

Abstract
Proline-, glutamic acid-, and leucine-rich protein 1 (PELP1), a coregulator of estrogen receptors alpha and beta, is a potential protooncogene implicated in several human cancers, including sexual hormone-responsive or sexual hormone-nonresponsive cancers. However, the functions of PELP1 in colorectal cancer remain unclear. In this study, western blot and bioinformatics revealed that PELP1 expression was higher in several colorectal cancer cell lines than in immortalized normal colorectal epithelium. PELP1 silencing by short hairpin RNA promoted the senescence and inhibited the proliferation, colony formation, migration, invasion, and xenograft tumor formation of the CRC cell line HT-29. Moreover, PELP1 silencing was accompanied by c-Src downregulation. c-Src upregulation partly alleviated the damage in HT-29 malignant behavior induced by PELP1 RNA interference. In conclusion, PELP1 exhibits an oncogenic function in colorectal cancer through c-Src upregulation.
1. Introduction
Colorectal cancer (CRC) is the second leading cause of cancer-related death in the developed countries [1, 2]. In China, the morbidity and mortality rates of CRC have rapidly increased over the past two decades; these rates are affected by many factors, including age, population, westernization lifestyle, and industrial pollution. Early-stage CRC lacks specific symptoms; thus, this disease is usually diagnosed at a relatively late stage. The five-year survival rate of early-stage CRC is 85% after surgical resection, whereas that of stage III CRC with lymph node metastasis is <50% after surgery [3]. Cancer results from the activation of numerous protooncogenes and the inhibition of tumor suppressors. CRC is characterized by the mutation of tumor suppressor genes, protooncogenes, DNA repair genes, growth factors and their receptor genes, apoptosis-related genes, and cell cycle checkpoint genes [4]. These diverse genes constitute a complicated regulated network implicated in CRC oncogenesis. However, interference with a single gene is not enough to cure cancer. Therefore, the master regulators of downstream effector genes involved in carcinogenesis must be urgently identified [5].
The specific functions of estrogen signaling in CRC remain unclear. Many studies implicated the dysregulation of estrogen receptors (ERs) in CRC [6–9]. However, the specific functions of ERα and ERβ in the cancerogenesis of colorectal epithelium remain controversial. Some researches [6, 8] implied that ERβ is the predominant form expressed in the normal colorectal epithelium and reduced in CRC. Thus, ERβ exerts protective effects against CRC; this finding is contradictory to the report that the ERβ protein is significantly upregulated in colorectal epithelial cells of carcinomas [10]. ERα is reportedly rarely expressed in normal colorectal epithelium and lacks functions in CRC carcinogenesis. However, several studies reported that the ERα gene is frequently hypermethylated in CRC [11, 12], suggesting that ERα hypermethylation can predict CRC progression. ERα upregulation in ERα-negative CRC cell lines can suppress cell growth [13]. The reversal of a hypermethylated inactive ERα can inhibit CRC in vitro and in vivo [14]. The estrogen-ER signaling pathway is evidently involved in CRC development. Therefore, comprehensive research must be conducted to elucidate the functions of ER in CRC.
Proline-, glutamic acid-, and leucine-rich protein 1 (PELP1) is a modulator of nongenomic actions of estrogen receptors (NMAR) and a coregulator of ER; PELP1 has been implicated in many physiological [15–17] and pathological processes [18]. Research has demonstrated that PELP1 is a protooncogene in hormone-responsive cancers, such as breast [19, 20], ovarian [21], endometrial [22, 23], and prostate cancers [24]. Moreover, some researchers determined the functions of PELP1 in hormone-nonresponsive cancers, such as brain tumor [25], lung cancer [26], and colorectal cancer [27, 28].
PELP1 is upregulated in CRC tissues [27, 28]. However, the exact function of PELP1 in CRC remains unknown. In the present study, we investigated the functions of PELP1 in several CRC cell lines. Bioinformatics and western blot showed that PELP1 expression was higher in CRC cell lines than in normal colorectal epithelium. PELP1 silencing by short hairpin RNA (shRNA) promoted the senescence and inhibited the proliferation, colony formation, migration, invasion, and xenograft tumor formation of the CRC cell line HT-29. Moreover, PELP1 silencing was accompanied by c-Src downregulation. c-Src upregulation partly recovered the oncogenic function of PELP1. These results demonstrated that PELP1 suppression can inhibit CRC in vitro and in vivo through c-Src downregulation.
2. Materials and Methods
2.1. Cell Line
The CRC cell lines COLO205, HT-29, SW-620, HCC-2998, and HCT-15 and normal colon epithelium cell line FHC were purchased from American Type Culture Collection (Manassas, VA). The five CRC cell lines were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. FHC was cultured in DMEM/F12 medium with the same supplement as the CRC cell lines. All cell lines were maintained at 37°C in a humidified atmosphere of 5% CO2 in an open-air incubator.
2.2. Bioinformatics
We performed a bioinformatics analysis on Oncomine (https://www.oncomine.org/resource/login.html) to analyze PELP1 expression in the CRC cell lines. We used a personal account to enter the Oncomine website. We input PELP1 on the search box to begin the search. After entering the page for PELP1 expression survey in almost all human cancer cell lines or tissues, we focused on CRC and obtained PELP1 expression data from the Lee et al. [29], Shankavaram et al. [30], and Garnett [31] cell line datasets. After data standardization, we presented PELP1 expression data in the five CRC cell lines as mean ± standard deviation (SD).
2.3. shRNA Interference of PELP1
HT-29 cells were seeded in 24-well plates (Corning) until they reached 50% to 60% confluence prior to transfection. Then, stable transfection was performed. Three shRNA sequences targeting the PELP1 gene were designed and synthesized by Shanghai Jima Pharmaceutical Technology, China. The three shRNA sequences of PELP1 and the negative control sequence are listed in Table 1. Lipofectamine LTX and Plus Reagent (Invitrogen) were used to transfect shRNA into HT-29 cells according to the manufacturer's protocol. The medium containing transfection reagents was replaced with RPMI1640 medium supplemented with 10% FBS at 18 h after the transfection. The cells were collected at 48 h after the transfection, processed in the following experiments, and then prepared for protein extraction. The silence efficiency of PELP1 was tested by western blot.
Table 1.
Interference sequence for shRNA.
| shRNA number | Sequence (3′-5′) |
|---|---|
| Negative control | GCAAGCTGACCCTGAAGTTGAGAACTTTGAAGTCCCAGTCGAACG |
| 1 | GGAGAAACAAAGGUUAUUUGAGAACTUUUAUUGGAAACAAAGAGG |
| 2 | CACAGUCUCACAUCGUUUAGAGAACTAUUUGCUACACUCUGACAC |
| 3 | CCAGGAGCTTGTTGAAGAAGAGAACTAAGAAGTTGTTCGAGGACC |
2.4. Western Blot
Total protein was extracted from HT-29-shRNA or HT-29-control cells using RIPA lysate buffer as described in our previous research [32]. After quantification by a bicinchoninic acid protein assay kit (Beyotime Biotechnology, China), equal amounts of proteins (20 μg to 25 μg) were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (PAGE) and processed for immunoblotting with a rabbit multiclonal antibody for PELP1 (diluted at 1 : 100, Santa Cruz) and c-Src (diluted at 1 : 500, Santa Cruz). A mouse polyclonal anti-β-actin antibody (diluted at 1 : 2000, Boster Biological Engineering, China) was used as an internal control. All protein bands were scanned using ChemiImager 5500 V2.03 software, and the integrated density values were calculated by a computerized image analysis system (Fluor Chen 2.0) and normalized with that of β-actin.
2.5. MTT
Cell viability and proliferation activity were determined with the MTT assay. HT-29-shRNA or HT-29-control cells were seeded in 96-well plates (Costar) at a density of 5,000 cells per well in complete medium (RPMI1640 supplemented with 10% FBS, 1% p/s) and then incubated for 12 h under standard conditions (37°C and 5% CO2). The total volume in each well was 200 μL. From the next day to the seventh day, 20 μL of MTT (5 mg/mL) was added into each well. After additional incubation for 4 h, the solution in each well was replaced with dimethyl sulfoxide (Sigma, USA) to solubilize formazan, the metabolic product of MTT. The plates were kept on a shaking mixer for 10 min to guarantee complete solubilization of formazan, and the optical density was recorded at 490 nm using a microplate luminometer. Results were expressed as means ± SD, and a growth curve was constructed. Data were analyzed by one-way ANOVA with the post hoc Tukey test applied for paired comparisons.
2.6. Plate Colony Forming Assay
Colony forming ability was examined by the plate colony formation assay. HT-29-shRNA or HT-29-control cells were seeded into six-centimeter plates (Costar) at a density of 3,000 cells per plate in complete medium and then incubated for approximately 2 weeks under standard conditions (37°C and 5% CO2). The cells were washed twice with phosphate-buffered saline (PBS) and then fixed with methanol for 15 min. After staining with 0.1% crystal violet for 20 min, the number of positive colonies with diameters exceeding 50 μm was counted under a light microscope with 100× magnification. The colony forming rate was calculated by dividing the number of positive colonies by the total number of cells seeded.
2.7. Transwell Small Chamber Invasion and Migration Assay
For the invasion assay, Transwell small chambers with 8 μm pore filters were coated with 12 μL of ice-cold Matrigel (7.5 mg/mL protein). In total, 50,000 HT-29-shRNA or HT-29-control cells were added to the upper chamber of these Matrigel chambers in 200 μL of serum-free RPMI 1640 medium. Then, the cells were placed in 24-well plates in 500 μL of RPMI 1640 medium containing 10% FBS. After 22 h of incubation, the cells were fixed with methanol and then stained with 0.1% crystal violet. Cotton tips were used to remove the cells that remained in the Matrigel or attached to the upper side of the filter. The number of cells on the lower side of the filter was counted under a light microscope.
The methods used for the migration assay were almost the same as for the invasion assay described above, except that no Matrigel was used to coat the well and the incubation time was 16 h.
2.8. β-Galactosidase (β-Gal) Staining
The cells were washed in PBS, fixed for 3 min to 5 min (room temperature) in 2% formaldehyde/0.2% glutaraldehyde (or 3% formaldehyde), washed again, and then incubated at 37°C (no CO2) with a fresh senescence-associated β-Gal stain solution containing 1 mg of 5-bromo-4-chloro-3-indolyl β-D-galactoside per mL (stock = 20 mg of dimethylformamide per mL)/40 mM citric acid/sodium phosphate, pH 6.0/5 mM potassium ferrocyanide/5 mM potassium ferricyanide/150 mM NaCl/2 mM MgCl2. Staining was evident within 2 h to 4 h and peaked within 12 h to 16 h. To detect lysosomal, β-Gal, the citric acid/sodium phosphate was pH 4.0.
2.9. Nude Mice Subcutaneous Xenograft Assay
Male BALB/c (nu/nu) mice aged 6 to 8 weeks were obtained from the Experimental Animal Center of Guangdong Province. The mice were housed and maintained in laminar flow cabinets under specific pathogen-free conditions according to the regulations and standards approved by the Animal Care and Ethics Committee of Shantou University Medical College.
To establish s.c. tumors, 1.5 × 106 HT-29-shRNA or HT-29-control cells were resuspended in 200 μL of RPMI1640 serum-free medium and injected via an 18-gauge needle into the s.c. space of both flanks of the mice. Tumor progression was documented once weekly by measurements using calipers, and tumor volumes were calculated by the following formula: length × width × height × 0.52 (in mm). The mice were given ethane anesthesia and then euthanized by cervical dislocation.
2.10. Stable Transfection of c-Src
The human c-Src coding region gene with a 376 bp sequence was amplified from homogenized HT-29 by RT-PCR. The sequences of the PCR primers used for c-Src in this study were as follows: sense, 5′-TGTTCGGAGGCTTCAACTCC-3′ and antisense, 5′-CAGTAGGCACCTTTCGTGGT-3′. The PCR products were cloned into a TA expression vector (Invitrogen, Carlsbad, CA, USA), and the sequence of the c-Src coding region was confirmed by sequencing. The resulting plasmids (pc-Src) were propagated in Escherichia coli and then purified through cesium chloride gradient. For gene transfection, HT-29-shRNA or HT-29-control cells were seeded in six-well plates at a concentration of 5 × 105 cells per well. HT-29-shRNA or HT-29-control cells approaching 80% to 90% confluence were transfected with 4 mg pc-Src with 10 mL of Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. The cells transfected with empty plasmid pcDNA3.1 (mock) were used as negative controls.
2.11. Quantitative RT-PCR
The cells or tissues were harvested with Trizol Reagent (Invitrogen, Carlsbad, CA, USA), and total RNA was isolated according to the manufacturer's instructions. cDNA synthesis was performed using the Superscript III RT-PCR kit (Invitrogen). Real-time PCR was carried out using a Cepheid SmartCycler II (Sunnyvale, CA, USA) with gene-specific real-time PCR primers. Results were normalized to GAPDH transcript levels, and the difference in fold expression was calculated using the ΔΔ CT method. The primers used for c-Src were as follows: sense, 5′-CTCTTCAGAGCCCTTGCTCA-3′ and antisense, 5′-ATTCACCCTCCCCCAAGGAA-3′. The length of the PCR products was 193 bp.
2.12. Statistics Analysis
Data from all quantitative assays were expressed as mean ± SD andwere analyzed using one-way ANOVA and independent-samples t-test. All statistical analyses were performed and visualized by GraphPad Prism 5.0. P < 0.05 was considered statistically significant.
3. Results
3.1. PELP1 Was Upregulated in CRC Cell Lines as Revealed by Western Blot and Bioinformatics
We performed western blot to test the protein expression of PELP1 in the CRC lines COLO205, HT-29, SW-620, HCC-2998, and HCT-15 and in the normal cell line FHC. As shown in Figure 1(a), PELP1 protein expression was higher in the CRC cell lines than in the immortalized colorectal epithelium. The bioinformatics data (Figure 1(b)) obtained from the Oncomine database also showed that PELP1 was upregulated in the CRC cell lines. These results implied that PELP1 served oncogenic functions in CRC.
Figure 1.
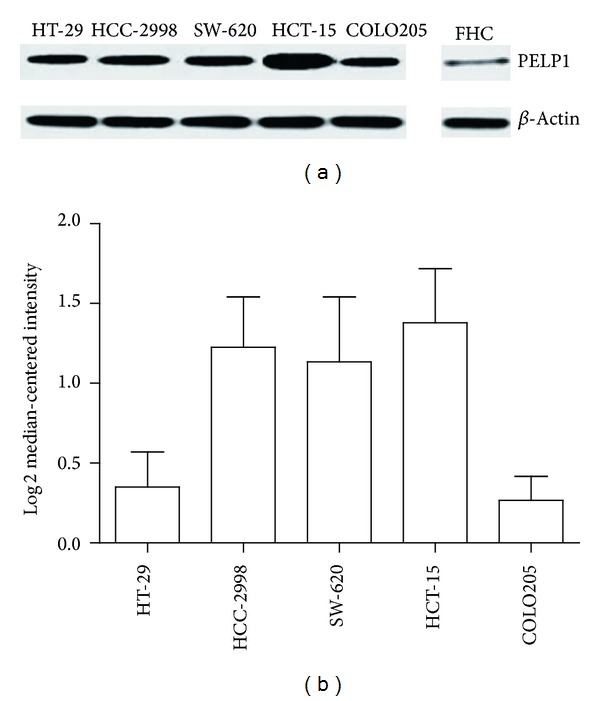
PELP1 expression was upregulated inCRC. (a) Western blot revealed that PELP1 protein expression was higher in the CRC cell lines HT-29, HCC-2998, SW-620, HCT-15, and COLO205 than in the normal colorectal epithelium FHC. (b) Informatics data suggested that PELP1 mRNA expression was increased in these five CRC cell lines.
3.2. PELP1 Downregulation by shRNA Inhibited CRC In Vitro
To identify the oncogenic function of PELP1 in CRC, we utilized shRNA to silence PELP1 expression in HT-29. After transfection shRNA #3 of PELP1 into HT-29, PELP1 expression decreased by 90% (Figure 2(a)). PELP1 silencing reduced the proliferation, colony formation (by 57.5%), migration (by 69.3%), and invasion (by 58%) abilities of HT-29 (Figures 2(b)–2(e)). β-Gal senescence assay demonstrated that PELP1 silencing promoted the senescence of HT-29 (Figure 2(f)). These results suggested that PELP1 downregulation inhibited the malignant behavior of CRC in vitro.
Figure 2.
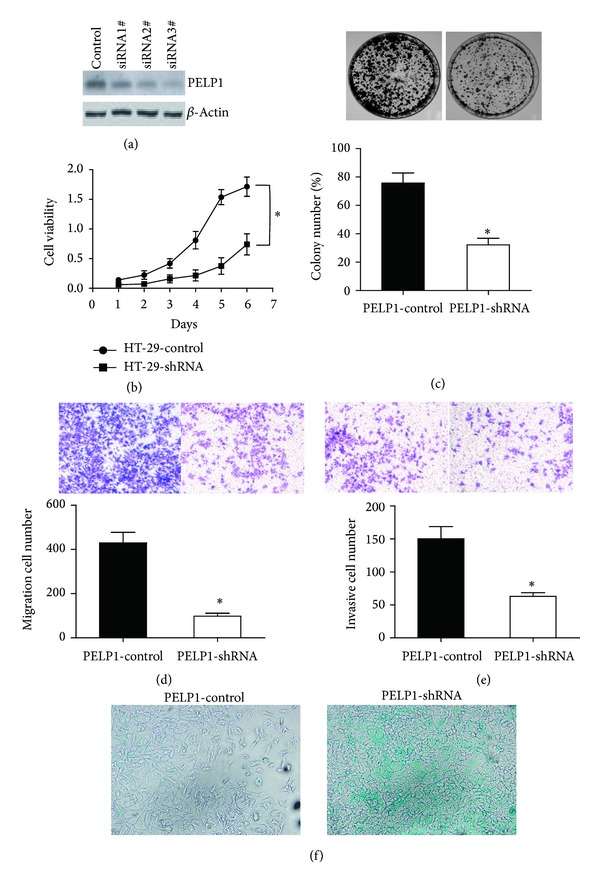
PELP1 downregulation inhibited the CRC cell line HT-29 in vitro. (a) After transfection with shRNA, PELP1 protein expression was decreased by 90%. shRNA #3 was selected for further investigations. (b) After PELP1 silencing, the cell viability of HT-29 was inhibited. (c) PELP1 silencing inhibited the colony formation ability of HT-29 by 57.5% (lower panel). A representative colony formation assay is shown (upper panel). (d) PELP1 silencing inhibited the migration ability of HT-29 by 69.3% (lower panel). A representative migration assay is shown (upper panel). (e) PELP1 silencing inhibited the invasion ability of HT-29 by 58% (lower panel). A representative invasive assay is shown (upper panel). (f) Senescence was induced by PELP1 silencing in HT-29. A representative β-Gal assay is shown.
3.3. PELP1 Downregulation by shRNA Inhibited Xenograft Formation Ability of CRC in Nude Mice
Xenograft formation ability reflects the malignant characteristic of cancer cells. We performed a subcutaneous xenograft formation experiment on nude mice to assess whether or not PELP1 can influence the xenograft formation ability of HT-29. After stable transfection with PELP1-shRNA in HT-29, HT-29-control and HT-29-shRNA cells were inoculated subcutaneously in both flanks of the nude mice. Results showed that PELP1-shRNA reduced the xenograft formation ability of HT-29 in nude mice (Figure 3).
Figure 3.
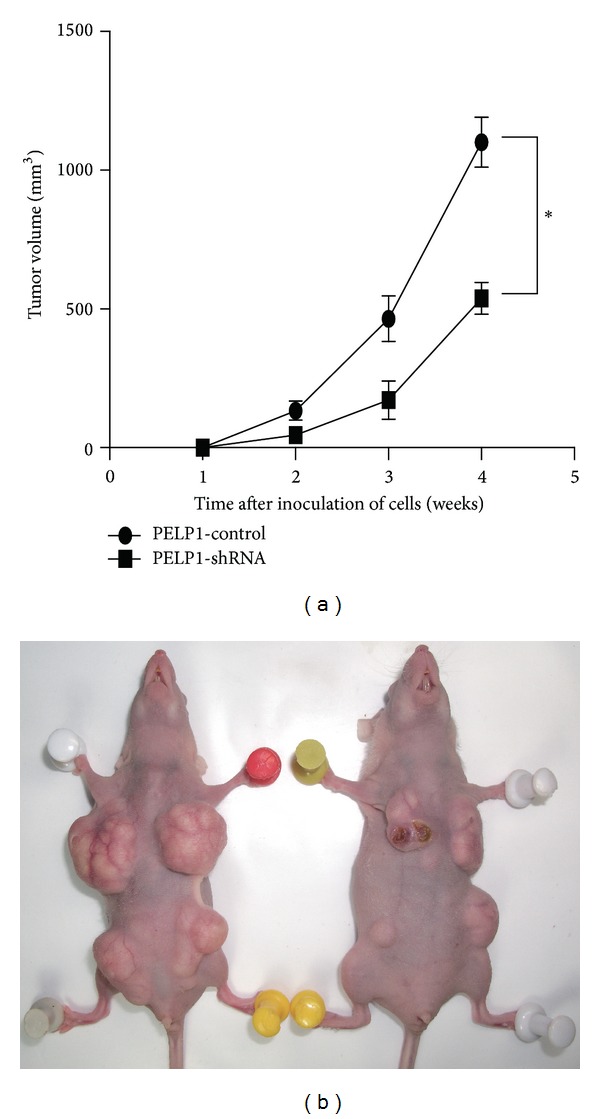
PELP1 downregulation inhibited the CRC cell line HT-29 in nude mice xenograft assay. PELP1 silencing inhibited CRC growth in nude mice (a). A representative nude mice xenograft assay is shown in (b).
3.4. Oncogenic Function of PELP1 in CRC Was Mediated by c-Src Upregulation
c-Src is a known protooncogene in colon cancer. Thus, we focused on the relationship between PELP1 and c-Src in CRC carcinogenesis. We tested c-Src expression by quantitative RT-PCR and western blot after PELP1 silencing by shRNA in HT-29 to explore the mechanism of PELP1 downregulation in suppressing CRC carcinogenesis. c-Src was reduced after PELP1 silencing at the mRNA (Figure 4(a)) and protein (Figure 4(b)) expression levels. To analyze whether or not c-Src participates in promoting the function of PELP1 in CRC, we upregulated the expression of c-Src after PELP1 silencing. As stated previously, PELP1 downregulation can inhibit the malignant behavior of CRC. Surprisingly, Src elevation can counteract the inhibition of CRC induced by PELP1 silencing (Figures 4(c)–4(g)). Therefore, the suppressing function of PELP1 downregulation in CRC carcinogenesis was mediated by c-Src inhibition.
Figure 4.
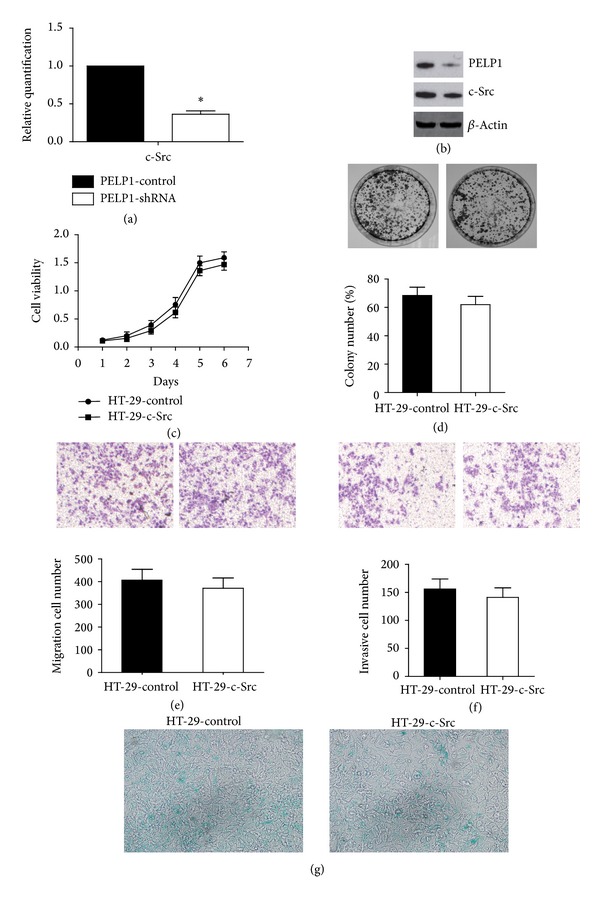
PELP1 silencing suppressed CRC through c-Src downregulation. (a) PELP1 silencing was accompanied by the downregulation of c-Src mRNA as determined by quantitative RT-PCR. (b) PELP1 silencing was accompanied by c-Src protein downregulation as determined by western blot. (c) Decreased cell viability induced by PELP1 silencing was recovered by c-Src upregulation in HT-29. (d) Decreased colony formation ability was recovered by c-Src upregulation in HT-29. (e) Decreased migration ability was recovered by c-Src upregulation in HT-29. (f) Decreased invasion ability was recovered by c-Src upregulation in HT-29. (g) Induced senescence by PELP1 silencing was inhibited after c-Src upregulation.
4. Discussion
In the present study, the oncogenic function of the ER coregulator PELP1 or NMAR was identified in CRC, a common and highly invasive type of cancer. We primarily clarified the oncogenic mechanism of PELP1 in relation to c-Src, an important oncogene. Although CRC cannot be completely regarded as a hormone-responsive cancer, emerging evidence implied that steroid hormones such as estrogen and androgen participate in CRC pathogenesis. However, the functions of ER and AR in CRC are complex and controversial because many complicated signaling pathways are involved in estrogen and androgen signaling. ERβ is usually regarded as a protective factor in CRC, and ERα is believed to have no participation in CRC [33]. Different from ERs, androgen receptors (ARs) serve no [34] suppressive [35] or promoting functions [36] in CRC. As a nuclear receptor coregulator, PELP1 can interact with ERs, ARs, glucocorticoid receptors, and progesterone receptors [37]. Therefore, we analyzed the functions of PELP1 in CRC to elucidate the complex signaling network involving ER and AR.
Two recent studies have evaluated the expression of PELP1 in CRC tissues by using immunohistochemistry [27, 28]. However, their findings were inconsistent. Tzelepi et al. [27] claimed that PELP1 expression decreases from normal colorectal epithelium to cancerous colorectal epithelium; by contrast, Grivas et al. [28] found that PELP1 protein expression in epithelial cells increases during colorectal tumorigenesis despite the fact that PELP1 overexpression in epithelial cells correlates with prolonged overall survival. Moreover, Grivas et al. [28] found that ERβ expression in epithelial cells is upregulated during colorectal tumorigenesis in male patients. They also found that ERβ expression correlates with increased risk of relapse, implying that ERβ serves an oncogenic function in male CRC patients. The present experimental results seemed to be partly consistent with those of Grivas et al. That is, bioinformatics and western blot assay demonstrated that the mRNA and protein expression levels of PELP1 increased in the CRC cell lines COLO205, HT-29, SW-620, HCC-2998, and HCT-15. Moreover, we found that the malignant behavior of CRC was inhibited in vitro and in vivo after PELP1 silencing by shRNA. These results suggested that PELP1 served an oncogenic function in CRC.
A decade ago, Peyton Rous described a filterable agent (i.e., virus) that can induce solid tumor formation in birds. After approximately 50 years, the Rous sarcoma virus was identified from Rous' filterable agent. Extensive research into the molecular biology and genetics of Rous sarcoma virus identified v-Src as a viral oncogene responsible for malignant transformation. Then, Bishop and Varmus [38] demonstrated that v-Src has a cellular counterpart, the first identified protooncogene c-Src. c-Src is a nonreceptor tyrosine kinase that is abnormally expressed in many human cancers and is linked with malignant biological behavior related to proliferation, adhesion, migration, invasion, and metastasis [39]. The oncogenic function of c-Src in CRC has been explored in different studies. The dysregulation of c-Src contributed to the initiation and development of CRC. In the present study, we linked PELP1 with c-Src in CRC. Several scholars have identified the fact that an interaction exists between PELP1 and c-Src. For example, Chakravarty et al. [40] found that estrogen-mediated extranuclear signaling promotes cytoskeleton reorganization through the ER-Src-PELP1-phosphoinositide 3-kinase-ILK1 pathway in breast cancer. PELP1 silencing can significantly inhibit c-Src activation. Dimple et al. [41] suggested that PELP1 downregulation reduces the proliferation and tumorigenic potential of ovarian cancer cells and affects the magnitude of c-Src and protein kinase B (AKT) signaling in a nude mouse model. Rajhans et al. [42] found that the PELP1-mediated induction of aromatase requires functional Src and phosphatidylinositol-3-kinase pathways. Conversely, c-Src phosphorylates PELP1 at the C terminal (tyrosine 920) domain [43]. However, PELP1 can function independent of c-Src. Kayahara et al. [44] found that PELP1 functionally interacts with both NH2- and COOH-terminal glucocorticoid receptor domains to modulate transactivation; they also found through inhibitor and c-Src knockdown studies that the function of PELP1 is independent of c-Src activity. In the present study, we found that the oncogenic function of PELP1 was partially mediated by c-Src in CRC. PELP1 silencing by shRNA downregulated c-Src expression. In addition, c-Src upregulation partly recovered the oncogenic function of PELP1. Our research suggested that the oncogenic effect of PELP1 in CRC was partly mediated by c-Src.
5. Conclusions
PELP1, a nuclear receptor coregulator, exerts oncogenic action in CRC. PELP1 silencing by shRNA promoted the senescence and inhibited the proliferation, colony formation, migration, invasion, and xenograft tumor formation of CRC. Moreover, PELP1 silencing was accompanied by c-Src downregulation. c-Src upregulation partly recovered the oncogenic function of PELP1. This study is the first to identify the oncogenic function of PELP1 in CRC, a hormone-nonresponsive cancer. Therefore, PELP1 can be regarded as a therapeutic target in hormone-nonresponsive cancers.
Acknowledgment
This work was supported by the Key Project of Hubei Province Education Department (D20122802).
Abbreviations
- PELP1:
Proline-, glutamic acid-, and leucine-rich protein 1
- c-Src:
Cellular counterpart of Rous sarcoma virus
- ER:
Estrogen receptor
- CRC:
Colorectal cancer.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Zhifeng Ning, Youzhi Zhang, and Hanwei Chen contributed equally to this work.
References
- 1.Gansler T, Ganz PA, Grant M, et al. Sixty years of ca: A cancer journal for clinicians. CA Cancer Journal for Clinicians. 2010;60(6):345–350. doi: 10.3322/caac.20088. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer Journal for Clinicians. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.O’Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. Journal of the National Cancer Institute. 2004;96(19):1420–1425. doi: 10.1093/jnci/djh275. [DOI] [PubMed] [Google Scholar]
- 4.Narayan S, Roy D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Molecular Cancer. 2003;2, article 41 doi: 10.1186/1476-4598-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lonard DM, O’Malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Molecular Cell. 2007;27(5):691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Rudolph A, Toth C, Hoffmeister M, et al. Expression of oestrogen receptor beta and prognosis of colorectal cancer. British Journal of Cancer. 2012;107:831–839. doi: 10.1038/bjc.2012.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He YQ, Sheng JQ, Ling XL, et al. Estradiol regulates miR-135b and mismatch repair gene expressions via estrogen receptor-β in colorectal cells. Experimental & Molecular Medicine. 2012;44:723–732. doi: 10.3858/emm.2012.44.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saleiro D, Murillo G, Benya RV, Bissonnette M, Hart J, Mehta RG. Estrogen receptor-beta protects against colitis-associated neoplasia in mice. International Journal of Cancer. 2012;131:2553–2561. doi: 10.1002/ijc.27578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasson RM, Briggs A, Carothers AM, et al. Estrogen receptor α or β loss in the colon of Min/+ mice promotes crypt expansion and impairs TGFβ and HNF3β signaling. Carcinogenesis. 2014;35(1):96–102. doi: 10.1093/carcin/bgt323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grivas PD, Tzelepi V, Sotiropoulou-Bonikou G, Kefalopoulou Z, Papavassiliou AG, Kalofonos H. Estrogen receptor α/β, AIB1, and TIF2 in colorectal carcinogenesis: do coregulators have prognostic significance? International Journal of Colorectal Disease. 2009;24(6):613–622. doi: 10.1007/s00384-009-0647-9. [DOI] [PubMed] [Google Scholar]
- 11.Jubb A, Bell S, Quirke P. Methylation and colorectal cancer. The Journal of Pathology. 2001;195:111–134. doi: 10.1002/path.923. [DOI] [PubMed] [Google Scholar]
- 12.Kondo Y, Issa JPJ. Epigenetic changes in colorectal cancer. Cancer and Metastasis Reviews. 2004;23(1-2):29–39. doi: 10.1023/a:1025806911782. [DOI] [PubMed] [Google Scholar]
- 13.Issa J-PJ, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nature Genetics. 1994;7(4):536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 14.Al-Azzawi F, Wahab M. Estrogen and colon cancer: current issues. Climacteric. 2002;5(1):3–14. [PubMed] [Google Scholar]
- 15.Wang J, Song S, Shi L, et al. Temporal expression of pelp1 during proliferation and osteogenic differentiation of rat bone marrow mesenchymal stem cells. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0075477.0075477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan Y, Zeng W, Song S, et al. Vitamin C induces periodontal ligament progenitor cell differentiation via activation of ERK pathway mediated by PELP1. Protein Cell. 2013;4:620–627. doi: 10.1007/s13238-013-3030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Zhu Q, Song S, et al. Increased PELP1 expression in rat periodontal ligament tissue in response to estrogens treatment. Journal of Molecular Histology. 2013;44:347–356. doi: 10.1007/s10735-013-9490-y. [DOI] [PubMed] [Google Scholar]
- 18.Girard BJ, Daniel AR, Lange CA, Ostrander JH. PELP1: a review of PELP1 interactions, signaling, and biology. Molecular and Cellular Endocrinology. 2013;8:00326–00322. doi: 10.1016/j.mce.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennani-Baiti IM. Integration of ERalpha-PELP1-HER2 signaling by LSD1 (KDM1A/AOF2) offers combinatorial therapeutic opportunities to circumventing hormone resistance in breast cancer. Breast Cancer Research. 2012;14, article 112 doi: 10.1186/bcr3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cortez V, Mann M, Tekmal S, et al. Targeting the PELP1-KDM1 axis as a potential therapeutic strategy for breast cancer. Breast Cancer Research. 2012;14, article R108 doi: 10.1186/bcr3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aust S, Horak P, Pils D, et al. The prognostic value of estrogen receptor beta and proline-, glutamic acid- and leucine-rich protein 1 (PELP1) expression in ovarian cancer. BMC Cancer. 2013;13:1471–2407. doi: 10.1186/1471-2407-13-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan J, Li X. PELP1/MNAR suppression inhibits proliferation and metastasis of endometrial carcinoma cells. Oncology Reports. 2012;28:2035–2042. doi: 10.3892/or.2012.2038. [DOI] [PubMed] [Google Scholar]
- 23.Vadlamudi RK, Balasenthil S, Broaddus RR, Gustafsson J-Å, Kumar R. Deregulation of estrogen receptor coactivator proline-, glutamic acid-, and leucine-rich protein-1/modulator of nongenomic activity of estrogen receptor in human endometrial tumors. Journal of Clinical Endocrinology and Metabolism. 2004;89(12):6130–6138. doi: 10.1210/jc.2004-0909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravindranathan P, Lee TK, Yang L, et al. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nature Communications. 2013;4, article 1923 doi: 10.1038/ncomms2912. [DOI] [PubMed] [Google Scholar]
- 25.Kefalopoulou Z, Tzelepi V, Zolota V, et al. Prognostic value of novel biomarkers in astrocytic brain tumors: nuclear receptor co-regulators AIB1, TIF2, and PELP1 are associated with high tumor grade and worse patient prognosis. Journal of Neuro-Oncology. 2012;106(1):23–31. doi: 10.1007/s11060-011-0637-y. [DOI] [PubMed] [Google Scholar]
- 26.Márquez-Garbán DC, Chen HW, Fishbein MC, Goodglick L, Pietras RJ. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids. 2007;72(2):135–143. doi: 10.1016/j.steroids.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tzelepi V, Grivas P, Kefalopoulou Z, Kalofonos H, Varakis JN, Sotiropoulou-Bonikou G. Expression of estrogen receptor co-regulators NCoR and PELP1 in epithelial cells and myofibroblasts of colorectal carcinomas: cytoplasmic translocation of NCoR in epithelial cells correlates with worse prognosis. Virchows Archiv. 2009;454(1):41–53. doi: 10.1007/s00428-008-0708-4. [DOI] [PubMed] [Google Scholar]
- 28.Grivas PD, Tzelepi V, Sotiropoulou-Bonikou G, Kefalopoulou Z, Papavassiliou AG, Kalofonos H. Expression of ERa,ERß and co-regulator PELP1/MNAR in colorectal cancer: prognostic significance and clinicopathologic correlations. Cellular Oncology. 2009;31(3):235–247. doi: 10.3233/CLO-2009-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JK, Havaleshko DM, Cho H, et al. A strategy for predicting the chemosensitivity of human cancers and its application to drug discovery. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(32):13086–13091. doi: 10.1073/pnas.0610292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shankavaram UT, Reinhold WC, Nishizuka S, et al. Transcript and protein expression profiles of the NCI-60 cancer cell panel: an integromic microarray study. Molecular Cancer Therapeutics. 2007;6(3):820–832. doi: 10.1158/1535-7163.MCT-06-0650. [DOI] [PubMed] [Google Scholar]
- 31.Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong H, Guo H, Xie L, et al. The metastasis-associated gene MTA3, a component of the Mi-2/NuRD transcriptional repression complex, predicts prognosis of gastroesophageal junction adenocarcinoma. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0062986.e62986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barzi A, Lenz AM, Labonte MJ, Lenz HJ. Molecular pathways: estrogen pathway in colorectal cancer. Clinical Cancer Research. 2013;19:5842–5848. doi: 10.1158/1078-0432.CCR-13-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Passarelli MN, Phipps AI, Potter JD, et al. Common single-nucleotide polymorphisms in the estrogen receptor beta promoter are associated with colorectal cancer survival in postmenopausal women. Cancer Research. 2013;73:767–775. doi: 10.1158/0008-5472.CAN-12-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slattery ML, Sweeney C, Murtaugh M, et al. Associations between vitamin D, vitamin D receptor gene and the androgen receptor gene with colon and rectal cancer. International Journal of Cancer. 2006;118(12):3140–3146. doi: 10.1002/ijc.21791. [DOI] [PubMed] [Google Scholar]
- 36.Mariani M, Zannoni GF, Sioletic S, et al. Gender influences the class III and V beta-tubulin ability to predict poor outcome in colorectal cancer. Clinical Cancer Research. 2012;18:2964–2975. doi: 10.1158/1078-0432.CCR-11-2318. [DOI] [PubMed] [Google Scholar]
- 37.Vadlamudi RK, Kumar R. Functional and biological properties of the nuclear receptor coregulator PELP1/MNAR. Nuclear receptor signaling. 2007;5, article e004 doi: 10.1621/nrs.05004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.GS M. The hunting of the Src. Nature Reviews Molecular Cell Biology. 2001;2:467–475. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 39.Zhang S, Yu D. Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends in Pharmacological Sciences. 2012;33(3):122–128. doi: 10.1016/j.tips.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chakravarty D, Nair SS, Santhamma B, et al. Extranuclear functions of ER impact invasive migration and metastasis by breast cancer cells. Cancer Research. 2010;70(10):4092–4101. doi: 10.1158/0008-5472.CAN-09-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dimple C, Nair SS, Rajhans R, et al. Role of PELP1/MNAR signaling in ovarian tumorigenesis. Cancer Research. 2008;68(12):4902–4909. doi: 10.1158/0008-5472.CAN-07-5698. [DOI] [PubMed] [Google Scholar]
- 42.Rajhans R, Nair HB, Nair SS, et al. Modulation of in situ estrogen synthesis by proline-, glutamic acid-, and leucine-rich protein-1: potential estrogen receptor autocrine signaling loop in breast cancer cells. Molecular Endocrinology. 2008;22(3):649–664. doi: 10.1210/me.2007-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheskis BJ, Greger J, Cooch N, et al. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids. 2008;73(9-10):901–905. doi: 10.1016/j.steroids.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 44.Kayahara M, Ohanian J, Ohanian V, Berry A, Vadlamudi R, Ray DW. MNAR functionally interacts with both NH2- and COOH-terminal GR domains to modulate transactivation. The American Journal of Physiology—Endocrinology and Metabolism. 2008;295(5):E1047–E1055. doi: 10.1152/ajpendo.90429.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]