

Abstract
The aim of this study was to evaluate the mechanisms of cytotoxicity of the sesquiterpene lactone 13-acetoxyrolandrolide, an NF-κB inhibitor that was previously isolated from Rolandra fruticosa. The effects associated with inhibition of the NFκB pathway included dose-dependent inhibition of the NF-κB subunit p65 (RelA) and inhibition of upstream mediators IKKβ and oncogenic K-Ras. The inhibitory concentration (IC50) of 13-acetoxyrolandrolide on K-Ras was 7.7 μM. The downstream effects of the inhibition of NF-κB activation were also investigated in vitro. After 24 h of treatment with 13-acetoxyrolandrolide, the mitochondrial transmembrane potential (ΔΨm) was depolarized in human colon cancer (HT-29) cells. The mitochondrial oxidative phosphorylation was also negatively affected and reduced levels of NAD(P)H were detected after 2 h of 13-acetoxyrolandrolide exposure. Furthermore, the expression of the pro-apoptotic protein caspase-3 increased in a concentration-dependent manner. Cell flow cytometry showed that 13-acetoxyrolandrolide induced cell cycle arrest at G1, suggesting that treated cells had undergone caspase-3-mediated apoptosis, suggesting negative effects on cancer cell proliferation. These results suggest that 13-acetoxyrolandrolide inhibits NF-κB and K-Ras as well as promotes cell death mediated through the mitochondrial apoptotic pathway.
Keywords: sesquiterpene, apoptosis, colon cancer, HT-29, K-Ras, reactive oxygen species, NF-κB, mitochondria
Introduction
The germacrane sesquiterpene lactone, 13-acetoxyrolandrolide, was previously isolated from Rolandra fruticosa (L.) Kuntze (Fig. 1) (Pan et al., 2010). This plant belongs to the tribe Vernonieae in the family Asteraceae that includes several species used in popular medicine for their anti-asthmatic, anti-helminthic and anti-rheumatic activities (Cabral Salles-de-Melo et al., 2010). R. fruticosa has a pantropical distribution and has been reported to have medicinal uses. The common name in Brazil is “vence tudo” and it is documented as having special significance in Candomblé, the Afro-Brazilian religion that originated in Bahia (Voeks, 1997). In an ongoing screening program for anticancer agents from natural sources, 13-acetoxyrolandrolide showed a cytotoxic effect (ED50 = 0.16 μM) in the HT-29 human colon cancer cell line and inhibited (IC50 = 7.1 μM) the transcription factor nuclear factor kappa B (NF-κB), which is a heterodimer consisting of p50 and p65 subunits that are encoded by the NFKB1 and RELA genes, respectively (Pan et al., 2010). The proliferation of cancer cells is dependent on the induction of anti-apoptotic proteins by NF-κB. There is evidence suggesting that the NF-κB pathway is responsible for regulating the survival and proliferation of cancer cells (Lerebours et al., 2008) and therefore the inhibition of NF-κB may have antitumor effects (Nakanishi and Toi, 2005). Anti-apoptotic effects are essential for survival and proliferation of malignant cells (Karin and Greten, 2005; Karin, 2006; Zhou et al., 2010). NF-κB prevents cancer cells from entering apoptosis and contributes to cancer progression in certain cancers, such as breast cancer, solid tumors and non-Hodgking’s lymphoma (Darnell, 2002; Cory and Cory, 2007). Moreover, NF-κB inhibition enhances the effect of existing therapeutic agents, particularly in drug-resistant cells (Sen et al., 2011). When inactive, NF-κB is found sequestered in the cytoplasm, bound to subunit IκB (Yin et al., 2009). After phosphorylation of IκB by the IκB kinase (IKK), NF-κB is subsequently released and is free to translocate from the cytoplasm into the nucleus where it transcribes target genes. This pathway is induced by inflammatory stimuli such as tumor necrosis factor-α (TNF-α) and other cytokinins, and also by growth factors such as the epidermal growth factor (EGF) (Karin, 2006). The epidermal growth factor receptor (EGFR) and the membrane associated Kirsten rat sarcoma protein (K-Ras) are part of a signaling transduction cascade which activate downstream several pathways, including the NF-κB pathway (Shubbert et al., 2007). The K-Ras protein plays a pivotal role in transducing a variety of extracellular signals that downstream induce transcription factors involved in cell growth and differentiation (Bharate et al., 2012). The K-Ras is frequently mutated in colon cancers, thus K-Ras may provide a possible target for preventing the progression of the disease (Karapetis et al., 2008). Recently, it has been suggested that oncogenic K-Ras promoted NF-κB activity in pancreatic cancer cells, mediated by glycogen synthase kinase-3 (GSK-3) and transforming growth factor β-activated kinase-1 (TAK-1) activity and downstream K-Ras induced IKK dependent NF-κB activation (Bang et al., 2013). Both TNF-α and EGF act on receptors located at the cell membrane to activate the NF-κB pathway downstream (Yin et al., 2009). Recently it has been shown that suppression of NF-κB activation induces mitochondrial dysfunction and promotes cell death (Rathore et al., 2012). Inducing cell death in cancer cells and inhibiting their proliferation is an approach for more effective chemotherapy and radiotherapy. Herein, the upstream K-Ras signaling pathway and NF-κB inhibitory effects of 13-acetoxyrolandrolide in HT-29 colon cancer have been investigated, the associated biochemical changes on the mitochondria were closely characterized and the apoptotic effects were examined.
Figure 1.

Structure of 13-acetoxyrolandrolide isolated from Rolandra fruticosa.
Materials and methods
The sesquiterpene lactone, 13-acetoxyrolandrolide (Fig. 1), was isolated and identified from the leaves of Rolandra fruticosa in a previous study (Pan et al., 2010). Rocaglamide was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY, USA). The Bradford protein assay kit, the Supersignal Femto LumiGLO kits and the human recombinant tumor necrosis factor-α (TNF-α) were obtained from Thermo Scientific (Rockford, IL, USA). Lithium dodecyl sulfate sample loading buffer (LDS), Nu-PAGE 10% SDS-PAGE Bis-Tris gel, SeeBlue® Plus2 Pre-Stained Standard Ladder, and Purelink RNase A were obtained from Invitrogen (Carlsbad, CA, USA). Primary antibodies (anti-NF-κBp65 and p50, anti-IKKα, anti-IKKβ, and anti-caspase-3) were purchased from Cell Signaling Technologies (Beverly, MA, USA). Anti-rabbit horseradish peroxidase (HRP)-conjugated antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). FeSO4 was obtained from Fischer Scientific Company (Fair Lawn, NJ). Daunorubicin was purchased from Tocris, Bristol, UK. Hydrogen peroxide was obtained from Fluka Biochemika, Steinhiem, Switzerland. Tris-buffered saline with Tween-20 buffer (TBS-T), vitamin C, propidium iodine (PI), and the fluorescent probe 2′,7′-dichlorfluorescein-diacetate (DCFH-DA) were obtained from Sigma Aldrich (St. Louis, MO, USA). The JC-1 mitochondrial membrane potential assay kit was purchased from Cayman Chemical Company (Ann Arbor, Michigan) and the Caspase–Glo3/7® kit was purchased from Promega (Madison, WI, USA). The reagents 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) and 1-methoxy-5-methylphenazidium methylsulfate (1-methoxy PMS) were purchased from Sigma Aldrich. The Ras GTPase Chemi ELISA kit was obtained from Active Motif (Carlsbad, CA, USA), and Epidermal Growth Factor (EGF) from Sigma Aldrich.
Cell line and culture
The HT-29 colon cancer cell line was purchased from American Type Culture Collection (ATCC), Manassas, VA. Cells were grown in RPMI-1640 medium, supplemented with 10% (v/v) FBS, penicillin (100 units/mL), streptomycin (100 μg/mL) and Fungizone® (0.25 μg/mL). The cells were maintained at 37°C and in an atmosphere with 5% CO2.
Immunoblotting
To determine the effects of 13-acetoxyrolandrolide on the NF-κB pathway, a previously published protocol was followed (Muñoz Acuña et al., 2012). In brief, cells were treated at different concentrations for 3 h. Cells were lysed using PhosphoSafe Lysis Buffer and analyzed by western blot analysis with primary (1:1000) and secondary (1:2000) antibodies. Protein concentrations were determined using a Bradford protein assay kit. Absorbance was measured using a Fluostar Optima plate reader (BMG Labtech Inc, Durham, NC). Samples were loaded with LDS sample loading buffer and resolved using Nu-PAGE 10% SDS-PAGE Bis-Tris gels together with SeeBlue® Plus2 Pre-Stained Standard. Electrophoresis was performed using SDS-PAGE running buffer in a Nu-PAGE XCell SureLock Module (Invitrogen, Grand Island, NY). Proteins were transferred to a polyvinyldiene fluoride (PVDF) membrane. The blots were then blocked at room temperature and probed using primary antibodies and BSA in TBS-T buffer. Conjugated antibodies were detected using the chemiluminescent substrate Supersignal Femto kit (Thermo Fisher Scientific, Waltham, MA) and relative band density was determined.
ROS assay
This method has previously been described (Kim et al., 2010). Briefly, HT-29 cells were seeded in 96-well plates and allowed to attach overnight. Then, the cells were treated with 13-acetoxyrolandrolide (0.028–280 μM), vitamin C (0.022–22.3 μM), and incubated at 37°C in 5% CO2 for 4.5 h. This was followed by treatment with H2O2 (1.25 mM) (the positive control), and FeSO4 (0.2 mM), and probed with the fluorescent probe DCFH-DA (5 mM) for 30 min at 37°C. Fluorescence was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm using a FLUOstar Optima fluorescence microplate reader (BMG Labtechnologies Inc., Durham, NC). Each experiment was carried out in triplicate and was representative of at least two different experiments.
K-Ras assay
A cell suspension of HT-29 cells (5×104cells/ml) was seeded on a 96-well plate and allowed to attach overnight. The cells were treated with 13-acetoxyrolandrolide and incubated for 3 h at 37°C, followed by removal of media and then washed three times with PBS and treated with EGF solution (5 ng/ml) for 2 min. The cells were lysed and protease inhibitor was added. The cell extracts were then aliquoted and stored at −80°C. Protein concentration was determined in each sample using a Bradford protein assay kit as described above. The K-Ras activity was tested by using Ras GTPase Chemi Elisa kit from Active Motif (Carlsbad, CA). Primary H-Ras antibody (1:500) and secondary antibody HRP-conjugated (1:5000) were added and incubated at room temperature for 1 h. Chemiluminescence solution was added, and the luminescence was detected using plate reader Fluostar Optima plate reader (BMG Labtech Inc, Durham, NC). Each data point is expressed as an average of triplicate readings from two independent experiments.
Mitochondrial transmembrane potential (ΔΨm) assay
The mitochondrial membrane potential FACS assay kit from Cayman Chemical Company (Ann Arbor, MI) was used to assess the mitochondrial transmembrane potential (ΔΨm) in colon cancer cells following a previously published protocol (Muñoz Acuña et al., 2012). In brief, cells were seeded on 10 cm plates and allowed to attach before treated with 13-acetoxyroladrolide for 24 h. After this time, the cells were harvested using Trypsin-EDTA (Gibco, Grand Island, NY), washed in phosphate-buffered saline (PBS), and re-suspended in assay buffer. The potentiometric dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazoylcarbocyanine iodide (JC-1) was used to stain the HT-29 cells. A volume of 50 μl of JC-1 stain (1 μM) was added to the cells, followed by incubation for 15 min at 37°C in 5% CO2. At a high ΔΨm, red fluorescent J-aggregates are formed in healthy cells; however, in apoptotic cells with a low ΔΨm, JC-1 remains in the monomeric form, which only exhibits green fluorescence. Analysis was performed using a BD FACS Canto II flow cytometer (BD Biosciences, San Jose, CA). Mitochondrial function was assessed, and J-aggregates were detected using an excitation wavelength of 520–570 nm, and emission of 570–610 nm, respectively.
XTT assay
To assess the intracellular levels of NAD(P)H the previously published protocol by Nakamura et al. 2003 was modified. HT-29 colon cancer cells were seeded (1×104cells/well) in a 96-well plate and allowed to attach. The cells were treated with 13-acetoxyrolandrolide (1.3 nM–100 μM) and incubated at 37°C and 5% CO2. Mitochondrial function was measured indirectly by measuring the formation of formazan dye produced by metabolically active cells at one-hour intervals over an 11-hour period. 100 μL of XTT and 1-methoxy PMS were prepared in RPMI media and added to each well to yield final concentrations of 0.25 mM for XTT and 0.01 μM for 1-methoxy PMS. The absorbance was measured at 485 nm and NAD(P)H depletion was calculated. Staurosporine (20 μM) was used as a positive control. Each sample was tested in triplicate and in two independent experiments.
Caspase-3/7 GLO assay
Caspase-3 and 7 activities were measured using the Caspase–Glo3/7® protocol following the manufacturer’s instructions (Promega Corp, Madison, WI). HeLa cells were treated with 13-acetoxyrolandrolide (0.08–2 μM) for 3 h. Paclitaxel (0.1 nM–1 μM) was used as a positive control. Caspase–Glo3/7 reagent was then added and luminescence was recorded using Fluostar Optima plate reader (BMG Labtech, Ortenberg, Germany) at 37°C and enzymatic activity was calculated. Each data point represents an average of triplicate readings.
Cell cycle analysis
The procedure was previously published (Muñoz Acuña et al., 2012). Briefly, HT-29 cells were seeded on 10 cm plates and treated with compound 13-acetoxyrolandrolide (10 μM) for 24 h. Cells were then washed with PBS and treated with trypsin EDTA, harvested, centrifuged, and re-suspended in PBS. Cells were fixed using ethanol (70%) and incubated on ice for 1 h, washed, re-suspended in ice-cold PBS and digested with RNase (20 mg/mL) for 1 h at 37°C. The cells were stained with PI (1.0 mg/mL; Sigma-Aldrich, St. Louis, MO) and protected from light until analyzed using a BD FACSCanto II at excitation wavelength 488 nm and emission wavelength 617 nm.
Statistical analysis
IC50 values for K-ras were calculated using Table Curve 2Dv4 (System Software Inc., San Jose, CA). For all experiments, mean values and standard deviations from triplicate samples of two independent experiments were calculated using Microsoft Excel 2010. To evaluate the statistical significance, the one-way analysis of variance (ANOVA) was performed. The statistical significance was evident if p < 0.05. The Tukey-Kramer’s test was employed to test the significance between treatments.
Results
NF-κB protein levels
The effects of 13-acetoxyrolandrolide on mediators of the NF-κB pathway and on activation of NF-κB were evaluated by western blot analysis (Fig. 2A and 2B). The attenuating effect on subunits p65 and p50 was concentration-dependent. Similarly, the expression of IKKβ, an upstream mediator in the canonical pathway, was found to be significantly down-regulated in a concentration-dependent manner in treated HT-29 cells. The results from the Western analysis indicated that 13-acetoxyrolandrolide down- regulated the upstream mediator IKKβ in colon cancer and inhibited the activation of NF-κB (Fig. 2A).
Figure 2.
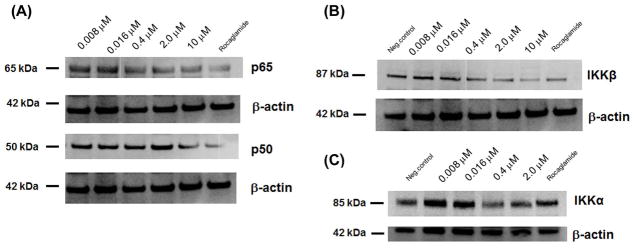
Immunoblot analysis showing that 13-acetoxyrolandrolide suppresses NF-κB activation after 3 h of incubation. Cells treated with rocaglamide were used as positive control. (A) Effect of 13-acetoxyrolandrolide on NF-κB p50 and p65. Subunit p65 (Rel) was down-regulated in a dose-dependent manner. (B) Effect of treatment on kinase IKKβ. IKKβ was significantly down-regulated in cells treated with 13-acetoxyrolandrolide. (C) Effect of treatment on kinase IKKα. β-Actin was used as an internal control.
Intracellular levels of ROS
Cancer cells are susceptible to oxidative stress and the oxidative pathway triggers cell death. The intracellular generation of ROS was measured and assessed by using a fluorescent probe DCFH-DA. The levels of ROS increased in HT-29 cells with increasing concentrations of 13-acetoxyrolandrolide after 4.5 h of treatment; however, the amounts of ROS were not significantly higher than the levels generated by hydrogen peroxide (H2O2), the positive control (Fig. 3). Vitamin C was used as negative control.
Figure 3.

Effect of 13-acetoxyrolandrolide on the ROS pathway in HT-29 cells. Cells were exposed for 4.5 h in the absence and presence of 13-acetoxyrolandrolide. Hydrogen peroxide (H2O2) was used as the positive control. ROS increase in treated cells was not significantly higher than H2O2, but higher than the negative control. Data is presented as mean ± SEM (*p < 0.01 and **p < 0.001 versus untreated cells).
K-Ras inhibition
The growth factor EGF was used to stimulate the K-Ras pathway. The inhibition of the upstream GTPase, K-Ras, was assessed in HT-29 colon cancer cells treated with the 13-acetoxyrolandrolide in a dose-dependent manner. The IC50 for 13-acetoxyrolandrolide was 7.7 μM when tested using the Ras GTPase Chemi ELISA assay.
Effects on the mitochondrial membrane potential
The percent of cells undergoing apoptosis due to changes in the mitochondrial membrane potential (ΨΔm) was examined by cytofluorometric analysis using JC-1 staining. More JC-1 monomers or apoptotic cells (64%) were detected in HT-29 cells treated with 13-acetoxyrolandrolide at 1 μM (Fig. 4). In untreated HT-29 cells, a higher percentage of JC-1 aggregates (78%) were observed (Fig. 4), suggesting the presence of a higher % of healthy cells.
Figure 4.

Effect of 13-acetoxyrolandrolide on the mitochondrial outer membrane potential (ΔΨm). HT-29 cells were treated with 13-acetoxyroladrolide (1 μM) for 24 h. The potentiometric dye JC-1was used to stain cells. Treatment of HT-29 cells with 13-acetoxyroladrolide at 1 μM (colored trace) caused 64% of JC-1 to remain as monomer, which suggests a loss of ΔΨm and the early stages of apoptosis. At high ΔΨm, red fluorescent J-aggregates are formed in healthy cells; however, in apoptotic cells with low ΔΨm, JC-1 remains in the monomeric form, which only exhibits green fluorescence.
Intracellular levels of NAD(P)H
The HT-29 colon cancer cells were exposed to 13-acetoxyrolandrolide to evaluate its effect on mitochondrial oxidative phosphorylation. The intracellular levels of NAD(P)H were monitored hourly for 11 hours following a previously published method (Nakamura et al., 2003). A decreased NAD(P)H generating capacity was detected in treated cells as early as 1 h after treatment (Fig. 5). In addition, the real-time XTT assay showed that between 2–3 h after treatment, the levels of NAD(P)H in the 13-acetoxyrolandrolide- and staurosporine- treated cells decreased to 60% (Fig. 5). This decrease was not detected in the untreated cells, where the levels of NAD(P)H were >90%. The effect on the intracellular NAD(P)H was concentration dependent and decreased levels of NAD(P)H were found with increasing concentrations of 13-acetoxyrolandrolide. The kinetic evaluation showed that the highest level of NAD(P)H was detected at 2 h. This was followed by a rapid drop and the lowest values of NAD(P)H were observed after 3 h of treatment (Fig. 6). Hence, the NAD(P)H difference between 13-acetoxyrolandrolide- treated and untreated cells was most significant at the fourth reading time-point (3 h). Thus, the effect of 13-acetoxyrolandrolide on the generation of NAD(P)H by the mitochondrial oxidative phosphorylation, was also time-dependent. The cells did not recover their capacity to generate intracellular NAD(P)H after treatment, and not until after 48 hours did the levels of NAD(P)H begin to increase due to cell doubling of the remnant cells (data not shown). Lower levels of NAD(P)H were also detected after 3 h of treatment with the positive control, staurosporine. The effect on the levels of NAD(P)H after 3 h of treatment was compared between 13-acetoxyrolandrolide and staurosporine. Even though cells treated with 13-acetoxyrolandrolide (800 nM) showed reduction to 60% of NAD(P)H when compared with 71% reduction of NAD(P)H in cells treated at a higher dose with the positive control, staurosporine (1.6 μM), the difference between these two groups was not statistically different (Fig. 6). Both 13-acetoxyrolandrolide and staurosporine shared a similar kinetic profile on the levels of NAD(P)H in the real time XTT-assay.
Figure 5.

Effect of 13-acetoxyrolandrolide on mitochondrial phosphorylation. Intracellular levels of NAD(P)H were measured in treated and untreated HT-29 cells over a 24 h period. HT-29 cells were treated with different concentrations of 13-acetoxyrolandrolide (0.0064, 0.032, 0.8, and 4.0 μM) and a positive control, staurosporine (1.6 μM). The real-time levels of intracellular NAD(P)H were affected in a dose- and time-dependent manner.
Figure 6.

Effect of 13-acetoxyrolandrolide on intracellular levels of NAD(P)H after 3 h of treatment. 13-Acetoxyrolandrolide (0.8 μM) and staurosporine (1.6 μM) were statistically compared with untreated cells. The results indicated that 13-acetoxyrolandrolide effectively depleted NAD(P)H when compared with untreated cells. The ability of 13-acetoxyrolandrilide at 0.8 μM in depleting NAD(P)H is comparable to that of the positive control staurosporine at 1.6 μM. Experiments performed in triplicate (*p < 0.05 versus not treated).
Caspase-3 activity
Caspase-3 activity was evaluated using western blot and Caspase 3/7 GLO® assay kit. Western blot analysis showed induced expression of caspase-3 and higher levels of the protein were detected in the cell lysates of HT-29 cells when treated with increasing concentration of acetoxyrolandrolide-13 (0.008–10 μM). The caspase-3 expression at the highest treated concentration of acetoxyrolandrolide (10 μM) was comparable to that of the positive control, rocaglamide, at the same concentration (Fig. 7). The enzymatic effect was dose-dependent when measured using the Caspase 3/7 GLO® assay kit (Fig. 8).
Figure 7.
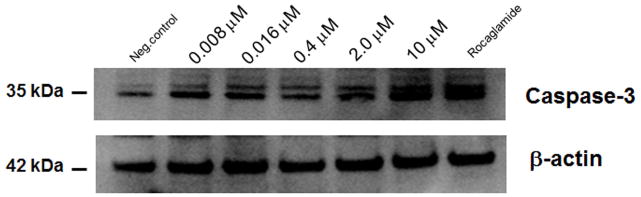
Effect of 13-acetoxyrolandrolide on levels of caspase-3 in HT-29 cells. Western blot analysis showed that caspase-3 was up-regulated in HT-29 cells when treated with 13-acetoxyrolandrolide (0.008, 0.016, 0.4, 2.0 and 10 μM). The effect was compared with a positive control, rocaglamide.
Figure 8.
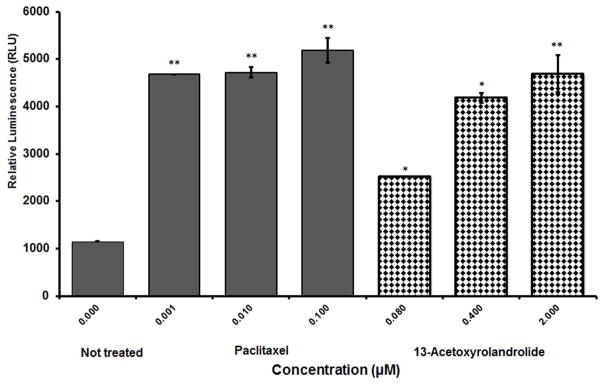
Caspase-3 inducing activity of 13-acetoxyrolandrolide detected using the Caspase–Glo3/7® assay. Caspase-3/7 activity was induced in a concentration- dependent manner after treatment with 13-acetoxyrolandrolide, in comparison with untreated HT-29 colon cancer cells. Experiments were performed in triplicate (*p < 0.05 and **p < 0.01 versus untreated cells).
Cell cycle analysis
The apoptotic effect and DNA fragmentation was determined using PI to stain DNA in HT-29 cells. The percentage of the cell population accumulated at the G1-phase in treated cells was compared to that of untreated HT-29 cells. In the untreated HT-29 cells, 55% of the population were detected at the G1 phase (Fig. 9A), compared to 68% for the treated cells (Fig. 9B).
Figure 9.
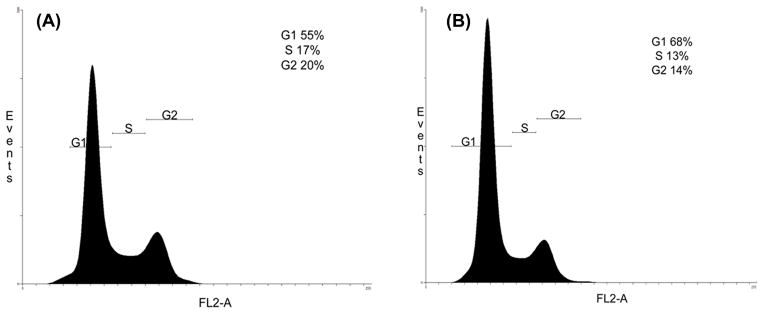
The effect of 13-acetoxyrolandrolide on the cell cycle progression in HT-29 cells. Cells were seeded on 10 cm plates, treated with 13-acetoxyrolandrolide (10 μM) for 24 h, and stained with PI (1.0 mg/mL) for analysis using BD FACSCanto. (A) A negative control of HT-29 cells shows 55% of cells in the G1 phase. (B) Treatment with 13-acetoxyrolandrolide showing 68% of HT-29 cells in the G1 phase.
Discussion
The transcription factor NF-κB is activated during inflammation and more recently there is increasing evidence showing that the activation of this genetic regulator and DNA binding protein is pivotal in the survival and proliferation of malignant cancer cells (Sato et al., 2011). Recent reports have shown that suppression of NF-κB activation negatively affects aerobic glycolysis, particularly glutamine metabolism, which induces mitochondrial dysfunction and promotes cell death (Rathore et al., 2012). In an attempt to characterize the effects of 13-acetoxyrolandrolide on the NF-κB pathway, the upstream mediators IKKα and IKKβ were analyzed by western blot analysis. Effects on the subunit p65 and p50 were also studied (Figs. 2A and 2B). A dose-dependent effect of 13-acetoxyrolandrolide on the NF-κB pathway was observed. The expression of IKKβ, an upstream mediator in the canonical pathway and an important activator of p65, was significantly down-regulated in a concentration-dependent manner in treated HT-29 cells. This suggested that treatment with 13-acetoxyrolandrolide interfered with NF-κB activation. Moreover, Western blot analysis confirmed and validated previous findings showing an attenuating effect on subunit p65 (RelA) in the nuclear extract of treated cells (Pan et al., 2010). According to previous reports, NF-κB prevented apoptosis (Darnell, 2002; Cory and Cory, 2007), thus the concentration-dependent inhibitory effect shown, suggested that 13-acetoxyrolandrolide displayed pro-apoptotic activity.
The apoptotic pathway can be initiated in cancer cells by different stimuli such as radiation, cytotoxic agents, oxidative stress and growth factor withdrawal, and two major pathways are known to induce programmed cell death, the extrinsic pathway mediated by the death FAS/TRAIL receptor, and the intrinsic mitochondrial pathway (Marini et al., 2005; Wu et al., 2011). Since the apoptotic pathway can also be triggered by the withdrawal of growth factors, such as EGF, and the generation of ROS (Efferth et al., 2007; Wu et al., 2011), the effect of 13-acetoxyrolandrolide on HT-29 cells was examined on both of these pathways. There was a small increase in the intracellular levels of ROS after 4.5 h of treatment. Levels of ROS were not significantly higher than the levels of ROS for hydrogen peroxide, the positive control, but were significantly higher than the levels observed for the untreated sample, negative control. This was attributed mainly to reflect the effects on the mitochondrial membrane potential and to a lesser extent to the activation of the oxidative stress pathway. The small increase in the intracellular levels of ROS suggested that oxidative stress was not responsible alone in initiating cell-death in colon cancer HT-29 cells treated with 13-acetoxyrolandrolide.
Second, growth factor EGF is the ligand of epidermal growth factor receptor (EGFR) on the cell surface and there has previously been reported anticancer activity against colon cancer when targeting EGFR. The growth factor EGF induces activity on the plasma membrane bound K-Ras protein, leading to down-stream signaling transduction. When tested on the EGF/K-Ras pathway, 13-acetoxyrolandrolide showed a moderate K-Ras inhibitory effect, interfering with the effect of the growth factor, EGF, on HT-29 colon cancer cells.
These findings suggested that “cross-talk” occurs between the EGF/K-Ras pathway and NF-κB pathway in HT-29 cells, possibly through MAPK kinase kinase 1 (MEKK1), which has also been reported to regulate down-stream IKKβ activity (Surh, 2003). According to previous studies, the EGF and MAPK pathway exerts a pro-survival mechanism in cancer cells probably by sequestering Bad, a Bcl-2 regulating protein, in the cytoplasm, thus preventing Bcl-2 from interacting with mitochondrial membrane associated proteins (Datta et al., 1997; Bonni et al., 1999). In addition, the inhibition of K-Ras was previously found to be associated with the dephosphorylation of Bad protein, which initiates the mitochondrial apoptotic pathway by forming a heterodimer with Bcl-2 and allowing the formation of pores on the outer membrane of the mitochondria (Hussain et al., 2012; Matsuzawa and Ichijo, 2001). Since mitochondrial metabolism is essential for EGF and K-Ras induced cell proliferation and tumorigenesis (Weinberg et al., 2010), 13-acetoxyrolandrolide was tested for its effect on the mitochondria in HT-29 colon cancer cells.
The permeability of the outer mitochondrial membrane is the point of no return in inducing cell death by the mitochondria intrinsic apoptotic pathway (Kang and Reynolds, 2009). To confirm the apoptotic effects of 13-acetoxyrolandrolide, the effect on the mitochondrial transmembrane potential (ψΔm) was investigated in HT-29 cells. When treated for 24 h with 13-acetoxyrolandrolide, the mitochondrial membrane potential (ψΔm) of HT-29 colon cancer cells was depolarized, suggesting that pro-apoptotic proteins and caspase activator might have been released.
The mitochondrial function and oxidative phosphorylation were measured over time using the XTT assay. Overall, the intracellular levels of NAD(P)H were assessed periodically at intervals of 1 h, over 12 h, 24 h and 48 h. Analysis of the mitochondrial oxidative phosphorylation revealed that the intracellular levels of NAD(P)H started to decrease after 2 h of exposure to 13-acetoxyrolandrolide. These findings indicated that the NAD(P)H generating capacity was either negatively affected and that the intracellular levels were depleted due to the increased permeability of the mitochondrial membrane induced by 13-acetoxyrolandrolide. The effect on the levels of NAD(P)H was concentration- and time-dependent. The effect on the mitochondria was assessed independently in two different experiments and it was found that both the oxidative phosphorylation and mitochondrial membrane potential (ψΔm) were negatively affected in treated cells, at 3 h and 24 h, respectively. This suggested that cells undergo early apoptosis after 2 h of treatment through the mitochondrial apoptotic pathway and after this time-point the treated cells do not recover their NAD(P)H generating capacity. However, a small increment of NAD(P)H was initiated after 48 h of treatment, due to the doubling of the remaining cells (data not shown). When cells were treated with 13-acetoxyrolandrolide at lower doses than staurosporine, the levels of NAD(P)H were reduced. This suggested that the sesquiterpene lactone displayed similar effect in reducing the mitochondrial oxidative phosphorylation when compared with the positive control, staurosporine. Levels of NAD(P)H affect the enzymatic activity of quinone reductase, an important detoxifying enzyme in cancer cells (Anusevicius et al., 2002) as previously reported for another sesquiterpene lactone, alantolactone, by inducing effect of on detoxifying enzymes in cancer cells (Seo et al., 2008; Khan et al., 2012). NAD(P)H is a critical substrate of NAD(P)H oxidase and has an important role in generating NAD+ for cellular metabolism and aerobic glycolysis in cancer cells (Lu et al., 2012). This not only confirmed previous findings showing that HT-29 cells are particularly susceptible to mitochondrial dysregulation (Shang et al., 2012), but it also showed the potential mechanism of action of 13-acetoxyrolandrolide.
Apoptosis requires the activation of caspases and of cytochrome c being released from the mitochondria (Liu et al., 1996; Slee et al., 1999). It was found that the presence of pro-apoptotic protein, caspase-3, increased in a concentration-dependent manner when cells were treated with 13-acetoxyrolandrolide. In addition, the enzymatic activity of caspase-3 increased when treated with 13-acetoxyrolandrolide in a concentration dependent manner (Figs. 8 and 9). Thus, the presence of increasing levels of caspase-3 was associated with increased enzymatic activity. This showed that caspase activation had occurred in response to NAD(P)H levels, and that HT-29 colon cancer cells undergo caspase-3-mediated cell death initiated by the intrinsic mitochondrial pathway.
The apoptotic effects of 13-acetoxyrolandrolide in colon cancer cells were evaluated using fluorescence cytometric analysis. The DNA content indicated that cell cycle arrest occurred at the G1-phase in treated cells. Cell cycle analysis showed an increase of the population of cells in G1-phase, which was found to be associated with apoptosis. This indicated a block of cell-cycle progression in colon cancer cells, confirming the antiproliferative effects of 13-acetoxyrolandrolide. In summary, the effect of K-Ras inhibition by 13-acetoxyrolandrolide resulted in IKKβ inhibition, negatively affecting NF-κB activation. It was also associated to increased permeability of the outer mitochondrial membrane, subsequently leading to mitochondrial dysfunction, which was followed by a depletion in NAD(P)H and caspase-3-induced apoptosis. The oncogenic KRAS mutations are commonly found in colon cancers (Nandan et al., 2008) and it has previously been shown that the oncogenic K-Ras protein induces mitochondrial metabolism (Telang et al., 2007). Targeting K-Ras and triggering the mitochondrial apoptotic pathway represents a new selective approach to target tumor cells. Moreover, this study showed that the agents that down-stream target transcription factor NF-κB, such as 13-acetoxyrolandrolide, increased the susceptibility of HT-29 colon cancer cells to undergo programmed cell death through the mitochondrial apoptotic pathway.
Several sesquiterpenes, originally identified from plants, are currently in clinical trials. The parthenolide analogue, dimethylaminoparthenolide is being evaluated in a Phase I clinical trial in leukemia patients that responds poorly to conventional chemotherapy (Ghantous et al., 2010). Although artemisinin has short half-life and poor bioavailability due to hepatocellular activity (Svensson et al., 1999), its derivatives are recommended by WHO to be used in antimalarial combination therapy against resistant strains of Plasmodium falciparum, and are being evaluated in patients with colorectal cancer (Ghantous et al., 2010). The pharmacokinetic properties of 13-acetoxyrolandrolide have not been evaluated in this study however the data presented from in vitro studies suggests that derivatives could be chemically optimized to increase the efficacy of current anticancer agents used to treat malignant cell resistant to chemotherapy, and prevent proliferation and metastasis of colon cancer. Through chemical optimization, bioavailability of the derivatives could also be improved. Many drugs that target mitochondria are derived from natural products e.g. resveratrol and betulinic acid (Fulda et al., 2010). Therefore future drug discovery programs that identify mitochondria-targeting agents from natural products based libraries may result in new antineoplastic agents.
Conclusion
Colon cancer is one of the most frequently diagnosed tumor malignancies and the incidence of the disease is 1.2 million per year (Jemal et al., 2011). The number of cases is rapidly increasing in areas of previously low risk due to changes in diet and life-style as well as to aging population (Jemal et al., 2011). No previous reports were found on a K-Ras inhibitor that induces cell death through the mitochondrial apoptotic pathway. To improve the prognosis of colon cancer and to lower the mortality rate, antiproliferative agents that promotes apoptotic cell death through K-Ras and mitochondrial changes, such as 13-acetoxyrolandrolide, could be developed to sensitize colon cancer cells and increase the efficacy of existing treatments.
Acknowledgments
We greatly acknowledge the financial support from program project grant P01 CA125066-S1 from the National Cancer Institute, NIH, Bethesda, MD to carry out this work.
Footnotes
Conflict of interest
The authors declare that no conflict of interest is disclosed.
References
- Anusevicius Z, Sarlauskas J, Cenas N. Two-electron reduction of quinones by rat liver NAD(P)H:quinone oxidoreductase: quantitative structure-activity relationships. Arch Biochem Biophys. 2002;404:254–262. doi: 10.1016/s0003-9861(02)00273-4. [DOI] [PubMed] [Google Scholar]
- Bang D, Wilson W, Ryan M, Yeh JJ, Baldwin AS. Function in pancreatic pancer via TAK1-TAB stabilization and regulation of non-canonical NF-κB. Cancer Discov. 2013;3:690–703. doi: 10.1158/2159-8290.CD-12-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharate SB, Singh B, Vishwakarma RA. Modulation of K-Ras signaling by natural products. Curr Med Chem. 2012;19:2273–2291. doi: 10.2174/092986712800229050. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Cabral Salles-de-Melo MR, Mendonça de Lucena R, Semir J, de Carvalho R, de Cassia Araujo Pereira R, Benko-Iseppon AM. Karyological features and cytotaxonomy of the tribe Vernonieae (Asteraceae) Plant Syst Evol. 2010;285:189–199. [Google Scholar]
- Cory AH, Cory JG. Understanding interactions between and among apoptosis inducing pathways in tumor cells. In Vivo. 2007;21:245–249. [PubMed] [Google Scholar]
- Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Efferth T, Giaisi M, Merling A, Krammer PH, Li-Weber M. Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS One. 2007;2:e693. doi: 10.1371/journal.pone.0000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9:447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov Today. 2010;15:668–678. doi: 10.1016/j.drudis.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Hussain AR, Ahmed SO, Ahmed M, Khan OS, Al Abdulmohsen S, Platanias LC, Al-Khan M, Yi F, Rasul A, Li T, Wang N, Gao H, Gao R, Ma T. Alantolactone induces apoptosis in glioblastoma cells via GSH depletion, ROS generation, and mitochondrial dysfunction. IUBMB Life. 2012;64:783–794. doi: 10.1002/iub.1068. [DOI] [PubMed] [Google Scholar]
- Kuraya KS, Uddin S. Cross-talk between NF-kappaB and the PI3-kinase/AKT pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis. PLoS One. 2012;7:e39945. doi: 10.1371/journal.pone.0039945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Kim JA, Lau EK, Pan L, Carcache de Blanco EJ. NF-kappaB inhibitors from Brucea javanica exhibiting intracellular effects on reactive oxygen species. Anticancer Res. 2010;30:3295–3300. [PMC free article] [PubMed] [Google Scholar]
- Kitada S, Krajewska M, Zhang X, Scudiero D, Zapata JM, Wang HG, Shabaik A, Tudor G, Krajewski S, Myers TG, Johnson GS, Sausville EA, Reed JC. Expression and location of pro-apoptotic Bcl-2 family protein BAD in normal human tissues and tumor cell lines. Am J Pathol. 1998;152:51–61. [PMC free article] [PubMed] [Google Scholar]
- Lerebours F, Vacher S, Andrieu C, Espie M, Marty M, Lidereau R, Bieche I. NF-kappa B genes have a major role in inflammatory breast cancer. BMC Cancer. 2008;8:41. doi: 10.1186/1471-2407-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Lu W, Hu Y, Chen G, Chen Z, Zhang H, Wang F, Feng L, Pelicano H, Wang H, Keating MJ, Liu J, McKeehan W, Wang H, Luo Y, Huang P. Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PLoS Biol. 2012;10:e1001326. doi: 10.1371/journal.pbio.1001326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini P, Schmid A, Jendrossek V, Faltin H, Daniel PT, Budach W, Belka C. Irradiation specifically sensitises solid tumour cell lines to TRAIL mediated apoptosis. BMC Cancer. 2005;5:5. doi: 10.1186/1471-2407-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa A, Ichijo H. Molecular mechanisms of the decision between life and death: regulation of apoptosis by apoptosis signal-regulating kinase 1. J Biochem. 2001;130:1–8. doi: 10.1093/oxfordjournals.jbchem.a002947. [DOI] [PubMed] [Google Scholar]
- Muñoz Acuña U, Wittwer J, Ayers S, Pearce CJ, Oberlies NH, Carcache de Blanco EJ. Effects of (5Z)-7-oxozeaenol on the oxidative pathway of cancer cells. Anticancer Res. 2012;32:2665–2671. [PMC free article] [PubMed] [Google Scholar]
- Nakamura J, Asakura S, Hester SD, de Murcia G, Caldecott KW, Swenberg JA. Quantitation of intracellular NAD(P)H can monitor an imbalance of DNA single strand break repair in base excision repair deficient cells in real time. Nucleic Acids Res. 2003;31:e104. doi: 10.1093/nar/gng105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- Nandan MO, McConnell BB, Ghaleb AM, Bialkowska AB, Sheng H, Shao J, Babbin BA, Robine S, Yang VW. Kruppel-like factor 5 mediates cellular transformation during oncogenic K-Ras-induced intestinal tumorigenesis. Gastroenterology. 2008;134:120–130. doi: 10.1053/j.gastro.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Lantvit DD, Riswan S, Kardono LBS, Chai H-B, Carcache de Blanco EJ, Farnsworth NR, Soejarto DD, Swanson SM, Kinghorn AD. Bioactivity-guided isolation of cytotoxic sesquiterpenes of Rolandra fruticosa. Phytochemistry. 71:635–640. doi: 10.1016/j.phytochem.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Rathore MG, Saumet A, Rossi JF, de Bettignies C, Tempe D, Lecellier CH, Villalba M. The NF-kappaB member p65 controls glutamine metabolism through miR-23a. Int J Biochem Cell Biol. 2012;44:1448–1456. doi: 10.1016/j.biocel.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Sato A, Kudo C, Yamakoshi H, Uehara Y, Ohori H, Ishioka C, Iwabuchi Y, Shibata H. Curcumin analog GO-Y030 is a novel inhibitor of IKKbeta that suppresses NF-kappaB signaling and induces apoptosis. Cancer Sci. 2011;102:1045–1051. doi: 10.1111/j.1349-7006.2011.01886.x. [DOI] [PubMed] [Google Scholar]
- Sen GS, Mohanty S, Hossain DM, Bhattacharyya S, Banerjee S, Chakraborty J, Saha S, Ray P, Bhattacharjee P, Mandal D, Bhattacharya A, Chattopadhyay S, Das T, Sa G. Curcumin enhances the efficacy of chemotherapy by tailoring p65NFkappaB-p300 crosstalk in favor of p53-p300 in breast cancer. J Biol Chem. 2011;286:42232–42247. doi: 10.1074/jbc.M111.262295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo JY, Lim SS, Kim JR, Lim JS, Ha YR, Lee IA, Kim EJ, Park JH, Kim JS. Nrf2-mediated induction of detoxifying enzymes by alantolactone present in Inula helenium. Phytother Res. 2008;22:1500–1505. doi: 10.1002/ptr.2521. [DOI] [PubMed] [Google Scholar]
- Shang LH, Li CM, Yang ZY, Che DH, Cao JY, Yu Y. Luffa echinata Roxb. induces human colon cancer cell (HT-29) death by triggering the mitochondrial apoptosis pathway. Molecules. 2012;17:5780–5794. doi: 10.3390/molecules17055780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- Svensson US, Sandström R, Carlborg O, Lennernäs H, Ashton M. High in situ rat intestinal permeability of artimisinin unaffected by multiple dosing and with no evidence of P-glycoprotein involvement. Drug Metab Disp. 1999;(27):227–232. [PubMed] [Google Scholar]
- Telang S, Lane AN, Nelson KK, Arumugam S, Chesney J. The oncoprotein H-RasV12 increases mitochondrial metabolism. Mol Cancer. 2007;6:77–90. doi: 10.1186/1476-4598-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeks RA. Sacred Leaves of Candomblé: African Magic, Medicine, and Religion in Brazil. University of Texas Press; Austin: 1997. p. xvii.p. 236. [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for K-Ras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CL, Huang AC, Yang JS, Liao CL, Lu HF, Chou ST, Ma CY, Hsia TC, Ko YC, Chung JG. Benzyl isothiocyanate (BITC) and phenethyl isothiocyanate (PEITC)-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of caspase-3, mitochondria dysfunction and nitric oxide (NO) in human osteogenic sarcoma U-2 OS cells. J Orthop Res. 2011;29:1199–1209. doi: 10.1002/jor.21350. [DOI] [PubMed] [Google Scholar]
- Yin Y, Chen X, Shu Y. Gene expression of the invasive phenotype of TNF-alpha-treated MCF-7 cells. Biomed Pharmacother. 2009;63:421–428. doi: 10.1016/j.biopha.2009.04.032. [DOI] [PubMed] [Google Scholar]
- Zhou R, Hu G, Gong AY, Chen XM. Binding of NF-kappaB p65 subunit to the promoter elements is involved in LPS-induced transactivation of miRNA genes in human biliary epithelial cells. Nucleic Acids Res. 2010;38:3222–3232. doi: 10.1093/nar/gkq056. [DOI] [PMC free article] [PubMed] [Google Scholar]