (Figure 4B).
FIGURE 4:
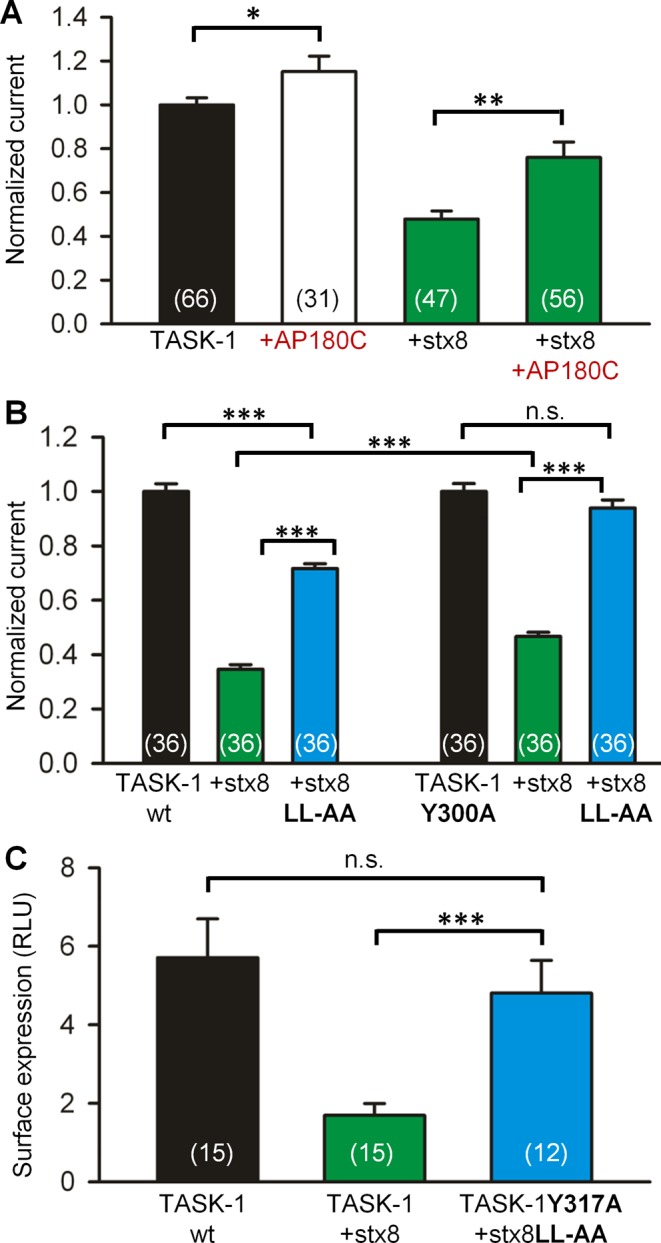
Clathrin-mediated endocytosis of TASK-1. (A) The effects of stx8 on TASK-1 currents in Xenopus oocytes with and without coexpression of AP180C, a suppressor of clathrin-mediated endocytosis. (B) The effects of mutating the dileucine-based endocytosis signal in stx8, the tyrosine-based endocytosis signal in TASK-1, or both on the amplitude of TASK-1 currents. For each batch of oocytes the currents were normalized to the currents measured with TASK-1 (or the mutant TASK-1Y300A) alone. (C) The effect of mutating the two endocytosis signals on the surface expression of HA-tagged TASK‑1 in Xenopus oocytes. The surface expression was measured using an antibody-based luminometric assay (Materials and Methods).
Very similar results were obtained in a surface expression assay in Xenopus oocytes (Figure 4C). Wild-type stx8 reduced the surface expression of wild-type TASK-1, but no significant effect of the SNARE protein on the surface expression of the channel was observed when the endocytosis signals of TASK-1 and stx8 were mutated. Because both the tyrosine-based and the leucine-based endocytosis signals interact with the AP‑2 adapter proteins of the clathrin coat (Traub, 2009; Mattera et al., 2011), our results raise the possibility that the endocytosis signals on TASK‑1 and stx8 may act in a cooperative manner to promote endocytosis of both proteins (see Discussion).
This idea was tested using an antibody uptake assay (Figure 5). The HA-tagged TASK-1 channels at the cell surface were decorated with a primary antibody at 4°C for 60 min. To initiate endocytosis, the cells were rewarmed to 37°C for 0, 15, and 30 min. At the end of the rewarming period, the cells were fixed with paraformaldehyde, and the channels at the cell surface were labeled with an Alexa Fluor 594–conjugated secondary antibody (red). Then the cells were permeabilized, and the internalized TASK-1 channels were labeled with an Alexa Fluor 488–conjugated secondary antibody (green). The green fluorescence signal in our assay represents the net uptake of the channels into the cells; channels recycled during the rewarming period would be included in the red fluorescence signal. Coexpression of stx8 led to a dramatic increase in the net uptake of HA-tagged TASK-1 channels, that is, a greater green/red ratio (Figure 5, A and B). In the presence of stx8, the increase in green fluorescence (indicating endocytosis) was detectable 15 min after rewarming; after 30 min, green fluorescence was increased about ninefold compared with control cells (not cotransfected with stx8). In contrast, coexpression of stx7 did not induce internalization of TASK-1 channels (Figure 5, A and B). Subsequently we mutated the endocytosis motifs of both TASK-1 and stx8 and repeated the antibody uptake assay. Consistent with our current measurements, coexpression of rTASK-1Y317A with stx8LL82,83AA was associated with a negligibly small antibody uptake (Figure 5, A and B). These data are consistent with the hypothesis that TASK-1 and stx8 are endocytosed in a cooperative manner.
FIGURE 5:

Analysis of endocytosis of TASK-1 using an antibody uptake assay. (A) COS-7 cells expressing HA epitope–tagged TASK-1 (or TASK-1 mutants) and stx8 (or stx8 mutants) were incubated with an anti-HA antibody at 4°C to label the channels at the cell surface and then warmed to 37°C to initiate internalization. After incubation at 37°C for 30 min, the anti-HA–labeled channels that remained on the surface were detected with a secondary antibody labeled with Alexa Fluor 594 (red). Then the cells were permeabilized, and the internalized channels were detected with a different secondary antibody, labeled with Alexa Fluor 488 (green). The measurements were carried out 48 h after transfection of TASK-1, TASK-1 mutants, stx8, stx8 mutants, or stx7. Note that rTASK‑1Y317A corresponds to hTASK-1Y300A. (B) Statistical evaluation of the antibody uptake assay under different conditions. The ratio between the fluorescence of internalized channels (green) and channels at the cell surface (red) was calculated at 0 and 30 min after heating to 37°C; number of cells is indicated in brackets. Scale bars, 50 μm.
TASK-1 and syntaxin-8 colocalize in an endosomal compartment
To visualize the subcellular compartment in which TASK-1 and stx8 are localized, we cotransfected constructs tagged with enhanced GFP (EGFP) and mCherry, respectively, in HeLa cells. Live-cell images showed that the channel and the SNARE protein mainly localized to vesicular structures throughout the cytoplasm (Figure 6, A–G). A relatively high number of these vesicles showed perinuclear localization, but some vesicles were also found in the periphery of the cells. A large fraction of the vesicles were labeled with both TASK-1 (green) and stx8 (red). The vesicles were highly mobile, as can be seen in Supplemental Video S1. Previous studies also reported localization of stx8 to early endosomes (Subramaniam et al., 2000; Kasai and Akagawa, 2001).
FIGURE 6:

Live-cell imaging of HeLa cells cotransfected with mCherry-tagged stx8 and EGFP-tagged hTASK-1. (A–G) Cotransfection of TASK-1 and stx8; the Pearson coefficient was 0.89 ± 0.02 (n = 7 cotransfection experiments, 37 cells). Plain arrows indicate colocalization; crossed arrows indicate lack of colocalization. (D–F) Higher magnifications of the region indicated by the square in C. (H–N) Cotransfection of TASK-1 and stx8Q179A; the Pearson coefficient was 0.86 ± 0.02 (n = 4 cotransfection experiments, 33 cells). (O–Q) Cotransfection of TASK-1 with stx8Δ216-233; the Pearson coefficient was 0.41 ± 0.03 (n = 4 transfection experiments, 33 cells). (K–M) Higher magnifications of the regions indicated by the square in J. All images were taken 48 h after transfection. Scale bars, 5 μm (A–C, G–I), 1 μm (D–F, K–M). (G, N) Intensity profiles of F and M (green line, EGFP; red line, mCherry). For calculating the Pearson coefficient, the entire cell was selected as region of interest.
The mutant stx8Q179A, which cannot be incorporated in the SNARE complex, was localized to apparently the same very mobile vesicular compartment (Supplemental Videos S2 and S3) and showed virtually the same colocalization with TASK-1 as wild-type stx8 (Figure 6, H–N); the Pearson coefficient was 0.86 ± 0.02. These findings are consistent with the idea that it is the unassembled SNARE protein that interacts with the channel. It should be noted that not all of the stx8 protein localized to intracellular vesicles; a fraction of stx8 and stx8Q179A localized to the surface membrane, as indicated by distinct labeling of the surface membrane in HeLa cells (Supplemental Figure S6). The stx8 mutant in which the transmembrane domain was removed (stx8Δ216-233) showed diffuse localization in the cytoplasm (Figure 6, O–Q) and had no significant effect on TASK-1 currents (Figure 3C).
To identify the intracellular compartment in which TASK-1 channels reside, we used the endosomal marker 2×FYVE (Gillooly et al., 2000, 2001; Subramaniam et al., 2000). We found that TASK-1 and 2×FYVE colocalized in a large fraction of the vesicular structures observed (Figure 7, A–G). In addition, we found partial colocalization TASK-1 with the marker of the early endosome, Rab5 (Figure 7, H–N). These findings suggest that the bulk of the heterologously expressed TASK-1 and stx8 proteins colocalized in an endosomal compartment.
FIGURE 7:

Live-cell imaging of HeLa cells cotransfected with fluorescence-labeled TASK-1 and the endosomal marker 2xFYVE or rab5. (A–G) Cotransfection of mCherry-tagged TASK-1 and EGFP-tagged 2xFYVE. Similar results were obtained in n = 3 transfections. Plain arrows indicate colocalization; crossed arrows indicate lack of colocalization. (H–N) Cotransfection of mCherry-tagged rab5 and EGFP-tagged TASK-1. (D–F, K–M) Higher magnifications of the regions indicated by the squares in C and J. (G, N) Intensity profiles of F and M (green line, EGFP; red line, mCherry/DsRed). All images were taken 48 h after transfection. Similar results were obtained in n = 3 transfections. Scale bars, 5 μm (A–C, H–J), 1 μm (D–F, K–M).
Analysis of the endocytosis of TASK-1 and stx8 by total internal reflection fluorescence microscopy
Finally, we used total internal reflection fluorescence (TIRF) microscopy at 30°C to test our hypothesis that TASK-1 channels and stx8 may be taken up cooperatively into an endosomal compartment. After transfection of EGFP-tagged clathrin light chain into HeLa cells, we observed relatively large static patches and rapidly appearing and disappearing small spots of 200–400 nm diameter (Supplemental Video S4), indicative of constitutive clathrin-mediated endocytosis. Transfection of mCherry-tagged stx8 showed some diffuse fluorescence and numerous small spots appearing and disappearing (Supplemental Video S5), which most likely represent the formation and budding of clathrin-coated pits and the fission of clathrin-coated vesicles. Transfection of TASK-1 showed similar dynamic spots (Supplemental Video S6).
Dynamic TIRF microscopy measurements after transfection with two labeled proteins required a change of filter sets and laser wavelength after each frame and therefore could only be carried out at a relatively low frame rate (0.2 Hz). Thus many of the short-lasting endocytic events were probably missed. Nevertheless, we succeeded in recording simultaneous endocytosis of TASK-1 channels and syntaxin-8 (Figure 8). Figure 8, A and C, shows TASK-1 and stx8 spots of ∼200 nm diameter appearing in consecutive frames taken at an interval of ∼5 s; colocalization of the two proteins was observed in two neighboring spots for at least 30 s. Figure 8, B and D, shows one spot with a shorter-lived colocalization of TASK-1 and stx8 (<5s). These data suggest that the channel and the SNARE protein were indeed endocytosed in the same vesicles.
FIGURE 8:

TIRF microscopy of TASK-1 and stx8. (A, B) HeLa cells transfected with EGFP-tagged hTASK-1 and mCherry-tagged stx8; scale bars, 5 μm. (C) Sequence of TIRF images taken from the cell shown in A; scale bars, 0.5 μm. The two bottom rows show the red and green channels (Materials and Methods). TASK-1 and stx8 are colocalized in diffraction-limited spots for at least 37 s. (D) Sequence of TIRF images taken from the cell shown in B; scale bars, 0.5 μm. The two bottom rows show the red and green channels (Materials and Methods). TASK-1 and stx8 are colocalized in diffraction-limited spots for at least 4.6 s.
DISCUSSION
We found that the acid-sensitive potassium channel TASK-1 interacts with the endosomal SNARE protein syntaxin-8 and tried to elucidate the functional consequences of this interaction. Potassium channels regulate many cellular functions. Activation or inhibition of potassium channels can change the membrane potential of electrically inexcitable cells or regulate the time course of the repolarization of action potentials in excitable cells; these changes in turn modulate Ca2+ influx into the cells and thus influence gene expression and the activity of various enzymes. The functional connection between potassium channels and SNARE proteins is not immediately obvious. In the exocytic and in the endocytic pathway, membrane proteins are shuttled between different intracellular compartments via transport vesicles. Membrane fusion of donor vesicles with the acceptor compartment, mediated by SNARE proteins, is the final and irreversible event in each step of intracellular traffic. Many SNARE proteins reside predominantly in specific compartments and may thus contribute the specificity and directionality of transport along different trafficking routes (Malsam et al., 2008; Südhof and Rothman, 2009). Recently, the concept has emerged that SNARE proteins may have physiological roles other than membrane fusion (Bezprozvanny et al., 1995; Leung et al., 2007; Hagalili et al., 2008; Singer-Lahat et al., 2008; Atlas, 2013; Bachnoff et al., 2013).
The evidence that TASK-1 robustly and specifically interacts with stx8 is based on the following observations: 1) The interaction between TASK-1 and stx8 was found under stringent conditions in membrane yeast-two-hybrid analysis. 2) The interaction was confirmed by coimmunoprecipitation of TASK-1 and stx8 in a cell line that endogenously expresses the two proteins. 3) Coexpression of stx8 with TASK-1 in oocytes and cultured mammalian cells caused a marked reduction of TASK-1 current amplitude, indicating functional interaction. 4) Coexpression of TASK-1 with stx7 and coexpression of TASK-3 with stx8 had no significant effect on current amplitude. 5) The surface expression of TASK-1 channels in Xenopus oocytes was markedly reduced by coexpression of stx8. 6) An antibody uptake assay in a mammalian cell line showed that the rate of endocytosis of TASK-1 channels was increased about ninefold after coexpression of stx8. 7) Analysis of chimeric TASK‑1/TASK-3 constructs showed that the C-terminus of TASK-1 was required (and sufficient) for functional interaction with stx8. 8) Yeast-two hybrid analysis and coexpression experiments in oocytes showed that the linker between the Hc helix and the SNARE helix of stx8 (amino acids 100–140) was essential for the interaction with TASK-1.
The linker region of stx8 is necessary but probably not sufficient for functional interaction of the SNARE protein with the channel, given that coexpression of stx8Δ216-233 (the mutant in which the transmembrane domain of stx8 was removed) had no significant effect on TASK-1 current. This finding suggests that the linker region needs to be localized near the membrane and perhaps at a defined distance from the surface membrane for the interaction with TASK-1 to take place.
We consider it very likely that TASK-1 interacts with the unassembled SNARE protein stx8, for the following reasons: 1) Overexpression of stx8 necessarily leads to an imbalance of the four SNARE proteins forming the endosomal SNARE complex and may cause the formation of isolated stx8 proteins; 2) overexpression of the other SNARE proteins of the endosomal SNARE complex, stx7, VAMP8, and vti1b, had no effect on TASK-1 currents; and 3) the stx8 mutant Q179A, which cannot form a SNARE complex, had the same effect on TASK-1 current amplitude and surface expression as wild-type stx8 and showed the same colocalization with TASK-1 in endosomes.
Several lines of evidence suggest that TASK-1 and stx8 are endocytosed together to the early endosomal compartment: 1) We observed a relatively weak but unequivocal expression of syntaxin-8 in the surface membrane of a mammalian cell line (Supplemental Figure S6), in agreement with previous work (Kasai and Akagawa, 2001). 2) Live-cell imaging showed extensive colocalization of TASK-1 and stx8 in an intracellular compartment, most likely corresponding to the early endosome, as indicated by colocalization with the endosomal markers 2×FYVE and rab5. The vesicles in which TASK-1 and stx8 were colocalized were highly mobile (Supplemental Videos S1–S3). 3) Using TIRF microscopy, we observed the formation of endocytic vesicles containing both TASK-1 and stx8. 4) In Xenopus oocytes, coexpression of stx8 had a smaller effect on the current carried by TASK‑1Y300A (in which the tyrosine-based endocytosis signal was removed) than on the current carried by wild-type TASK-1 channels. 5) Conversely, the current carried by wild-type TASK-1 channels was less reduced by coexpression of stx8LL82,83AA (in which the leucine-based endocytosis signal was removed) than by coexpression of wild-type stx8. 6) When TASK-1Y300A was coexpressed with stx8LL82,83AA, the effect of the SNARE protein on TASK-1 current and surface expression of TASK-1 was abolished. 7) The antibody uptake assay showed that endocytosis of the channels was almost completely abolished when both endocytic motifs were mutated (coexpression of TASK‑1Y300A and stx8LL82,83AA in COS-7 cells). The latter finding suggests that the two endocytic motifs, which both interact with the adaptor protein complex AP-2, may promote endocytosis in a cooperative manner.
There are some precedent cases in which cooperative binding of two membrane proteins to a coat protein complex has been observed. Springer and Schekman (1998) found that the yeast SNARE proteins Bet1p and Bos1p interact with the monomeric G-protein Sar1p and that both Sar1p and one of the SNARE proteins are required for COPII coat formation from isolated endoplasmic reticulum membranes (Springer and Schekman, 1998). Haucke and De Camilli (1999) found that the adaptor protein complex AP-2 interacts with both synaptotagmin (a Ca2+-dependent SNARE regulator carrying dilysine-based endocytosis signals) and SV2a (a transmembrane protein of synaptic vesicles carrying YxxΦ endocytosis signals); the binding of synaptotagmin to AP-2 was enhanced in the presence of peptides containing the YxxΦ signals of SV2a (Haucke and De Camilli, 1999). Kornfeld and coworkers found that dileucine-based and tyrosine-based sorting signals interact with the AP-1 adaptor complex in such a way that binding of one sorting signal facilitates the binding of the other sorting signal (Lee et al., 2008). Owen and coworkers showed that AP‑2 exhibits tighter binding to a membrane harboring both dileucine- and tyrosine-based sorting signals (Jackson et al., 2010). All of these results were obtained in vitro using sophisticated biochemical assays. The present study shows that in intact cells the potassium channel TASK-1 (with its YAEV motif) and the SNARE protein stx8 (with its DRRQNLL motif) are internalized via clathrin-mediated endocytosis in a cooperative manner. Although there is emerging evidence that different subtypes of clathrin-coated vesicles may control the internalization of distinct cargoes (Lakadamyali et al., 2006; Doherty and McMahon, 2009; Loerke et al., 2009; Mettlen et al., 2010), it is not clear to what extent cargo molecules determine the fate of a transport vesicle. It is tempting to speculate that in vivo the joint endocytosis of TASK-1 and unassembled stx8 may provide the basis for the subsequent steps in the intracellular transport of the channel. SNARE molecules are important determinants of vesicular identity (Jahn and Scheller, 2006; McNew, 2008), and the endowment of a transport vesicle with components of the endosomal SNARE complex may influence its itinerary. After dissociation from TASK-1, the Qc-SNARE protein stx8 may participate in the formation of endosomal SNARE complexes, and in this way stx8 may contribute to the sorting of the channel to specific intracellular compartments.
Previous studies of the interaction between SNARE proteins and channels mainly focused on the effects of the neuronal Qa-SNARE protein stx1A on the gating of calcium channels and potassium channels in neurons (Bezprozvanny et al., 1995, 2000; Leung et al., 2005; Chang et al., 2011; Etzioni et al., 2011; Weiss and Zamponi, 2012). In a few studies, the effects of stx1A on the trafficking of potassium channels were also investigated. For example, the rate of endocytosis of ATP-sensitive potassium channels was found to be increased after coexpression of stx1A (Chen et al., 2011). Furthermore, it was reported that coexpression of stx1A reduced the surface expression of the voltage-activated potassium channel Kv2.1 (Leung et al., 2003) but increased the surface expression of Kv1.1 (Feinshreiber et al., 2009). However, the mechanisms underlying these changes in surface expression have not been investigated, and no putative sorting signals have been identified. As far as we know, there are no previous studies on the effects of endosomal SNARE proteins on the intracellular traffic of potassium channels.
In conclusion, our results suggest that the SNARE protein stx8 reaches its destination (the endosomal compartment) via the surface membrane. We propose that TASK-1 and the endosomal SNARE protein stx8 interact at the surface membrane and are transported to the early endosome in a cooperative manner via clathrin-mediated endocytosis. Thus, stx8 has functions other than mediating membrane fusion; these functions are most likely mediated by the unassembled form of stx8 and are independent of other endosomal SNARE proteins. Our results provide some evidence for the idea that the endocytosis of SNARE proteins may be modulated by the presence of specific cargo proteins like TASK-1, and that, conversely, the endocytosis of membrane proteins may be modulated by unassembled SNARE proteins.
MATERIALS AND METHODS
Animal studies
For experiments involving Xenopus oocytes, adult female African clawed frogs (Xenopus laevis) were used. The frogs were anesthetized by putting them in water containing 1 g/l tricaine. Stage V oocytes were obtained from the ovarian lobes. Anesthesia and operation were carried out in accordance with the principles of German legislation with approval of the animal welfare officer of the Medical Faculty of Marburg University under the governance of the Regierungspräsidium Giessen (the regional veterinary heath authority).
Molecular cloning and mutagenesis
Supplemental Table S1 summarizes the constructs used in this study. In most experiments with human TASK-1, we used a mutant in which the amino acids at positions 2 and 3, KR, were replaced by NQ (Zuzarte et al., 2009; NQTASK-1). This mutant shows higher surface expression and higher current amplitude than wild type (Zuzarte et al., 2009), resulting in better signal-to-noise ratio. Otherwise, the results obtained with NQTASK-1 were identical to those obtained with wild-type TASK-1 (Schiekel et al., 2013). We also used chimeras of TASK-1 and TASK‑3 (Renigunta et al., 2006) in which the C-terminus of TASK-1 was replaced by the C-terminus of TASK‑3 (T1/T3 mutant) or vice versa (T3/T1 mutant). Chimeras were generated using an overlap extension PCR method. QuikChange (Agilent Technologies, Santa Clara, CA) was used to introduce point mutations. For surface luminescence and antibody uptake measurements, TASK‑1 containing an external HA epitope tag was used (Zuzarte et al., 2009). All DNA constructs were verified by sequencing.
Split-ubiquitin membrane yeast two-hybrid assay
We used the split-ubiquitin variant (Molecular Biotechnology, Göttingen, Germany) of the yeast-two-hybrid system to identify proteins that interact with hTASK-1. This assay allows the use of full-length integral membrane proteins as baits; it screens for interactions with integral membrane proteins and membrane-associated proteins. Identification of positive clones, recovery of library plasmids, and identification of prey sequences were carried out following the manufacturer's guidelines. Protein–protein interaction was determined by growth on plates lacking the amino acids leucine, tryptophan, histidine, and adenine (–LWHA). Positive colonies were further verified using the second marker β-galactosidase. The yeast strains expressing the baits were transformed with a human brain cDNA library (Molecular Biotechnology) for screening or with prey vectors for specific interaction studies. The nucleotide sequence of the screened positive clones was determined by sequencing, and the identity of the encoded putative interacting proteins was determined by a database search (BLASTX).
Cell culture
HeLa, COS-7, and A549 cells were cultured in high-glucose DMEM, 10% fetal calf serum (FCS; Life Technologies, Paisley, United Kingdom), and 1% penicillin/streptomycin (PAA Laboratories, Pasching, Austria). CHO cells were cultured in MEM alpha with glutamine (Thermo Fisher Scientific, Waltham, MA), 10% FCS, and 1% penicillin/streptomycin (PAA Laboratories). For live-cell imaging and TIRF microscopy, the cells were seeded in 35-mm glass-bottom dishes (ibidi, Martinsried, Germany) and transfected 24 h later with the indicated constructs using jetPRIME reagent (Polyplus, Illkirch, France) or Fugene6 (Invitrogen, Darmstadt, Germany) according to the manufacturers’ protocols and further maintained in the same cell culture medium without phenol red. For each method we used the cell line that appeared most suitable. CHO cells were preferred for patch-clamp experiments with transfected channels because they express very few endogenous channels that might contaminate the measurements. HeLa cells were preferred for imaging because they are optimal for detecting membrane localization and the early endosome; they show a “ring” of surface membrane (unlike COS cells, which are very flat) and have numerous clearly visible endosomal vesicles. The antibody uptake assay (empirically) worked best with COS-7 cells.
RT-PCR
The total RNA of A549 cells was isolated by using the HighPure RNA kit (Roche, Mannheim, Germany). The RNA was reverse transcribed into cDNA by using Superscript II reverse transcriptase (Invitrogen). RT-PCR was performed with AmpliTaq Gold DNA polymerase (Applied Biosystems, Darmstadt, Germany) using the following intron-spanning primers. Glyceraldehyde-3-phosphate dehydrogenase: sense, 5′‑CATCACCATCTTCCAGGAGCGA-3′, and antisense, 5′‑GTCTTCTGGGTGGCAGTGATGG-3′. TASK-1: sense, 5′-GCTCCTTCTACTTCGCCATC‑3′, and antisense, 5′‑CAGGTACCTCACCAAGGTGT‑3′. Stx8: sense, 5′-TCGCCCTTTTGAAGGACTTA-3′, and antisense, 5′- CCATTTGTTTTTGGCGACTT-3′. Stx7: sense, 5′-CAGCAGATTATCAGCGCAAA-3′, and antisense, 5′-CATATGCAATGTGGGCAAAA-3′. VAMP8: sense, 5′‑GGAGCAAGCAGGAAGTGAAC-3′, and antisense, 5′‑TGGCAAAGAGCACAATGAAG-3′. Vti1b: sense, 5′‑ATCACTGGCTGGAGAAGGTG‑3′, and antisense, 5′-ATCTCTGCCAGCGTTTCATT-3′. PCRs were performed with the following program: (95°C, 10 min) × 1 cycle, (95°C, 40 s; 55°C, 40 s; 72°C, 1 min) × 30 cycles, and (72°C, 5 min) × 1 cycle. The identity of all PCR products was confirmed by sequencing.
Coimmunoprecipitation
Immunoprecipitations were carried out using the Dynabeads Protein G Immunoprecipitation Kit (Invitrogen) according to the manufacturer's instructions. In one set of experiments, we used HeLa cells expressing myc-tagged stx8 and GFP-tagged TASK-1; the cells were lysed with Triton X-100, and the lysate was probed with a mouse anti-myc antibody (Santa Cruz Biotechnology, Dallas, TX) coupled to Protein G Dynabeads using the cross-linking agent BS3 (Invitrogen). In another set of experiments, we used A549 cells endogenously expressing TASK-1 and stx8; the cells were lysed using a buffer containing 50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% Na deoxycholate, and 10 μl/ml protease-inhibitor cocktail (Sigma, Taufkirchen, Germany), and the lysate was probed with an anti–TASK-1 antibody (APC-024; Alomone, Jerusalem, Israel) coupled to protein G Dynabeads using the cross-linking agent BS3 (Invitrogen). Antibody-coated beads were then incubated with the cell lysates under rotation for 6 h at 4°C. Dynabeads coated with antigen–antibody complex were washed extensively (4×) with the wash buffer supplied by the manufacturer. The proteins on Dynabeads were eluted by boiling at 95°C for 10 min in SDS sample loading buffer and separated on a 10–12% SDS–PAGE under reducing conditions. Proteins were transferred to nitrocellulose membrane and probed with either mouse anti-myc antibody (1:1000; Santa Cruz Biotechnology), rabbit anti-GFP antibody (1:1000; Abcam, Cambridge, United Kingdom), anti–TASK-1 (1:1000; Alomone), anti-stx8 (1:1000; BD Biosciences, Heidelberg, Germany), or anti-stx7 (1:1000; Acris Antibodies, Herford, Germany). The membrane was washed and visualized with IRDye LICOR-secondary antibodies (1:5000; LI-COR Biosciences, Bad Homburg, Germany). Immunoreactivity was detected using an infrared imaging system (Odyssey Sa; LI-COR Biosciences).
Measurements of TASK-1 currents in transfected CHO cells
Electrophysiological measurements were performed with CHO cells 24 h after transfection with rat TASK-1 or human NQTASK-1, either alone or together with human stx7 or stx8. The cells were superfused at room temperature with a bath solution containing (mM): 135 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 0.33 NaH2PO4, 10 glucose, and 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES); the pH was adjusted to 7.4 with NaOH. Patch-clamp experiments were performed in the whole-cell configuration using pipettes pulled from borosilicate glass capillaries. The patch pipettes (resistance, 3–6 MΩ) were filled with an “intracellular” solution containing (mM): 60 KCl, 65 K glutamate, 5 ethylene glycol tetraacetic acid, 3.5 MgCl2, 2 CaCl2, 3 K2ATP, 0.2 Na2GTP, and 5 HEPES; the pH was adjusted to 7.2 with KOH. Steady-state current–voltage relations were obtained by applying slow voltage ramps (40 mV/s) between −120 and +40 mV. The liquid junction potential between the patch electrode and the bath solution (∼−8 mV) was not compensated.
Voltage-clamp measurements with Xenopus oocytes
For heterologous expression in Xenopus oocytes, cRNA was transcribed in vitro from Nhe1-linearized plasmids containing the cDNA of interest using T7 RNA polymerase (mMessage mMachine T7 Kit; Ambion, Austin, TX). cRNA quality was determined by gel electrophoresis and ultraviolet spectroscopy. Defolliculated Xenopus oocytes were injected with nuclease-free water containing cRNA coding for TASK-1 (0.76 ng/oocyte), T1/T3 chimeras (0.76 ng/oocyte), or TASK-3 (0.05 ng/oocyte) alone or together with stx8, stx7, and stx8/stx7 chimeras, VAMP8, vti1B, or AP180C (all 6 ng/oocyte). Oocytes were incubated at 19°C for 24–48 h in ND96 solution containing (mM) 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES (pH 7.4–7.5), supplemented with 100 μg/ml gentamicin and 2.5 mM sodium pyruvate. Two-microelectrode voltage-clamp measurements with ramp-shaped voltage commands were performed with a TurboTec‑10 C amplifier (npi electronic, Tamm, Germany), and data were recorded at a sampling rate of 120 Hz. The oocytes were placed in a small-volume perfusion chamber and superfused with ND96 solution. For quantification of current amplitude under different experimental conditions, the currents were measured at 0 mV. The experiments were carried out at room temperature (20–23°C).
Surface expression analysis in Xenopus oocytes
Surface expression of HA-tagged TASK-1 channels in Xenopus oocytes was analyzed 2 d after injection with the cRNA for TASK-1 (0.76 ng/oocyte) alone or together with stx8 (6 ng/oocyte) as described previously (Zuzarte et al., 2009). Oocytes were incubated for 30 min in ND96 solution containing 1% bovine serum albumin (BSA) at 4°C to block nonspecific binding of antibodies. Subsequently, oocytes were incubated for 60 min at 4°C with rat monoclonal anti-HA antibody (clone 3F10, 0.1 mg/ml; Roche) in 1% BSA/ND96, washed six times at 4°C with 1% BSA/ND96, and incubated with peroxidase-conjugated, affinity-purified F(ab)2 fragment goat anti-rat immunoglobulin G antibody (0.8 mg/ml; Jackson ImmunoResearch, Newmarket, UK) in 1% BSA/ND96 for 60 min. Oocytes were washed thoroughly, initially in 1% BSA/ND96 (at 4°C for 60 min) and then in ND96 without BSA (at 4°C for 15 min). Individual oocytes were placed in 20 μl of SuperSignal Elisa Femto solution (Pierce Protein Biology Products, Rockford, IL), and chemiluminescence was quantified in a luminometer (Lumat LB9507; Berthold Technologies, Bad Wildbad, Germany). The luminescence produced by uninjected oocytes was used as reference signal (negative control).
Antibody uptake assay
COS-7 cells were seeded in glass-bottom Petri dishes (35-mm diameter) and transfected with HA-tagged TASK-1 alone or together with stx8 or stx7. Fluorescence imaging experiments with COS-7 cells were performed 24 h after transfection. Cells were blocked with 1× phosphate-buffered saline (PBS) containing 5% FCS (Life Technologies) for 30 min at room temperature. Channels at the cell surface were labeled with mouse anti-HA antibody (1:1000; Sigma-Aldrich) at 4°C for 60 min. The cells were then washed three times with PBS at 4°C to remove unbound antibody. Subsequently, the labeled channels were allowed to internalize for 0, 15, or 30 min at 37°C. The cells were then fixed for 10 min at 4°C in PBS containing 4% paraformaldehyde. The channels remaining at the surface were labeled with a saturating concentration of Alexa Fluor 594–conjugated goat anti-mouse secondary antibody (1:1000; Molecular Probes, Darmstadt, Germany). After cell permeabilization with 0.1% Triton X-100 for 5 min at room temperature, internalized channels were labeled with Alexa Fluor 488–conjugated goat anti-mouse secondary antibody (1:5000; Molecular Probes). Then the cells were washed extensively with PBS buffer and covered with the antifade agent Mowiol 4-88 (Carl Roth, Karlsruhe, Germany; containing 25 μg/μl DABCO). The cells were visualized using an Olympus IX71 microscope equipped with a 60× objective (PlanApo 60×/1.40 Oil; Olympus, Hamburg, Germany), a cooled 12-bit charge-coupled device (CCD) camera (SensiCam QE, PCO, Kelheim, Germany), and the corresponding filters for the fluorescent dyes. Images were acquired with Image-Pro Plus 4.5 (Media Cybernetics, Rockville, MD) and analyzed with NIS Elements AR 4 software (Nikon, Düsseldorf, Germany). Channel endocytosis was quantified by taking the ratio between the fluorescence of the Alexa Fluor 488–coupled secondary antibody (green, internalized channels) and the fluorescence of the Alexa Fluor 594–coupled secondary antibody (red, channels at the cell surface). The background green fluorescence measured after application of Mowiol 4-88 was very low (between 0.1 and 1% of the red fluorescence; see Figure 5B); it varied between different Petri dishes, probably due to the presence of a very small fraction of damaged cells.
Live-cell imaging
HeLa cells were analyzed 24–48 h after transfection using an inverted Nikon Eclipse Ti microscope equipped with a 100× objective (Plan Apo VC 100× Oil DIC N2; Nikon). EGFP and mCherry/DsRed channels were acquired simultaneously using a dual camera port system composed of custom-made excitation/emission filters, a dual-band beam splitter, and two cooled 14-bit electron-multiplying CCD cameras (DU-885; Andor Technology, Belfast, UK). For recording live-cell image sequences, cells were maintained at 37°C by means of a stage heater (ibidi, Martinsried, Germany) with a temperature control system (TC 20; npi, Tamm, Germany) and an objective heater (PeCon, Erbach, Germany). Images and sequences were acquired and analyzed with NIS Elements AR 4 software (Nikon).
Total internal reflection microscopy
HeLa cells were analyzed 48 h after transfection using an inverted Leica AF 6000 LX TIRF microscope equipped with a 100× objective (HCX PL APO 100× 1.47 Oil; Leica) and a 12-bit CCD camera (DFC350FXR2; Leica); the temperature was 28–30°C. The penetration depth of the evanescent wave was 70 and 90 nm, respectively, for the lasers exciting EGFP and mCherry. The exposure time was set individually for each cell and channel to obtain the best signal-to-noise ratio. Image sequences were acquired every 0.5–5 s for ∼10 min with LAS AF software (Leica Microsystems, Wetzlar, Germany), and data were postprocessed using NIS Elements AR 4 software.
Statistics
Data are reported as means ± SEM. Statistical significance was determined using Student's t test. In the figures, statistically significant differences from control values are marked by asterisks: *p < 0.05; **p < 0.01; ***p < 0.001.
Supplementary Material
Acknowledgments
We thank Doris Wagner and Kirsten Ramlow for excellent technical support. Some of the constructs used were generously supplied by Harald Stenmark (Oslo University Hospital, Oslo, Norway; 2×FYVE-EGFP, syntaxin-7), Wanjin Hong (Institute of Molecular and Cell Biology, Singapore; VAMP8), Harvey McMahon (MRC Laboratory of Molecular Biology, Cambridge, United Kingdom; AP180C), and Ralf Jacob (Institute of Cytobiology, Marburg, Germany; clathrin light chain). This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB593-TP4 and FOR1086-TP7 to J.D.) and the P. E. Kempkes Foundation (to V.R.).
Abbreviations used:
- AP180C
C‑terminus of adaptor protein 180
- GFP
green fluorescent protein
- HA
hemagglutinin
- myc
myelocytomatosis oncogene–derived dekapeptide
- SNARE
soluble N-ethylmaleimide–factor attachment protein receptor
- stx
syntaxin
- TASK-1
TREK-related acid-sensitive K+ channel
- VAMP2
vesicle-associated membrane protein 2
- vti1b
vesicle transport through interaction with target-SNARE analogue 1b
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-10-0592) on April 17, 2014.
REFERENCES
- Atlas D. The voltage-gated calcium channel functions as the molecular switch of synaptic transmission. Annu Rev Biochem. 2013;82:607–635. doi: 10.1146/annurev-biochem-080411-121438. [DOI] [PubMed] [Google Scholar]
- Bachnoff N, Cohen-Kutner M, Trus M, Atlas D. Intra-membrane signaling between the voltage-gated Ca2+-channel and cysteine residues of syntaxin 1A coordinates synchronous release. Sci Rep. 2013;3:1620. doi: 10.1038/srep01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandulik S, Penton D, Barhanin J, Warth R. TASK1 and TASK3 potassium channels: determinants of aldosterone secretion and adrenocortical zonation. Horm Metab Res. 2010;42:450–457. doi: 10.1055/s-0029-1243601. [DOI] [PubMed] [Google Scholar]
- Bayliss DA, Barrett PQ. Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol Sci. 2008;29:566–575. doi: 10.1016/j.tips.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnia R, Munro S. Organelle identity and the signposts for membrane traffic. Nature. 2005;438:597–604. doi: 10.1038/nature04397. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Zhong P, Scheller RH, Tsien RW. Molecular determinants of the functional interaction between syntaxin and N-type Ca2+ channel gating. Proc Natl Acad Sci USA. 2000;97:13943–13948. doi: 10.1073/pnas.220389697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilan F, Nacfer M, Fresquet F, Norez C, Melin P, Martin-Berge A, Costa de Beauregard MA, Becq F, Kitzis A, Thoreau V. Endosomal SNARE proteins regulate CFTR activity and trafficking in epithelial cells. Exp Cell Res. 2008;314:2199–2211. doi: 10.1016/j.yexcr.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Bilan F, Thoreau V, Nacfer M, Derand R, Norez C, Cantereau A, Garcia M, Becq F, Kitzis A. Syntaxin 8 impairs trafficking of cystic fibrosis transmembrane conductance regulator (CFTR) and inhibits its channel activity. J Cell Sci. 2004;117:1923–1935. doi: 10.1242/jcs.01070. [DOI] [PubMed] [Google Scholar]
- Bittner S, et al. TASK1 modulates inflammation and neurodegeneration in autoimmune inflammation of the central nervous system. Brain. 2009;132:2501–2516. doi: 10.1093/brain/awp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20:427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Chang N, Liang T, Lin X, Kang Y, Xie H, Feng ZP, Gaisano HY. Syntaxin-1A interacts with distinct domains within nucleotide-binding folds of sulfonylurea receptor 1 to inhibit beta-cell ATP-sensitive potassium channels. J Biol Chem. 2011;286:23308–23318. doi: 10.1074/jbc.M111.217950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C, Liang T, Kang Y, Lin X, Xie H, Feng ZP, Gaisano HY. Syntaxin-1A inhibits KATP channels by interacting with specific conserved motifs within sulfonylurea receptor 2A. J Mol Cell Cardiol. 2011a;51:790–802. doi: 10.1016/j.yjmcc.2011.08.011. [DOI] [PubMed] [Google Scholar]
- Chao CC, Mihic A, Tsushima RG, Gaisano HY. SNARE protein regulation of cardiac potassium channels and atrial natriuretic factor secretion. J Mol Cell Cardiol. 2011b;50:401–407. doi: 10.1016/j.yjmcc.2010.11.018. [DOI] [PubMed] [Google Scholar]
- Chen PC, Bruederle CE, Gaisano HY, Shyng SL. Syntaxin 1A regulates surface expression of beta-cell ATP-sensitive potassium channels. Am J Physiol Cell Physiol. 2011;300:C506–C516. doi: 10.1152/ajpcell.00429.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- Dai XQ, et al. The voltage-dependent potassium channel subunit Kv2.1 regulates insulin secretion from rodent and human islets independently of its electrical function. Diabetologia. 2012;55:1709–1720. doi: 10.1007/s00125-012-2512-6. [DOI] [PubMed] [Google Scholar]
- Decher N, et al. Knock-out of the potassium channel TASK-1 leads to a prolonged QT interval and a disturbed QRS complex. Cell Physiol Biochem. 2011;28:77–86. doi: 10.1159/000331715. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M. TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 1997;16:5464–5471. doi: 10.1093/emboj/16.17.5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzioni A, Siloni S, Chikvashvilli D, Strulovich R, Sachyani D, Regev N, Greitzer-Antes D, Hirsch JA, Lotan I. Regulation of neuronal M-channel gating in an isoform-specific manner: functional interplay between calmodulin and syntaxin 1A. J Neurosci. 2011;31:14158–14171. doi: 10.1523/JNEUROSCI.2666-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenburger BH, Jensen JB, Dickson EJ, Suh BC, Hille B. Phosphoinositides: lipid regulators of membrane proteins. J Physiol. 2010;588:3179–3185. doi: 10.1113/jphysiol.2010.192153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Sutton RB, Brunger AT, Jahn R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci USA. 1998;95:15781–15786. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinshreiber L, Chikvashvili D, Michaelevski I, Lotan I. Syntaxin modulates Kv1.1 through dual action on channel surface expression and conductance. Biochem. 2009;48:4109–4114. doi: 10.1021/bi9002088. [DOI] [PubMed] [Google Scholar]
- Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000;19:4577–4588. doi: 10.1093/emboj/19.17.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillooly DJ, Simonsen A, Stenmark H. Cellular functions of phosphatidylinositol 3-phosphate and FYVE domain proteins. Biochem J. 2001;355:249–258. doi: 10.1042/0264-6021:3550249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagalili Y, Bachnoff N, Atlas D. The voltage-gated Ca2+ channel is the Ca2+ sensor protein of secretion. Biochem. 2008;47:13822–13830. doi: 10.1021/bi801619f. [DOI] [PubMed] [Google Scholar]
- Haucke V, De Camilli P. AP-2 recruitment to synaptotagmin stimulated by tyrosine-based endocytic motifs. Science. 1999;285:1268–1271. doi: 10.1126/science.285.5431.1268. [DOI] [PubMed] [Google Scholar]
- Heitzmann D, et al. Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J. 2008;27:179–187. doi: 10.1038/sj.emboj.7601934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson LP, Kelly BT, McCoy AJ, Gaffry T, James LC, Collins BM, Honing S, Evans PR, Owen DJ. A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell. 2010;141:1220–1229. doi: 10.1016/j.cell.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs—engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Kasai K, Akagawa K. Roles of the cytoplasmic and transmembrane domains of syntaxins in intracellular localization and trafficking. J Cell Sci. 2001;114:3115–3124. doi: 10.1242/jcs.114.17.3115. [DOI] [PubMed] [Google Scholar]
- Lakadamyali M, Rust MJ, Zhuang X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell. 2006;124:997–1009. doi: 10.1016/j.cell.2005.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Doray B, Govero J, Kornfeld S. Binding of cargo sorting signals to AP-1 enhances its association with ADP ribosylation factor 1-GTP. J Cell Biol. 2008;180:467–472. doi: 10.1083/jcb.200709037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MT, Mishra A, Lambright DG. Structural mechanisms for regulation of membrane traffic by rab GTPases. Traffic. 2009;10:1377–1389. doi: 10.1111/j.1600-0854.2009.00942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Leung YM, Kang Y, Gao X, Xia F, Xie H, Sheu L, Tsuk S, Lotan I, Tsushima RG, Gaisano HY. Syntaxin 1A binds to the cytoplasmic C terminus of Kv2.1 to regulate channel gating and trafficking. J Biol Chem. 2003;278:17532–17538. doi: 10.1074/jbc.M213088200. [DOI] [PubMed] [Google Scholar]
- Leung YM, Kang Y, Xia F, Sheu L, Gao X, Xie H, Tsushima RG, Gaisano HY. Open form of syntaxin-1A is a more potent inhibitor than wild-type syntaxin-1A of Kv2.1 channels. Biochem J. 2005;387:195–202. doi: 10.1042/BJ20041625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung YM, Kwan EP, Ng B, Kang Y, Gaisano HY. SNAREing voltage-gated K+ and ATP-sensitive K+ channels: tuning beta-cell excitability with syntaxin-1A and other exocytotic proteins. Endocr Rev. 2007;28:653–663. doi: 10.1210/er.2007-0010. [DOI] [PubMed] [Google Scholar]
- Limberg SH, et al. TASK-1 channels may modulate action potential duration of human atrial cardiomyocytes. Cell Physiol Biochem. 2011;28:613–624. doi: 10.1159/000335757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerke D, Mettlen M, Yarar D, Jaqaman K, Jaqaman H, Danuser G, Schmid SL. Cargo and dynamin regulate clathrin-coated pit maturation. PLoS Biol. 2009;7:e57. doi: 10.1371/journal.pbio.1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malsam J, Kreye S, Söllner TH. Membrane fusion: SNAREs and regulation. Cell Mol Life Sci. 2008;65:2814–2832. doi: 10.1007/s00018-008-8352-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mant A, Williams S, O'Kelly I. Acid sensitive background potassium channels K2P3.1 and K2P9.1 undergo rapid dynamin-dependent endocytosis. Channels (Austin) 2013;7:288–292. doi: 10.4161/chan.25120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maritzen T, Koo SJ, Haucke V. Turning CALM into excitement: AP180 and CALM in endocytosis and disease. Biol Cell. 2012;104:588–602. doi: 10.1111/boc.201200008. [DOI] [PubMed] [Google Scholar]
- Mathie A, Rees KA, El Hachmane MF, Veale EL. Trafficking of neuronal two pore domain potassium channels. Curr Neuropharmacol. 2010;8:276–286. doi: 10.2174/157015910792246146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera R, Boehm M, Chaudhuri R, Prabhu Y, Bonifacino JS. Conservation and diversification of dileucine signal recognition by adaptor protein (AP) complex variants. J Biol Chem. 2011;286:2022–2030. doi: 10.1074/jbc.M110.197178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- McNew JA. Regulation of SNARE-mediated membrane fusion during exocytosis. Chem Rev. 2008;108:1669–1686. doi: 10.1021/cr0782325. [DOI] [PubMed] [Google Scholar]
- McNew JA, Parlati F, Fukuda R, Johnston RJ, Paz K, Paumet F, Sollner TH, Rothman JE. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 2000;407:153–159. doi: 10.1038/35025000. [DOI] [PubMed] [Google Scholar]
- Mettlen M, Loerke D, Yarar D, Danuser G, Schmid SL. Cargo- and adaptor-specific mechanisms regulate clathrin-mediated endocytosis. J Cell Biol. 2010;188:919–933. doi: 10.1083/jcb.200908078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuth SG, Budde T, Kanyshkova T, Broicher T, Munsch T, Pape HC. Contribution of TWIK-related acid-sensitive K+ channel 1 (TASK1) and TASK3 channels to the control of activity modes in thalamocortical neurons. J Neurosci. 2003;23:6460–6469. doi: 10.1523/JNEUROSCI.23-16-06460.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucadyil TJ, Schmid SL. Conserved functions of membrane active GTPases in coated vesicle formation. Science. 2009;325:1217–1220. doi: 10.1126/science.1171004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzke C, et al. The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc Res. 2007;75:59–68. doi: 10.1016/j.cardiores.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Rajan S, Wischmeyer E, Liu GX, Preisig-Müller R, Daut J, Karschin A, Derst C. TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histidine as pH sensor. J Biol Chem. 2000;275:16650–16657. doi: 10.1074/jbc.M000030200. [DOI] [PubMed] [Google Scholar]
- Renigunta V, et al. The retention factor p11 confers an endoplasmic reticulum-localization signal to the potassium channel TASK-1. Traffic. 2006;7:168–181. doi: 10.1111/j.1600-0854.2005.00375.x. [DOI] [PubMed] [Google Scholar]
- Schiekel J, Lindner M, Hetzel A, Wemhöner K, Renigunta V, Schlichthörl G, Decher N, Oliver D, Daut J. The inhibition of the potassium channel TASK-1 in rat cardiac muscle by endothelin-1 is mediated by phospholipase C. Cardiovasc Res. 2013;97:97–105. doi: 10.1093/cvr/cvs285. [DOI] [PubMed] [Google Scholar]
- Singer-Lahat D, Chikvashvili D, Lotan I. Direct interaction of endogenous Kv channels with syntaxin enhances exocytosis by neuroendocrine cells. PLoS One. 2008;3:e1381. doi: 10.1371/journal.pone.0001381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Daut J, Schwappach B. Membrane proteins as 14-3-3 clients in functional regulation and intracellular transport. Physiology (Bethesda) 2011;26:181–191. doi: 10.1152/physiol.00042.2010. [DOI] [PubMed] [Google Scholar]
- Springer S, Schekman R. Nucleation of COPII vesicular coat complex by endoplasmic reticulum to Golgi vesicle SNAREs. Science. 1998;281:698–700. doi: 10.1126/science.281.5377.698. [DOI] [PubMed] [Google Scholar]
- Subramaniam VN, Loh E, Horstmann H, Habermann A, Xu Y, Coe J, Griffiths G, Hong W. Preferential association of syntaxin 8 with the early endosome. J Cell Sci. 2000;113:997–1008. doi: 10.1242/jcs.113.6.997. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang BL, Gee HY, Lee MG. The cystic fibrosis transmembrane conductance regulator's expanding SNARE interactome. Traffic. 2011;12:364–371. doi: 10.1111/j.1600-0854.2011.01161.x. [DOI] [PubMed] [Google Scholar]
- Traub LM. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat Rev Mol Cell Biol. 2009;10:583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- Weiss N, Zamponi GW. Regulation of voltage-gated calcium channels by synaptic proteins. Adv Exp Med Biol. 2012;740:759–775. doi: 10.1007/978-94-007-2888-2_33. [DOI] [PubMed] [Google Scholar]
- Zuzarte M, Heusser K, Renigunta V, Schlichthörl G, Rinné S, Wischmeyer E, Daut J, Schwappach B, Preisig-Müller R. Intracellular traffic of the K+ channels TASK-1 and TASK-3: role of N- and C-terminal sorting signals and interaction with 14-3-3 proteins. J Physiol. 2009;587:929–952. doi: 10.1113/jphysiol.2008.164756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.