

The RING-domain E3 ubiquitin ligase deltex3L regulates ubiquitination of ESCRT-0 and endosomal sorting of CXCR4 by inhibiting the activity of the HECT-domain E3 ubiquitin ligase AIP4.
Abstract
G protein–coupled receptor (GPCR) sorting into the degradative pathway is important for limiting the duration and magnitude of signaling. Agonist activation of the GPCR CXCR4 induces its rapid ubiquitination and sorting to lysosomes via the endosomal sorting complex required for transport (ESCRT) pathway. We recently reported that ESCRT-0 ubiquitination is linked to the efficiency with which CXCR4 is sorted for lysosomal degradation; however mechanistic insight is lacking. Here we define a novel role for the really interesting new gene–domain E3 ubiquitin ligase deltex-3-like (DTX3L) in regulating CXCR4 sorting from endosomes to lysosomes. We show that DTX3L localizes to early endosomes upon CXCR4 activation and interacts directly with and inhibits the activity of the E3 ubiquitin ligase atrophin-1 interacting protein 4. This serves to limit the extent to which ESCRT-0 is ubiquitinated and is able to sort CXCR4 for lysosomal degradation. Therefore we define a novel role for DTX3L in GPCR endosomal sorting and reveal an unprecedented link between two distinct E3 ubiquitin ligases to control the activity of the ESCRT machinery.
INTRODUCTION
G protein–coupled receptors (GPCRs) mediate the cellular responses of most hormones and neurotransmitters. GPCR signaling is under tight regulatory control, and any perturbations may be deleterious (Hanyaloglu and von Zastrow, 2008; Marchese et al., 2008). In general, upon ligand binding, GPCRs are rapidly phosphorylated and internalized through clathrin-coated pits via a β-arrestin–dependent pathway to early endosomes (Moore et al., 2007). On endosomes, GPCRs are sorted into either a recycling or a degradative pathway (Hanyaloglu and von Zastrow, 2008; Marchese et al., 2008). GPCRs that sort into the recycling pathway restore the cell surface complement of receptors accessible to ligand, thereby contributing to functional recovery of hormonal responsiveness. GPCRs that sort into the degradative pathway are targeted for lysosomal degradation, leading to loss in total receptor complement and long-term attenuation of signaling. Proper endosomal sorting is necessary to ensure appropriate GPCR signaling, but mechanistic insight is lacking.
Direct modification of GPCRs by ubiquitin is required for targeting many GPCRs into the degradative pathway (Shenoy, 2007; Hislop and von Zastrow, 2011; Marchese and Trejo, 2013). This has been well characterized for the chemokine receptor CXCR4, in which ubiquitination of carboxyl-terminal lysine residues is necessary for its sorting to lysosomes (Marchese and Benovic, 2001; Bhandari et al., 2009). Sorting of ubiquitinated CXCR4 from early endosomes to lysosomes occurs via the endosomal-sorting complex required for transport (ESCRT) pathway (Marchese et al., 2003a; Malik and Marchese, 2010; Malerod et al., 2007; Valiathan and Resh, 2008). The ESCRT pathway comprises four multisubunit protein complexes (ESCRT-0–III) and the AAA Vps4 complex, which act in a concerted manner to direct ubiquitinated cargo into intraluminal vesicles (ILVs) of multivesicular bodies (MVBs), which then fuse with lysosomes, where degradation occurs (Gruenberg and Stenmark, 2004; Piper and Katzmann, 2007; Hurley, 2008; Raiborg and Stenmark, 2009; Henne et al., 2013). Initial recognition of ubiquitinated cargo such as CXCR4 for targeting into ILVs is mediated by ESCRT-0 (Raiborg et al., 2002; Bilodeau et al., 2003; Katzmann et al., 2003; Marchese et al., 2003a; Slagsvold et al., 2006). ESCRT-0 comprises two subunits—hepatocyte growth factor receptor tyrosine kinase substrate (HRS) and signal-transducing adaptor molecule (STAM)—and is localized to endosomal membranes in part via its ability to bind selectively to phosphatidylinositol 3-phosphate (PI3P), an endosomally enriched phospholipid, via the FYVE domain in HRS (Raiborg et al., 2001; Stenmark et al., 2002). Both ESCRT-0 subunits HRS and STAM have ubiquitin-binding domains (UBDs; Bilodeau et al., 2002; Raiborg et al., 2002; Prag et al., 2007; Ren and Hurley, 2010; Henne et al., 2011), and because ESCRT-0 can exist as heterotetrameric complexes on endosomal membranes, it is possible that ESCRT-0 might bind several ubiquitin moieties simultaneously (Mayers et al., 2011). On binding and recruitment of cargo, ESCRT-0 then recruits ESCRT-I, followed by other elements of the sorting machinery, which ultimately leads to sorting of the ubiquitinated cargo into ILVs (Raiborg and Stenmark, 2009; Henne et al., 2011).
A common feature of ubiquitin-binding proteins is that they are also subject to monoubiquitination (Polo et al., 2002; Hicke and Dunn, 2003) via a mechanism referred to as coupled monoubiquitination that involves their own UBD (Woelk et al., 2006). Both ESCRT-0 subunits HRS and STAM are subject to monoubiquitination. HRS ubiquitination is predicted to inhibit its sorting activity, possibly by inducing an autoinhibitory conformation such that the ubiquitin moiety is believed to interact with its own UBD in either a cis- or a trans-acting manner, thus preventing ESCRT-0 from recognizing ubiquitinated cargo and performing its sorting function (Hoeller et al., 2006). Activated CXCR4 induces ubiquitination of HRS and STAM, suggesting that CXCR4 regulates its own down-regulation by regulating the ubiquitination status of the sorting machinery. We showed that when HRS ubiquitination is blocked, CXCR4 sorting to lysosomes is enhanced (Malik and Marchese, 2010). However, this process remains poorly understood.
The E3 ubiquitin ligase atrophin-1 interacting protein 4 (AIP4) mediates ubiquitination of ESCRT-0 and CXCR4 (Marchese et al., 2003b). AIP4 and its mouse orthologue, Itch, belong to the Nedd4 family of homologous to the E6AP carboxy terminus (HECT)–domain E3 ubiquitin ligases and mediate ubiquitination of a diverse set of proteins involved in discrete cellular processes (Rotin and Kumar, 2009). AIP4 is believed to exist in an autoinhibitory conformation that may be relieved by either phosphorylation (Gallagher et al., 2006) or protein–protein binding (Mund and Pelham, 2009). AIP4 interacts with and regulates the stability or activity of other E3 ligases. AIP4 interacts with deltex-1 (DTX1), a really interesting new gene (RING)–domain E3 ubiquitin ligase, and mediates its ubiquitination and lysosomal degradation, thereby controlling ligand-independent Notch signaling (Chastagner et al., 2006). AIP4 also interacts with Cbl-c, also a RING-domain E3 ubiquitin ligase, which act synergistically in mediating ubiquitination and lysosomal degradation of the epidermal growth factor receptor (EGFR; Courbard et al., 2002). Whether these ligases regulate AIP4-dependent ubiquitination and/or endosomal sorting of CXCR4 remains to be determined.
Here we show for the first time that the RING-domain E3 ubiquitin ligase deltex-3-like (DTX3L) regulates CXCR4 trafficking to lysosomes. On CXCR4 activation, DTX3L colocalizes with CXCR4 on early endosomes, where it interacts with AIP4 to regulate the ubiquitination status of ESCRT-0. Our data suggest that DTX3L regulates ESCRT-0 ubiquitination and its sorting activity by controlling AIP4 ligase activity, thereby titrating the amount of CXCR4 that is sorted for lysosomal degradation.
RESULTS
DTX3L mediates CXCR4 degradation
The deltex (DTX) family of ubiquitin ligases comprises three closely related proteins (DTX1–3) and a distantly related protein known as deltex-3-like (DTX3L). Members of the DTX family of proteins are mainly known for their role in Notch signaling (Matsuno et al., 1995, 1998, 2002; Takeyama et al., 2003; Yamada et al., 2011). DTX ubiquitin ligases have a highly conserved carboxyl-terminal RING domain (Takeyama et al., 2003). DTX1 and DTX2 possess a central proline-rich region (PRR) and a basic N-terminal region that binds to intracellular Notch ankyrin repeats. In contrast, DTX3 and DTX3L contain a unique N-terminus and lack the central proline-rich region; however, DTX3 and DTX3L are highly homologous to the other DTX family proteins within the C-terminal RING domain (Takeyama et al., 2003). To examine whether the DTX proteins regulate lysosomal trafficking of CXCR4, we performed a small interfering RNA (siRNA) screen to determine whether these ligases are involved in agonist-induced degradation of CXCR4. HeLa cells were transiently transfected with siRNA directed against DTX family members, and degradation of endogenous CXCR4 was examined by immunoblot analysis in cells treated with vehicle (phosphate-buffered saline [PBS] plus 0.1% bovine serum albumin [BSA]) or 10 nM CXCL12 for 3 h. Consistent with our previously published data (Marchese et al., 2003b), siRNA directed against AIP4 inhibits agonist-induced CXCR4 degradation (Figure 1). Agonist-induced degradation of CXCR4 was significantly inhibited in cells transfected with siRNA directed against DTX3L (Figure 1). However, siRNA directed against DTX1-3 had no effect on CXCR4 degradation (unpublished data). These data indicate that DTX3L mediates CXCR4 degradation.
FIGURE 1:

A role for DTX3L in CXCR4 degradation. (A) HeLa cells transfected with siRNA directed against control, AIP4, or DTX3L were treated with vehicle (PBS + 0.1% BSA) or 10 nM CXCL12 for 3 h. Whole-cell lysates were analyzed for the levels of endogenous CXCR4 and the indicated proteins by immunoblotting (IB). (B) CXCR4 levels normalized to actin were determined by densitometric analysis. Data represent the average percentage CXCR4 degraded in CXCL12-treated vs. vehicle-treated cells. Representative immunoblots are shown in A, and B represents quantification of eight independent experiments. Error bars represent SEM. Data were analyzed by a one-way ANOVA (p < 0.0001), followed by Bonferroni's posthoc test. CXCR4 degradation in AIP4 (p < 0.001) and DTX3L (p < 0.001) siRNA–treated cells was significantly different from siRNA control (siCtrl).
To gain insight into the role that DTX3L has in CXCR4 trafficking, we next examined the distribution of endogenous DTX3L in HeLa cells treated with CXCL12 by confocal immunofluorescence microscopy. In vehicle-treated cells, DTX3L distribution is mainly diffuse, although a fraction also shows a punctate distribution (Figure 2, A and B). In cells treated with CXCL12, the number of DTX3L puncta significantly increased compared with vehicle-treated cells (Figure 2, A and B). To determine the identity of these DTX3L-containing vesicles, we costained HeLa cells for the early endosomal and late endosomal/lysosomal markers early endosomal antigen 1 (EEA1) and lysosome-associated membrane protein 2 (LAMP2), respectively. There was a significant increase in the number of DTX3L puncta that colocalized with EEA1 upon CXCL12 treatment compared with vehicle, as determined by calculating the Pearson product–moment correlation coefficient, suggesting that DTX3L is recruited to early endosomes (Figure 2, A and C). The amount of DTX3L colocalizing with LAMP2 was not affected by CXCL12 treatment, indicating that DTX3L localization to lysosomes is not dependent upon CXCR4 activation (Figure 2, A and D).
FIGURE 2:

DTX3L localizes to early endosomes upon CXCR4 activation. (A) HeLa cells were treated with vehicle or CXCL12 for 30 min. Cells were then fixed, permeabilized, and incubated with antibodies directed against DTX3L and the early endosomal marker EEA1 or the lysosomal marker LAMP2. Inset represents 3× enlargement of the boxed region. Yellow in the merged image indicates colocalization between DTX3L and EEA1 or LAMP2. Differential interference contrast (DIC) images are shown. Equal acquisition settings (gain and intensity) were used between parallel samples within each experiment. Representative images from four independent experiments. (B) CXCR4 activation increases DTX3L puncta number in HeLa cells. DTX3L puncta were counted using ImageJ software. Data represent the average puncta count from 35–45 cells from four independent experiments. Error bars represent SEM (not visible). (C, D) Pearson product–moment correlation coefficient calculated to determine the level of colocalization between DTX3L and EEA1 (C) and LAMP2 (D). Data were analyzed by Student's t test.
Because CXCR4 is sorted from early endosomes to lysosomes, where it is degraded (Marchese and Benovic, 2001; Bhandari et al., 2007), and given that DTX3L localizes to early endosomes upon CXCR4 activation, it is possible that DTX3L regulates CXCR4 sorting from early endosomes to lysosomes. To determine this, we examined the distribution of endogenous CXCR4 by confocal immunofluorescence microscopy in control or DTX3L siRNA–transfected cells treated with vehicle or CXCL12 for 3 h. In vehicle-treated cells, CXCR4 was localized mainly to the plasma membrane in both control and DTX3L siRNA-treated cells (unpublished data), similar to what we previously reported (Malik and Marchese, 2010). On agonist treatment, in control siRNA–treated cells, CXCR4 significantly colocalized with the late endosomal/lysosomal marker LAMP2 and only partially colocalized with the early endosomal marker EEA1 (Figure 3). In contrast, in DTX3L siRNA–treated cells, CXCR4 significantly colocalized with EEA1 and not LAMP2, indicating that trafficking from endosomes to lysosomes was impaired in DTX3L-depleted cells (Figure 3). Consistent with this, there were significantly more CXCR4 puncta in DTX3L-depleted cells than with control (Figure 3C), indicating that CXCR4 was not degraded. Taken together, these data suggest that DTX3L is necessary for sorting CXCR4 from early endosomes to lysosomes.
FIGURE 3:

DTX3L regulates CXCR4 sorting from early endosomes to lysosomes. (A) HeLa cells transfected with siRNA directed against control or DTX3L were treated with vehicle (PBS + 0.1% BSA) or 10 nM CXCL12 for 3 h. Cells were then fixed, permeabilized, and incubated with antibodies directed against CXCR4, DTX3L, early endosomal marker EEA1, or late endosomal/lysosomal marker LAMP2. Inset represents 3× enlargement of the boxed region. Yellow in the merged image indicates colocalization between CXCR4 and EEA1 or LAMP2. DIC images are shown. Equal acquisition settings (gain and intensity) were used between parallel samples within each experiment. Representative images are from four independent experiments. (B) Pearson product–moment correlation coefficient determined using the ImageJ plug-in Colocalization Finder. Data represent the average from four independent experiments. Data were analyzed by a one-way ANOVA (p < 0.001), followed by Bonferroni's posthoc test. (C) CXCR4 puncta counted using the ImageJ function Analyze Particles. Data represent the average puncta count from 45–60 cells from three independent experiments. Error bars represent SEM. Data were analyzed by Student's t test.
DTX3L interacts directly with AIP4
Because DTX3L colocalizes with CXCR4 on early endosomes and regulates CXCR4 endosomal sorting, we hypothesized that DTX3L may interact with key endosomal proteins to regulate CXCR4 lysosomal trafficking. Because AIP4 may be found in complex with other E3 ligases, including DTX1 (Chastagner et al., 2006; Courbard et al., 2002), and regulates CXCR4 endosomal sorting (Marchese et al., 2003b), we next examined whether AIP4 interacts with DTX3L. Endogenous AIP4 was observed in immunoprecipitates of endogenous DTX3L from HeLa cells but not immunoglobulin G (IgG) isotype control (Figure 4A). A glutathione S-transferase (GST) fusion protein of full-length AIP4 (GST-AIP4), but not GST alone, interacts with endogenous DTX3L in HeLa cells (Supplemental Figure S1) and with recombinant histidine (His)-DTX3L, in a concentration-dependent manner, indicating that the interaction is direct (Figure 4B).
FIGURE 4:

AIP4 and DTX3L interact directly and form a complex in HeLa cells. (A) Cleared lysates from HeLa cells (500 μg) were incubated with either anti-IgG control or anti-DTX3L antibodies to immunoprecipitate endogenous DTX3L. (B) Equimolar amounts of His-tagged DTX3L (1–10 nM) were incubated with equal amounts of purified GST-AIP4 (100 nmol). Samples were resolved by 10% SDS–PAGE and analyzed by immunoblotting for DTX3L or GST. Input represents 10% of HIS-DTX3L used in the binding reaction. Data represent the fold change in GST-AIP4 binding to HIS-DTX3L. Data are representative of three independent experiments. Data were analyzed by one-way ANOVA, followed by Bonferroni's posthoc test. Data show a significant increase in His-DTX3L binding to GST-AIP4 with increasing His-DTX3L concentration. (C) HeLa lysates (500 μg) were treated with CXCL12 (10 nM) and incubated with either anti-IgG control or anti-DTX3L antibodies to immunoprecipitate endogenous DTX3L. Samples were resolved by 10% SDS–PAGE and analyzed by immunoblotting with the indicated antibodies. The amount of AIP4 binding was quantitated by densitometric analysis from four independent experiments and then analyzed by one-way ANOVA, followed by followed by Bonferroni's posthoc test for significance (p < 0.05). Error bars represent the SEM. (D) HeLa cells transfected with FLAG-tagged AIP4 were treated with 10 nM CXCL12 for 0–60 min. Cells were fixed, permeabilized, and incubated with antibodies directed against FLAG-AIP4, DTX3L, and EEA1. Representative images are from three independent experiments. DIC images are shown. Inset, 3× enlargement of the boxed region. Equal acquisition settings (gain and intensity) were used between parallel samples within each experiment. (E) Immunoblot showing level of FLAG-AIP4 expression over endogenous AIP4. (F) FLAG-AIP4 and DTX3L show strong colocalization, as determined by calculating the Pearson product–moment correlation coefficient. (G, H) Puncta were counted using the particle analysis software of ImageJ. Data represent the average FLAG-AIP4 (G) or DTX3L (H) puncta per cell from four independent experiments. Data were analyzed by one-way ANOVA, followed by Bonferroni's posthoc test.
We next examined whether the interaction between DTX3L and AIP4 is regulated by CXCR4 activation. Treatment of cells with CXCL12 significantly enhanced the interaction between endogenous DTX3L and AIP4 and was maximal after 15 min of CXCL12 treatment (Figure 4C). Further, FLAG-tagged AIP4 showed significant colocalization with endogenous DTX3L on EEA1-labeled early endosomes upon CXCL12 treatment (Figure 4, D and F). The levels of FLAG-AIP4 puncta were maximal between 15 and 30 min of agonist treatment and returned toward basal levels by 60 min (Figure 4, D and G), whereas DTX3L puncta remained associated with early endosomes for up to 60 min (Figure 4, D and H). FLAG-AIP4 was transiently expressed approximately twofold to threefold over endogenous AIP4 (Figure 4E). Taken together, these data indicate that DTX3L interacts directly with AIP4 upon CXCR4 activation on early endosomes.
DTX3L regulates the ubiquitination of ESCRT-0 subunits HRS and STAM-1
We previously showed that AIP4 controls CXCR4 endosomal sorting in part by regulating ubiquitination of ESCRT-0 (Marchese et al., 2003b). ESCRT-0 subunits HRS and STAM1 also coimmunoprecipitated with DTX3L in addition to AIP4 (Figure 4A). We next assessed whether DTX3L regulates ESCRT-0 ubiquitination in HeLa cells. As shown in Figure 5A, DTX3L depletion leads to significant increase in HRS ubiquitination or hyperubiquitination (∼2.5-fold) compared with control. We also observed a modest increase in STAM-1 ubiquitination (∼1.6-fold) in cells depleted of DTX3L compared with control (Figure 5B). DTX3L was not detected in the immunoprecipitates of HRS (Figure 5A) or STAM-1 (Figure 5B) because the ubiquitination experiments were performed under denaturing conditions. Taken together, these data indicate that DTX3L negatively regulates ESCRT-0 ubiquitination. Hyperubiquitination of HRS or STAM does not change their levels in cells; therefore DTX3L does not regulate their stability by promoting their degradation (Figure 5, A and B). We previously proposed that HRS ubiquitination by AIP4 induces an autoinhibitory conformation that may sterically inhibit HRS and, hence, ESCRT-0 from interacting with ubiquitinated CXCR4 (Malik and Marchese, 2010).
FIGURE 5:

DTX3L regulates ESCRT-0 ubiquitination and complex integrity. (A, B) DTX3L regulates the ubiquitination status of ESCRT-0 subunits HRS and STAM-1. HeLa cells were transfected with siRNA directed against control or DTX3L plus FLAG-ubiquitin and T7-HRS (A) or T7-STAM-1 (B). Denatured cleared lysates (300 μg) were incubated with an anti-T7 polyclonal antibody for 1 h and then protein G agarose for 1 h. Bound proteins were eluted in 2× sample buffer and resolved by 7% SDS–PAGE and analyzed by immunoblotting with the indicated antibodies. Immunoblots are representative of three independent experiments. Data were quantified by densitometric analyses and followed by Student's t test. Ubiquitination of T7-HRS or T7-STAM-1 in control siRNA–treated samples was normalized to 1 and compared with ubiquitination in the DTX3L siRNA–treated samples. Error bars represent SEM. (C, D) DTX3L regulates the integrity of ESCRT-0. HeLa cells were transfected and siRNA directed against control or DTX3L. Cleared cell lysates (300 μg) were incubated with control IgG and HRS (C) or STAM-1 (D) antibodies. Bound proteins were eluted in 2× sample buffer, resolved by 7% SDS–PAGE, and analyzed by immunoblotting with the indicated antibodies. Immunoblots are representative of three or four independent experiments. Data were quantified by densitometric analysis, and control siRNA–treated samples were normalized to 1 for either HRS or STAM-1 and compared with binding in the DTX3L siRNA–treated samples. Data were analyzed by Student's t test.
It is also possible that hyperubiquitination may affect the integrity of the ESCRT-0 complex by preventing HRS and STAM from interacting with each other. To examine this, we determined the ability of HRS and STAM-1 to interact with each other in DTX3L-depleted cells by coimmunoprecipitation. HeLa cells were transiently transfected with siRNA directed against DTX3L or control, followed by reciprocal coimmunoprecipitation and immunoblotting of endogenous HRS and STAM-1. As shown in Figure 5, C and D, coimmunoprecipitation of HRS and STAM-1 significantly decreased in DTX3L siRNA–transfected cells compared with control siRNA, suggesting that when DTX3L is depleted, HRS and STAM-1 are unable to interact with each other. To determine whether this also leads to a reduced amount of ESCRT-0 on early endosomes, we examined the distribution of endogenous HRS and STAM-1 by confocal immunofluorescence microscopy. There was reduced HRS (Figure 6, A and B) and STAM-1 (Figure 6, D and E) punctate staining in DTX3L-depleted cells compared with control. Both HRS (Figure 6C) and STAM-1 (Figure 6F) showed a high level of colocalization with DTX3L in control siRNA–treated cells. Although DTX3L depletion resulted in HRS and STAM-1 hyperubiquitination and decreased localization to endosomes, this did not lead to their degradation because in DTX3L-depleted cells their levels were unchanged compared with control, as determined by immunoblotting (Figure 5, A–D).
FIGURE 6:

DTX3L regulates the distribution of HRS and STAM1. (A, D) HeLa cells transfected with either control or DTX3L siRNA were treated with 10 nM CXCL12 for 30 min. Cells were fixed, permeabilized, and incubated with antibodies directed against DTX3L, HRS, (A) and STAM-1 (D). Inset, 3× enlargement of the boxed region. Yellow in the merged image indicates colocalization between HRS (A) or STAM-1 (D) and DTX3L. DIC images are shown. Equal acquisition settings (gain and intensity) were used between parallel samples within each experiment. (B, E) HRS and STAM-1 puncta are reduced in DTX3L siRNA–treated cells. Puncta were counted using the particle analysis software of ImageJ. Data represent the average HRS (B) or STAM-1 (E) puncta per cell from four and three independent experiments, respectively. For the HRS puncta analysis, 110–145 cells were examined, and for the STAM-1 puncta analysis, 45–50 cells were examined. Data were analyzed by Student's t test. DTX3L shows strong colocalization with HRS (C) and STAM-1 (F) in control siRNA–treated cells, as determined by calculating the Pearson product–moment correlation coefficient.
Because ESCRT-0 is localized to early endosomes in part via interaction between the FYVE domain of HRS and endosomally enriched phospholipid PI3P (Raiborg et al., 2001; Stenmark et al., 2002), it is possible that DTX3L regulates the steady-state levels of PI3P to control ESCRT-0 endosomal localization. To examine this, we transfected cells with a fluorescent-tagged FYVE-domain fusion construct, similar to an approach used by others to monitor PI3P levels in cells by confocal microscopy (Gillooly et al., 2000). As shown in Supplemental Figure S2, in DTX3L-depleted cells, yellow fluorescent protein (YFP)–2×FYVE was localized to early endosomes as compared with control cells. In contrast, wortmannin treatment, which blocks the production of PI3P at the dose used, abolished endosomal localization of YFP-2×FYVE in both control and DTX3L siRNA–treated cells and to a lesser degree EEA1 (Supplemental Figure S2). These data indicate that DTX3L does not regulate the steady-state levels of PI3P. When taken together, the data suggest that DTX3L controls the ubiquitination status of HRS and STAM-1 and the ability of ESCRT-0 to localize to early endosomes and sort CXCR4 for lysosomal degradation. Because DTX3L regulates ESCRT-0 ubiquitination and function, it may have a general role in endosomal sorting, but this remains to be determined.
DTX3L attenuates the E3 ubiquitin ligase activity of AIP4
Next we sought to elucidate the mechanism by which DTX3L regulates the ubiquitination status of ESCRT-0. Because AIP4 and DTX3L interact, we next determined whether DTX3L regulates the E3 ubiquitin ligase activity of AIP4. To examine this, we performed an in vitro ubiquitination assay using purified AIP4, His-DTX3L, and their catalytically inactive mutants (AIP4-C830A; DTX3L-3C/A). The catalytically inactive mutants are unable to load ubiquitin directly (AIP4-C830A) or indirectly (DTXL3L-3C/A) and therefore serve as negative controls. As shown in Figure 7A, AIP4 (lane 6, top) and DTX3L (lane 8, middle) self-ubiquitinate, which is a common feature of E3 ubiquitin ligases. Conversely, the AIP4-C830A (Figure 7A, lane 7, top) and the DTX3L-3C/A (Figure 7A, lane 9, middle) mutants were unable to self-ubiquitinate, confirming that they are indeed catalytically inactive. These data also reveal that although AIP4 and DTX3L interact, they are unable to ubiquitinate each other. AIP4 was unable to ubiquitinate the catalytically inactive DTX3L-3C/A (Figure 7A, lane 11, middle), and DTX3L was unable to ubiquitinate AIP4-C830A (Figure 7A, lane 12, top).
FIGURE 7:
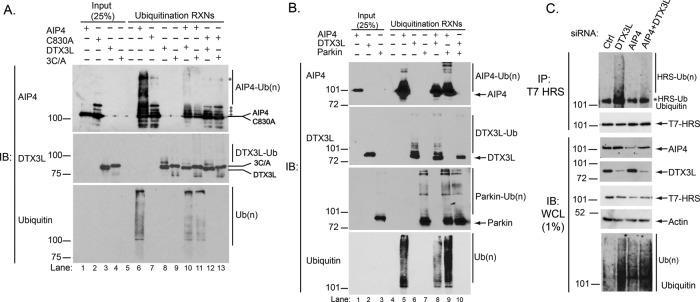
DTX3L inhibits the E3 ubiquitin ligase activity of AIP4. (A) Purified AIP4, AIP4-C830A, HIS-DTX3L, or HIS-DTX3L-RING domain mutant (3C/A) were incubated with ATP, E1, E2 (UbcH5c), and ubiquitin in a final volume of 20 μl at room temperature for 1½ h. Reactions were stopped by the addition of 20 μl of 2× sample buffer. Proteins were resolved by 7% SDS–PAGE and analyzed by immunoblotting with the indicated antibodies. The C830A cleavage from GST was not complete, resulting in detection of multiple bands in both the input and reaction lanes (indicated with asterisks) that correspond to uncleaved and degraded products of GST-C830A, as determined by immunoblot analysis with an anti-GST antibody (unpublished data). A high–molecular weight nonspecific band is also noted with an asterisk. Data are representative of six independent experiments. (B) Purified AIP4, HIS-DTX3L, or MBP-Parkin were incubated with ATP, E1, E2 (UbcH5c), and ubiquitin in a final volume of 20 μl at room temperature for 1½ h. Reactions were stopped by the addition of 20 μl of 2× sample buffer. Proteins were resolved by 7% SDS–PAGE and analyzed by immunoblotting with the indicated antibodies. Data are representative of four independent experiments. (C) Hyperubiquitination of HRS in DTX3L-depleted cells is suppressed by AIP4 depletion. HeLa cells were transfected with siRNA directed against control, DTX3L, or AIP4 plus FLAG-ubiquitin and T7-HRS. Samples were processed as described in the legend to Figure 5, A and B. Data are representative of four independent experiments.
When AIP4 and DTX3L were coincubated, there was a large decrease in ubiquitination of AIP4 compared with AIP4 alone (Figure 7A, lane 10 vs. lane 6, top). There was also a small decrease in DTX3L ubiquitination when DTX3L was coincubated with AIP4 (Figure 7A, lane 10 vs. lane 8, middle). To confirm these results, we immunoblotted samples for ubiquitin (Figure 7A, bottom). The anti-ubiquitin immunoblot also shows less AIP4 self-ubiquitination when AIP4 was coincubated with DTX3L or DTX3L-3C/A. The anti-ubiquitin (P4D1) antibody recognizes AIP4 self-ubiquitination but not self-ubiquitination of DTX3L, likely because it does not have high affinity for chain linkages produced by DTX3L. Using N- and C-terminal truncations of full-length DTX3L, we were able to deduce the AIP4-binding region on DTX3L to lie within the N-terminal region beyond the DTX3L RING domain (Supplemental Figure S1B). This suggests that binding of AIP4 to the unique N-terminal region of DTX3L seems to be sufficient to prevent AIP4 ligase activity in vitro. Overall these data indicate that DTX3L inhibits AIP4 ligase activity independent of its catalytic activity, suggesting that the interaction of DTX3L with AIP4 inhibits its activity.
To further ensure that the decrease in AIP4 self-ubiquitination seen in the presence of DTX3L was not due to sequestration of ubiquitin, we performed similar in vitro ubiquitination experiments using the E3 ubiquitin ligase Parkin. As shown in Figure 7B, when AIP4 is incubated with Parkin, AIP4 self-ubiquitination is not inhibited compared with when it is incubated with DTX3L. In fact, AIP4 self-ubiquitination may be more efficient in the presence of Parkin due to the presence of high–molecular weight species not present when it is incubated alone (Figure 7B, lane 8 vs. lane 5, top). This was further supported by the ubiquitin blot (Figure 7B, bottom). DTX3L self-ubiquitination was somewhat decreased in the presence of wild-type AIP4 (Figure 7, A, lane 10, and B, lane 8) but was substantially decreased in the presence of Parkin (Figure 7B, lane 10). The reason for this remains to be investigated. Parkin self-ubiquitination was not impaired by AIP4 or DTX3L (Figure 7B), indicating specificity between AIP4 and DTX3L. Taken together, these data further support that DTX3L inhibits AIP4 ubiquitin ligase activity.
To determine whether DTX3L regulates AIP4 ubiquitin ligase activity in cells, we examined the ubiquitination status of HRS, an AIP4 substrate (Marchese et al., 2003b), in HeLa cells depleted of DTX3L or AIP4 alone or together. As shown in Figure 7C, hyperubiquitination of HRS observed when DTX3L was depleted alone was suppressed when DTX3L and AIP4 were depleted simultaneously. These data are consistent with an inhibitory effect of DTX3L on AIP4 ligase activity in cells. DTX3L heterodimerizes with the related E3 ubiquitin ligase DTX1, leading to enhanced DTX1 and/or DTX3L ubiquitin ligase activity (Takeyama et al., 2003). In addition, AIP4 interacts with and mediates polyubiquitination of DTX1, thereby regulating the degradation of DTX1 in lysosomes (Chastagner et al., 2006). Our data here suggest that it is the interaction per se rather than ubiquitination of AIP4 by DTX3L that negatively affects AIP4 activity. AIP4 may exist in an autoinhibitory conformation, and typically phosphorylation or protein binding renders it active (Gallagher et al., 2006; Mund and Pelham, 2009). It is unclear how DTX3L binding to AIP4 may inhibit its activity, although it is conceivable that DTX3L may somehow prevent AIP4 from undergoing a conformational change. It is also possible that DTX3L inhibits AIP4 activity because it affects E2 binding to AIP4 and/or E2 ubiquitin loading. Further investigation will be required to determine this, but regardless of the mechanism, our data indicate that DTX3L attenuates AIP4 ligase activity in cells.
DISCUSSION
Endosomal sorting is essential for regulating GPCR signaling. In this study we defined a novel role for the RING-domain E3 ubiquitin ligase DTX3L in regulating endosomal sorting of the GPCR CXCR4. We showed that DTX3L controls CXCR4 sorting from early endosomes to lysosomes by regulating the ubiquitination status of ESCRT-0 subunits HRS and STAM-1. DTX3L does not directly mediate ubiquitination of HRS and STAM-1, but regulates their ubiquitination by inhibiting the HECT-domain E3 ubiquitin ligase AIP4. Our data are consistent with a model in which, upon CXCR4 activation, endogenous DTX3L is recruited to early endosomes and interacts directly with AIP4 and antagonizes AIP4 ligase activity, thereby limiting the extent to which ESCRT-0 subunits are ubiquitinated (Figure 8). This maintains ESCRT-0 on early endosomes such that it is able to interact with ubiquitinated CXCR4 and sort it for lysosomal degradation. We propose that DTX3L regulates ESCRT-0 sorting activity and controls the amount of CXCR4 that is targeted for lysosomal degradation by serving as an endogenous inhibitor of AIP4 (Figure 8). It is possible that DTX3L regulation of ESCRT-0 may be a general mechanism to regulate sorting of ubiquitinated cargo.
FIGURE 8:
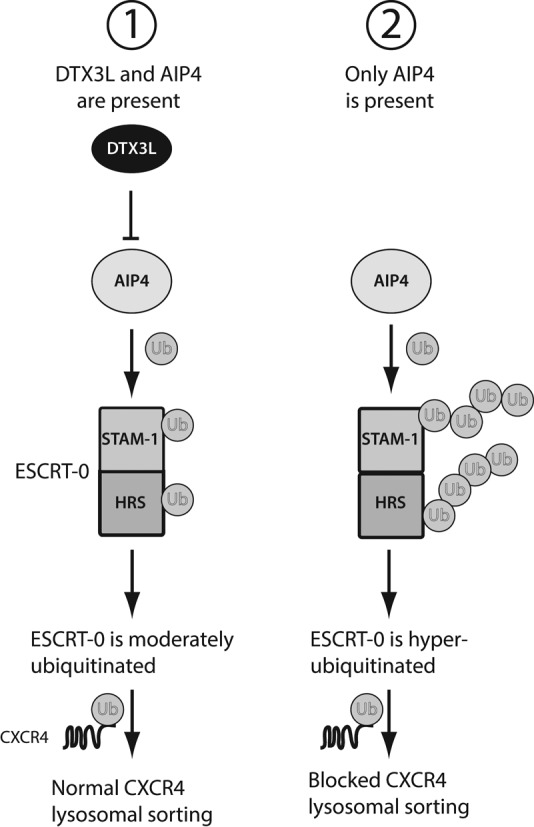
Proposed model for how ESCRT-0 ubiquitination regulates CXCR4 lysosomal sorting. 1) When both DTX3L and AIP4 are present, CXCR4 undergoes rapid agonist-induced lysosomal sorting and degradation. ESCRT-0 is moderately ubiquitinated as AIP4 activity is suppressed by DTX3L, and ESCRT-0 is able to efficiently sort ubiquitinated CXCR4 for lysosomal degradation. 2) When DTX3L levels are reduced by siRNA, AIP4 activity is NOT suppressed and it is able to hyperubiquitinate ESCRT-0. This renders ESCRT-0 nonfunctional because ESCRT-0 subunits HRS and STAM are unable to associate with each other and are no longer localized to endosomes; therefore ESCRT-0 is unable to sort ubiquitinated CXCR4 for lysosomal degradation.
DTX3L (also known as B lymphoma and BAL–associated protein [BBAP]) was first identified as a binding partner to the gene B aggressive lymphoma (BAL; Takeyama et al., 2003). BAL is not a DTX3L substrate, but when BAL1 interacts with DTX3L, it mediates its nuclear localization, where DTX3L has a role in the DNA damage response pathway by mediating monoubiquitination of histone H4 (Yan et al., 2009, 2013). To the best of our knowledge, our study is the first report to describe a role for DTX3L in membrane trafficking. Our data show that DTX3L is mainly cytosolic, but upon CXCR4 activation, DTX3L colocalizes with early endosome marker EEA1 (Figure 2). DTX3L also colocalizes with the late endosome and lysosomal marker LAMP2, but this colocalization does not change upon CXCR4 activation (Figure 2). These data are consistent with DTX3L being recruited to early endosomes upon CXCR4 activation and regulating sorting of CXCR4 to lysosomes. DTX3L may also have a role on late endosomes or lysosomes, but this remains to be investigated. Accordingly, in DTX3L-depleted cells, CXCR4 accumulates on early endosomes and does not traffic to lysosomes (Figure 3). Therefore we have defined a novel role for DTX3L in mediating endosomal sorting of CXCR4.
The recruitment of DTX3L to early endosomes may occur in concert with AIP4. On CXCR4 activation, FLAG-AIP4 and DTX3L localize to early endosomes at similar time points (Figure 4, C and D). We show here that DTX3L interacts directly with AIP4 (Figure 4B) and they exist as a complex in cells that also includes ESCRT-0 subunits HRS and STAM-1 (Figure 4A). Therefore it is possible that AIP4 and, hence, DTX3L are recruited to early endosomes together via ESCRT-0. However, this is unlikely because in the wortmannin-treated cells, DTX3L was still present on early endosomes, whereas, conversely, the positive control YFP-2×FYVE (an ESCRT-0 reporter) showed almost no endosomal localization (Supplemental Figure S2). It remains to be determined how AIP4 and DTX3L are recruited to endosomes. DTX3L remained associated with early endosomes for up to 60 min after agonist treatment, whereas AIP4 showed very little endosomal association at this time point (Figure 4, D–F). Therefore, although AIP4 and DTX3L are recruited to early endosomes together, the retention of DTX3L on early endosomes is likely mediated by other factors. As a consequence, it is conceivable that DTX3L has additional roles at later steps in the sorting process.
Our study links the role of DTX3L on CXCR4 endosomal sorting to ESCRT-0 ubiquitination. In DTX3L-depleted cells, both HRS and STAM-1 are hyperubiquitinated (Figure 5, A and B). ESCRT-0 binds to ubiquitinated cargo such as CXCR4 via the multiple ubiquitin-binding domains (UBDs) in HRS and STAM-1 (Bilodeau et al., 2002; Raiborg et al., 2002; Prag et al., 2007; Ren and Hurley, 2010; Henne et al., 2011). Monoubiquitination of HRS is predicted to induce an autoinhibitory conformation that prevents ESCRT-0 from interacting with ubiquitinated receptors, thereby inhibiting its endosomal sorting function (Hoeller et al., 2006). Here we show that ubiquitination also affects ESCRT-0 complex integrity and localization (Figures 5, C and D, and 6). Hyperubiquitination of HRS and STAM-1 does not lead to their degradation because their levels do not change in DTX3L-depleted cells, and therefore ubiquitination likely regulates endosomal localization of ESCRT-0 (Figure 5). ESCRT-0 localizes to early endosomes in part via the PI3P-binding FYVE domain of HRS (Raiborg et al., 2001). However, loss of ESCRT-0 from endosomes is not due to a decrease in the steady-state endosomal levels of PI3P, as depletion of DTX3L does not affect YFP-2×FYVE domain puncta, used here as a reporter to monitor PI3P levels in cells (Supplemental Figure S2). However, it is possible that multiple ubiquitin moieties and/or polyubiquitin chains attached to HRS and/or STAM-1 could render ESCRT-0 less able to bind to PI3P, leading to its removal from endosomal membranes and its destabilization. Our data contrast with what has been observed in yeast cells, where ubiquitination of Vps27, the orthologue of HRS, does not affect ESCRT-0 sorting activity, as sorting of ubiquitinated cargo remains intact when Vps27 ubiquitination is impaired (Stringer and Piper, 2011). A recent study suggested that HRS ubiquitination is linked to impaired lysosomal sorting of EGFR, which is similar to what we observe here for CXCR4 (Sun et al., 2013). It is possible that yeast and mammalian ESCRT-0 are differentially regulated by ubiquitination, possibly by additional factors that have an effect on its sorting activity.
Our data indicate that DTX3L regulates ESCRT-0 ubiquitination by inhibiting the activity of AIP4. Hyperubiquitination of HRS in DTX3L-depleted cells is suppressed when AIP4 is also depleted (Figure 7C). DTX3L likely inhibits AIP4 via an interaction, as both active and catalytically inactive DTX3L inhibit AIP4 ligase activity (Figure 7A). Taken together, these data suggest that DTX3L may serve as an endogenous inhibitor of AIP4. DTX3L therefore may have an effect on other AIP4 substrates. CXCR4 is also ubiquitinated by AIP4, and we also observed increased ubiquitination of CXCR4 in DTX3L-depleted cells (Supplemental Figure S3). Typically, increased ubiquitination of CXCR4 should promote its degradation and not inhibit it; however, because ESCRT-0 function is attenuated in DTX3L-depleted cells, CXCR4 is unable to be sorted to lysosomes and therefore accumulates on early endosomes (Figure 3). This reinforces the fact that endosomal sorting represents a critical step in targeting GPCRs for lysosomal degradation.
AIP4 has been shown to interact with other RING-domain E3 ubiquitin ligases, including DTX1, Cbl-c, and RNF11. AIP4 interacts with Cbl-c, and they act synergistically in mediating ubiquitination and lysosomal degradation of EGFR (Courbard et al., 2002). Further, although AIP4 interacts with RNF11, to our knowledge, the functional significance of this interaction remains unknown (Kitching et al., 2003). Recently, RNF11 was physically linked to ESCRT-0 and lysosomal degradation of EGFR, but the mechanism remains to be determined (Kostaras et al., 2013). Furthermore, AIP4 interacts with DTX1 and mediates its ubiquitination and lysosomal degradation (Chastagner et al., 2006). DTX3L also interacts with DTX1, resulting in greater activity of each E3 ubiquitin ligase (Takeyama et al., 2003). In contrast, our data reveal that AIP4 and DTX3L have less activity when they interact, which affects ubiquitination of AIP4 substrates (Figure 7, A and C). Whether DTX3L has substrates relevant to GPCR endosomal sorting remains to be determined. We did observe an overall increase in the total level of ubiquitination in cells depleted of DTX3L, as assessed by immunoblotting of whole-cell lysates (Figures 5, A and B, and 7C and Supplemental Figure S3). This could be linked to enhanced ubiquitination of AIP4 substrates in addition to HRS and CXCR4, which we report here, but this remains to be examined. Alternatively, DTX3L may have a general inhibitory effect on cellular ubiquitination, but this seems unlikely, given that DTX3L does not inhibit DTX1 activity (Takeyama et al., 2003). To the best of our knowledge, histone H4 is the only identified DTX3L substrate (Yan et al., 2009).
ESCRT-0 ubiquitination is also regulated by DUBs. AMSH and USP8 are endosomally localized DUBs that regulate the ubiquitination status of HRS or STAM (Berlin et al., 2010; Sierra et al., 2010). Depletion of USP8 by siRNA enhances HRS ubiquitination and inhibits CXCR4 degradation (Berlin et al., 2010). Depletion of AMSH by siRNA does not affect CXCR4 degradation (Malik and Marchese, 2010; Sierra et al., 2010), but expression of AMSH dominant-negative constructs enhances HRS or STAM ubiquitination and inhibits CXCR4 degradation (Sierra et al., 2010). It is possible that DTX3L could be regulating these DUBs, which, when DTX3L is depleted, would be less active, leading to hyperubiquitination of ESCRT-0. However, in AMSH- or USP8-depleted cells, HRS and STAM-1 still colocalize with endosomes (Berlin et al., 2010; Sierra et al., 2010), in contrast to when DTX3L is depleted (Figure 6). AMSH and USP8 have been linked to later stages of endosomal sorting (Clague et al., 2012), but whether this could have an effect on ESCRT-0 endosomal localization remains to be determined. Of interest, DTX3L appears to have the opposite effect on ESCRT-0 ubiquitination than β-arrestin-1 (also known as arrestin-2; Malik and Marchese, 2010). β-Arrestin-1 interacts with ESCRT-0 subunit STAM-1 to promote HRS ubiquitination (Malik and Marchese, 2010). Therefore there may a balance between DTX3L and β-arrestin action on AIP4 that regulates the ubiquitination status of ESCRT-0 and thereby dictates the amount of CXCR4 sorted for degradation.
This study reveals for the first time a novel role for the RING-domain E3 ubiquitin ligase DTX3L in endocytic trafficking. This is distinct from its other known function in the DNA damage response pathway (Yan et al., 2009, 2013). DTX3L has been linked to diffuse large B-cell lymphomas (DLBCLs; Juszczynski et al., 2006). Of interest, high levels of CXCR4 are also found in multiple lymphomas, including DLBCL (Arai et al., 2000), and, recently, CXCR4 expression levels were shown to correlate with distinct DLBCL subtypes (Deutsch et al., 2013). In future studies, it will be interesting to examine whether DTX3L via CXCR4 may be linked to DLBCL and other CXCR4-dependent diseases.
MATERIALS AND METHODS
Cell lines, antibodies, and reagents
HeLa cells were from the American Type Culture Collection (Manassas, VA). Cells were maintained in DMEM (Hyclone Laboratories (Logan, UT)) supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories). The mouse monoclonal antibodies directed against HRS (c-19) and ubiquitin (P4D1) and the goat polyclonal directed against DTX3L (N-16) were from Santa Cruz Biotechnology (Santa Cruz, CA). The CXCR4 (2B11), EEA1, and isotype control antibodies were from BD Biosciences (San Jose, CA). The T7 goat polyclonal antibody was from Abcam (Cambridge, MA). The STAM-1 and HRS rabbit polyclonal antibodies were from the ProteinTech group (Chicago, IL). The anti-LAMP2 (H4B4) antibody was from the Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA). The Itch (AIP4) rabbit monoclonal antibody was from Abcam. The mouse monoclonal anti-FLAG antibody conjugated to horseradish peroxidase (M2-HRP) was from Sigma-Aldrich (St. Louis, MO). The actin antibody was from MP Biomedicals (Aurora, OH). Alexa Fluor 635–conjugated goat anti-mouse, Alexa Fluor 633–conjugated goat anti-rat, Alexa Fluor 488–conjugated goat anti-mouse, Alexa Fluor 555–conjugated donkey anti-goat, and Alexa Fluor 488–conjugated goat anti-rabbit antibodies were from Molecular Probes (Eugene, OR). Mounting medium for immunofluorescence microscopy was from Vector Laboratories (Burlingame, CA). Stromal cell-derived factor-1α (SDF-1α; CXCL12) and epidermal growth factor were from PeproTech (Rocky Hill, NJ). The control luciferase, DTX1-3, DTX3L (GGAGAAAGGAGGCGAAUUA; GENOME SMARTpool 1 Cat No. d-007143-01), and AIP4 (GGUGACAAAGAGCCAACAGAG) siRNAs were from Dharmacon RNA Technologies (Lafayette, CO).
DNA constructs
The cDNA for DTX3L in pCMV-SPORT6 (clone ID 4339554) was obtained from Thermo Scientific (Pittsburgh, PA). The DTX3L cDNA was amplified by PCR, and the product was subcloned into the NotI and XbaI sites of 3×FLAG-pCMV-10 (Sigma-Aldrich), the BamHI and XhoI sites of pGEX-6P-1 (GE Healthcare, Piscataway, NJ) or the BamHI and XhoI sites of pET-21a(+) (a kind gift from Katherine Knight, Loyola University Chicago, Chicago, IL). To generate the N-terminal (NT) and C-terminal (CT) truncations of DTX3L, primers were made based on the DTX3L truncations previously published (Takeyama et al., 2003). Briefly, the NT truncation of DTX3L spans amino acids 1–464, and the CT truncation covers amino acids 528–740. Full-length GST-DTX3L was used as a template in the PCR, and PCR products were subcloned into the BamHI and XhoI sites of pGEX-6P-1. The DTX3L-3C/A mutant was designed based on a DTX1 mutant previously reported (Chastagner et al., 2006). Briefly, cysteine residues 561, 596, and 599 within the RING domain were mutated to alanine residues using two-step PCR and subcloned into pET-21a(+) as described for wild-type DTX3L. The FLAG-tagged AIP4-C830A mutant was made by digesting myc-AIP4-C830A-pRK5 with XhoI and BamHI and subcloning and replacing the equivalent fragment in 3×FLAG-pCMV-10-AIP4. To make GST-AIP4-C830A, the FLAG-AIP4-C830A cDNA was used as a template for PCR and the product was subcloned into the BamHI and NotI sites of pGEX-6P-1. YFP-2×FYVE was made by amplifying amino acid residues 147–223 from HRS by two-step PCR and subcloning in tandem into the XhoI and EcoRI sites of pEYFP-C1 (Clontech, Mountain View, CA). The YFP-2×FYVE construct was expressed in HeLa cells and shown to overlap with EEA1 staining (unpublished data). The cDNAs encoding FLAG-ubiquitin, HA-CXCR4, GST-AIP4, FLAG-AIP4, T7-STAM-1, and T7-HRS were described previously (Bhandari et al., 2009; Malik and Marchese, 2010).
Confocal fluorescence microscopy
HeLa cells plated onto coverslips coated with poly-l-lysine (0.1 mg/ml; Sigma-Aldrich) were serum starved with DMEM supplemented with 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, for 3 h at 37°C. Cells were treated with 10 nM CXCL12 or vehicle for 0–3 h, followed by fixation with 3.7% paraformaldehyde and permeabilization with 0.05% (wt/vol) saponin, as described previously (Malik et al., 2012). Cells were incubated with antibodies directed against CXCR4, DTX3L, EEA1, LAMP2, HRS, STAM-1, or FLAG for 1 h at 37°C, followed by a 30-min incubation at 37°C with Alexa Fluor–conjugated secondary antibodies. We tested several antibodies directed against AIP4 but were unable to detect endogenous AIP4 by immunofluorescence microscopy, and therefore we transiently expressed FLAG-AIP4 to approximately twofold over endogenous levels, as determined by immunoblotting, to study AIP4 distribution in HeLa cells. Cells were washed and mounted on glass slides using mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Slides were viewed using a Zeiss LSM 510 laser-scanning confocal microscope equipped with a Plan-Apo 63×/1.4 numerical aperture oil lens objective (Carl Zeiss, Jena, Germany). Images were acquired using a 1.4-megapixel, cooled extended spectral range RGB digital camera set at 512 × 512 resolution. Equal acquisition settings (gain and intensity) were used between parallel samples within each experiment. Images were analyzed using ImageJ software (version 1.37v; National Institutes of Health, Bethesda, MD) and Photoshop (CS4; Adobe, San Jose, CA). The Pearson product–moment correlation coefficient was determined using the ImageJ plug-in Colocalization Finder. Pixels were restrained to the minimum ratio of 75% to reduce noise from channel bleedthrough. Analysis results in values ranging from 0 to 1, where 0 represents no colocalization and 1 represents absolute colocalization. Puncta analysis was performed using the Analyze Particles macro in ImageJ. Images (8-bit) were manually thresholded to minimum (130–150) and maximum (250) values to exclude background noise. Particles counted were restrained to a size of 0.05–1.0 and circularity of 0.5–1.0.
Degradation assays
CXCR4 degradation was assessed by immunoblot analysis, as previously described (Marchese and Benovic, 2001). HeLa cells grown in six-well plates were transfected with 50 pmol of siRNA directed against DTX family members, AIP4, or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or luciferase using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), as previously described (Marchese et al., 2003b). The next day, cells were passed into 12-well plates and allowed to grow for another 24 h. Cells were washed with DMEM and then treated with 50 μg/ml cyclohexamide for 15 min at 37°C, followed by treatment with vehicle (0.5% BSA in PBS) or 10 nM CXCL12 for 3 h. After treatment, cells were washed once with PBS and collected in 300 μl of 2× sample buffer (8% SDS, 10% glycerol, 0.7 M β-mercaptoethanol, 37.5 mM Tris HCl, pH 6.5, 0.003% bromophenol blue). Equal amounts of samples were analyzed by 10% SDS–PAGE and immunoblotting with antibodies directed against CXCR4, DTX3L, AIP4, actin, or tubulin. Receptor degradation was determined by densitometric analysis of similar enhanced chemiluminescent exposures from multiple experiments and calculated as the percentage receptor degraded in CXCL12-treated cells compared with vehicle.
Protein purification
Escherichia coli BL21 cells transformed with cDNA encoding GST fusion proteins in pGEX-6p1 or His-tagged proteins in pET-21a(+) were grown overnight in Luria broth (Fisher Scientific, Pittsburgh, PA) containing 100 μg/ml ampicillin. Cultures were diluted (3.7%) the following morning and grown to an OD600 of 0.35–0.40 at 37°C. Protein expression was induced with 0.1–0.3 mM isopropyl-1-thio-β-d-galactopyranoside (Sigma-Aldrich) for 1–3 h at 18°C. Cells were then pelleted by centrifugation and resuspended in 0.7 ml of lysis buffer (20 mM Tris-Cl, pH 7.4, 150 mM NaCl, 0.1% Triton X-100, 1 mM dithiothreitol [DTT], and 10 μg/ml each of leupeptin, aprotinin, and pepstatin-A). DTT was omitted when purifying His-tagged proteins. Cells were lysed by sonication and clarified by centrifugation. For GST fusion protein purifications, clarified lysates were incubated with 25 μl of a 50% slurry of glutathione–Sepharose 4B (GE Healthcare) resin overnight while rocking at 4°C for 15–17 h. Samples were washed three times, and equal amounts (100 nmol) of each immobilized fusion protein were incubated with cleared HeLa lysates transiently transfected without or with 1 μg FLAG-AIP4 (500 μg of lysate for endogenous DTX3L binding or 100 μg for the overexpression of FLAG-AIP4). Samples were incubated at 4°C for 15–17 or 1 h, respectively, followed by immunoblot analysis. For His-tagged protein purification, clarified lysates were incubated with 20 μl of a 50% slurry of Talon Metal Affinity Beads (Clontech) resin overnight while rocking at 4°C. Samples were quickly washed three times and resuspended in 100 μl of lysis buffer. To determine direct binding of GST-AIP4 to His-DTX3L, increasing concentrations of His-DTX3L (5-10 nM) were incubated with equal amounts of GST-AIP4 (100 nmol) for 1 h at 4°C, followed by immunoblot analysis.
To cleave the GST tag from GST-AIP4 and GST-C830A, immobilized proteins were incubated with 10 U of PreScission Protease (GE Healthcare) for 12 h while rocking at 4°C. For His-tagged proteins, proteins were eluted in lysis buffer containing 100 mM imidazole while rocking at 4°C. Elution fractions were dialyzed in dialysis buffer (50 mM Tris-Cl, pH 7.4) overnight and concentrated using Spectra/Gel absorbent (Spectrum Labs, Rancho Dominguez, CA). Samples were snap frozen and stored at −80°C until use.
In-cell and in vitro ubiquitination assays
For HRS and STAM ubiquitination, HeLa cells grown on 6-cm dishes were transfected with 1 μg of FLAG-ubiquitin and 4 μg of T7-HRS or T7-STAM-1 and control siRNA or siRNA directed against DTX3L, AIP4, or the combination (25 nM final concentration for each siRNA) using Lipofectamine 2000, essentially as previously described (Marchese et al., 2003b). The next day, cells were washed twice with ice-cold PBS and collected into 200 μl of denaturing ubiquitination buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% SDS, 1% Triton X-100, 5 mM EDTA, 20 mM N-ethylmaleimide, and protease inhibitors [10 μg/ml each aprotinin, leupeptin, and pepstatin A; Roche, Indianapolis, IN]). Samples were boiled for 5 min, sonicated, and diluted with 1.8 ml of dilution buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 5 mM EDTA). Samples were cleared by centrifugation, and protein concentrations were determined using the BCA Protein Assay Kit (Pierce, Rockford, IL). A total of 300 μg of supernatant was incubated with 2 μg of an anti-T7 goat polyclonal antibody directed overnight while rocking at 4°C. Samples were then incubated with 20 μl of a 50% slurry of Protein G agarose (Roche) for 1 h while rocking at 4°C. Bound proteins were eluted in 25 μl of 2× sample buffer, resolved by 7% SDS–PAGE, and then analyzed by immunoblotting with antibodies directed against the FLAG and T7 epitopes, DTX3L, or actin. For CXCR4 ubiquitination, we essentially followed a protocol identical to one previously described (Marchese et al., 2003b; Marchese, 2009).
In vitro ubiquitination assays were performed using GST-purified/cleaved AIP4 or AIP4-C830A-HECT mutant and His-DTX3L or DTX3L-3C/A RING mutant alone or in combination. Briefly, purified E3s (500 ng) were incubated alone or together with E1 (0.5 μg), E2 (UbcH5; 0.5 μg), and ubiquitin (2.5 μg) and ATP in a final volume of 20 μl for 1½ h at room temperature. Reactions were stopped by the addition of 20 μl of 2× sample buffer. For the Parkin experiments, purified MBP-Parkin, AIP4, and His-DTX3L were allowed to react alone and in combination. Samples were separated by 7% SDS–PAGE and analyzed by immunoblotting with antibodies directed against DTX3L, AIP4, Parkin, and anti-ubiquitin (P4D1).
Coimmunoprecipitation studies
HeLa cells plated in 10-cm dishes were serum starved for 4 h and then treated for 0–60 min with 10 nM CXCL12. Cells were placed on ice, washed once with ice-cold PBS, and lysed in immunoprecipitation buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, protease inhibitors [10 μg/ml each aprotinin, leupeptin, and pepstatin A; Roche]). Lysates were cleared by centrifugation at 13,000 rpm for 10 min in a 5417r-Eppendorf microcentrifuge. Clarified lysates (500 μg) were incubated with an antibody (2 μg) directed against DTX3L or goat IgG control for 16 h at 4°C. Then 20 μl of a 50% slurry of protein G agarose was added, and samples were incubated for an additional 1 h. Immunoprecipitated proteins were denatured by the addition of 20 μl of 2× sample buffer, resolved by 10% SDS–PAGE, and analyzed by immunoblotting using antibodies directed against AIP4, DTX3L, and actin. For the endogenous coimmunoprecipitation experiments, lysates were collected in a similar manner but left untreated. Specifically, for the ESCRT-0 coimmunoprecipitation, HeLa cells were transfected with control siRNA or DTX3L siRNA (25 nM final concentration) for 24 h before immunoprecipitation of cleared lysates (300 μg) with 2 μg of either anti-HRS or mouse IgG control and anti-STAM-1 or rabbit IgG control for 1 h at 4°C. Then 20 μl of a 50% slurry of protein A agarose was added, and samples were incubated for an additional 1 h. Immunoblots were probed with antibodies against AIP4, DTX3L, HRS, STAM1, or actin. STAM2 reagents do not allow unambiguous identification in the immunoprecipitates. Data were quantified by densitometric analysis. For the time-course coimmunoprecipitation analysis, vehicle values were set to 1, and treatment times were represented as a fraction of vehicle. In the ESCRT-0 coimmunoprecipitation, control siRNA values were set to 1, and DTX3L siRNA samples were calculated as a fraction of the control siRNA.
Statistical analysis
Data were analyzed by Student's t test and one-way analysis of variance (ANOVA) using Prism 4.0 for Macintosh (GraphPad Software, San Diego, CA)
Supplementary Material
Acknowledgments
We thank Rohit Malik for making the YFP-2×FYVE construct. MBP-Parkin was kindly provided by Vsevolod Gurevich (Vanderbilt University, Nashville, TN). This work was supported by National Institutes of Health Grant GM106727 to A.M. J.H. is supported by an American Heart Association predoctoral fellowship (13PRE14280030.)
Abbreviations used:
- AIP4
atrophin-1 interacting protein 4
- DTX3L
deltex-3-like
- EEA1
early endosomal antigen 1
- ESCRT
endosomal sorting complex required for transport
- GPCR
G protein–coupled receptor
- HECT
homologous to the E6AP carboxy terminus
- HRS
hepatocyte growth factor receptor tyrosine kinase substrate
- ILV
intraluminal vesicle
- LAMP2
lysosome-associated membrane protein 2
- MVB
multivesicular body
- RING
really interesting new gene
- STAM
signal-transducing adaptor molecule
- UBD
ubiquitin-binding domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-10-0612) on April 30, 2014.
J.H. designed, performed, and analyzed the experiments and prepared the manuscript; A.M. contributed to project design, data analysis, and manuscript preparation.
REFERENCES
- Arai J, Yasukawa M, Yakushijin Y, Miyazaki T, Fujita S. Stromal cells in lymph nodes attract B-lymphoma cells via production of stromal cell-derived factor-1. Eur J Haematol. 2000;64:323–332. doi: 10.1034/j.1600-0609.2000.90147.x. [DOI] [PubMed] [Google Scholar]
- Berlin I, Higginbotham KM, Dise RS, Sierra MI, Nash PD. The deubiquitinating enzyme USP8 promotes trafficking and degradation of the chemokine receptor 4 at the sorting endosome. J Biol Chem. 2010;285:37895–37908. doi: 10.1074/jbc.M110.129411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari D, Robia SL, Marchese A. The E3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol Biol Cell. 2009;20:1324–1339. doi: 10.1091/mbc.E08-03-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari D, Trejo J, Benovic JL, Marchese A. Arrestin-2 interacts with the ubiquitin-protein isopeptide ligase atrophin-interacting protein 4 and mediates endosomal sorting of the chemokine receptor CXCR4. J Biol Chem. 2007;282:36971–36979. doi: 10.1074/jbc.M705085200. [DOI] [PubMed] [Google Scholar]
- Bilodeau PS, Urbanowski JL, Winistorfer SC, Piper RC. The Vps27p Hse1p complex binds ubiquitin and mediates endosomal protein sorting. Nat Cell Biol. 2002;4:534–539. doi: 10.1038/ncb815. [DOI] [PubMed] [Google Scholar]
- Bilodeau PS, Winistorfer SC, Kearney WR, Robertson AD, Piper RC. Vps27-Hse1 and ESCRT-I complexes cooperate to increase efficiency of sorting ubiquitinated proteins at the endosome. Eur J Cell Biol. 2003;163:237–243. doi: 10.1083/jcb.200305007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chastagner P, Israel A, Brou C. Itch/AIP4 mediates deltex degradation through the formation of K29-linked polyubiquitin chains. EMBO Rep. 2006;7:1147–1153. doi: 10.1038/sj.embor.7400822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Coulson JM, Urbe S. Cellular functions of the DUBs. J Cell Sci. 2012;125:277–286. doi: 10.1242/jcs.090985. [DOI] [PubMed] [Google Scholar]
- Courbard JR, Fiore F, Adelaide J, Borg JP, Birnbaum D, Ollendorff V. Interaction between two ubiquitin-protein isopeptide ligases of different classes, CBLC and AIP4/ITCH. J Biol Chem. 2002;277:45267–45275. doi: 10.1074/jbc.M206460200. [DOI] [PubMed] [Google Scholar]
- Deutsch AJ, Steinbauer E, Hofmann NA, Strunk D, Gerlza T, Beham-Schmid C, Schaider H, Neumeister P. Chemokine receptors in gastric MALT lymphoma: loss of CXCR4 and upregulation of CXCR7 is associated with progression to diffuse large B-cell lymphoma. Mod Pathol. 2013;26:182–194. doi: 10.1038/modpathol.2012.134. [DOI] [PubMed] [Google Scholar]
- Gallagher E, Gao M, Liu YC, Karin M. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc Natl Acad Sci USA. 2006;103:1717–1722. doi: 10.1073/pnas.0510664103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000;19:4577–4588. doi: 10.1093/emboj/19.17.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol. 2004;5:317–323. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Dev Cell. 2011;21:77–91. doi: 10.1016/j.devcel.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Henne WM, Stenmark H, Emr SD. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a016766. a016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Hislop JN, von Zastrow M. Role of ubiquitination in endocytic trafficking of G-protein-coupled receptors. Traffic. 2011;12:137–148. doi: 10.1111/j.1600-0854.2010.01121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeller D, et al. Regulation of ubiquitin-binding proteins by monoubiquitination. Nat Cell Biol. 2006;8:163–169. doi: 10.1038/ncb1354. [DOI] [PubMed] [Google Scholar]
- Hurley JH. ESCRT complexes and the biogenesis of multivesicular bodies. Curr Opin Cell Biol. 2008;20:4–11. doi: 10.1016/j.ceb.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszczynski P, Kutok JL, Li C, Mitra J, Aguiar RC, Shipp MA. BAL1 and BBAP are regulated by a gamma interferon-responsive bidirectional promoter and are overexpressed in diffuse large B-cell lymphomas with a prominent inflammatory infiltrate. Mol Cell Biol. 2006;26:5348–5359. doi: 10.1128/MCB.02351-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann DJ, Stefan CJ, Babst M, Emr SD. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol. 2003;162:413–423. doi: 10.1083/jcb.200302136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitching R, Wong MJ, Koehler D, Burger AM, Landberg G, Gish G, Seth A. The RING-H2 protein RNF11 is differentially expressed in breast tumours and interacts with HECT-type E3 ligases. Biochim Biophys Acta. 2003;1639:104–112. doi: 10.1016/j.bbadis.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Kostaras E, Sflomos G, Pedersen NM, Stenmark H, Fotsis T, Murphy C. SARA and RNF11 interact with each other and ESCRT-0 core proteins and regulate degradative EGFR trafficking. Oncogene. 2013;32:5220–5232. doi: 10.1038/onc.2012.554. [DOI] [PubMed] [Google Scholar]
- Malerod L, Stuffers S, Brech A, Stenmark H. Vps22/EAP30 in ESCRT-II mediates endosomal sorting of growth factor and chemokine receptors destined for lysosomal degradation. Traffic. 2007;8:1617–1629. doi: 10.1111/j.1600-0854.2007.00630.x. [DOI] [PubMed] [Google Scholar]
- Malik R, Marchese A. Arrestin-2 interacts with the endosomal sorting complex required for transport machinery to modulate endosomal sorting of CXCR4. Mol Biol Cell. 2010;21:2529–2541. doi: 10.1091/mbc.E10-02-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Soh UJ, Trejo J, Marchese A. Novel roles for the E3 ubiquitin ligase atrophin-interacting protein 4 and signal transduction adaptor molecule 1 in G protein-coupled receptor signaling. J Biol Chem. 2012;287:9013–9027. doi: 10.1074/jbc.M111.336792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A. Ubiquitination of chemokine receptors. Methods Enzymol. 2009;460:413–422. doi: 10.1016/S0076-6879(09)05221-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem. 2001;276:45509–45512. doi: 10.1074/jbc.C100527200. [DOI] [PubMed] [Google Scholar]
- Marchese A, Chen C, Kim YM, Benovic JL. The ins and outs of G protein-coupled receptor trafficking. Trends Biochem Sci. 2003a;28:369–376. doi: 10.1016/S0968-0004(03)00134-8. [DOI] [PubMed] [Google Scholar]
- Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell. 2003b;5:709–722. doi: 10.1016/s1534-5807(03)00321-6. [DOI] [PubMed] [Google Scholar]
- Marchese A, Trejo J. Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell Signal. 2013;25:707–716. doi: 10.1016/j.cellsig.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno K, Diederich RJ, Go MJ, Blaumueller CM, Artavanis-Tsakonas S. Deltex acts as a positive regulator of Notch signaling through interactions with the Notch ankyrin repeats. Development. 1995;121:2633–2644. doi: 10.1242/dev.121.8.2633. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Eastman D, Mitsiades T, Quinn AM, Carcanciu ML, Ordentlich P, Kadesch T, Artavanis-Tsakonas S. Human deltex is a conserved regulator of Notch signalling. Nat Genet. 1998;19:74–78. doi: 10.1038/ng0598-74. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Ito M, Hori K, Miyashita F, Suzuki S, Kishi N, Artavanis-Tsakonas S, Okano H. Involvement of a proline-rich motif and RING-H2 finger of Deltex in the regulation of Notch signaling. Development. 2002;129:1049–1059. doi: 10.1242/dev.129.4.1049. [DOI] [PubMed] [Google Scholar]
- Mayers JR, Fyfe I, Schuh AL, Chapman ER, Edwardson JM, Audhya A. ESCRT-0 assembles as a heterotetrameric complex on membranes and binds multiple ubiquitinylated cargoes simultaneously. J Biol Chem. 2011;286:9636–9645. doi: 10.1074/jbc.M110.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- Mund T, Pelham HR. Control of the activity of WW-HECT domain E3 ubiquitin ligases by NDFIP proteins. EMBO Rep. 2009;10:501–507. doi: 10.1038/embor.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper RC, Katzmann DJ. Biogenesis and function of multivesicular bodies. Annu Rev Cell Dev Biol. 2007;23:519–547. doi: 10.1146/annurev.cellbio.23.090506.123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo S, Sigismund S, Faretta M, Guidi M, Capua MR, Bossi G, Chen H, De Camilli P, Di Fiore PP. A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature. 2002;416:451–455. doi: 10.1038/416451a. [DOI] [PubMed] [Google Scholar]
- Prag G, Watson H, Kim YC, Beach BM, Ghirlando R, Hummer G, Bonifacino JS, Hurley JH. The Vps27/Hse1 complex is a GAT domain-based scaffold for ubiquitin-dependent sorting. Dev Cell. 2007;12:973–986. doi: 10.1016/j.devcel.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H. HRS sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol. 2002;4:394–398. doi: 10.1038/ncb791. [DOI] [PubMed] [Google Scholar]
- Raiborg C, Bremnes B, Mehlum A, Gillooly DJ, D'Arrigo A, Stang E, Stenmark H. FYVE and coiled-coil domains determine the specific localisation of Hrs to early endosomes. J Cell Sci. 2001;114:2255–2263. doi: 10.1242/jcs.114.12.2255. [DOI] [PubMed] [Google Scholar]
- Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- Ren X, Hurley JH. VHS domains of ESCRT-0 cooperate in high-avidity binding to polyubiquitinated cargo. EMBO J. 2010;29:1045–1054. doi: 10.1038/emboj.2010.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol. 2009;10:398–409. doi: 10.1038/nrm2690. [DOI] [PubMed] [Google Scholar]
- Shenoy SK. Seven-transmembrane receptors and ubiquitination. Circ Res. 2007;100:1142–1154. doi: 10.1161/01.RES.0000261939.88744.5a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra MI, Wright MH, Nash PD. AMSH interacts with ESCRT-0 to regulate the stability and trafficking of CXCR4. J Biol Chem. 2010;285:13990–14004. doi: 10.1074/jbc.M109.061309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slagsvold T, Marchese A, Brech A, Stenmark H. CISK attenuates degradation of the chemokine receptor CXCR4 via the ubiquitin ligase AIP4. EMBO J. 2006;25:3738–3749. doi: 10.1038/sj.emboj.7601267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H, Aasland R, Driscoll PC. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett. 2002;513:77–84. doi: 10.1016/s0014-5793(01)03308-7. [DOI] [PubMed] [Google Scholar]
- Stringer DK, Piper RC. A single ubiquitin is sufficient for cargo protein entry into MVBs in the absence of ESCRT ubiquitination. J Cell Biol. 2011;192:229–242. doi: 10.1083/jcb.201008121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Hedman AC, Tan X, Schill NJ, Anderson RA. Endosomal type Igamma PIP 5-kinase controls EGF receptor lysosomal sorting. Dev Cell. 2013;25:144–155. doi: 10.1016/j.devcel.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeyama K, Aguiar RC, Gu L, He C, Freeman GJ, Kutok JL, Aster JC, Shipp MA. The BAL-binding protein BBAP and related Deltex family members exhibit ubiquitin-protein isopeptide ligase activity. J Biol Chem. 2003;278:21930–21937. doi: 10.1074/jbc.M301157200. [DOI] [PubMed] [Google Scholar]
- Valiathan RR, Resh MD. Differential control of CXCR4 and CD4 downregulation by HIV-1 Gag. Virol J. 2008;5:23. doi: 10.1186/1743-422X-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woelk T, Oldrini B, Maspero E, Confalonieri S, Cavallaro E, Di Fiore PP, Polo S. Molecular mechanisms of coupled monoubiquitination. Nat Cell Biol. 2006;8:1246–1254. doi: 10.1038/ncb1484. [DOI] [PubMed] [Google Scholar]
- Yamada K, Fuwa TJ, Ayukawa T, Tanaka T, Nakamura A, Wilkin MB, Baron M, Matsuno K. Roles of Drosophila deltex in Notch receptor endocytic trafficking and activation. Genes Cells. 2011;16:261–272. doi: 10.1111/j.1365-2443.2011.01488.x. [DOI] [PubMed] [Google Scholar]
- Yan Q, Dutt S, Xu R, Graves K, Juszczynski P, Manis JP, Shipp MA. BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol Cell. 2009;36:110–120. doi: 10.1016/j.molcel.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Xu R, Zhu L, Cheng X, Wang Z, Manis J, Shipp MA. BAL1 and its partner E3 ligase, BBAP, link Poly(ADP-ribose) activation, ubiquitylation, and double-strand DNA repair independent of ATM, MDC1, and RNF8. Mol Cell Biol. 2013;33:845–857. doi: 10.1128/MCB.00990-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.