

Ubp3 is an antisilencing factor. Accordingly, loss of Upb3 leads to lower RNAPII occupancy in heterochromatic regions and suppression of unequal recombination in rDNA. However, ubp3Δ mutants have a shortened replicative life span, suggesting that recombination frequency is not directly correlated with aging.
Abstract
Ubp3 is a conserved ubiquitin protease that acts as an antisilencing factor in MAT and telomeric regions. Here we show that ubp3∆ mutants also display increased silencing in ribosomal DNA (rDNA). Consistent with this, RNA polymerase II occupancy is lower in cells lacking Ubp3 than in wild-type cells in all heterochromatic regions. Moreover, in a ubp3∆ mutant, unequal recombination in rDNA is highly suppressed. We present genetic evidence that this effect on rDNA recombination, but not silencing, is entirely dependent on the silencing factor Sir2. Further, ubp3∆ sir2∆ mutants age prematurely at the same rate as sir2∆ mutants. Thus our data suggest that recombination negatively influences replicative life span more so than silencing. However, in ubp3∆ mutants, recombination is not a prerequisite for aging, since cells lacking Ubp3 have a shorter life span than isogenic wild-type cells. We discuss the data in view of different models on how silencing and unequal recombination affect replicative life span and the role of Ubp3 in these processes.
INTRODUCTION
In eukaryotes, transcription by RNA polymerase II (RNAPII) is highly influenced by chromatin. In general, eukaryotic chromosomes are organized into transcriptionally active euchromatin and repressed heterochromatin. Placement of nucleosomes—relative binding sites of transcription factors or RNAPII—has a great effect on transcriptional initiation. Thus, simply by impeding access to binding sites of transcription factors, nucleosomes can inhibit transcriptional preinitiation complex formation. In euchromatin, histone acetylation is associated with transcriptional activity, and it has been shown that acetylated histones destabilize their interactions with DNA as well as nucleosomes (Lee et al., 1993; Wang and Hayes, 2008). Even though there are cases in which deacetylation can activate gene transcription (De Nadal et al., 2004), hypoacetylated histones are considered as repressive to transcription. For instance, histone H4 acetylated on lysine 16 (H4K16ac) is found on chromatin throughout the genome except in transcriptionally silent heterochromatin (Suka et al., 2001; Smith et al., 2002).
The domains of chromatin-mediated silencing in Saccharomyces cerevisiae are found at the subtelomeric regions, within the ribosomal DNA repeats (rDNAs) and at the cryptic mating-type loci HMR and HML (Rusche et al., 2003). HMR and HML comprise genes (a1, a2 and α1, α2, respectively) that encode transcriptional regulators that are controlled by flanking cis-acting elements called silencers. Origin recognition complex, Rap1, and Abf1 bind these silencers and initiate formation of silent chromatin by recruiting Sir2, Sir3, and Sir4 (Strahl-Bolsinger et al., 1997). H4K16ac is deacetylated by Sir2, which now, being hypoacetylated, has a higher affinity for the Sir complex. The spreading of Sir complexes propagates through iterative cycles of H4K16 deacetylation by Sir2. Limiting levels of Sir proteins, histone acetylation, and the presence of the histone variant H2A.Z counteract improper spreading of silent chromatin into euchromatin (Suka et al., 2002; Meneghini et al., 2003). A similar mode of action takes place at telomeres, resulting in silent chromatin that gradually decreases with distance from the telomere. In rDNA, the scaffold for Sir2 recruitment is different from telomeres and mating-type loci. Here the nucleolar protein Net1 and the phosphatase Cdc14 recruit Sir2, and together they form the regulator of nucleolar silencing and telophase exit (RENT) complex (Huang and Moazed, 2003). To maintain stringent control of transcription in heterochromatin, all of these silencing factors are required. The importance of maintaining heterochromatin integrity should not be underestimated. For example, in eukaryotes, transcription-associated recombination (TAR) is a fundamental process that is important for DNA integrity. Kobayashi and Ganley (2005) proposed a model of TAR in rDNA in which RNAPII transcription of noncoding RNA (ncRNA) induces recombination, which can lead to accumulation of extrachromosomal DNA and loss of rDNA. In extension, these events may lead to premature aging (Sinclair and Guarente, 1997). Consistent with this, in mutants or cells devoid of any of the RENT subunits, multiple severe phenotypes have been observed, including premature aging, rearrangement and/or loss of genetic material, and disease (Burhans and Weinberger, 2012).
Deubiquitinating enzymes (DUBs) catalyze the reversal of ubiquitination of target proteins. DUBs play a key role in various biological processes, including protein degradation, cell cycle control, stress response, DNA repair, immune response, signal transduction, gene regulation, endocytosis, and vesicle trafficking, and constitute one of the largest classes of proteases and are crucial players in the ubiquitin–proteasome system (UPS). DUBs also process ubiquitin precursors and are responsible for the reversal of ubiquitination to prevent degradation or modify substrate activity (Amerik and Hochstrasser, 2004). In yeast, several ubiquitin-specific processing proteases (UBPs) have been identified. UBP3, the yeast homologue of human USP10, encodes a 101.9-kDa DUB that, together with its cofactor, Bre5, forms a complex that can regulate 1) anterograde and retrograde transport between endoplasmic reticulum and Golgi (Cohen et al., 2003a, b), 2) DNA repair (Mao and Smerdon, 2010), and 3) protein aggregate clearance (Oling et al., 2014).
Ubp3 has also been shown to participate in and regulate transcription. For instance, Ubp3 has a positive role in activating osmoresponsive genes (Sole et al., 2011) and properly inducing PHO5 (Kvint et al., 2008). In addition, Ubp3 physically interacts with TFIID (Auty et al., 2004), Tbp1 (Chew et al., 2010), and RNAPII (Kvint et al., 2008). Both Tbp1 and RNAPII can be saved from 26S proteasomal degradation by Ubp3 (Kvint et al., 2008; Chew et al., 2010). Moreover, loss of Ubp3 increases silencing at silent mating-type loci and telomeric DNA regions (Moazed and Johnson, 1996).
In this work we report that Ubp3 acts as an antisilencing factor at all three loci that display silencing in S. cerevisiae based on growth assays on selective media using reporter genes. Using chromatin immunoprecipitation (ChIP) assays, we demonstrate that RNAPII occupancy is diminished in heterochromatic DNA in cells lacking Ubp3. The results suggest that the reduction in RNAPII levels residing in rDNA in ubp3∆ mutants may be caused by increased Net1 bound to rDNA. Furthermore, in line with previously published data suggesting that the frequency of unequal recombination in rDNA is regulated by RNAPII activity (Kobayashi et al., 2004; Kobayashi and Ganley, 2005), unequal crossover was mitigated in a ubp3∆ mutant. Of interest, ubp3∆ mutants display shortened replicative life span, raising questions about whether and to what extent increased silencing and lower recombination frequency in rDNA suppress aging.
RESULTS
ubp3∆ mutants are silenced more than wild-type cells in all heterochromatic regions
Moazed and Johnson (1996) demonstrated that in ubp3∆ mutants, expression of mRNA-encoding genes at the MAT locus (HML and HMR) and in telomeric regions are negatively regulated. This is consistent with our data showing that ubp3∆ mutants carrying an inserted URA3 allele at HMR (Ehrenhofer-Murray et al., 1999) grow poorly on media lacking uracil compared with wild-type cells (Figure 1A). Ubp3 forms a complex with Bre5 (Cohen et al., 2003a), and a number of reports show that cells lacking BRE5 display an identical phenotype to ubp3∆ mutants (Cohen et al., 2003a; Kvint et al., 2008). Therefore we tested whether BRE5 also had an effect on silencing of a URA3 gene integrated proximal to telomere VII-L (Gottschling et al., 1990; Kaufman et al., 1997). Indeed, a bre5∆ mutant phenocopied cells lacking UBP3 with regard to silencing of telomeric regions (Figure 1B). Moreover, the ubp3∆ bre5∆ double mutant displayed an identical growth pattern to the individual mutants (Figure 1B). The third region that resembles heterochromatin in S. cerevisiae is the tandemly repeating rDNA on chromosome XII (Figure 1C). To test whether UBP3 also has an effect on silencing here, we deleted UBP3 in a strain carrying a URA3 gene integrated in rDNA (JS306; Smith and Boeke, 1997). Similar to the effects on telomeric and mating-type loci, cells lacking UBP3 displayed a near-complete loss of growth on plates lacking uracil (Figure 1D), and this could be reversed if a copy of UBP3 was introduced on a CEN plasmid (Figure 1E), indicating that the increased silencing was specific for UBP3. Together these data show that Ubp3 affects gene silencing in a negative way in the heterochromatic regions (of telomeres, rDNA, and MAT locus) of S. cerevisiae.
FIGURE 1:

Ubp3 antagonizes silencing in telomeric regions, mating-type loci, and rDNA. (A) Ubp3 suppresses silencing at HMR. Silencing was measured by monitoring growth of 10-fold serial dilutions of cells plated on SC medium with and without uracil. (B) Ubp3 and Bre5 suppress silencing. Silencing was measured by monitoring growth of 10-fold serial dilutions of cells plated on medium lacking uracil or containing 5-flouroorotic acid (5-FOA). SC medium was used as a plating control. (C) Physical structure of the tandemly repeating rDNA. Bottom, a single unit depicted in detail. Each unit contains an RNAPI-transcribed 35S precursor rRNA and a divergently RNAPIII-transcribed 5S rRNA (blue). Each unit is divided by a nontranscribed unit (NTS), which is divided in two (NTS1 and NTS2, respectively) by the 5S gene. Also shown is the RFB,  , the autonomously replicating sequence (rARS,
, the autonomously replicating sequence (rARS,  ), and the bidirectional E-pro promoter (green). The site of insertion of the silencing reporter is also shown (mURA; JS306). (D) Spot test of JS306 cells (wild type and ubp3∆ mutants) on SC plates with or without uracil. (E) Growth on SC medium with or without uracil of ubp3∆ mutants carrying an empty vector or a plasmid containing UBP3.
), and the bidirectional E-pro promoter (green). The site of insertion of the silencing reporter is also shown (mURA; JS306). (D) Spot test of JS306 cells (wild type and ubp3∆ mutants) on SC plates with or without uracil. (E) Growth on SC medium with or without uracil of ubp3∆ mutants carrying an empty vector or a plasmid containing UBP3.
RNAPII occupancy in heterochromatic regions is reduced in cells lacking Ubp3
To understand how Ubp3 affects transcription of silenced genes, we measured RNAPII abundance at the promoter and in the coding sequence of a URA3 gene inserted in rDNA (JS306; Smith and Boeke, 1997) by ChIP assay. Previously, it was shown that heterochromatin influences DNA shearing (Teytelman et al., 2009). Therefore we tested whether our DNA shearing method was sufficient to generate properly sized DNA fragments for the ChIP assay. We used Southern blotting, and the result verified that the technique we used (see Materials and Methods and Supplemental Materials and Methods) was adequate (Supplemental Figure S1). ChIP analysis showed a clear reduction in RNAPII occupancy both at the URA3 promoter and in the open reading frame in the ubp3∆ mutant compared with an isogenic wild-type strain (Figure 2A). Similarly, reduced levels of RNAPII were detected in the telomeric region (Figure 2B) and HML (Figure 2C). Moreover, RNAPII occurrence outside of the URA3 locus proximal to the telomere (i.e., in the truncated, promoter-less ADH4 gene) was also reduced in ubp3∆ cells compared with the wild-type strain (Figure 2B), indicating that not only are potentially transcribed DNA or promoter regions affected by Ubp3, but heterochromatin per se is less accessible to RNAPII. Indeed, when we examined the distribution of RNAPII at the right arm of chromosome VI, we found that in ubp3∆ mutants, there was less RNAPII present than for wild-type cells (Figure 2D). This was also true in rDNA. In fact, RNAPII levels are quite scarce across the entire rDNA region in ubp3∆ mutants compared with wild-type cells (Figure 2E). Accordingly, measures of ncRNA levels expressed from the E-pro promoter by reverse transcription PCR (RT-PCR) using strand-specific primers showed a clear reduction in a ubp3∆ mutant (Supplemental Figure S2A). PCR coamplifications with oligonucleotide pairs for ACT1 and CUP1 were used for normalizations. In addition, RT-PCR measures of expression of the mURA3 gene showed similar results (Supplemental Figure S2A). Overall our results suggest that in ubp3∆ mutants, increased silencing is caused by a reduction in RNAPII occupancy in heterochromatic regions. Note that overall levels of RNAPII in whole-cell extracts are not different between wild-type cells and ubp3∆ mutants (Kvint et al., 2008), that RNAPII was equally well immunoprecipitated from wild-type and ubp3∆ mutant ChIP extracts (Supplemental Figure S2B), and that RNAPII occupancy in the actively transcribed gene ACT1 did not differ between wild-type cells and ubp3∆ mutants (Supplemental Figure S2C), suggesting that no obstructive factors influenced the immunoprecipitation differentially (between wild-type and ubp3∆ cells) and that Ubp3 predominantly affects RNAPII occupancy in heterochromatin.
FIGURE 2:

RNAPII occupancy in cells lacking Ubp3 is lower than in wild type cells. (A) Relative RNAPII occupancy in the promoter (PtrpURA) and the open reading frame (ORF) of the mURA construct in wild type and ubp3∆. RNAPII levels in wild type are set to 1. (B) Schematic diagram of the chromosomal location of a URA3 gene integrated at the telomeric region of chromosome VII (Gottschling et al., 1990). Relative RNAPII levels detected in the promoter and the ORF of the URA3 gene integrated at the telomeric region of chromosome VII as depicted in wild type and ubp3∆. (C) Schematic diagram of the HML locus at chromosome III. Detected levels of RNAPII at different positions at HML. Wild-type levels are set to 1. (D) Physical structure of the right end of chromosome VI (TEL VI-R). Relative RNAPII occupancy at TEL VI-R region in the ubp3∆ mutant and wild type. Wild-type levels are set to 1. (E) Physical structure of a single unit of tandemly repeating rDNA. RNAPII levels across an rDNA repeat in a ubp3∆ mutant (black bars) and a wild-type strain (gray area topped with black line). RNAPII levels in wild type are set to 1. Values are calculated as (RNAPII/input)/(RNAPIICup1/inputCup1). PCR products analyzed in the ChIP assays are depicted below each physical structure presented (B, C, and E). The numbered PCR products in E are the same as in Huang and Moazed (2003). Data are represented as the mean (± SD) of at least two experiments.
Sir2 distribution is altered in heterochromatic regions in cells devoid of UBP3
Because ubp3∆ mutants consistently display reduced levels of RNAPII attached to rDNA, telomeric regions, and MAT, we examined whether Sir protein levels were also affected, given that Sir proteins are refractory to transcription. At MAT and telomeres, Sir3/Sir4 recruits Sir2, whereas in rDNA, Sir2 is recruited by the RENT complex (Strahl-Bolsinger et al., 1997; Huang and Moazed, 2003). Sir proteins were monitored by ChIP using tandem affinity purification (TAP)–tagged strains (Sir2-TAP and Sir3-TAP). At the right arm of chromosome VI, Sir2 and Sir3 levels were slightly higher in ubp3∆ mutants than with wild type (Figure 3, A and B), whereas at HML there was approximately two times as much Sir2 and Sir3 in the ubp3∆ mutant (Figure 3, C and D). Next we examined rDNA. In rDNA, not Sir3 but the RENT complex recruits Sir2 (Huang and Moazed, 2003), and thus, as expected, we detected very low levels of Sir3 here in both wild type and ubp3∆ (Figure 3D). Sir2 was previously shown to support transcriptional silencing in rDNA (Fritze et al., 1997). However, we detected, if anything, less Sir2 protein at rDNA in the ubp3∆ mutant than with wild-type (Figure 3E). Of importance, there was no difference in total levels of Sir2 or Sir3 between wild-type cells and ubp3∆ mutants, as measured by Western blotting of whole-cell extracts (Supplemental Figure S3).
FIGURE 3:

Sir2 and Sir3 occupancies in wild-type and ubp3∆ mutants. (A) Relative Sir2-TAP occupancy at indicated places of the right end of chromosome VI (Figure 2D) in the ubp3∆ mutant and wild-type cells. (B) Relative Sir3-myc occupancy at the indicated places of the right end of chromosome VI in the ubp3∆ mutant and wild-type cells. (C) Representative graphs showing relative Sir2-TAP occupancy at the indicated places in the HML locus (Figure 2C) in the ubp3∆ mutant and wild-type cells, respectively. (D) Relative Sir3-myc occupancy at the indicated places in the HML locus and rDNA (Figure 2, C and E). (E) Relative Sir2-TAP levels across an rDNA repeat in a wild-type strain and ubp3∆ mutant. Physical structure of a single unit of the tandemly repeating rDNA, with PCR products analyzed in the ChIP assay indicated as horizontal bars below. (F) RNAPII levels across an rDNA repeat in wild type (set to 1) and ubp3∆, sir2∆, and ubp3∆ sir2∆ mutants. Values are calculated as (Sir2-TAP or Sir3-myc/input) divided by (Sir2-TAP or Sir3-mycCup1/inputCup1) or (RNAPII/input)/(RNAPIICup1/inputCup1). Data are represented as the mean (± SD) of at least three experiments.
To test the epistasis between UBP3 and SIR2, we measured RNAPII occupancy in rDNA in sir2∆ mutants by ChIP. Consistent with previous reports, the levels of RNAPII cross-linked to rDNA in sir2∆ mutants were higher than with wild-type cells (Figure 3F). Of interest, in the ubp3∆ sir2∆ double mutant, RNAPII occupancy was lower than in a sir2∆ single mutant (Figure 3F), and, accordingly, ubp3∆ sir2∆ double mutants grew poorly on media lacking uracil (Supplemental Figure S4A). As expected and previously shown (Fritze et al., 1997), a sir2∆ mutant lost its capacity to silence the reporter gene in rDNA (Supplemental Figure S4B). This suggests that Ubp3 partly functions in a Sir2-independent pathway in allowing RNAPII access to rDNA.
In addition to blocking access to DNA by RNAPII, Sir2 has also been proposed to hamper the transcriptional activity of RNAPII by deacetylating lysine 16 of histone 4 (H4K16) in heterochromatin (Cesarini et al., 2012). Hence we tested whether there were any differences in H4K16 acetylation (H4K16Ac) in these regions (Supplemental Figure S5). The levels of H4K16Ac did not differ significantly between wild type and ubp3∆ mutants at the right arm of chromosome VI. When SIR2 was knocked out, however, H4K16Ac levels increased as expected in both wild-type and ubp3∆ mutant cells (Supplemental Figure S5A). Conversely, at HML, the degree of H4K16 acetylation was drastically reduced in a ubp3∆ mutant compared with a wild-type strain (Supplemental Figure S5B). The ratio of H4K16Ac/H4 in the ubp3∆ mutant was ∼30% of wild-type levels, whereas in the sir2∆ mutant the ratio was significantly higher than in wild-type cells (Supplemental Figure S5B). In rDNA there was no difference in H4K16Ac levels between wild-type and ubp3∆ mutants, but levels were high in cells devoid of SIR2, as expected (Supplemental Figure S5C). Thus the increase in silencing seen in ubp3∆ mutants is likely mediated by the effects on Sir2 levels and H4K16 acetylation in HML but not in telomeric regions or rDNA.
Net1 and Fob1 levels at rDNA are altered in ubp3∆ mutants
In rDNA, Fob1 facilitates Net1/Cdc14 (RENT) binding, which in turn recruits Sir2 (Straight et al., 1999). Therefore we next investigated the abundance of the RENT complex in rDNA by ChIP using C-terminally Myc-tagged Net1. In agreement with previous findings (Huang and Moazed, 2003), we observed a pattern with two peaks (NTS1 and NTS2) of intense Net1 occupancy (Figure 4A). In ubp3∆ mutants, we discovered that Net1 levels in rDNA were higher than in wild-type cells (Figure 4A). This increase was also observed when Net1 was TAP tagged (unpublished data). Next we measured Fob1 levels bound to rDNA. As previously reported (Huang and Moazed, 2003; Ha et al., 2012), Fob1 amassed around the replication fork barrier (RFB) in NTS1 (Figure 4B). In ubp3∆ mutants this accumulation was somewhat enhanced (Figure 4B). The elevated levels of Net1 (and Fob1) in rDNA could thus be a reason for the reduced RNAPII presence/activity in cells devoid of UBP3.
FIGURE 4:
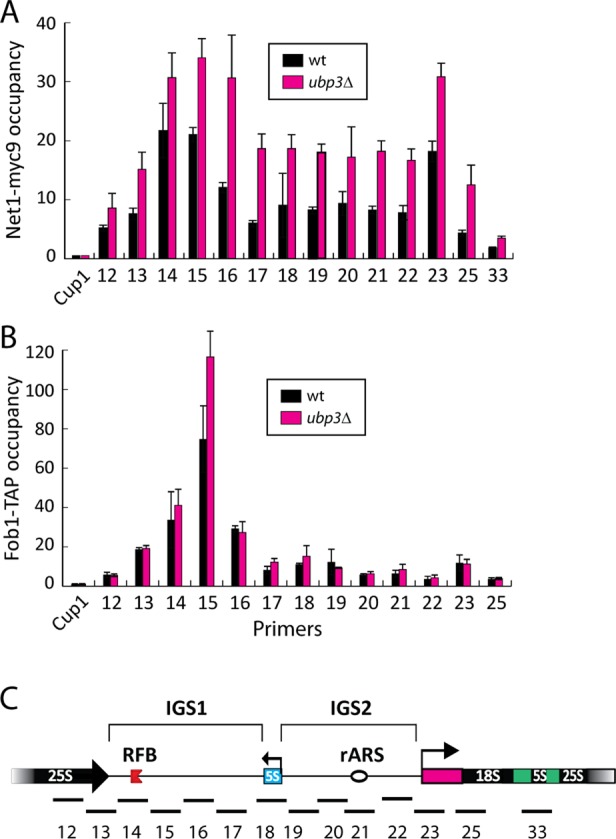
Net1 and Fob1 are abundant at rDNA in ubp3∆ mutants. (A) Net1-myc9 occupancy across an rDNA repeat in wild-type strain and a ubp3∆ mutant. (B) Fob1-TAP occupancy across an rDNA repeat in a wild-type strain and a ubp3∆ mutant. (C) Physical structure of a unit of the tandemly repeating rDNA. PCR products analyzed in the ChIP assay are indicated as bars below. Values are calculated as (Net1-myc or Fob1-TAP/input)/(Net1-myc or Fob1-TAPCup1/inputCup1). Data are represented as the mean (± SD) of at least three experiments.
Ubp3 suppresses unequal recombination in rDNA in a Sir2-dependent manner
It was reported that transcription by RNAPII from the E-pro promoter in the nontranscribed spacer in rDNA (Figure 1C) has a positive effect on unequal recombination between sister chromatids (Kobayashi et al., 2004; Kobayashi and Ganley, 2005). Consequently, in cells lacking Sir2, hyperrecombination in rDNA occurs (Kaeberlein et al., 1999), and ncRNA expression from the E-pro promoter is elevated (Cesarini et al., 2012). In contrast, FOB1 is required for recombination in rDNA (Kobayashi and Horiuchi, 1996). However, Fob1 appears to counteract its own recombination activity by recruiting Net1 and Sir2 to RFB, which promotes silencing and rDNA stability. Therefore we tested whether a strain lacking UBP3 had an altered frequency of unequal crossover as assayed by marker loss using a strain (JD83) with an ADE2 gene inserted in rDNA (Kaeberlein et al., 1999). As predicted, due to the increased silencing and reduced expression of ncRNA (from E-pro), ubp3∆ mutants displayed significantly decreased recombination in rDNA (Figure 5). In addition, the rDNA copy number is not affected in cells lacking Ubp3 and thus this cannot be the cause of the lower recombination rate (Supplemental Figure S6). The reduced recombination frequency could be reversed if a copy of UBP3 was introduced on a CEN plasmid (Figure 5), indicating that the reduced recombination frequency was specific for UBP3. However, in cells devoid of both Sir2 and Ubp3 the recombination frequency was quite similar to that for a sir2∆ single mutant (Figure 5). Thus SIR2 is epistatic to UBP3 with regard to unequal recombination in rDNA. In summary, our results suggest that frequency of recombination is not necessarily directly proportional to RNAPII transcription in rDNA (Figure 5 and Supplemental Figure S2A). It is worth noting that often in the analysis of this type of screen it is not entirely clear which colonies are exactly half red/half white. Frequently, the percentage of red to white does not match the expected theoretical outcome (i.e., 50/50, 25/75, etc.). Sometimes colonies have a mosaic pattern, or even 40% white/60% red sectors or vice versa. In the literature, however, the overall trend is obvious, in that there is a significant difference between wild-type and sir2∆ mutant strains, but the degree of variance can differ.
FIGURE 5:
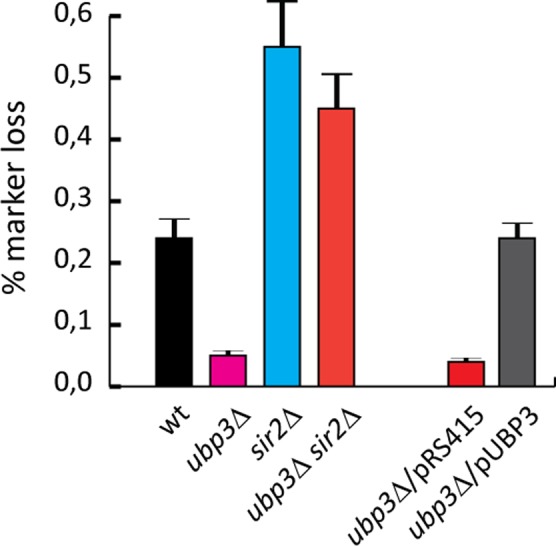
ubp3∆ suppresses unequal recombination in rDNA in a Sir2-dependent manner. Unequal recombination is represented as percentage marker loss, calculated as the ratio of half-sectored colonies to the total number of colonies, excluding entirely red colonies. See Materials and Methods for further details.
ubp3∆ mutants are short lived compared with wild-type cells
Numerous studies on rDNA integrity report a correlation between disrupted silencing, increased recombination, and shortened replicative life span (RLS). For instance, sir2∆ mutants have a significantly shorter life span than wild-type cells, whereas fob1∆ mutants have an extended life span. FOB1 is partly epistatic to SIR2 for life span, since a sir2∆ fob1∆ double mutant has a life span similar to that of wild-type cells (Defossez et al., 1999). In addition, in sir2∆ fob1∆ double mutants, recombination frequencies are similar to those in wild-type cells, whereas in a fob1∆ single mutant, recombination is low (Kobayashi and Horiuchi, 1996; Smith et al., 2009). To test how Ubp3 affects replicative life span, we tested our ubp3∆ mutant strains. Surprisingly, the augmented silencing and the decreased recombination frequency taking place in ubp3∆ mutants did not cause an extended replicative life span. Instead, the life span of ubp3∆ mutants was ∼70% of that of wild-type cells (Figure 6A). Moreover, no difference in life span was observed when a ubp3∆ sir2∆ double mutant was compared with a sir2∆ single mutant (Figure 6A). Thus SIR2 is epistatic to UBP3 in determining RLS.
FIGURE 6:
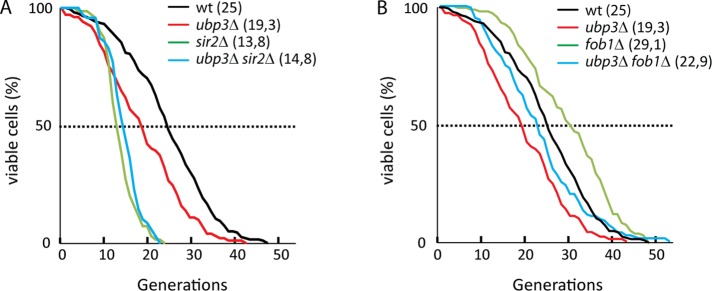
Ubp3 is required for longevity. (A) Replicative life span of wild type (black, n = 204) and ubp3∆ (red, n = 100), sir2∆ (green, n = 70), and ubp3∆ sir2∆ (blue, n = 70) mutants. (B) Replicative life span of wild type (black, n = 204) and ubp3∆ (red, n = 100), fob1∆ (green, n = 106), and ubp3∆ fob1∆ (blue, n = 119) mutants. Average life spans are shown in brackets.
Next we measured RLS in cells devoid of FOB1. As expected, cells lacking FOB1 had an extended life span compared with wild-type cells (Figure 6B). When UBP3 was deleted in a fob1∆ mutant, RLS approached that of wild-type cells. Thus it is likely that reduced transcriptional activity and diminished recombination frequency, as are phenotypes of cells devoid of UBP3 or FOB1, are not a prerequisite for extended life span. Instead, one interpretation is that Ubp3 takes part in another process affecting RLS that overrides the beneficial effects that silencing and recombination in rDNA have on RLS.
In aging cells, gradual decline in protein quality control and accumulation of protein aggregates occur (Heeren et al., 2004; Erjavec et al., 2007). Ubp3 forms a complex with Bre5 (Cohen et al., 2003a), and a number of reports shown that bre5∆ and ubp3∆ mutants display similar phenotypes, such as deubiquitinating Sec23 (a COPII subunit; Cohen et al., 2003a) and 6- azauracil sensitivity (Kvint et al., 2008). Furthermore, ubp3∆ and bre5∆ mutants have identical phenotypes regarding clearance of protein aggregates (Oling et al., 2014). Therefore we tested RLS in cells devoid of BRE5 and found that they also have a shortened life span compared with wild-type cells (Supplemental Figure S7).
DISCUSSION
In this study we elucidated how silencing is altered in cells lacking Ubp3. We confirmed, as described by Moazed and Johnson (1996), that ubp3∆ mutants display more silencing at the MAT loci and in telomeric regions. In addition, we found that cells lacking UBP3 are also more silenced in rDNA. There are several hypotheses explaining how and why heterochromatin is silent. One idea is that the Sir complexes (or RENT complexes at rDNA) and hypoacetylated histones sterically hinder access to specific DNA sequences recognized by transcription factors (e.g., RNAPII). However, there are several sequence-specific factors that are permitted access, such as homologous or site-specific recombination enzymes and retrotransposon integration factors (Holmes and Broach, 1996; Zou et al., 1996). In ubp3∆ mutants, the levels of H4K16Ac and Sir2 associated with heterochromatin were altered differentially in different heterochromatic regions. At HML the ratio of H4K16Ac/H4 was much lower in cells devoid of UBP3 than with wild-type cells. Similarly, Sir2 levels at HML were almost doubled in the ubp3∆ mutant. Thus in HML the elevated levels of Sir2 and deacetylated H4K16 may indeed hinder transcription factor access (i.e., RNAPII, and more so in cells lacking Ubp3). In contrast, in rDNA and in telomeric regions, the H4K16Ac/H4 ratio was not significantly affected, whereas Sir2 levels were slightly lower (in the ubp3∆ mutant) or largely unchanged, respectively. Thus Sir2 and/or hypoacetylated H4K16 cannot explain the increased silencing in ubp3∆ mutants at telomeres and in rDNA.
In a study on heterochromatin, it was shown that general transcription factors TATA-binding protein (TBP) and RNAPII assembled at silent promoters in HMR but that no initiation ensued, suggesting that Sir-generated heterochromatin suppresses transcription at a subsequent step (Sekinger and Gross, 2001). Later this model was reinforced when the same lab showed that TFIIH and the Ser5-phosphorylated isoform of RNAPII could be detected at Sir-mediated silent promoters, suggesting that RNAPII was poised to start transcription (Gao and Gross, 2008). However, another lab published data showing that none of TFIIB, TFIIE, or RNAPII occupy silenced promoters; only an activator (Ppr1) was able to associate at the promoters (Chen and Widom, 2005). Thus how silent chromatin inhibits RNAPII-dependent transcription is obscure in many regards. With this in mind, note that many reports and different labs use different strain backgrounds in their studies on gene silencing, which could potentially lead to indistinct results. In this study two different strain backgrounds (BY strains and W303) were used (see Supplemental Table S1), and the results did match.
In this work we present data that match those of Gross and coworkers (Sekinger and Gross, 2001; Gao and Gross, 2008). We find experimental support for their model and that it is applicable to all silent loci in S. cerevisiae. Essentially, increased silencing in ubp3∆ mutants is likely to be caused by lower levels of RNAPII in these heterochromatic regions, suggesting that RNAPII is indeed active in wild-type cells in these regions. In addition, Sir3 and RENT occupancy is generally higher in ubp3∆ mutants in MAT and rDNA, respectively, suggesting that silent chromatin is not saturated completely with silencing factors in wild-type cells. Thus we propose that RNAPII is in fact present and active in heterochromatic DNA in wild-type cells and that this is dependent on Ubp3 (Figures 1, A, C, and E, and 2, A–E). Whereas the effect on silencing by Ubp3 in MAT and rDNA may be explained by elevated levels of silencing factors (i.e., Sir3 and Net1), in telomeric regions our data are not sufficient for any interpretations. However, it is possible that a countered RNAPII (i.e., such as an RNAPII trying to transcribe in dense heterochromatin in telomeric regions) is likely to be ubiquitinated and thus prematurely terminated, as previously suggested (Kvint et al., 2008). Although these data demonstrate that in wild-type cells RNAPII can assemble and engage in transcription at MAT, telomeric regions, and rDNA, we also found that Ubp3 is required for nonequal recombination in rDNA in a Sir2-dependent manner, whereas sir2∆ mutants are dependent on UBP3 for full derepression of a URA3 allele in rDNA. This contrasts with the observation that recombination is directly proportional to the levels of transcriptional activity in the nontranscribed spacers in rDNA, as was previously proposed (Kobayashi et al., 2004; Kobayashi and Ganley, 2005). However, Sir2 may have a second function in inhibiting recombination that is partly independent of its role in silencing.
Replicative aging has been associated with loss of silencing and increased nonequal recombination in rDNA and vice versa (Burhans and Weinberger, 2007). Of interest, our data demonstrate that ubp3∆ mutants age faster than wild-type cells despite increased silencing and reduced recombination in rDNA. One possible explanation for this could be that ubp3∆ mutants have delayed clearance of protein aggregates and premature accumulation of Hsp70 aggregates during aging (Oling et al., 2014). Proper clearance of protein aggregates is a requirement to combat premature aging (Heeren et al., 2004; Erjavec et al., 2007; Kruegel et al., 2011). Furthermore, deletion of FOB1 did not fully suppress the reduced life span of a ubp3∆ mutant. This further implies that Ubp3 has another function central for sustaining longevity that bypasses the potential benefits that increased silencing and reduced recombination have on senescence (e.g., corrupt protein quality control). It also suggests either that loss of FOB1 further decreases recombination in ubp3∆ mutants (potentially causing RLS extension) or that Fob1 also affects life span extension via a pathway other than rDNA recombination.
Cells devoid of Sir2 have a shortened life span (Kaeberlein et al., 1999). The replicative life span of a ubp3∆ sir2∆ double mutant is similar to that of a sir2∆ single mutant. Thus, in cells lacking both UBP3 and SIR2, life span correlates with higher recombination frequency. In contrast, the life span of cells devoid of FOB1 and SIR2 is longer than that in a sir2∆ mutant but shorter than that in a fob1∆ mutant. However, it has been proposed that this is due to Sir2 having other functions important for longevity that cannot be compensated for by a deletion of FOB1. For example, Sir2 is required for asymmetric segregation of aberrant proteins during mitosis (Aguilaniu et al., 2003) and has been linked to defense against reactive oxidative species (ROS; Erjavec et al., 2007), both of which have been connected to aging. In addition, as mentioned, UBP3 and BRE5 have been implicated in protein quality control, and thus impairment of this process by loss of these genes could have a negative effect on RLS (Oling et al., 2014). Similar to ubp3∆ mutants, we found that cells lacking Bre5 age faster than wild-type cells (Supplemental Figure S7), although large-scale analysis of single-gene deletions found that bre5∆ cells had increased life span (Kaeberlein et al., 2005), a discrepancy we cannot explain. Taken together, the effect on life span by the loss of both UBP3 and SIR2 may be due to a combination of rDNA recombination defects, deficiency in dealing with ROS, and insufficient protein quality control.
MATERIALS AND METHODS
Yeast strains and procedures
Strains used in this work are derived from S288C and W303 genetic backgrounds (Supplemental Table S1). S. cerevisiae strains were grown and manipulated by using standard techniques (Sherman, 1991). Deletion mutants were constructed either by PCR-mediated knockout or sporulation. Transformants and dissected spores were verified by PCR. For growth in rich media, strains were grown in yeast extract/peptone/dextrose (YPD) with 2% glucose. Cells grown in synthetic defined medium were grown with yeast nitrogen base plus ammonium sulfate and 2% glucose.
ChIP
ChIP assays were performed essentially as previously described (Kristjuhan et al., 2002). Briefly, cultures were cross-linked with 1% formaldehyde for 15 min at room temperature. Cross-linking was quenched with 200 mM glycine for 5 min. Cells were pelleted by centrifugation at 4500 rpm for 4 min at 4°C and then washed once with phosphate-buffered saline and then resuspended in FA lysis buffer (50 mM Tris, pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na deoxycholate, 1× protease inhibitors). After disrupting cells with glass beads, the chromatin was sonicated using the Biorupter UCD-200, Diagenode system (power, high; time, 30 s on/30 s off for 15 min) to yield DNA fragments of ∼100–700 base pairs. The resulting extract was centrifuged twice at 14,000 rpm to remove debris. The immunoprecipitate (extract, protein G [or A] beads and antibodies) was washed at 4°C after incubation (>4 h) as follows: 1 × 10 min in FA lysis buffer, 3 × 10 min in FA500 (FA lysis with 500 mM NaCl), 3 × 10 min in ChIP wash buffer (10 mM Tris, pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% Na deoxycholate, 1 mM EDTA), and twice in TES (10 mM Tris, pH 7.5, 1 mM EDTA, 100 mM NaCl). The precipitate was eluted in 100 μl of elution buffer (50 mM Tris, pH 8.0, 10 mM EDTA, 1% SDS) at 30°C. DNA–protein cross-links were reversed by incubating at 65°C for 6 h, and the DNA was isolated using a PCR Purification Kit (Qiagen, Valencia, CA). 4H8 (anti-Rpb1 antibody) was from Upstate Biotechnology (Lake Placid, NY), and the H3C antibody was a gift from Claes Gustafsson (University of Gothenburg). Immunoglobulin G Sepharose 6 Fast Flow from GE Healthcare (Piscataway, NJ) was used to immunoprecipitate (IP) TAP-tagged proteins. Coprecipitated DNA was analyzed in triplicate by quantitative PCR using SYBR green and the ABI Prism 7000 Sequence detection system (Applied Biosystems, Foster City, CA) or a Bio-Rad Q5 system (Bio-Rad, Hercules, CA). Primer sequences are available upon request. To exclude detection from the ura3-1 allele in PKY090, a specific primer for URA3 was used (Supplemental Figure S8). In general, values were normalized to inputs, and as an internal control, the CUP1 gene was used unless otherwise stated. Relative fold enrichment was determined by calculating the ratio (IP/input)/(CUP1IP/CUP1input) unless otherwise stated. All values expressed in bar graphs are results from at least two independent ChIP experiments and three independent PCR assays.
rDNA instability assay
The marker loss assay was performed as previously described (Kaeberlein et al., 1999). A strain (JD83) with a single ADE2 marker gene inserted in one rDNA copy was used (Kaeberlein et al., 1999). Strains were grown to mid log phase in YPD medium, diluted, and plated onto synthetic complete (SC) solid medium (250–350 cells/plate). Colonies were grown at 30°C for 3 d and then transferred to 4°C for 4 d, at which point half-sector (red/white) colony formation was detected visually. Experiments were performed at least three times per strain. At least 30,000 colonies were scored per strain. The unequal sister chromatin crossover rate was calculated by dividing the number of half-red/half-white colonies by the total number of colonies (excluding fully red colonies).
Replicative life span assay
A micromanipulator (Singer Instruments, Watchet, United Kingdom) was used to measure replicative life span (Kaeberlein et al., 1999). Briefly, cells were grown overnight in YPD, diluted, plated, and allowed to recover on YPD plates before being assayed for RLS, which was performed independently at least twice for each strain.
Supplementary Material
Acknowledgments
We thank Steven Buratowski, Claes Gustafsson, Jesper Svejstrup, Bruce Stillman, Maria Falkenberg, Matt Kaeberlein, and Eulalia de Nadal for antibodies, reagents, plasmids, and strains. We thank Thomas Nystrom for support and Jay Uhler for invaluable help with Southern blottings. This work was supported by grants from the Carl Tryggers Foundation and the Wilhelm and Martina Lundgrens Research Foundation to K.K.
Abbreviations used:
- ChIP
chromatin immunoprecipitation
- RENT
regulator of nucleolar silencing and telephase exit
- RLS
replicative life span
- TAR
transcription-associated recombination
- UPS
ubiquitin–proteasome system
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-10-0591) on April 23, 2014.
REFERENCES
- Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003;299:1751–1753. doi: 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Auty R, Steen H, Myers LC, Persinger J, Bartholomew B, Gygi SP, Buratowski S. Purification of active TFIID from Saccharomyces cerevisiae. Extensive promoter contacts and co-activator function. J Biol Chem. 2004;279:49973–49981. doi: 10.1074/jbc.M409849200. [DOI] [PubMed] [Google Scholar]
- Burhans WC, Weinberger M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007;35:7545–7556. doi: 10.1093/nar/gkm1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burhans WC, Weinberger M. DNA damage and DNA replication stress in yeast models of aging. Subcell Biochem. 2012;57:187–206. doi: 10.1007/978-94-007-2561-4_9. [DOI] [PubMed] [Google Scholar]
- Cesarini E, D'Alfonso A, Camilloni G. H4K16 acetylation affects recombination and ncRNA transcription at rDNA in Saccharomyces cerevisiae. Mol Biol Cell. 2012;23:2770–2781. doi: 10.1091/mbc.E12-02-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Widom J. Mechanism of transcriptional silencing in yeast. Cell. 2005;120:37–48. doi: 10.1016/j.cell.2004.11.030. [DOI] [PubMed] [Google Scholar]
- Chew BS, Siew WL, Xiao B, Lehming N. Transcriptional activation requires protection of the TATA-binding protein Tbp1 by the ubiquitin-specific protease Ubp3. Biochem J. 2010;431:391–399. doi: 10.1042/BJ20101152. [DOI] [PubMed] [Google Scholar]
- Cohen M, Stutz F, Belgareh N, Haguenauer-Tsapis R, Dargemont C. Ubp3 requires a cofactor, Bre5, to specifically de-ubiquitinate the COPII protein, Sec23. Nat Cell Biol. 2003a;5:661–667. doi: 10.1038/ncb1003. [DOI] [PubMed] [Google Scholar]
- Cohen M, Stutz F, Dargemont C. Deubiquitination, a new player in Golgi to endoplasmic reticulum retrograde transport. J Biol Chem. 2003b;278:51989–51992. doi: 10.1074/jbc.C300451200. [DOI] [PubMed] [Google Scholar]
- Defossez PA, Prusty R, Kaeberlein M, Lin SJ, Ferrigno P, Silver PA, Keil RL, Guarente L. Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol Cell. 1999;3:447–455. doi: 10.1016/s1097-2765(00)80472-4. [DOI] [PubMed] [Google Scholar]
- De Nadal E, Zapater M, Alepuz PM, Sumoy L, Mas G, Posas F. The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature. 2004;427:370–374. doi: 10.1038/nature02258. [DOI] [PubMed] [Google Scholar]
- Ehrenhofer-Murray AE, Kamakaka RT, Rine J. A role for the replication proteins PCNA, RF-C, polymerase epsilon and Cdc45 in transcriptional silencing in Saccharomyces cerevisiae. Genetics. 1999;153:1171–1182. doi: 10.1093/genetics/153.3.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erjavec N, Larsson L, Grantham J, Nystrom T. Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p. Genes Dev. 2007;21:2410–2421. doi: 10.1101/gad.439307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritze CE, Verschueren K, Strich R, Easton Esposito R. Direct evidence for SIR2 modulation of chromatin structure in yeast rDNA. EMBO J. 1997;16:6495–6509. doi: 10.1093/emboj/16.21.6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Gross DS. Sir2 silences gene transcription by targeting the transition between RNA polymerase II initiation and elongation. Mol Cell Biol. 2008;28:3979–3994. doi: 10.1128/MCB.00019-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling DE, Aparicio OM, Billington BL, Zakian VA. Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell. 1990;63:751–762. doi: 10.1016/0092-8674(90)90141-z. [DOI] [PubMed] [Google Scholar]
- Ha CW, Sung MK, Huh WK. Nsi1 plays a significant role in the silencing of ribosomal DNA in Saccharomyces cerevisiae. Nucleic Acids Res. 2012;40:4892–4903. doi: 10.1093/nar/gks188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeren G, Jarolim S, Laun P, Rinnerthaler M, Stolze K, Perrone GG, Kohlwein SD, Nohl H, Dawes IW, Breitenbach M. The role of respiration, reactive oxygen species and oxidative stress in mother cell-specific ageing of yeast strains defective in the RAS signalling pathway. FEMS Yeast Res. 2004;5:157–167. doi: 10.1016/j.femsyr.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Holmes SG, Broach JR. Silencers are required for inheritance of the repressed state in yeast. Genes Dev. 1996;10:1021–1032. doi: 10.1101/gad.10.8.1021. [DOI] [PubMed] [Google Scholar]
- Huang J, Moazed D. Association of the RENT complex with nontranscribed and coding regions of rDNA and a regional requirement for the replication fork block protein Fob1 in rDNA silencing. Genes Dev. 2003;17:2162–2176. doi: 10.1101/gad.1108403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kaufman PD, Kobayashi R, Stillman B. Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor-I. Genes Dev. 1997;11:345–357. doi: 10.1101/gad.11.3.345. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Ganley AR. Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science. 2005;309:1581–1584. doi: 10.1126/science.1116102. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Horiuchi T. A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells. 1996;1:465–474. doi: 10.1046/j.1365-2443.1996.d01-256.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Horiuchi T, Tongaonkar P, Vu L, Nomura M. SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell. 2004;117:441–453. doi: 10.1016/s0092-8674(04)00414-3. [DOI] [PubMed] [Google Scholar]
- Kristjuhan A, Walker J, Suka N, Grunstein M, Roberts D, Cairns BR, Svejstrup JQ. Transcriptional inhibition of genes with severe histone h3 hypoacetylation in the coding region. Mol Cell. 2002;10:925–933. doi: 10.1016/s1097-2765(02)00647-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruegel U, et al. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 2011;7:e1002253. doi: 10.1371/journal.pgen.1002253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvint K, Uhler JP, Taschner MJ, Sigurdsson S, Erdjument-Bromage H, Tempst P, Svejstrup JQ. Reversal of RNA polymerase II ubiquitylation by the ubiquitin protease Ubp3. Mol Cell. 2008;30:498–506. doi: 10.1016/j.molcel.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- Mao P, Smerdon MJ. Yeast deubiquitinase Ubp3 interacts with the 26 S proteasome to facilitate Rad4 degradation. J Biol Chem. 2010;285:37542–37550. doi: 10.1074/jbc.M110.170175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell. 2003;112:725–736. doi: 10.1016/s0092-8674(03)00123-5. [DOI] [PubMed] [Google Scholar]
- Moazed D, Johnson D. A deubiquitinating enzyme interacts with SIR4 and regulates silencing in S. cerevisiae. Cell. 1996;86:667–677. doi: 10.1016/s0092-8674(00)80139-7. [DOI] [PubMed] [Google Scholar]
- Oling D, Eisele F, Kvint K, Nystrom T. Opposing roles of Ubp3-dependent deubiquitination regulate replicative life span and heat resistance. EMBO J. 2014;33:747–761. doi: 10.1002/embj.201386822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- Sekinger EA, Gross DS. Silenced chromatin is permissive to activator binding and PIC recruitment. Cell. 2001;105:403–414. doi: 10.1016/s0092-8674(01)00329-4. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Smith DL, Jr, Li C, Matecic M, Maqani N, Bryk M, Smith JS. Calorie restriction effects on silencing and recombination at the yeast rDNA. Aging Cell. 2009;8:633–642. doi: 10.1111/j.1474-9726.2009.00516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Avalos J, Celic I, Muhammad S, Wolberger C, Boeke JD. SIR2 family of NAD(+)-dependent protein deacetylases. Methods Enzymol. 2002;353:282–300. doi: 10.1016/s0076-6879(02)53056-1. [DOI] [PubMed] [Google Scholar]
- Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997;11:241–254. doi: 10.1101/gad.11.2.241. [DOI] [PubMed] [Google Scholar]
- Sole C, Nadal-Ribelles M, Kraft C, Peter M, Posas F, de Nadal E. Control of Ubp3 ubiquitin protease activity by the Hog1 SAPK modulates transcription upon osmostress. EMBO J. 2011;30:3274–3284. doi: 10.1038/emboj.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 1997;11:83–93. doi: 10.1101/gad.11.1.83. [DOI] [PubMed] [Google Scholar]
- Straight AF, Shou W, Dowd GJ, Turck CW, Deshaies RJ, Johnson AD, Moazed D. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell. 1999;97:245–256. doi: 10.1016/s0092-8674(00)80734-5. [DOI] [PubMed] [Google Scholar]
- Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet. 2002;32:378–383. doi: 10.1038/ng1017. [DOI] [PubMed] [Google Scholar]
- Suka N, Suka Y, Carmen AA, Wu J, Grunstein M. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol Cell. 2001;8:473–479. doi: 10.1016/s1097-2765(01)00301-x. [DOI] [PubMed] [Google Scholar]
- Teytelman L, Ozaydin B, Zill O, Lefrancois P, Snyder M, Rine J, Eisen MB. Impact of chromatin structures on DNA processing for genomic analyses. PLoS One. 2009;4:e6700. doi: 10.1371/journal.pone.0006700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hayes JJ. Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol Cell Biol. 2008;28:227–236. doi: 10.1128/MCB.01245-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Ke N, Kim JM, Voytas DF. The Saccharomyces retrotransposon Ty5 integrates preferentially into regions of silent chromatin at the telomeres and mating loci. Genes Dev. 1996;10:634–645. doi: 10.1101/gad.10.5.634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.