

Abstract
Purpose
Inflammatory response in injured brain parenchyma after traumatic brain injury (TBI) is crucial to its pathologic process. In order to follow the microglia activation and neuroinflammation after TBI, herein, we performed PET imaging in a rat TBI model using 18F-labeled DPA-714, a ligand of 18 KDa translocator protein (TSPO).
Methods
TBI was induced in male SD rats by controlled cortical impact (CCI). The success of the TBI model was confirmed by magnetic resonance imaging (MRI). Automated synthesis of [18F]DPA-714 was carried out using a slightly modified TRACERLab FX-FN module. In vivo PET imaging was performed at different time points after surgery by an Inveon small animal PET scanner. The specificity of [18F]DPA-714 was confirmed by displacement study with an unlabeled competitive TSPO ligand, PK11195. Ex vivo autoradiography as well as immunofluorescence staining was carried out to confirm the in vivo PET results.
Results
Both in vivo T2-weighted MR images and ex vivo TTC staining results revealed successful establishment of the TBI model. Compared with the sham group, [18F]DPA-714 uptake was significantly higher in the injured brain area on PET images. Increased lesion-to-normal brain ratio of [18F]DPA-714 in TBI rats was observed at day 2 after surgery, peaked around day 6 (2.65 ± 0.36) and then decreased gradually to nearly normal level at day 28. Displacement study using PK11195 confirmed the specific binding of [18F]DPA-714 to TSPO. Ex vivo autoradiography was consistent with in vivo PET results. The immunofluorescence staining showed a time course of TSPO expression after TBI and the temporal and spatial distribution of microglia in damaged brain area.
Conclusion
TSPO targeted PET using [18F]DPA-714 as the imaging probe can be used to dynamically monitor inflammatory response after TBI in a non-invasive manner. This method will not only facilitate a better understanding of inflammation process after traumatic brain injury, but also provide a useful in vivo monitoring strategy for anti-inflammation therapy of TBI.
Keywords: Traumatic brain injury (TBI), Translocator protein (TSPO), [18F]DPA-714, PET, molecular imaging
Introduction
Traumatic brain injury (TBI) is one of the leading causes of mortality and morbidity for people under the age of 45, which can result in temporal or long-term, even lifelong physical, cognitive, and behavioral problems [1]. It causes structural damage and functional deficits due to both primary and secondary injuries. The primary injury is the mechanical disruption of brain tissue while the secondary injury is a cascade of metabolic, cellular and molecular events afterwards, which is a result of biochemical derangements such as glutamate excitoxicity, enhanced level of free radicals, neuroinflammation and apoptosis [2]. Among them, neuroinflammation represents a key pathological response to brain injury and is characterized by glial cell activation including microglia and astrocyte, peripheral leukocyte and macrophage infiltration, and upregulation of pro-inflammatory mediators [3]. The inflammatory reaction after TBI is a double-edged sword and the detrimental effects of neuroinflammation on the injured brain have been well recognized [4]. However, it has also been proven clearly that beneficial effects can be achieved if neuroinflammation is controlled in a regulated manner and for defined periods of time [5]. Hence, accurate monitoring the dynamics of neuroinflammaion after TBI would be very helpful to design effective therapeutic interventions to avoid secondary injury.
As a primary safeguard of brain homogenesis, microglia undergo a shift of morphology as well as functional property once the brain is under pathological conditions including traumatic injury [6]. Activated microglia are involved in tissue repair, amplification of inflammatory effects and neuronal degeneration, and functional state of microglia can, to some extent, reflect the status of immune reaction in brain [5, 7]. Consequently, neuroinflammation in several central nervous disease models, such as brain tumor [8], stroke [9], Alzheimer’s disease [10], has been evaluated by positron emission tomography (PET) using 18 KDa translocator protein (TSPO) targeted imaging probes. The reason to choose TSPO as the microglia marker is that the majority of the TSPO-positive cells in injured brain are microglia. Moreover, TSPO is expressed at low levels in the healthy brain but is remarkably up-regulated in response to injury and inflammation [11].
There have been several TSPO ligands labeled with radioisotopes for either autoradiography or in vivo PET of neuroinflammation [12–15]. Compared with other radio-ligands of TSPO, [18F]DPA-714 has the advantage of high binding affinity and target-to-background ratio [16, 17]. Moreover, the physical property of F-18 (t1/2 = 109.8 min) make it highly suitable for in vivo PET imaging. The pilot clinical study in healthy humans confirmed the in vivo stability and biodistribution, and acceptable effective dose estimation of [18F]DPA-714, supporting further clinical trials and preclinical evaluation with this PET imaging [18].
Therefore, here we performed a longitudinal imaging study using [18F]DPA-714 for non-invasive and real-time evaluation of neuroinflammation in a rat TBI model. We also performed magnetic resonance imaging (MRI) and [18F]FDG PET to monitor the anatomical and metabolic changes of the injury sites. These imaging studies are expected to shed more light on better understanding of inflammation process after traumatic brain injury and provide a useful in vivo monitoring method for anti-inflammation therapy of TBI.
Materials and Methods
General materials
All reagents were of analytical grade and obtained from commercial sources. [18F]F− radionuclide was obtained from the NIH Clinical Center’s cyclotron facility by proton irradiation of 18O-enriched water. [18F]FDG was purchased from the Nuclear Pharmacy of Cardinal Health and reconstituted with sterile saline.
Synthesis of [18F]DPA-714
Automated synthesis of 18F-DPA-714 was carried out using a slightly modified TRACERLab FX-FN module (GE Medical Systems). In brief, [18F]fluoride (typically 200–400 μL, 103–133 mCi) was trapped by the Chromafix® cartridge (Macherey-Nagel, prepared by washing with 2 mL of ethanol and then rinsing with 4 mL of water) under vacuum was eluted and transferred to the reaction vessel with a solution containing K2CO3 (3 mg in 300 μL of pure water), acetonitrile (500 μL), and 10 mg of Kryptofix-222 (K222, 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane). The reaction mixture was then evaporated to dryness. Then tosylate substrate (3–4 mg) was dissolved in dimethyl sulfoxide (0.6 mL) and transferred to the dry 18F-labeled KF-K222 complex and allowed to react at 165 °C for 5 min. After completion, the reaction mixture was diluted with semi-preparative HPLC solvent (2 mL) and passed through a Sep-Pak® light Alumina N cartridge (Waters, prepared by washing with 4 mL of ethanol and then rinsing with 8 mL of water). The crude solution was injected into a Phenomenex Luna 5 μm C18 semi-preparative reversed-phase HPLC column (250 × 10 mm), with a mobile phase of H2O and acetonitrile (55/45, v/v) at a flow rate of 4.0 mL/min. The retention time (tR) of [18F]DPA-714 was determined to be 21 min. The final formulation of the tracer was performed using a Sep-Pak® Plus C18-based system. The yield was 24 ± 3% with a specific activity of 41–107 GBq/μmol (n = 8). The excess ethanol was removed through rotary evaporation and concentrated to 250–350 μL. The tracer was diluted with PBS for final formulation and animal study.
Animal model of traumatic brain injury
All animal studies were conducted in accordance with the principles and procedures outlined in the NIH Guide for the Care and Use of Laboratory Animals [19] and were approved by the Institutional Animal Care and Use Committee of the Clinical Center (National Institutes of Health).
TBI was induced in adult male Sprague-Dawley rats (body weight: 300–400 g) by controlled cortical impact (CCI). Briefly, rats were anesthetized in a ventilated chamber by isoflurane (3.5% for induction and 2% for maintenance) and then placed in a stereotaxic device. A warm pad was used to keep body temperature at 37 °C during the surgery. After drilling a hole on the left area of skull (2.8 mm left and 3.2 mm back to bregma), a rounded impact tip with a diameter of 5 mm was used to contuse the exposed dura mater. The tip penetration depth was 3.0 mm and the velocity was 5 m/s. Following CCI, the burr hole was covered with bone flip, sealed with Jet Denture Repair Professional Package (Lang Dental Manufacturing Co., Inc., IL). Sham-treated controls were treated similarly without activating the impactor.
Magnetic resonance imaging (MRI)
All MRI scans were performed on a 7.0 T small animal scanner (Bruker, Pharmascan) with a body coil. Rats were anesthetized by isofluorane and kept warm by a circulating water pad. T2-weighted images were obtained by a RARE sequence. The parameters were as follows: TR = 3000 ms, effective TE = 70 ms, NEX = 4, matrix size = 256 × 256, FOV = 4 × 4 cm, slice thickness = 1 mm. The total acquisition time was approximately 8 minutes for each rat.
Positron emission tomography and data analysis
[18F]FDG PET was performed on days 1, 5, 9 and 15 after TBI surgery (n = 3/group). A total of fourteen rats with TBI were used for static [18F]DPA-714 imaging study before and on days 1, 2, 6, 10, 16, and 28 (n = 4–6/group) after traumatic injury. Another four rats were used as sham control. All PET acquisitions were performed with an Inveon small animal PET scanner (Siemens Preclinical Solutions). For static PET, a single dose of either [18F]FDG (about 18.5 MBq (500 μCi)) or [18F]DPA-714 (about 25.9 MBq (700 μCi)) was injected into animals via tail vein under isoflurane anesthesia. One hour after tracer injection, a 15-min static PET scan was performed with a heating pad to keep animal body temperature at 37 °C. For [18F]DPA-714 displacement study, one hour dynamic acquisition was performed on rats at day 6 after TBI (n = 3) with 5 mg/kg unlabeled PK11195 (mixture of R-PK11195 and R,S-PK11195, Adipogen Catalog No. AG-CR1-0008) administered via tail vein at 30 min later after tracer injection. All the images were reconstructed using a two-dimensional ordered-subset expectation maximum algorithm (2D OSEM). No scatter and attenuation correction was performed during imaging reconstruction.
Imaging analysis was performed with Inveon Research Workplace (Siemens Preclinical Solution). With fused PET/MRI images as guidance, three-dimensional ellipsoidal regions of interest (ROIs) were manually defined on the region of lesions in the ipsilateral hemisphere. Another ROI with the same shape was drawn on the corresponding region in the contralateral brain sphere as normal background signals.
2, 3, 5-Triphenyl-2H-tetrazolium chloride (TTC) staining
Slices of 1-mm thickness adjacent to those used for autoradiography were obtained for TTC staining as previously described [20]. All slices were stained with 2 % TTC solution and put in dark for 20 min at 37 °C.
Ex vivo autoradiography
Ex vivo autoradiography was performed on days 2, 6, 10 and 20 after TBI surgery (n = 3 for each time point). Around 25.9 MBq (700 μCi) of [18F]DPA-714 was administered via tail vein into each rat under anesthesia. The rats were sacrificed at 1 h postinjection and the brains were carefully harvested. Axial sections with thickness of 1 mm were obtained for autoradiography study. The tissue slices was exposed to a high-efficiency storage phosphor screen overnight and developed in a Cyclone Plus Storage Phosphor System (PerkinElmer, MA). The autoradiographs were analyzed with Optiquant acquisition and analysis software 5.0 (Perkin Elmer, MA). With TTC as reference, all autoradiographs were scaled with the same minimum and maximum thresholds to optimize the visualization of the lesion sites. Then ROIs were outlined over the lesion sites on PET images at the ipsilateral sphere for quantification. Corresponding ROIs drawn at the contralateral normal brain tissue were used to determine the lesion-to-normal brain ratio.
Immunofluorescence staining
Brain cryosections with a thickness of 8 μm were obtained using a Ultrapro 5000 cryostat (Vibratome). All the sections were fixed with Z-fix solution for 15 min, and then the non-specific binding sites were blocked by PBS containing 1% bovine serum albumin (BSA) for 30 min. Slices were incubated with primary antibodies at 4 °C overnight and then with secondary antibodies at room temperature for another 60 min. After each step, slices were washed gently 3 times with PBS containing 0.05 % tween 20 (PBST) for 5 min. For different staining targets, the antibodies and concentrations were as follows: rabbit anti-rat PBR (TSPO) antibody (1:50; Santa Cruz; sc-20120), mouse anti-rat CD68 antibody (1:100; Abcam; ab955) and goat anti-rat GFAP antibody (1:100; Abcam; ab53554); Cy3-conjugated donkey anti-rabbit secondary antibody (1:200; Jackson ImmunoResearch Laboratories), Dylight 488-conjugated donkey anti-rabbit secondary antibody (1:200; Jackson ImmunoResearch Laboratories) and Cy3-conjugated donkey anti-goat secondary antibody (1:200, Jackson ImmunoResearch Laboratories). All tissue slices were mounted with medium containing 4′, 6-diamidino-2-phenylindole (DAPI), and then observed by an epifluorescence microscope (X81; Olympus).
Statistical analysis
All data were expressed as means plus or minus standard deviation (means ± SD). Statistical analysis was performed with SPSS software (version 18.0; SPSS, Inc., Chicago, IL, USA). One-way analysis of variance (ANOVA) was used for data comparison between different groups and Student’s t test was used for two group comparison. A P value < 0.05 was considered statistically significant.
Results
Metabolic changes in the brain
From day 1 after surgery, a localized region at cortex area with high signal intensity was observed on coronal T2-weighted MRI of rat brain. This region is consistent with the injured site of TBI within several days. Therefore, T2-weighted MRI could provide an anatomic reference for PET images (Fig. 1a). In order to evaluate the glucose metabolism of injured brain, [18F]FDG PET was performed on days 1, 5, 9 and 15 after surgery. Fig. 1b shows representative coronal PET images of [18F]FDG uptake in TBI rat brain at different time points. We also fused MRI and FDG PET images (Fig. 1c) to facilitate data analysis. An obvious [18F]FDG uptake defect can be seen over the traumatic area at day 1 with a lesion-to-normal brain ratio of 0.86 ± 0.07 in the TBI group vs. a ratio of 0.98 ± 0.01 in the sham group (p < 0.05). However, the uptake ratio of [18F]FDG increased significantly at both day 5 (1.27 ± 0.07; p < 0.05, compared with the sham group) and day 9 (1.25 ± 0.09; p < 0.05, compared with sham group) and then decreased to 1.01± 0.03 at day 15 (p > 0.05, compared with sham group) (Fig. 1d).
Fig. 1.
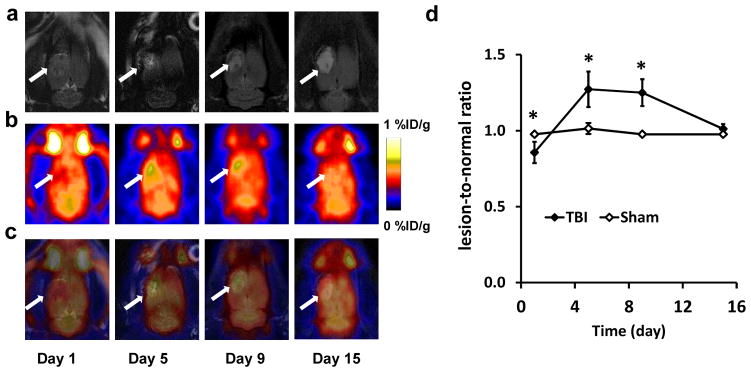
a) Representative coronal T2-weighted MRIs and b) PET images using [18F]FDG at different time points after TBI. The lesion area is indicated by white arrow. c) Fused PET and MRI images at different time points after TBI. d) Quantification of [18F]FDG uptake in the brain tissue after surgery over time, expressed as lesion-to-normal ratio ( *p < 0.05).
Imaging of TSPO expression with [18F]DPA-714 PET
A longitudinal PET study using [18F]DPA-714 as imaging probe was performed to evaluate the dynamics of TSPO expression in brain parenchyma after traumatic injury. Beginning at day 2 after surgery, increased tracer accumulation was observed over the injury site and reached a very high level at day 6. Then, the signal intensity decreased gradually with time. At day 28, no apparent localized tracer uptake was observed (Fig. 2a).
Fig. 2.

[18F]DPA-714 PET images of TSPO expression. a) Representative coronal PET images of [18F]DPA-714 uptake at lesion area after TBI surgery. White arrows indicate the damaged regions. b) Quantification of [18F]DPA-714 uptake at different time points, expressed as lesion-to-normal ratio. The highest tracer uptake appeared at day 6, compared with control group. c) Quantification of [18F]DPA-714 uptake over time after TBI surgery. The uptake ratio increased significantly from day 2 to day 16 compared with sham group (*p < 0.05. **p < 0.01)
Quantitative result of [18F]DPA-714 uptake was presented in Fig. 2b. The lesion-to-normal brain ratio of tracer uptake was 1.68 ± 0.15 at day 2, which was significantly higher than that of the control group (1.02 ± 0.08; p < 0.01). The [18F]DPA-714 uptake in the injury region peaked at day 6 with a ratio of 2.65 ± 0.36 (p < 0.001). Afterwards, the tracer uptake ratio continued to drop but was still significantly higher than that of normal rats at day 10 and day 16 (2.17 ± 0.17 and 1.70 ± 0.75, respectively; p < 0.01). At day 28 after TBI surgery, the ratio was reduced to 1.33 ± 0.15, which was slightly higher than that of normal rats but without significant difference (p > 0.05). The change of uptake ratio of [18F]DPA-714 in rat brains with TBI and sham treatment over time was presented in Fig. 2c. A slight fluctuation of [18F]DPA-714 uptake in the sham group but no significant difference was observed. The [18F]DPA-714 uptakes of TBI group at both day 1 and day 28 were not significantly different from those of the sham group.
In order to further confirm the specific binding of [18F]DPA-714 to TSPO in brain tissue, a dynamic displacement study was performed using unlabeled PK11195 on TBI rats at day 6 after surgery (Fig. 3). The uptake of [18F]DPA-714 at the injured brain area reached a plateau in several minutes after injection in both groups. After a bonus injection of PK11195 at 30 min, the tracer uptake in the lesion region dropped quickly to a plateau of a much lower level. A slight decrease of [18F]DPA-714 uptake was also seen in the normal brain tissue but with much less extent than that in the lesion region. As shown in Fig. 3b, before displacement, an apparent localized tracer accumulation was observed over injured brain site. After PK11195 displacement, the signal intensity dropped dramatically, indicating the binding specificity of [18F]DPA-714 to TSPO (0.15 ± 0.02 vs. 0.36 ± 0.12 %ID/g; p < 0.05) (Fig. 3c). At the same time, the [18F]DPA-714 uptake in the surrounding non-brain tissues showed no apparent changes before and after PK11195 displacement.
Fig. 3.

a) [18F]DPA-714 displacement study using 5 mg/kg PK11195 at day 6 after TBI. Time-activity curves of ROI placed on the lesion and normal brain area with or without PK11195 displacement, expressed as %ID/g of tissue. Arrow indicates the time point of PK11195 injection during one-hour dynamic PET acquisition. b) Representative coronal [18F]DPA-714 PET images of the brain acquired before and after PK11195 displacement. c) [18F]DPA-714 uptake (%ID/g) of the lesion with and without PK11195 displacement at 1 h post-injection (*p < 0.05).
Ex vivo autoradiography
To further investigate the specific binding of [18F]DPA-714 to inflammatory brain tissue, ex vivo autoradiography was performed on healthy rats and TBI rats at days 2, 6, 10 and 20 (n = 3/group) after surgery. Typical axial autoradiographs and the corresponding TTC staining pictures were shown in Fig. 4a. Increased uptake was localized within the traumatic injury area, as evidenced by TTC staining. The quantification of [18F]DPA-714 binding showed a similar trend to that of in vivo PET imaging. The uptake ratio increased from day 2, with the highest ratio at day 6, and then decreased gradually within several days (Fig. 4b).
Fig. 4.

a) Representative in vitro autoradiographs of [18F]DPA-714 uptake in brain tissue and corresponding TTC staining on adjacent slices from the same rats at different time points after surgery. b) Quantification of [18F]DPA-714 uptake ratio based on autoradiograph results after TBI compared with control group. *P < 0.05. **P < 0.01.
Immunohistologic staining
To further evaluate the TSPO expression in traumatically injured brain tissues, we performed immunofluorescence staining on tissue sections acquired at different time points after surgery. The staining results demonstrated a similar time course of TSPO expression with both in vivo PET and ex vivo autoradiography results. The peak of TSPO expression appeared at day 6 after TBI as indicated by strong red fluorescence signal (Figs. 5 and 6).
Fig. 5.

Immunofluorescence staining of brain tissues harvested at different time points after TBI surgery with anti-TSPO (red) and anti-CD68 (green) antibodies. Nuclei were counterstained with DAPI (blue).
Fig. 6.

Immunofluorescence staining of brain slices at different time points after TBI surgery with anti-TSPO (red) and anti-GFAP (green) antibodies. Nuclei were counterstained with DAPI (blue).
In order to delineate the cell source of TSPO overexpression, we co-stained the brain tissue sections with either CD68 (Fig. 5) and GFAP (Fig. 6). Compared with normal brain tissue, a few number of CD68 positive cells appeared from day 2 within the lesion site. At day 6, the injured area was full of CD68 positive cells with an amoeba shape, which were morphologically identified as activated microglia. There were still many CD68 positive cells with a visually smaller size in the damaged brain tissue at days 10 and 16. At early stage (from day 2 to day 6) after surgery, the majority of fluorescence signals from TSPO and CD68 staining overlaid, indicating that TSPO expression is mainly from activated microglia in the injured brain. In the late stage, especially at day 16 after surgery, only partial overlay was observed (Fig. 5). As shown in Fig. 6, GFAP positive cells appeared at day 10 after surgery and showed overlay with TSPO positive cells, indicating that the localized astrocytes may also contribute to the expression of TSPO.
Discussion
As a promising marker of neuroinflammation, TSPO is highly expressed in activated microglia. The low level expression of TSPO in normal brain tissues provides a decent contrast to outstand the inflammatory area. Therefore, [18F]DPA-714 has been used for PET imaging of neuroinflammation in different models [9, 17, 21]. However, to the best of our knowledge, no application of this particular tracer in TBI model has been reported. In this study, we used [18F]DPA-714, to dynamically monitor neuroinflammation in rat brain suffering from traumatic injury. The injury induced neuroinflammation led to local accumulation of [18F]DPA-714. Moreover, the longitudinal imaging revealed the time frame of microglia activation in a real-time and non-invasive manner.
As the most dominant PET imaging tracer, [18F]FDG has also been used to evaluate the cerebral glucose metabolism in rodent CNS disease models [13, 22, 23]. Consistent with these studies, we found a low-uptake area in the early phase after damage due to disturbance of blood circulation and normal brain function. It is well established that the localized microglia and infiltrated peripheral immune cells have an elevated glycolysis after activation due to the oxidative burst [24]. Consequently, we observed high signal intensity from the disarranged brain parenchyma on [18F]FDG brain map at day 5 and day 9. At day 15 after surgery, there was no significant difference in [18F]FDG uptake ratio between the TBI group and the sham group. This was likely due to a combination of reduced inflammatory response and self-recovery of brain tissue, as evidenced by the immunofluorescence staining results of GFAP, an astrocyte cell maker [25]. However, the time frame of [18F]FDG uptake did not agree with the previous report which claimed a lower metabolic ratio even at day 7 after injury [13]. The discrepancy could be explained by the use of different TBI models which may cause different levels of injury severity and inflammatory response pattern. Besides, we used Sprague-Dawley rats while Wistar rats were used in their study [13]. The species difference might also partially account for the discrepancy [26].
PET imaging using other TSPO ligands such as [11C]PK11195 [12] and [18F]FE-DAA1106 [13], demonstrated the feasibility to observe the inflammatory changes after traumatic injury. In vitro autoradiography using [3H]PK11195 showed an significant increase of TSPO expression after traumatic injury began from day 3 and lasted at least 14 days [15]. In vivo PET imaging using [11C]PK11195 showed increased binding in TBI rats at day 10 after surgery [12]. However, the imaging contrast is not ideal, which may be due to the short physical half-life of 11C and thus inadequate clearance of the tracer as well as the relatively high nonspecific binding of [11C]PK11195 [21].
PET imaging with [18F]FEDAA1106 in TBI models demonstrated that the inflammatory response to the injury peaked at 1 week after impact [13]. DPA-714 is another TSPO ligand with high binding affinity and specificity [27]. Moreover, [18F]DPA-714 could be produced with higher yield than [18F]FEDAA1106 [28].
With [18F]DPA-714 PET, we found that significantly increased signal intensity at the lesion area appeared from day 2 and remained high level until day 16. The images at day 6 witnessed a peak of [18F]DPA-714 uptake ratio between lesion and normal brain tissue. This time frame of TSPO expression was in line with the results using other imaging probes [12, 13]. The specificity of [18F]DPA-714 uptake was confirmed by the displacement study using PK11195 as the competitor. It is of note the blockage was not complete, which was similar to the previous reports [27]. There may be several explanations behind this phenomenon such as the existence of different subtypes of TSPO [27] or not identical binding site for DPA-714 and PK11195 [29]. It could also be induced by the nonspecific leakage and retention of [18F]DPA-714 due to edema and blood-brain-barrier (BBB) disruption after traumatic injury [30].
Due to the excellent anatomical resolution, T2 weighted MRI was performed to provide a method for lesion confirmation and guidance for PET quantification. It is still very challenging to outline PET ROIs based on the fused PET/MRI images due to the use of separate scanning systems. We are confident that the PET results did reflect the trend of TSPO level changes over time as results were consistent with the ex vivo autoradiography data.
The increased [18F]DPA-714 accumulation also correlated well with the increased TSPO level evaluated by immunofluorescence staining (Fig. 5 and 6). We co-stained the tissue section with CD68 specific antibody and found that only a few CD68+ cells can be seen in the injured foci at day 2 and the number increased greatly at day 6 and day 10. It has been reported that resident activated microglia is the main cell type that contributes to the elevated TSPO expression in damaged brain parenchyma [31]. Study using chimeric green fluorescent protein-expressing mice showed that resident activated microglia is the predominant cell type in the acute and subacute phase after stroke [32]. Therefore, it is reasonable to establish a causal correlation between microglia activation and quantitative analysis of PET images using [18F]DPA-714 as the imaging probe. However, it has been also reported that peripheral macrophages and reactive astrocytes can also express TSPO in a different profile [33, 34]. Indeed, GFAP positive astrocytes were observed in injured brain tissue after day 10 and these cells also showed TSPO overexpression. Based on these results, we speculated that increased PET signal at the early stage is more related to activated microglia. At late stage, the increased [18F]DPA-714 uptake in the lesion site resulted from TSPO overexpression of microglia and other neuroinflammation related cells. Further investigation is still needed to fully delineate of the contributions of different cell types to the outcome of PET.
Conclusion
In this study, we successful applied [18F]DPA-714 PET imaging to visualize neuroinflammation reaction induced by traumatic injury in a rat model. The longitudinal PET imaging reflected the time frame of TSPO overexpression from activated microglia and astrocytes in a real-time and non-invasive manner. This method will facilitate better understanding of inflammation process after traumatic brain injury, and provide a useful in vivo monitoring strategy for anti-inflammation therapy of TBI.
Acknowledgments
This work was supported by the National Basic Research Program of China (973 program, 2013CB733803, 2013CB733802), and the Intramural Research Program (IRP) of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH).
Footnotes
Conflict of interest statement:
The authors declare that they have no conflict of interest.
References
- 1.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–8. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Ceulemans AG, Zgavc T, Kooijman R, Hachimi-Idrissi S, Sarre S, Michotte Y. The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J Neuroinflammation. 2010;7:74. doi: 10.1186/1742-2094-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 5.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–8. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 6.Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11:775–87. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–40. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 8.Winkeler A, Boisgard R, Awde AR, Dubois A, Theze B, Zheng J, et al. The translocator protein ligand [18F]DPA-714 images glioma and activated microglia in vivo. Eur J Nucl Med Mol Imaging. 2012;39:811–23. doi: 10.1007/s00259-011-2041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin A, Boisgard R, Theze B, Van Camp N, Kuhnast B, Damont A, et al. Evaluation of the PBR/TSPO radioligand [18F]DPA-714 in a rat model of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:230–41. doi: 10.1038/jcbfm.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ji B, Maeda J, Sawada M, Ono M, Okauchi T, Inaji M, et al. Imaging of peripheral benzodiazepine receptor expression as biomarkers of detrimental versus beneficial glial responses in mouse models of Alzheimer’s and other CNS pathologies. J Neurosci. 2008;28:12255–67. doi: 10.1523/JNEUROSCI.2312-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scarf AM, Kassiou M. The translocator protein. J Nucl Med. 2011;52:677–80. doi: 10.2967/jnumed.110.086629. [DOI] [PubMed] [Google Scholar]
- 12.Folkersma H, Foster Dingley JC, van Berckel BN, Rozemuller A, Boellaard R, Huisman MC, et al. Increased cerebral (R)-[11C]PK11195 uptake and glutamate release in a rat model of traumatic brain injury: a longitudinal pilot study. J Neuroinflammation. 2011;8:67. doi: 10.1186/1742-2094-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu I, Inaji M, Maeda J, Okauchi T, Nariai T, Ohno K, et al. Glial cell-mediated deterioration and repair of the nervous system after traumatic brain injury in a rat model as assessed by positron emission tomography. J Neurotrauma. 2010;27:1463–75. doi: 10.1089/neu.2009.1196. [DOI] [PubMed] [Google Scholar]
- 14.Venneti S, Lopresti BJ, Wang G, Slagel SL, Mason NS, Mathis CA, et al. A comparison of the high-affinity peripheral benzodiazepine receptor ligands DAA1106 and (R)-PK11195 in rat models of neuroinflammation: implications for PET imaging of microglial activation. J Neurochem. 2007;102:2118–31. doi: 10.1111/j.1471-4159.2007.04690.x. [DOI] [PubMed] [Google Scholar]
- 15.Raghavendra Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic brain injury leads to increased expression of peripheral-type benzodiazepine receptors, neuronal death, and activation of astrocytes and microglia in rat thalamus. Exp Neurol. 2000;161:102–14. doi: 10.1006/exnr.1999.7269. [DOI] [PubMed] [Google Scholar]
- 16.Chauveau F, Van Camp N, Dolle F, Kuhnast B, Hinnen F, Damont A, et al. Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J Nucl Med. 2009;50:468–76. doi: 10.2967/jnumed.108.058669. [DOI] [PubMed] [Google Scholar]
- 17.Doorduin J, Klein HC, Dierckx RA, James M, Kassiou M, de Vries EF. [11C]-DPA-713 and [18F]-DPA-714 as new PET tracers for TSPO: a comparison with [11C]-(R)-PK11195 in a rat model of herpes encephalitis. Mol Imaging Biol. 2009;11:386–98. doi: 10.1007/s11307-009-0211-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arlicot N, Vercouillie J, Ribeiro MJ, Tauber C, Venel Y, Baulieu JL, et al. Initial evaluation in healthy humans of [18F]DPA-714, a potential PET biomarker for neuroinflammation. Nucl Med Biol. 2012;39:570–8. doi: 10.1016/j.nucmedbio.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academy Press; 2010. [Google Scholar]
- 20.Baskaya MK, Dogan A, Temiz C, Dempsey RJ. Application of 2,3,5-triphenyltetrazolium chloride staining to evaluate injury volume after controlled cortical impact brain injury: role of brain edema in evolution of injury volume. J Neurotrauma. 2000;17:93–9. doi: 10.1089/neu.2000.17.93. [DOI] [PubMed] [Google Scholar]
- 21.Boutin H, Prenant C, Maroy R, Galea J, Greenhalgh AD, Smigova A, et al. [18F]DPA-714: direct comparison with [11C]PK11195 in a model of cerebral ischemia in rats. PLoS One. 2013;8:e56441. doi: 10.1371/journal.pone.0056441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai W, Guzman R, Hsu AR, Wang H, Chen K, Sun G, et al. Positron emission tomography imaging of poststroke angiogenesis. Stroke. 2009;40:270–7. doi: 10.1161/STROKEAHA.108.517474. [DOI] [PubMed] [Google Scholar]
- 23.Radu CG, Shu CJ, Shelly SM, Phelps ME, Witte ON. Positron emission tomography with computed tomography imaging of neuroinflammation in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2007;104:1937–42. doi: 10.1073/pnas.0610544104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen DL, Schuster DP. Positron emission tomography with [18F]fluorodeoxyglucose to evaluate neutrophil kinetics during acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286:L834–40. doi: 10.1152/ajplung.00339.2003. [DOI] [PubMed] [Google Scholar]
- 25.Tardy M, Fages C, Le Prince G, Rolland B, Nunez J. Regulation of the glial fibrillary acidic protein (GFAP) and of its encoding mRNA in the developing brain and in cultured astrocytes. Adv Exp Med Biol. 1990;265:41–52. doi: 10.1007/978-1-4757-5876-4_4. [DOI] [PubMed] [Google Scholar]
- 26.Galan X, Llobera M, Ramirez I. Lipoprotein lipase in developing rat tissues: differences between Wistar and Sprague-Dawley rats. Biol Neonate. 1993;64:295–303. doi: 10.1159/000244002. [DOI] [PubMed] [Google Scholar]
- 27.James ML, Fulton RR, Vercoullie J, Henderson DJ, Garreau L, Chalon S, et al. DPA-714, a new translocator protein-specific ligand: synthesis, radiofluorination, and pharmacologic characterization. J Nucl Med. 2008;49:814–22. doi: 10.2967/jnumed.107.046151. [DOI] [PubMed] [Google Scholar]
- 28.Kuhnast B, Damont A, Hinnen F, Catarina T, Demphel S, Le Helleix S, et al. [18F]DPA-714, [18F]PBR111 and [18F]FEDAA1106-selective radioligands for imaging TSPO 18 kDa with PET: automated radiosynthesis on a TRACERLAb FX-FN synthesizer and quality controls. Appl Radiat Isot. 2012;70:489–97. doi: 10.1016/j.apradiso.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 29.James ML, Selleri S, Kassiou M. Development of ligands for the peripheral benzodiazepine receptor. Curr Med Chem. 2006;13:1991–2001. doi: 10.2174/092986706777584979. [DOI] [PubMed] [Google Scholar]
- 30.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen MK, Baidoo K, Verina T, Guilarte TR. Peripheral benzodiazepine receptor imaging in CNS demyelination: functional implications of anatomical and cellular localization. Brain. 2004;127:1379–92. doi: 10.1093/brain/awh161. [DOI] [PubMed] [Google Scholar]
- 32.Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2005;196:290–7. doi: 10.1016/j.expneurol.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Venneti S, Lopresti BJ, Wang G, Hamilton RL, Mathis CA, Klunk WE, et al. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiol Aging. 2009;30:1217–26. doi: 10.1016/j.neurobiolaging.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maeda J, Higuchi M, Inaji M, Ji B, Haneda E, Okauchi T, et al. Phase-dependent roles of reactive microglia and astrocytes in nervous system injury as delineated by imaging of peripheral benzodiazepine receptor. Brain Res. 2007;1157:100–11. doi: 10.1016/j.brainres.2007.04.054. [DOI] [PubMed] [Google Scholar]