

Abstract
Diversity in the forestomach microbiome is one of the key features of ruminant animals. The diverse microbial community adapts to a wide array of dietary feedstuffs and management strategies. Understanding rumen microbiome composition, adaptation, and function has global implications ranging from climatology to applied animal production. Classical knowledge of rumen microbiology was based on anaerobic, culture-dependent methods. Next-generation sequencing and other molecular techniques have uncovered novel features of the rumen microbiome. For instance, pyrosequencing of the 16S ribosomal RNA gene has revealed the taxonomic identity of bacteria and archaea to the genus level, and when complemented with barcoding adds multiple samples to a single run. Whole genome shotgun sequencing generates true metagenomic sequences to predict the functional capability of a microbiome, and can also be used to construct genomes of isolated organisms. Integration of high-throughput data describing the rumen microbiome with classic fermentation and animal performance parameters has produced meaningful advances and opened additional areas for study. In this review, we highlight recent studies of the rumen microbiome in the context of cattle production focusing on nutrition, rumen development, animal efficiency, and microbial function.
Keywords: cattle, metabolism, microbiome, nutrition, rumen
Introduction
Ruminants are important contributors of meat, milk, fiber, and draft worldwide. There are more than 3.5 billion domesticated ruminants including cattle, sheep, goats, and buffalo (http://faostat.fao.org/). Distinct from many herbivores, the pregastric fermentation of ruminants highlights the benefit of a symbiotic host–microbe relationship as the rumen microbiome facilitates utilization of a diverse array of plant materials. Considering cellulose is the most abundant organic molecule on earth, the ability to effectively ferment cellulolytic materials uniquely positions ruminants to utilize vast resources globally without directly competing with humans. The rumen microbiome has also been cited as a resource of novel enzymes for cellulosic biofuel production.1,2
Continuous fermentation in the rumen is driven by a diverse and competitive microbiome consisting of bacteria, archaea, protozoa, and fungi.3 Regardless of the substrates provided by the diet, volatile fatty acids (VFA) and microbial crude protein (MCP), the principal products of fermentation, are responsible for addressing a large portion of host energy and protein requirements.4 While VFA can diffuse directly across the ruminal epithelium, MCP is absorbed in the small intestine as amino acids, dipeptides, and tripeptides.5,6 Ammonia released from ruminal protein degradation can be utilized for microbial growth in the rumen, absorbed through the ruminal wall to be detoxified in the liver to urea, and subsequently recycled throughout the body or excreted in urine as urea.
Fermentation end products released through eructation, including CO2 and methane, represent a loss of energy and contribute to greenhouse gases. Hydrogen was considered the main energy source for methane released by Archaea, but recent findings suggest that a new class of methanogens also metabolizes methylamines.7 The adaptive nature of the rumen microbiome allows ruminants to convert a wide array of low- and high-quality feedstuffs into high-quality MCP via fermentation.4 However, countless nutritional strategies potentially create an equal number of unique microbiomes. Description of these microbiomes will enable a greater understanding of the host–microbe relationship and its impact on animal performance.
Historical understanding of rumen microbiology was based on culture-based techniques pioneered by Robert Hungate.8,9 Successful simulation of anaerobic conditions in vitro facilitated significant discoveries expanding the knowledge of the rumen microbiome through improved descriptions of bacterial species such as Streptococcus bovis10 and Megasphaera elsdenii.11 In isolated cultures, substrates and products of bacterial strains were described in detail, providing the foundational understanding of their function. Advent of nucleic acid-based molecular technologies has ushered in a new culture-independent perspective of microbial ecology unbiased by the culturing aptitude of microbial species. With molecular techniques, bacterial species long-accepted as having prominent roles in ruminal function were detected at relatively low levels. For example, in fiber-adherent rumen fractions, the well-described cellulolytic genus Ruminococcus has not been observed in quantities above 2%,12–14 suggesting a lesser role in cellulose degradation than originally believed. Although cultured species may not be the most prevalent, they may serve as markers for uncultured bacteria occupying the same functional niche in the rumen. The most comprehensive studies will use a combination of techniques to enable comparisons against decades of culture-based knowledge.
Ruminant nutrition has traditionally focused on measuring performance, voluntary intake, fermentation parameters, rate of passage, diet digestibility, and nitrogen metabolism to describe a diet’s ability to meet animal requirements. Each of these measures is inseparably linked with the rumen microbial community (Fig. 1). Feeding strategies, additives, and supplements are used to optimize animal performance, yet for many the mode of action is unknown. Rumen microbiology research historically has failed to correlate differences in bacterial populations, function, or phylogeny to meaningful responses in the animal. Utilization of new, high-throughput techniques allows microbial communities to be described in greater resolution than ever before. With nucleic acid-based, high-throughput approaches, a microbial understanding of proven nutritional strategies can set the foundation for new advancements in ruminant production.
Figure 1.

The link between nutrition, metabolism, and the rumen microbiome. Diet composition and dry matter intake directly impact the rumen microbiome composition. Changes in microbiome composition affect bacteria within functional niches responsible for feedstuff degradation. Bacteria within each functional niche possess many enzymes to fulfill their role, and the examples are involved in fiber degradation. Fermentation of feedstuffs results in production of volatile fatty acids (VFA), methane (CH4), carbon dioxide (CO2), ammonia (NH3), and microbial crude protein (MCP). While CH4 and CO2 are released by eructation, VFA and NH3 are absorbed by rumen epithelium tissue to be transported to the liver. Fatty acids (FA) and MCP are taken up by the small intestine and also taken to the liver. Hepatic tissues convert NH3 to urea to be recycled to the gastrointestinal tract. Additionally, propionate is converted to glucose and distributed with amino acids and lipoproteins (LP) to extrahepatic tissues. Overall growth in tissue mass leads to animal production and increases in food products.
The multitude of steps required for analysis and the various methods to accomplish each step make comparisons between studies challenging. Each rumen functions as its own system, and no two rumen systems are identical. Moreover, the substrates provided (ie, the ration) directly influence the microbiome structure. Our review of the literature will include pertinent details of methodology, analysis, and experimental design.
16S rRNA Sequencing Methodology and Analysis Basics
Extraction of nucleic acids is the first sample processing step, but it also represents the first opportunity for bias to affect the outcome. Obtaining the greatest possible amount of DNA is desirable to increase the representativeness of the sample. Although there are many commercial kits available, most published studies include modifications for best results. Further discussion on extraction methodology is beyond the scope of this paper, but comparisons have been described by Henderson et al.15 and Villegas-Rivera et al.16
After extraction of DNA, amplification of a phylogenetic marker gene is used to identify microbiota. The small subunit of the 16S rRNA gene is most commonly chosen because of its highly conserved and variable regions, extensive database of reference sequences, and precedent set by previous work. Sequencing of 16S amplicons is currently limited by the capability of available sequencing platforms. Therefore, several variable regions are typically selected from the 16S rRNA gene which contains about 1500 bp. Particular regions may be more informative depending on the microbiome of interest, and several reviews of different 16S primers have been conducted.17,18 High-throughput sequencing platforms are rapidly improving and likely will be capable of sequencing the length of the entire 16S rRNA gene in the near future. The Roche 454 FLX platform has been more commonly used in rumen microbiome sequencing projects to date (Table 1) because of longer read lengths, but additional throughput capacity and decreased cost of Illumina platforms will likely increase their use moving forward.19
Table 1.
Summary of bovine rumen metagenomic publications.
| TITLE | YEAR | SEQUENCING PLATFORM | MAJOR FINDING | REFERENCE |
|---|---|---|---|---|
| “Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases” | 2009 | 454 GS20 | - Significant differences exist in rumen metabolic potential between animals on the same diet. - Many unique glycoside hydrolases are present in the rumen microbiome. |
39 |
| “Rumen bacterial diversity associated with changing from bermudagrass hay to grazed winter wheat diets” | 2010 | 454 GS FLX | - Switching from hay to wheat diet affects bacterial composition and diversity structure in the rumen microbiome. | 38 |
| “Rumen microbial population dynamics during adaptation to a high-grain diet” | 2010 | ABI 3700 | - M. elsdenii, S. bovis, S. ruminantium, and P. bryantii increased with additional grain in the diet while fibrolytic bacteria decreased. | 51 |
| “Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing” | 2010 | 454 GS FLX | - Abundance of the phylum Bacteroidetes increased with addition of distillers grains to growing diet. | 72 |
| “Microbiome analysis of dairy cows fed pasture or total mixed ration diets” | 2011 | 454 GS FLX | - Pasture diet increased Prevotella abundance regardless of fraction. - Protozoal community was unaffected by diet or rumen fraction. |
76 |
| “Metagenomic discovery of biomass-degrading genes and genomes from cow rumen” | 2011 | Illumina Hiseq 2000 | - 88% of carbohydrate-active candidate genes were greater than 25% dissimilar to deposited genes. | 2 |
| “Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools” | 2012 | 454 GS FLX | - Significant changes in phylogenetic composition of the rumen microbiome occurred during development, but the metabolic potential remained stable. | 46 |
| “Nitrogen metabolism and rumen microbial enumeration in lactating cows with divergent residual feed intake fed high-digestibility pasture” | 2012 | 454 GS FLX | - Archaea, bacteria, protozoa, and fungal communities were largely similar between inefficient and efficient cows. - Differences were observed for 3 bacterial families and two protozoa species. |
69 |
| “High throughput whole rumen metagenome profiling using untargeted massively parallel sequencing” | 2012 | Illumina GAIIx | - There is greater variation in the rumen metagenome between animals than replicates from the same rumen. | 105 |
| “Composition and similarity of bovine rumen microbiota across individual animals” | 2012 | 454 GS FLX | - Although significant heterogeneity exists between the rumen microbiome of animals on a similar diet, phylogenetic comparisons indicate more similarities. | 47 |
| “Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen” | 2012 | 454 GS FLX | - Compared with the bacterial community, archeal and fungal communities were more consistent in the liquid and solid fractions. | 26 |
| “Comparative survey of rumen microbial communities and metabolites across one caprine and three bovine groups using barcoded pyrosequencing and 1H-NMR spectroscopy” | 2012 | 454 GS FLX | - Microbiome composition is affected by diet, host animal breed, and may be associated with rumen metabolites. | 106 |
| “Perturbation dynamics of the rumen microbiota in response to exogenous butyrate” | 2012 | 454 GS FLX | - Exogenous butyrate infusion increased Ruminobacter and Treponema populations within the rumen microbiome. | 107 |
| “Phage-bacteria relationships and CRISPR elements revealed by a metagenomic survey of the rumen microbiome” | 2012 | 454 GS FLX, Illumina GAIIx | - Rumen bacteriophages are diverse, unique to the rumen, and likely influence the microbial community. | 108 |
| “The bacterial community composition of the bovine rumen detected using pyrosequencing of 16S rRNA genes” | 2012 | 454 GS FLX | - The core rumen microbiome is affected by rumen development and diet. | 109 |
| “The effect of brown midrib corn silage and dried distillers’ grains with solubles on milk production, nitrogen utilization and microbial community structure in dairy cows” | 2012 | 454 GS FLX | - Inclusion of dried distillers grains increased Bacteroidetes:Firmicutes ratio and nitrogen and neutral detergent fiber digestion. | 110 |
| “Evaluation of the ruminal bacterial diversity of cattle fed diets containing citrus pulp pellets” | 2012 | 454 GS FLX | - Greater inclusion of citrus pulp pellets impacted the rumen microbiome and increased bacilli bacteria. | 111 |
| “Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities” | 2013 | 454 GS FLX | - Simultaneous pyrosequencing of bacterial, archaeal, and eukaryotic DNA can efficiently describe the entire rumen microbiome and elucidate potential relationships between microorganisms. | 25 |
| “Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis” | 2013 | 454 GS FLX | - Minor bacteria in the epimural community were most affected by acidosis challenge and may indicate acidosis susceptibility. | 57 |
| “Investigating the effect of two methane-mitigating diets on the rumen microbiome using massively parallel sequencing” | 2013 | Illumina Hiseq 2000 | - Identified contigs associated with lower methane production were observed in rumen fluid and feces. | 112 |
| “Effect of post-extraction algal residue supplementation on the rumen microbiome of steers consuming low-quality forage” | 2013 | 454 GS FLX | - Dietary inclusion of post-extraction algal residue increased proportion of Firmicutes families Ruminococcaceae, Lachnospiraceae, and Clostridiaceae. | 41 |
| “Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16S rRNA gene pyrosequencing” | 2013 | 454 GS FLX | - Fibrobacteraceae and Ruminococcaceae decreased in relative abundance with increasing concentrate in a forage-based diet. | 82 |
| “Effect of dietary forage sources on rumen microbiota, rumen fermentation and biogenic amines in dairy cows” | 2013 | 454 GS FLX | - Forage source within TMR affected abundance of Prevotella, Rikenellaceae, Selenomonas, and Ruminococcaceae. | 80 |
| “Impact of subacute ruminal acidosis (SARA) adaptation of rumen microbiota in dairy cattle using pyrosequencing” | 2013 | 454 GS FLX | - Induction of SARA decreased Prevotella, Treponema, and alpha diversity and may be linked to increased ruminal lipopolysaccharides. | 113 |
| “Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer” | 2013 | 454 GS FLX | - Distinct microbiomes exist in segments of the gastrointestinal tract and likely correspond to the physiological function of each segment. | 114 |
| “The effects of a probiotic yeast on the bacterial diversity and population structure in the rumen of cattle” | 2013 | 454 GS FLX | - High yeast treatment decreased Prevotella abundance and ruminal ammonia concentration while increasing propionate production. | 115 |
| “Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets” | 2013 | 454 GS FLX | - High starch and oil treatment significantly altered the microbiome by increasing Prevotellaceae and decreasing Ruminococcaceae. | 86 |
| “Relationship between the rumen microbiome and residual feed intake-efficiency of Brahman bulls stocked on bermudagrass pastures” | 2014 | 454 GS FLX | - Inefficient bulls had greater Prevotella, and lower stocking intensity increased microbiome diversity and richness. | 68 |
| “Microbial biodiversity of the liquid fraction of rumen content from lactating cows” | 2014 | 454 GS FLX | - Supplementation of lyophilized and dried yeast increased Bacillus abundance but did not impact fermentation. | 116 |
| “Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential” | 2014 | 454 GS FLX | - The rumen microbiome is established prior to consumption of solid food, but solid food intake determines microbiome composition. | 117 |
| “Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency” | 2014 | 454 GS FLX | - Firmicutes:Bacteroidetes was positively associated with milk fat content while many taxa were related to residual feed intake phenotype. | 118 |
| “Effect of feeding dried distillers grains with solubles on ruminal biohydrogenation, intestinal fatty acid profile, and gut microbial diversity evaluated through DNA pyro-sequencing” | 2014 | 454 GS FLX | - Greater inclusion of DDG with solubles corresponded to higher biohydrogenation of dietary unsaturated fatty acids and a decrease in phylum Fibrobacteres. - Diet did not affect abundance of biohydrogenating species. |
119 |
| “Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves” | 2014 | 454 GS FLX | - Three-week-old calves had greater Prevotella in the ruminal epithelium-attached community compared to digesta, and had region-specific microbiomes in the gastrointestinal tract. | 120 |
Barcoding enabled evaluation of many microbiomes on a single run. Referred to as multiplexing, barcoding involves adding a short string of nucleotides to the beginning of all amplicons from a single sample before sequencing.20 Output reads from sequencing can then be sorted based on the barcodes, which is known as demultiplexing. Raw sequence reads must be quality filtered to some degree before downstream analysis. Filtering typically involves removing reads based on length, quality score, ambiguous bases, homopolymers, and chimeric sequences.21 Denoising is also commonly employed on Roche 454 runs to model and remove noise from the sequencing run output.22,23 Preprocessing of reads can be the most computationally intensive step, and it will need to be improved and streamlined to facilitate analysis of the consistently larger data sets.
High-quality sequence reads from each sample are initially clustered into operational taxonomic units (OTUs) at a defined level of similarity. An OTU of sequences at 97% similarity is commonly used as a proxy for species. A single sequence from an OTU is selected to compare to a database reference alignment for taxonomic assignment. The primary databases utilized include Greengenes, SILVA, and RDP. Comparing sequences identified as a particular bacterial taxa with the total sequences would result in the relative abundance of the bacteria of interest. The relative abundance based on OTU number or taxonomy can then be used for many methods of downstream analysis to compare treatments, richness, and diversity. While this information provides a snapshot of the rumen microbiome, it does not evaluate the community dynamics causing shifts within the microbiome. Figure 2 depicts the workflow of rumen microbiome projects using a variety of approaches. For greater detail of sequence analysis methodology, we direct the reader to a recent review by Di Bella et al.19
Figure 2.

Rumen microbiome project workflow.
Many methods of downstream analysis have ecology origins and may be unfamiliar to animal scientists. Measures of diversity are foundational principles in microbial ecology that allow changes in community composition to be detected. While richness refers to a count of observed species or OTUs, evenness describes the similarity of species’ population sizes in a community. Commonly used richness estimators include Chao1 and abundance-based coverage estimator (ACE). The combination of richness and evenness represents the diversity of the microbiome. Alpha diversity represents the diversity within a sample and is often described using the Shannon index. In contrast, beta diversity describes diversity differences between samples and is frequently expressed with UniFrac distance, Bray–Curtis metric, or Morisita–Horn metric. A more complete description of individual metrics and ecology principles can be found in a book by Magurran.24
Pyrosequencing the Whole Rumen Microbiome
In addition to bacteria, the rumen microbiome includes archaea and eukaryotes. While most research has focused on either bacterial or archaeal species, Kittelmann et al.25 characterized the entire microbiome populations of cattle fed various diets. A mixture of barcoded amplicons was used from all three domains for simultaneous pyrosequencing. While 1,000 reads per sample were sufficient to describe protist and fungal diversity, 5,000 reads were not sufficient to describe the bacterial community using OTUs defined at 97% similarity. Results from pyrosequencing of archaea were compared with denaturing gradient gel electrophoresis (DGGE); while pyrosequencing provided greater resolution at 10,000 reads per sample, only 0.63% of the total sequences were not captured by DGGE. Although DGGE effectively reveals shifts in the community, pyrosequencing allows for evaluation of phylogenetic relationships in the community without creation of a clone library. Pairwise comparisons across all kingdoms using Spearman’s rank correlation indicated a positive association of Methanobrevibacter ruminantium clade with the Fibrobacteraceae family suggesting a possible functional relationship. Simultaneous pyrosequencing and complete taxonomic analysis of the rumen microbiome may provide a broader perspective of symbiotic and antagonistic microbial relationships in the rumen.
A similar approach utilized pyrosequencing and Sanger sequencing to characterize rumen bacteria, archaea, and fungi of beef cows.26 Of the more than 4,000 observed bacterial OTUs, only 29% were shared with the NCBI, SILVA, and RDP repositories. The primary bacterial taxa included Prevotella, Oscillibacter, Coprococcus, unclassified Ruminococcaceae, and Butyrivibrio representing about 40% of all bacteria taxa observed. However, 90% of the archaeal OTUs were previously observed in the public databases. Describing complex interactions across the rumen microbiome domains can contribute to our understanding of the observed physiological performance differences in response to nutrition or management. With the dearth of ruminal protist and fungal pyrosequencing efforts, additional research is needed to ensure database sequences reflect all species found in the rumen.
Microbiome Changes within the Rumen
The rumen is divided into interconnected sacs by pillars that along with the reticulum consist of one large fermentation chamber.27 Feed particle size is reduced over time through rumination and fermentation.28,29 Small dense particles settle in the ventral sacs while fibrous materials float toward the dorsal sacs. Variation in particle composition and pH within the rumen has been correlated to changes in the bacterial community.30,31 Microbial populations in various compartments of the reticulorumen were estimated using terminal restriction fragment length polymorphism (TRFLP) community fingerprinting.32 Actinobacteria and Bacteroidetes phyla were more prevalent in the reticulum compared with the rumen (12 vs. 7% and 19 vs. 12%, respectively) in steers fed a feedlot ration. Similar work using fingerprint analysis to compare five sampling locations within the rumen of three dairy cows reported a high degree of community similarity (95.3%).33 Although inherent variation in the rumen microbiome of an individual animal may be limited, a consistent sampling technique will still improve the ability to detect microbiome differences among animals or treatments.
Although rumen fistulas allow convenient sample collection, surgery cost may limit the number of animals sampled and impact statistical power. An alternative route of ruminal sample collection uses an oral stomach tube to obtain mixed ruminal fluid. A comparison of bacterial communities in cattle and sheep via DGGE fingerprinting did not reveal significant differences between samples collected using a stomach tube versus fistula.34 This finding was recently confirmed in post-weaned dairy calves as collection method did not affect the observed microbial community or molar proportion of VFA.35 However, evaluating fiber-adherent bacterial populations neither may be feasible using a stomach tube nor may correspond to other areas of the rumen. Microbial populations may be subject to diurnal variation depending on feeding schedule and diet composition. Nevertheless, time (samples collected at −3, +3, and +9 hours relative to feeding) did not affect DGGE banding patterns observed for three dairy cows fed once daily, even though fermentation parameters were affected by time.33 In contrast, analysis of bacterial community fingerprints in two dairy cows on a similar diet fed twice per day and monitored over four feeding cycles (sampling at 0, 2, 4, 6, 9, and 12 hours)31 indicated greater changes in the liquid communities over time than in solid samples. A significant effect of time on the relative abundance of Fibrobacter succinogenes and Ruminococcus albus was also observed in dairy cows sampled four hours after feeding and two hours before feeding.36 Greater community resolution by pyrosequencing may be needed to more accurately describe diurnal variation on different diets.
Larue et al.37 used DGGE and automated ribosomal intergenic spacer analysis (ARISA) to first report different bacterial communities present in liquid and solid fractions. This finding has also been supported using high-throughput methods.38,39 The rumen microbiome of beef heifers on a concentrate diet was evaluated using DGGE and real-time PCR; although cluster analysis of banding patterns did not elucidate a sample fraction effect, Ruminococcus spp., F. succinogenes, and Selenomonas ruminantium were observed in greater relative abundance in the solid fraction than in the liquid fraction.40 Dairy cows on a total mixed ration (TMR) had greater proportions of Butyrivibrio fibrisolvens, Prevotella ruminicola, and S. bovis in the liquid fraction, whereas Eubacterium ruminantium, F. succinogenes, and R. albus were greater in the solid fraction.36 Pyrosequencing of ruminal samples from beef steers on a low-quality forage (LQF) diet indicated genera Prevotella and Treponema represented a greater proportion of sequences in solid samples, but Paludibacter and Succiniclasticum were greater in the liquid fraction.41 The bacteria more predominantly observed in the solid fraction likely have a role in the degradation of cellulolytic compounds. Identifying specific taxa more prevalent in the fibrous fraction of the rumen may provide insight into the functional role of uncultured species.
Host Effect on the Rumen Microbiome
While many have noted rumen microbial populations are animal specific, Weimer et al.30 demonstrated the host effect on community composition. Ruminal contents from two cows consuming a similar diet with divergent fermentation profiles (ruminal pH = 6.9 vs. 6.1; total VFA concentration = 57 vs. 77 mM) were completely exchanged. Samples collected before and after the exchange indicated the pre-exchange rumen pH and VFA levels returned to before transfer levels within 24 hours after the exchange. Moreover, based on ARISA fingerprinting technique, it was observed that the bacterial community returned to prior structure within 14 days for one cow and within 61 days for the other. Using 454 sequencing and full-length 16S clone library revealed significant variation in the composition of the fiber-adherent microbiome of three steers.39 Community fingerprint analysis of ruminal samples taken from dairy cows at different times and separated into two fractions indicated the variation between animals was greater than variation between fraction or time point.31,33 Mechanisms to describe the relationship between the host and microbiome are not well defined. Individual selective preferences, feeding intake patterns, rumination time, and drinking behavior are likely to be critical in the regulation of the microbial community composition.
Development of the Rumen Microbiome
In current management systems, young dairy calves are fed milk replacer and are progressively transitioned to solid feed (ie, “starter rations”) to decrease labor and improve ruminal development. A wide variety of dietary strategies have been researched over the years seeking to obtain optimal performance gains and ruminal development. While a consensus feeding regimen has not been reached, a recent review by Khan et al.42 details the progress in this area. Inclusion of chopped forage in starter rations increases ruminal epithelium development and rumination.43,44 The rumen microbiome of growing calves is a key component of the developing rumen and is altered by management and nutrition.40
Pyrosequencing techniques were used to evaluate the rumen microbiome changes in dairy calves at one to three days, two months, six months, and two years of age.45 Ruminal fluid samples were collected using a stomach tube from five calves in each age group. Defining OTU at 97% similarity, rarefaction curves indicated 11,000 reads per sample were sufficient to describe ruminal bacteria for calves at one to three days and two months of age but insufficient for cattle at six months and two years of age. As expected, greater bacteria populations were present in older calves and cows compared with newborn and two-month old calves. In one-to-three-day-old calves, Firmicutes was the dominant phylum. Specifically, Streptococcus was more abundant at day 1 and then rapidly declined to less than 0.1% at two months of age. Relative abundance of phylum Bacteroidetes increased with age; within the phylum, genus Bacteroides was most abundant at one to three days but decreased with age as relative abundance of Prevotella increased.
Because the rumen bacterial community in newborn calves is not similar to mature cattle, a dramatic change from primary colonization to mature animal must occur. From day 1 to 3, there was a distinct decrease in aerobic and facultative anaerobes coupled with an increase in bacteria associated with obligatory anaerobic function. Ruminococcus flavefaciens, a cellulolytic species, was detectable in the rumen at one day of age, suggesting possible alternate functions, dependence on other bacteria to meet nutritional requirements, or potential environmental contamination. Some ruminococci can grow on glucose, albeit at very slow rates, and prefer cellobiose.28
Diversity indices, OTU counts, and similarity within group increased with age. Although only five different animals were used for each age group, the mature ruminal environment was a more homogeneous, restricted niche compared with the rumen of a newborn calf. Compared with newborn calves, a greater dependence on products of microbial fermentation by mature cows to address nutritional demands may dictate a narrower range of functionality within the microbiome. Furthermore, newborn calves have an esophageal groove that shunts milk from the esophagus to the omasum, hence, limiting ruminal digestion. Understanding the natural progression of the rumen microbiome and its function is necessary to evaluate changes in the microbial environment caused by diet or management.
Using pyrosequencing and shotgun sequencing, additional research determined the temporal change of the rumen microbiome of dairy calves on a milk replacer diet at 14 and 42 days of age.46 Three calves were sampled at each time point, and more than 10,000 16S reads were obtained for each sample. Confirming previously discussed findings, the relative abundance of Bacteroidetes increased from 46% at 14 days to 75% in 42 days calves. Therefore, significant decreases were observed for the Firmicutes and Proteobacteria phyla. At 14 days, the main genera detected included Prevotella, Bacteroides, Oscillibacter, Paraprevotella, Butyricimonas, and Pelistega; however, by 42 days the main genera had shifted to Bacteroides, Prevotella, Porphyromonas, Butyricimonas, and Coprococcus.
Richness estimates were greater at 14 days suggesting a more heterogeneous and transient genera compared with the older calves. Considering fewer sequence reads from the younger calves classified at the genus level (52 vs. 69%), there are likely more species in the early rumen microbiome yet to be observed. Whole genome shotgun sequences revealed more than 8,000 protein families of which 3,000 were observed in all samples. Evaluating the protein families with the COG classification indicated a highly similar metabolic potential within the microbiome at different ages. However, several glycoside hydrolase families with cellulase activities were more abundant in 42-day-old calves. The TonB-dependent receptor was observed in the greatest abundance at 42 days, was present in high levels within the genome of Bacteroides species, and is involved in the degradation of polysaccharides. Although fiber was absent from the diet, at least 60 glycoside hydrolase families were observed, and beta-galactosidases represented one of the most abundant protein families. While the calf rumen microbiome may possess surprising metabolic potential, it is clearly a community with great phylogenetic fluctuation.
Rumen Microbiome Similarity Across Individuals
To adequately describe a microbiome of interest, an understanding of the sequencing depth and diversity present in a microbial community is required. Variation in ruminal ecology of 16 dairy cows on a common diet was evaluated using pyrosequencing and real-time PCR.47 At a depth of 9,500 reads per sample, almost 5,000 total OTUs were identified. Each sample had an average of 1,800 OTUs; however, only 157 OTUs were identified in each rumen sample, which aligned to 32 genera. Real-time PCR estimates, a more sensitive quantification technique, were compared with the relative abundance observed in pyrosequencing. Across all samples, greater than 0.87 Pearson correlation was observed between pyrosequencing and real-time PCR-estimated relative abundances for the four evaluated species. Under these rumen conditions and sequencing depth, pyrosequencing was accurate for quantitative abundance of F. succinogenes S85, M. elsdenii T81, and R. amylophilus H18, but Succinivibrio dextrinosolvens 22b was underrepresented by pyrosequencing.
Overall, pairwise Bray–Curtis index revealed a similarity of only 51% among samples, but the weighted UniFrac metric estimated average phylogenetic similarity among samples at 82%. This suggests that although OTUs vary between animals, communities share OTUs phylogenetically related below the OTU-defined similarity level of 97% (genus, order, etc.). These results underscored the taxonomic variation present among animals consuming a 30% roughage and 70% concentrate diet. A relatively small, core microbiome (32 genera) indicates that additional taxa may not have significant functional contributions or that the functional similarity between animals is greater than the observed taxonomic similarity.
Transitional Effects on the Rumen Microbiome in Beef Cattle
Dietary transitions during the animal’s life directly impact ruminal microbial composition. The segmented nature of the beef industry represents necessary time points of adaptation for the rumen microbiome. Ensuring proper development and adaptation of the rumen benefits animal productivity and well-being. When cattle transfer production phases in the beef industry, the accompanying dietary change requires the rumen microbiome to adjust.48,49 After weaning, calves are classically stocked on available forage, which is often winter wheat in the southern US.50
Using pyrosequencing, Pitta et al.38 evaluated the transition of the microbiome in 14 yearling cattle fed bermudagrass hay (11% crude protein (CP)) followed by wheat pasture (20% CP). Across solid, liquid, and whole ruminal digesta samples, an average sequencing depth of about 2,500 reads per sample was obtained. With a total of 149 bacterial genera identified, the liquid fraction of the hay diet contained the greatest number of bacteria, whereas the whole digesta fraction from the wheat diet only had 118 genera that aligned at greater than 95% similarity. Rarefaction curve, ACE, and Chao1 estimated greater diversity in all three sample factions of the hay diet compared with the wheat diet.
Shannon alpha diversity index analysis indicated the average bacterial alpha diversity was greater for steers on the hay diet regardless of fraction (6.3 vs. 5.5, at 97% similarity level). Prevotella was the most abundant genus observed and was greater in the liquid fraction from both diets. Rikenella was detected in greater abundance in the hay diet relative to wheat and was the second most abundant genus. Considering the hay diet contained greater neutral detergent fiber (NDF; 68 vs. 44%) and was less digestible (57 vs. 80%), greater species richness may be required to degrade additional cellulose in harvested C4 grasses compared with grazed C3 grasses.
After the stocker phase, beef cattle are transitioned to a high-concentrate ration at a feedlot in a stepwise manner. The effect of this process on the rumen microbiome was evaluated using a multifaceted approach including TRFLP, real-time PCR, and 16S library sequencing.51 The step-up diets consisted of dietary hay-to-grain ratios of 80:20, 60:40, 40:60, and 20:80. An additional four steers remained on prairie hay for the duration of the experiment. Results of TRFLP indicated that the first major change in the rumen environment occurred after the shift from diet 60:40 to diet 40:60, and the change in bacterial community was even more pronounced on the 20:80 diet. When published, only 30–50% of the terminal restriction fragments could be annotated to phylogenetic assignments, and only 115 bacterial genera were identified.
16S libraries were constructed from two steers on the 20:80 and two steers on the prairie hay treatments. The library from steers on prairie hay had 398 different OTUs, whereas the library from steers on the concentrate diet (20:80) had 315 OTUs. However, only 24 OTUs were shared among libraries. Steers on the prairie hay diet had a significantly greater number of bacteria from the Fibrobacteres phylum, but fewer bacteria from the Firmicutes and Bacteroidetes phyla. However, there was a greater proportion of unclassified bacteria from the sequence library of steers on prairie hay compared with the high-concentrate diet (33 vs. 10%) suggesting previous sequences deposited in the database could have biased the taxonomic assignments.32 Database bias decreases with additional sequence deposits, and previously unclassified bacteria would likely be classified upon reanalysis of the sequence data. Additionally, Firmicutes was well represented under both dietary conditions suggesting the phylum is a core bacterial component of the rumen.
Results from PCR analysis indicated an increase in M. elsdenii, S. bovis, S. ruminantium, and Prevotella bryantii during adaptation to a high-concentrate diet.51 Culture-based research has demonstrated that M. elsdenii utilizes lactic acid released in the rumen contributing to stabilization of rumen pH and prevention of acidosis.11,52 S. bovis is an amylolytic, facultative anaerobe known to increase with the addition of starch to the diet or when ruminal pH decreases.53,54 Although S. bovis increased two-fold on the first step-up diet, populations decreased on the remaining three diets suggesting the step-up diets were effective in adaptation to a high-concentrate diet. As expected, B. fibrisolvens and F. succinogenes populations decreased with addition of concentrate to the diet. Although both have fibrolytic capabilities, F. succinogenes decreased more rapidly during adaptation compared with B. fibrisolvens. The 40-fold decrease of F. succinogenes is similar to other published observations.55 B. fibrisolvens can also utilize maltose and sucrose,56 and significant decreases were not evident until a 20-fold reduction occurred on the 20:80 diet. Although TRFLP only provides a broad perspective of a microbial community, using it in tandem with sequencing of 16S libraries and real-time PCR created a complete description of how the rumen microbiome adapts to high-grain, concentrate diets.
Recent work by Petri et al.57 focusing on bacteria attached to rumen epithelium (“epimural community”) also evaluated how the rumen microbiome adapts from a forage to a high-grain diet. After adaptation to the high-grain diet, an acidotic challenge induced by feed intake restriction followed by a single dosage of dry-rolled barley was also performed. Ruminal epithelial samples and pH were monitored during each dietary phase, and pyrosequencing, DGGE, and real-time PCR were used to describe microbiome changes.
Forage and mixed forage diets had more similar epimural communities than the high-grain diet using clustering analysis of DGGE results. Of the six bacterial species quantified by real-time PCR, only F. succinogenes was affected by diet with the greatest amount of animals fed the forage diet. Although average abundance values varied, treatment responses were consistent for Prevotella, F. succinogenes, M. elsdenii, and S. bovis using PCR and pyrosequencing approaches. Pyrosequencing results yielded more than 3,000 reads per sample with an average of 149 OTUs defined at 97% similarity. However, no dietary effect on species diversity or richness was observed in the epimural community.
Firmicutes were the predominant phylum on all treatments representing greater than 65% in relative abundance, but it was unaffected by diet transitions. In contrast, candidate division TM7 phylum was greater on forage-based diets whereas Actinobacteria was greater on the high-grain and acidotic challenge diets. Butyrivibrio was greater on forage-based diets, and B. fibrisolvens and Ruminococcus were associated with higher pH and less time with pH below 5.8, 5.5, and 5.2. Succinovibrio, Eubacterium cellulosolvens incertae sedis, Roseburia, Atopobium, and Succiniclasticum were mostly undetected on forage diets but were greater on the high-grain diet. During the acidotic challenge, Streptococcus and Lactobacillus increased significantly affirming their role in lactate production during acute acidosis. Desulfocurvus, a sulfate-reducing bacterium, was observed only during the acidotic challenge where it increased up to almost 1% of epithelial bacteria; this genus has been observed in marine environments, and some strains have the ability to utilize lactate and pyruvate in the presence of sulfur.58,59
Initial research on the rumen epimural microbiome using DGGE suggested this community may remain more consistent through dietary changes compared with the solid and liquid bacterial communities.60 The epithelium-attached bacteria in the rumen are likely associated with fermentation end products and VFA absorption; thus, defining this community may reveal strategies the microbiome uses to adapt to significant dietary changes.
Transitional Effects on the Rumen Microbiome in Dairy Cattle
Distinct phases in dairy management require adaptation on the part of the cow especially during the transition from late-pregnancy through lactation. This time prepartum and postpartum corresponds to changes in diet composition, nutritional requirements, and rapid mobilization of body stores to provide energy for milk production.61 Additionally, the normal decrease in voluntary dry matter intake immediately before parturition increases the susceptibility to postpartum acidosis because of the substantial increase in fermentable starches. This transition period, complicated by the stress of parturition, is when most metabolic problems are observed in dairy cows. Therefore, it is important to understand the changes in the rumen microbiome during this critical period.
Rumen microbiomes of seven cows were evaluated during the transition period at seven time points from −21 to +21 days postpartum.62 Real-time PCR and sequenced TRFLP libraries were used to describe the bacterial community and compare it to seven mid-lactation cows serving as the control. Based on TRFLP analysis, phylum Firmicutes represented more than 50% of the restriction fragments and significantly increased from 57% at day −7 to 68% at +21; the specific families responsible for this increase included Lactobacillaceae, Streptococcaceae, and incertae sedis XI. Phylum Bacteroidetes decreased in relative abundance from 14% at day −21 to 5% at day +21 because of decreasing prevalence of Rikenellaceae family. Similarly, steers transitioning from moderate-quality hay to high-quality wheat pasture38 also had a decrease in Rikenella.
Real-time PCR results of bacteria extensively evaluated in culture-based studies may not represent a large portion of ruminal bacteria, but can serve as an indicator of bacteria with similar functions within the rumen. As expected, with an increase in dietary energy postpartum, Prevotella brevis, P. ruminicola, R. amylophilus, Anaerovibrio lipolytica, S. bovis, S. ruminantium, and Lactobacillus spp. increased. However, elevated levels of F. succinogenes were observed postpartum until day 14, and M. elsdenii remained essentially unchanged during the transition period.
As the primary lactate-utilizer in the rumen, M. elsdenii was expected to increase with the rise in lactate (produced by S. bovis and Lactobacillus spp.) because of additional inclusion of rapidly fermentable carbohydrates in the postpartum diet. A decrease in M. elsdenii would limit conversion of lactate to propionate potentially exacerbating the negative energy balance that characterizes the postpartum period.61 While other research on the bacteria during the transition period is limited, Mohammed et al.63 did not observe major changes in the rumen microbiome pre- and postpartum using ARISA. There was no relationship between community fingerprinting and community composition to severity of observed acidosis. A greater understanding of the microbiome during the transition period is still needed to improve rumen function via nutritional management. This could help reduce incidence of acidosis and metabolic disorders.
Animal Efficiency and the Rumen Microbiome
Beef and dairy cattle production are tied to the animal’s ability to efficiently convert feedstuffs to a usable product eg, meat and milk. Residual feed intake (RFI) is a tool used to select animals that perform similarly to their counterparts with less feed intake. Improvements in efficiency can be quantified with RFI, and association with greater diet digestion suggests that rumen microbiome may contribute to differences in observed RFI.64 Initially, DGGE banding patterns revealed different bacterial phylotypes between steers with divergent RFI phenotypes with greater similarity among steers with a negative RFI (efficient).65 S. dextrinosolvens and Robinsoniella peoriensis were associated with animals with a positive RFI (inefficient) after sequencing of DGGE bands.66 Seven other bands were identified as Prevotella and were observed in steers with both negative and positive RFI. In similar work, Prevotella was also observed in greater relative abundance in animals with positive RFI with greater overall differences observed in cattle on a high-concentrate diet and grazing animals.67,68
Rius et al.69 fed 16 dairy cows selected for negative or positive RFI a similar diet of fresh ryegrass and evaluated intake, digestion, N retention, and the microbial environment. Although no differences were observed in intake, pH, VFA profile, or urinary N output, cows with negative RFI had greater digestion of N and OM in addition to higher ruminal ammonia levels. After pyrosequencing, bacterial communities visualized by principal component analysis (PCA) analysis did not reveal a relationship between RFI phenotype and the rumen microbiome; however, several bacterial taxa were associated with RFI. The relative abundance of Lachnospiraceae was greater in cows with negative RFI (28 vs. 24%), whereas Fibrobacteraceae and Prevotellaceae were greater in cows with positive RFI. Overall, there was no observed difference detected in the microbial community as it relates to changes in ruminal fermentation.
Using community fingerprinting techniques (DGGE), Hernandez-Sanabria et al.70 also found inconclusive differences between positive and negative RFI groups. Difficulty in determining the association between RFI and the rumen microbiome may stem from RFI phenotypes changing significantly from a forage to a concentrate diet.71 Moreover, the rumen microbiome may be a less significant biological underpinning of RFI phenotype in groups of genetically diverse cattle. A combination of high-throughput methods, greater sequencing depth, and a larger number of animals may be required to more clearly discern subtle differences in the rumen microbiome associated with efficient and inefficient animals.
Effect of Corn Co-Products on the Rumen Microbiome
Increased ethanol production in the US has resulted in greater utilization of the subsequent co-product, distillers grains, in beef cattle rations. Distillers grains are the unfermented grain residue that contains more protein and NDF compared with the unprocessed grain. Callaway et al.72 evaluated bacterial diversity in the rumen of cows consuming increasing levels of dried distillers grains (DDG) at 0, 25, and 50% replacing a commercial concentrate feed. Pooled samples from two steers on each diet were pyrosequenced to describe the ruminal bacteria populations. Analysis of samples collected before the experiment indicated the presence of 74 genera with Prevotella as the most abundant. Averages across three high-concentrate diets resulted in detection of more than 400 species. For diets containing 50% DDG, Prevotella and Bacteroides increased 152 and 276% in relative abundance whereas Succinivibrio decreased 406% compared with the 0% DDG diet. Moreover, the trend indicated a decrease in Firmicutes:Bacteroidetes with increasing DDG inclusion because of greater Prevotella and Bacteroides. The proteolytic ability of Prevotella and Bacteroides has been previously reported;73,74 the response to DDG may be explained by the increase in dietary CP with DDG addition. Reported changes in the rumen microbiome of steers consuming 50% DDG coincided with a decrease in ruminal pH. However, pH values (6.58–7.18) were atypical for high-concentrate diets, and the unknown composition of the commercial feed prevents accurate diet comparisons.
Corn gluten feed (CGF), a co-product of the corn milling, is produced after the germ, starch, and gluten have been removed from the corn kernel. Similar to DDG, CGF is higher in protein and fiber than corn. Using a replicated 4 × 4 Latin square, the effect of increasing wet CGF (0, 11, 23, and 34%) was evaluated in lactating dairy cows in diets with equivalent NDF and CP.36 After 25 days of diet adaptation, ruminal samples were collected eight times over three days and pooled by cow and period. Increasing CGF elicited a change in ruminal fermentation including a linear increase in propionate and valerate in addition to a decrease in acetate, isovalerate, and pH.75 Quantitative PCR was used to determine the response of well-known taxa, but no linear or quadratic effects were observed for the nine taxon of interest. The sum of the species only represented from 6 to 16% of the total bacteria in the rumen determined using a universal primer. Inter-animal variation accounted for 10–55% of the random variance; thus, unexplained variance and between animal variation were both important contributors to the overall variance.36 Although relative abundance of the evaluated species was stable, changes in bacterial function and many unobserved populations could be related to the documented shifts in fermentation.
Effects of Forage and TMRs on the Microbiome
There are two main nutritional strategies utilized in the dairy industry: a seasonal, forage-based diet or a concentrate-based TMR. Maintaining a healthy and efficient rumen microbiome is a key component for optimizing feedstuff utilization and improving milk production. As the TMR forage:concentrate ratio is changed to meet lactation requirements, microbiome composition is also altered (Fig. 3). Using pyrosequencing, de Menezes et al.76 compared the effect of grazing or feeding a TMR on the liquid and solid rumen bacterial community structure. Four dairy cows were used in a crossover design with two weeks for diet adaptation. At the phylum level, Bacteroidetes and Firmicutes represented more than 80% of the total sequences for all samples. There was 10.5% dissimilarity between bacterial populations on pasture and TMR mostly attributed to differences in Bacteroidetes and Firmicutes; the relative abundance of Firmicutes was greatest in the TMR liquid fraction and lowest in the TMR solid fraction suggesting a more defined niche compared with the forage diet. However, the 14.9% dissimilarity between liquid and solid fractions was because of differences in Fibrobacteres and Actinobacteria. The solid fraction favored Fibrobacteres, but Actinobacteria was more predominant in the liquid fraction. Prevotellaceae, Lachnospiraceae, and Ruminococcaceae were the most abundant families. Prevotellaceae was more prevalent on pasture regardless of fraction. Prevotella strains have been shown to produce propionate;77 thus, increased Prevotella abundance could be a factor in reducing methane production as reported in a companion experiment.78 Combined, Fibrobacteraceae and Spirochaetaceae families did not contribute 10% of sequences, but were associated with the solid fraction regardless of diet suggesting a role in fiber degradation.
Figure 3.
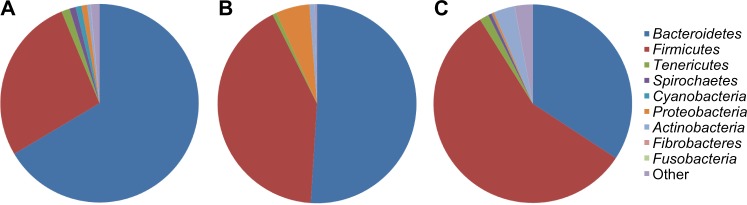
Variation in rumen microbiome of dairy cattle phyla composition. (A) 45:55 forage:concentrate TMR,80 (B) 30:70 forage:concentrate TMR,47 and (C) subacute ruminal acidosis condition.113
Similar work evaluated the rumen bacterial community using real-time PCR to quantify cultured and uncultured species in liquid and solid fractions of Jersey cows fed a forage diet and Holstein cows fed a TMR.79 Universal primers were used to measure total bacteria, and species-specific primers amplified informative regions of the 16S gene. Jersey cows had 1.7 × 108 (16S rRNA copies/μg DNA) total bacteria, whereas Holstein cows had 5.1 × 108, but there was no difference between liquid and solid fractions. Although the genus Prevotella accounted for more than 25% of absolute abundance in all samples, the well-described P. ruminicola represented only about 1% of absolute abundance. However, the genus Prevotella primers may have caused an overestimation because the primers matched non-Prevotella sequences obtained from the rumen in the RDP database. Although there was no effect of diet or sample fraction on the relative abundance of Prevotella, absolute abundance increased in the liquid fraction of concentrate-fed Holstein cows. Diet effects were confounded with breed.
As forage is a primary component of the diet of most ruminants, recent research implemented pyrosequencing to determine effects of different forage sources included in a TMR on the rumen microbiome.80 The TMR consisted of a forage:concentrate ratio of 45:55 with approximately 20% of the diet consisting of alfalfa or corn stover. While forage source did not affect dry matter intake or milk yield and composition, milk efficiency (milk yield/dry matter intake) was greater with the alfalfa treatment. As expected, the corresponding in situ results indicated a greater rate of organic matter and CP degradation for alfalfa compared with corn stover. This coincided with greater MCP and N conversion (milk protein yield/CP intake) as well as decreased urea N loss via urine for the alfalfa treatment.81 At a depth of more than 11,000 sequences per sample, 2,690 and 2,523 OTUs were observed with the corn stover and alfalfa diets, respectively. Accordingly, a greater Shannon alpha diversity index was observed with the corn stover diet, further supporting the tenant of increased diversity when less digestible diets are fed. Bacteroidetes was the predominant phyla observed followed by Firmicutes; this is in agreement with previous work where the observed ruminal pH was also about 6.5.41,47,82 Greater relative abundance of Prevotella and Selenomonas genera was observed on the alfalfa diet and may explain the greater concentrations of propionate and butyrate observed. Alternatively, Paraprevotella, unclassified Rikenellaceae, unclassified Ruminococcaceae, Anaerotruncus, and Papillibacter genera were greater with the corn stover diet.80 Higher quality forages are not only important dietary contributors to maintain high milk production but also impact the rumen microbiome.
Effect of Dietary Lipids on the Rumen Microbiome
Dietary lipids are included to increase the energy density of diets fed to high-producing ruminants, dairy cows in peak lactation, and beef steers on finishing diets. Unless protected, dietary lipids undergo microbial lipolysis and biohydrogenation in the rumen.83 Increasing fish oil, a highly unsaturated source of fatty acids (FAs), inclusion in dairy cattle decreased the proportion of B. fibrisolvens and Psuedobutyrivibrio, but Propionibacterium acnes increased significantly at high levels of supplementation (150 and 300 g/day fish oil).84 The final FA profile of the digesta leaving the rumen is correlated to FA profile in meat and milk. Moreover, some microbes are sensitive to high levels of polyunsaturated fatty acids (PUFA); growth of cellulolytic bacteria in vitro was decreased by the presence of PUFA.85 Utilizing culture-independent methods to describe microbial interactions with dietary lipids will enhance our fundamental understanding of ruminal lipid metabolism and overall effects on fermentation.
Pyrosequencing was used recently to determine the effect of starch level and inclusion of sunflower oil on the rumen microbiome.86 Diets consisted of either high or low levels of starch (34 and 20% starch, respectively) and inclusion of sunflower oil (5 and 0%). At an average depth of 7,000 sequences per sample, the mean observed OTUs was 2,297 defined at 95% similarity; numerically, observed OTUs were greatest in the low starch diet (2,939) and least on the high starch diet with sunflower oil (1,729). The Shannon alpha diversity index decreased in the high starch diet compared with the low starch, and the reduction was similar to responses observed by Pitta et al.38 and Fernando et al.51 with increasing starch. Chao1 and ACE richness estimates were greater than observed OTUs, but significantly decreased with starch inclusion. Based on PCA of the bacterial communities, greater dissimilarity was observed for cows on high starch diets.
Across all diets, more than 80% of the bacteria were represented by the phyla Bacteroidetes and Firmicutes. Although phyla Fibrobacteraceae and Spirochetes were quantitatively minor in relative abundance, both decreased significantly with inclusion of high starch similar to previous work. While the decrease in Spirochetes was similar to responses observed by de Menezes et al.76 Fibrobacteraceae increased with the TMR (higher starch diet). Additionally, similar to Pitta et al.38 Rikenella was observed at a greater relative abundance on the low starch diets potentially because of limited affinity for amylose and sunflower oil. Within high starch diets, inclusion of sunflower oil decreased Ruminococcaceae incertae sedis, Oscillibacter, Fastidiosipila, and Bifidobacterium, but increased Prevotella substantially. The increase in Prevotella may reflect an opportunistic increase after the addition of oil because it may have hindered growth of other bacteria. Others have identified terminal restriction fragments of bacteria associated with biohydrogenation intermediates as Prevotella and Lachnospiraceae incertae sedis.87 Beyond well-described cultured bacteria, continued elucidation of the rumen microbiome’s effect on lipids and unsaturated FAs will inform potential effects of diet on the FA composition of food products derived from ruminants.
Utilization of LQF by the Rumen Microbiome
The dynamic and adaptive qualities of the rumen microbiome allow ruminants to utilize many industrial co-products including DDG, corn gluten, cottonseed meal (CSM), and hulls. Similar to any fermentation process, a change in substrates alters resulting end products. Within the cow-calf phase of beef production, spring calving herds often have access to LQF (less than 7% CP) during mid to late gestation when energy requirements increase. Insufficient N does not maintain efficient fermentation and ultimately leads to performance losses. Small amounts of high protein co-products are often used to increase intake and utilization of LQF via improvements in forage digestibility.88–90 Moreover, less frequent supplementation (two days per week) also improves forage utilization via nitrogen recycling, but may favor hyper ammonia-producing bacteria in the rumen.91,92 While the physiological response to protein supplementation is well documented with increases in ruminal ammonia, passage rate, and nitrogen retention, there is a scarcity of literature related to the rumen microbiome on a LQF diet. Next-generation pyrosequencing has been used to describe rumen ecology on forage grazing, feedlot concentrate, and dairy TMR type rations, but the effect of an exclusive LQF diet on the microbiome has yet to be evaluated with pyrosequencing.
Chemical treatment of low-quality straws disrupt the cellulolytic structure within the cell wall and promotes greater forage utilization, but understanding of the microbial response to forage treatment is limited. Nguyen et al.93 evaluated the effect of urea and lime-treated rice straw on intake, digestion, fermentation, and the rumen microbial environment of swamp buffalo in a 4 × 4 Latin square design. A 2 × 2 treatment arrangement consisted of ad libitum access to rice straw or urea and lime-treated rice straw with additional provision of a 15% CP supplement containing 0 or 4% urea. Ruminal fluid was collected to estimate cellulolytic, amylolytic, and proteolytic bacteria using the roll-tube technique as well as to measure methanogen and cellulolytic populations with real-time PCR. No response was observed to treatment for amylolytic or proteolytic bacteria. However, an increase in cellulolytic bacteria was observed with increased provision of degradable intake protein.
Total bacteria populations were estimated by real-time PCR and ranged from 3.3 to 4.9 × 1012 copies/mL of ruminal contents. Of the three cellulolytic species evaluated, only increases in R. albus corresponded to increased ruminal ammonia and organic matter digestion; the smallest population was observed with untreated rice straw without urea and the greatest population with the treated straw with urea included in the supplement (3.8 × 109 and 3.2 × 1010, respectively). Additional research is needed to determine consistent responses of cellulolytic species to supplemental N and importance of cellulolytic species to overall diet digestion.
Recent advances in algal biofuel production could create a supply of the co-product, post-extraction algal residue (PEAR), to be utilized as a protein supplement in cow-calf operations.94 The high protein level (18% CP) of PEAR is similar to other supplements; however, the high ash content (44%) is unique. Drewery94 observed similar improvements in forage intake and digestion in response to isonitrogenous levels of PEAR and CSM while the corresponding changes in rumen microbiome were also surveyed using pyrosequencing.41 The 5 × 5 Latin square included three levels of PEAR supplementation as well as a CSM and negative control treatment provided to steers consuming oat straw ad libitum.
Sequencing yielded about 8,000 reads per sample and an average of 698 OTUs defined at 97% similarity. Weighted PCoA analysis clearly indicated a separation between the liquid and solid fractions, but no dietary effects were observed. Bacteroidetes was the predominant phylum for all treatments ranging from 66 to 83%. Within the liquid fraction, the relative abundance of Bacteroidetes decreased with additional PEAR supplementation whereas Firmicutes increased. Prevotella was the most prevalent genus and increased in the solid fraction. Three families corresponded to the increase in Firmicutes with additional PEAR including Ruminococcaceae, Lachnospiraceae, and Clostridiaceae. It is noteworthy that despite significant differences in fermentation and degradation between the negative control and the CSM positive control treatments, the rumen microbiome was essentially unchanged. Further work is needed to describe relative abundance and the possible relationship to dynamic metabolic potential.
Importance of Prevotella
In many studies, Prevotella has been observed as the dominant bacterial species in the rumen under various dietary conditions.14,38,95 Moreover, three ruminal species of the Prevotella genus account for as much as 70% of the rumen bacterial population.14,47 The Prevotella group was originally classified as succinate-producing bacteria,96 and the required nutrient hemin allows it to be sorted into two subspecies. The four characterized rumen Prevotella species include P. ruminicola, P. bryantii, Prevotella albensis, and P. brevis.97,98 Cultivated Prevotella strains display highly varied genetic divergence99 suggesting uncultured Prevotella strains also have diverse functions. While different degrees of polysaccharide-degrading ability have been described in four species,100 large clusters of Prevotella-related sequences have been associated with the fiber fraction of ruminal digesta12 and also the liquid fractions.38 Even though Prevotella can be the most abundant species observed, cultured species only accounted for a small fraction of the total Prevotella population.14,97
The whole genome of a P. ruminicola and P. bryantii strain was sequenced to evaluate their genetic similarity.101 The genome size and number of genes were similar between the strains, but the G + C content varied from 48 to 39%. Moreover, of the approximately 40 local syntenic blocks (groups of four or more genes), only 14 syntenic blocks were shared between the species. Most of the syntenic blocks were associated with polysaccharide metabolism and transport enzymes.101 Sequenced ribosomal intergenic spacer clone libraries revealed Prevotella was more abundant in animals fed forage only diet compared with those fed forage and concentrate diet.37 Although cultured species are unable to ferment cellulose, P. ruminicola did improve forage cellulose digestion when co-cultured with cellulolytic F. succinogenes and R. flavefaciens in vitro.102 This synergistic effect is likely because of the ability to catabolize xylan and pectin in the cell wall.100,103 Considering Prevotella is often the dominant genus within rumen, additional research is required on uncultured species to understand their function and importance to the rumen microbiome.
Metagenomic Perspective of the Rumen
Beyond descriptions of bacterial species richness and abundance, the rumen is viewed as a highly evolved and diverse reservoir of microbial functions with an unmatched ability to degrade plant material. The impact of a greater understanding of the rumen’s metabolic capabilities would extend beyond animal production and the cellulosic biofuel industry. Additionally, elucidating the functional roles of uncultured bacteria will facilitate more meaningful interpretation of pyrosequencing-based results.
Substantial variation among rumen microbiomes of steers fed a common diet was described by Brulc et al.39 Their broader interests sought to ascertain differences in metabolic potential to degrade cellulose in addition to defining microbial community structure. Within three steers on an identical diet, the number of OTUs (97% similarity) per sample ranged from 161 to 259 based on full-length 16S libraries with a total of 510 unique OTUs. Furthermore, ACE and Chao1 curves at 97% similarity indicate approximately 2,000 full-length 16S sequences are sufficient to describe OTUs present. Nonmetric multidimensional scaling indicated the phylogenetic makeup of the full-length 16S libraries differed from the pyrosequenced 16S sequences.
Pyrosequencing was used to evaluate genes present in the rumen metagenome related to fiber degradation. A wide diversity of glucoside hydrolases belonging to 35 families was present. Alternatively, only three families of carbohydrate-binding modules and three dockerin modules were detected. This finding contrasts previous belief that fibrous plant material degradation is linked specifically to hydrolysis of the main chains of cellulose and hemicellulose; rather, the wealth of enzymes that breakdown the side chains (galactans and arabinans104) of these polymers appears to be important for initial colonization of fiber.
More recent efforts have utilized newer sequencing platforms to delve deeper into the rumen microbiome. More than 268 gigabases or 1.5 billion read pairs were obtained from sequencing a single sample of the bacterial community attached to switchgrass.2 De novo assembly of these reads resulted in more than 2.5 million predicted open reading frames at an average of 542 bp and 55% predicted full-length genes. Within the full-length genes exclusively, 27,000 candidate genes were identified that were predicted to contain at least catalytic domain of interest or carbohydrate-binding module. This is more than five times greater the number of candidate carbohydrate genes from any previous study.
While Brulc et al.39 observed a greater number of genes encoding oligosaccharide-degrading enzymes, Hess et al.2 observed significantly more genes encoding endoglucanases, endohemicellulases, and debranching enzymes. Comparing the candidate enzyme genes to the NCBI non-redundant database indicated that only 12% of the candidate enzyme genes had greater than 75% similarity to deposited genes.2 A random subset of the candidate genes (n = 233) was selected, and 68% were correctly identified through specific primer design. The biochemical activity of 90 candidate genes predicted to contain a glycoside hydrolase family 3, 5, 8, 9, 10, 26, or 48 domains or a carbohydrate-binding molecule was evaluated. Enzymatic activity was tested on 10 different substrates relevant to carbohydrate catabolism and revealed 57% of tested proteins had activity against at least one substrate. Moreover, the large quantity of reads allowed a draft genome for 15 species to be assembled. Overall, results indicated the benefits of deep sequencing of the rumen metagenome to further define and discover metabolic potential in the rumen.
Conclusions and New Frontiers
Our primary objective in this review was to provide a concise overview of rumen microbiome research over the last 15 years as it relates to nutrition and metabolism. The significant wealth of metabolic capabilities within the rumen microbiome may be the key to unlocking greater animal production efficiency and industrial fermentation. Ever advancing nucleic acid-based technology has redefined our ability to describe the rumen microbiome and created new opportunities to investigate the complex relationships and niches within the microbial community. Limited functional knowledge of the uncultured majority in the rumen places importance on assembling genomes for many of these microbes and is currently being undertaken by Hungate 1000 (www.hungate1000.org.nz/) and FibRumBa database (www.jcvi.org/rumenomics/) efforts.
While genome assemblies describe the metabolic potential of microbes, more research will focus on expressed genes in a microbiome (metatranscriptome) using RNA-seq. Increased sequencing efforts will be supported by continued platform development with longer reads at a lower costs allowing more widespread use of the technology. Greater rates of data generation by high-throughput methods will require additional bioinformatics tools to process data and obtain meaningful (functional) results. Considering that diet provides substrates to the microbiome, fundamental nutrition concepts will remain critical to experimental design and interpretation of results. Further defining the communication between the host and microbiome could help elucidate many driving factors affecting shifts within the microbial community. The rumen microbiome is the distinguishing factor of all ruminants, and improved understanding of this complex community will lead to more efficient food production in the future.
Footnotes
ACADEMIC EDITOR: JT Efird, Associate Editor
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
This paper was subject to independent, expert peer review by a minimum of two blind peer reviewers. All editorial decisions were made by the independent academic editor. All authors have provided signed confirmation of their compliance with ethical and legal obligations including (but not limited to) use of any copyrighted material, compliance with ICMJE authorship and competing interests disclosure guidelines and, where applicable, compliance with legal and ethical guidelines on human and animal research participants.
Author Contributions
JCM wrote the first draft of the manuscript. JCM, TAW, and JJL contributed to the writing of the manuscript. JCM, TAW, and JJL jointly developed the structure and arguments for the paper. JCM, TAW, and JJL made critical revisions and approved the final version. All authors reviewed and approved the final manuscript.
FUNDING: Authors disclose no funding sources.
REFERENCES
- 1.Rubin EM. Genomics of cellulosic biofuels. Nature. 2008;454(7206):841–5. doi: 10.1038/nature07190. [DOI] [PubMed] [Google Scholar]
- 2.Hess M, Sczyrba A, Egan R, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331(6016):463–7. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 3.Mackie RI, Aminov RI, White BA, McSweeney CS. Molecular ecology and diversity in gut microbial ecosystems. In: Conje PB, editor. Ruminant Physiology: Digestion, Metabolism, Growth and Reproduction. New York: CABI Publishing; 2000. pp. 61–77. [Google Scholar]
- 4.Russell JB, O’Connor JD, Fox DG, Van Soest PJ, Sniffen CJ. A net carbohydrate and protein system for evaluating cattle diets: I. Ruminal fermentation. J Anim Sci. 1992;70(11):3551–61. doi: 10.2527/1992.70113551x. [DOI] [PubMed] [Google Scholar]
- 5.Webb KE. Intestinal absorption of protein hydrolysis products: a review. J Anim Sci. 1990;68(9):3011–22. doi: 10.2527/1990.6893011x. [DOI] [PubMed] [Google Scholar]
- 6.Bergman EN. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. 1990;70(2):567–90. doi: 10.1152/physrev.1990.70.2.567. [DOI] [PubMed] [Google Scholar]
- 7.Poulsen M, Schwab C, Jensen BB, et al. Methylotrophic methanogenic. Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat Commun. 2013;4:1428. doi: 10.1038/ncomms2432. [DOI] [PubMed] [Google Scholar]
- 8.Hungate RE. Rumen and its Microbes. New York: Academic Press; 1966. [Google Scholar]
- 9.Krause DO, Nagaraja TG, Wright ADG, Callaway TR. Board-invited review: rumen microbiology: leading the way in microbial ecology. J Anim Sci. 2013;91(1):331–41. doi: 10.2527/jas.2012-5567. [DOI] [PubMed] [Google Scholar]
- 10.Russell JB, Hino T. Regulation of lactate production in Streptococcus bovis: a spiraling effect that contributes to rumen acidosis. J Dairy Sci. 1985;68(7):1712–21. doi: 10.3168/jds.s0022-0302(85)81017-1. [DOI] [PubMed] [Google Scholar]
- 11.Counotte GHM, Prins RA, Janssen RHAM, deBie MJA. Role of Megasphaera elsdenii in the fermentation of DL-[2–13C]lactate in the rumen of dairy cattle. Appl Environ Microbiol. 1981;42(4):649–55. doi: 10.1128/aem.42.4.649-655.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koike S, Kobayashi Y. Fibrolytic rumen bacteria: their ecology and functions. Asian Austral J Anim Sci. 2009;22:131–8. [Google Scholar]
- 13.Krause DO, Dalrymple BP, Smith WJ, Mackie RI, McSweeney CS. 16S rDNA sequencing of Ruminococcus albus and Ruminococcus flavefaciens: design of a signature probe and its application in adult sheep. Microbiology. 1999;145(7):1797–807. doi: 10.1099/13500872-145-7-1797. [DOI] [PubMed] [Google Scholar]
- 14.Stevenson D, Weimer P. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl Microbiol Biotechnol. 2007;75(1):165–74. doi: 10.1007/s00253-006-0802-y. [DOI] [PubMed] [Google Scholar]
- 15.Henderson G, Cox F, Kittelmann S, et al. Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities. PLoS One. 2013;8(9):e74787. doi: 10.1371/journal.pone.0074787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villegas-Rivera G, Vargas-Cabrera Y, González-Silva N, et al. Evaluation of DNA extraction methods of rumen microbial populations. World J Microbiol Biotechnol. 2013;29(2):301–7. doi: 10.1007/s11274-012-1183-2. [DOI] [PubMed] [Google Scholar]
- 17.Soergel DAW, Dey N, Knight R, Brenner SE. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012;6(7):1440–4. doi: 10.1038/ismej.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Youssef N, Sheik CS, Krumholz LR, Najar FZ, Roe BA, Elshahed MS. Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl Environ Microbiol. 2009;75(16):5227–36. doi: 10.1128/AEM.00592-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Bella JM, Bao Y, Gloor GB, Burton JP, Reid G. High throughput sequencing methods and analysis for microbiome research. J Microbiol Methods. 2013;95(3):401–14. doi: 10.1016/j.mimet.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Parameswaran P, Jalili R, Tao L, et al. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res. 2007;35(19):e130. doi: 10.1093/nar/gkm760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;6(12):e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosen MJ, Callahan BJ, Fisher DS, Holmes SP. Denoising PCR-amplified metagenome data. BMC Bioinformatics. 2012;13:283. doi: 10.1186/1471-2105-13-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quince C, Lanzen A, Davenport R, Turnbaugh P. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12(1):38–55. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magurran AE. Measuring Biological Diversity. Malden, MA: Blackwell Science Ltd; 2004. [Google Scholar]
- 25.Kittelmann S, Seedorf H, Walters WA, et al. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS One. 2013;8(2):e47879. doi: 10.1371/journal.pone.0047879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fouts DE, Szpakowski S, Purushe J, et al. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen. PLoS One. 2012;7(11):e48289. doi: 10.1371/journal.pone.0048289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sellers AF, Stevens CE. Motor functions of the ruminant forestomach. Physiol Rev. 1966;46(4):634–61. doi: 10.1152/physrev.1966.46.4.634. [DOI] [PubMed] [Google Scholar]
- 28.Russell JB. Rumen Microbiology and its Role in Ruminant Nutrition. Ithaca, NY: Cornell University; 2002. [Google Scholar]
- 29.Russell JB, Rychlik JL. Factors that alter rumen microbial ecology. Science. 2001;292(5519):1119–22. doi: 10.1126/science.1058830. [DOI] [PubMed] [Google Scholar]
- 30.Weimer PJ, Stevenson DM, Mantovani HC, Man SLC. Host specificity of the ruminal bacterial community in the dairy cow following near-total exchange of ruminal contents. J Dairy Sci. 2010;93(12):5902–12. doi: 10.3168/jds.2010-3500. [DOI] [PubMed] [Google Scholar]
- 31.Welkie DG, Stevenson DM, Weimer PJ. ARISA analysis of ruminal bacterial community dynamics in lactating dairy cows during the feeding cycle. Anaerobe. 2010;16(2):94–100. doi: 10.1016/j.anaerobe.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Fernando SC. Meta-functional Genomics of the Bovine Rumen [dissertation] Stillwater, OK: Oklahoma State University; 2008. [Google Scholar]
- 33.Li M, Penner GB, Hernandez-Sanabria E, Oba M, Guan LL. Effects of sampling location and time, and host animal on assessment of bacterial diversity and fermentation parameters in the bovine rumen. J Appl Microbiol. 2009;107(6):1924–34. doi: 10.1111/j.1365-2672.2009.04376.x. [DOI] [PubMed] [Google Scholar]
- 34.Lodge-Ivey SL, Browne-Silva J, Horvath MB. Technical note: bacterial diversity and fermentation end products in rumen fluid samples collected via oral lavage or rumen cannula. J Anim Sci. 2009;87(7):2333–7. doi: 10.2527/jas.2008-1472. [DOI] [PubMed] [Google Scholar]
- 35.Terré M, Castells L, Fàbregas F, Bach A. Short communication: Comparison of pH, volatile fatty acids, and microbiome of rumen samples from preweaned calves obtained via cannula or stomach tube. J Dairy Sci. 2013;96(8):5290–4. doi: 10.3168/jds.2012-5921. [DOI] [PubMed] [Google Scholar]
- 36.Mullins CR, Mamedova LK, Carpenter AJ, et al. Analysis of rumen microbial populations in lactating dairy cattle fed diets varying in carbohydrate profiles and Saccharomyces cerevisiae fermentation product. J Dairy Sci. 2013;96(9):5872–81. doi: 10.3168/jds.2013-6775. [DOI] [PubMed] [Google Scholar]
- 37.Larue R, Yu Z, Parisi VA, Egan AR, Morrison M. Novel microbial diversity adherent to plant biomass in the herbivore gastrointestinal tract, as revealed by ribosomal intergenic spacer analysis and rrs gene sequencing. Environ Microbiol. 2005;7(4):530–43. doi: 10.1111/j.1462-2920.2005.00721.x. [DOI] [PubMed] [Google Scholar]
- 38.Pitta DW, Pinchak E, Dowd SE, et al. Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets. Microb Ecol. 2010;59(3):511–22. doi: 10.1007/s00248-009-9609-6. [DOI] [PubMed] [Google Scholar]
- 39.Brulc JM, Antonopoulos DA, Miller ME, et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci USA. 2009;106(6):1948–53. doi: 10.1073/pnas.0806191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petri RM, Forster RJ, Yang W, McKinnon JJ, McAllister TA. Characterization of rumen bacterial diversity and fermentation parameters in concentrate fed cattle with and without forage. J Appl Microbiol. 2012;112(6):1152–62. doi: 10.1111/j.1365-2672.2012.05295.x. [DOI] [PubMed] [Google Scholar]
- 41.McCann JC. Effect of Post-extraction Algal Residue Supplementation on the Rumen Microbiome of Steers Consuming Low-quality Forage [MS thesis] College Station, TX: Texas A&M University; 2013. [Google Scholar]
- 42.Khan MA, Weary DM, von Keyserlingk MAG. Invited review: effects of milk ration on solid feed intake, weaning, and performance in dairy heifers. J Dairy Sci. 2011;94(3):1071–81. doi: 10.3168/jds.2010-3733. [DOI] [PubMed] [Google Scholar]
- 43.Castells L, Bach A, Araujo G, Montoro C, Terré M. Effect of different forage sources on performance and feeding behavior of Holstein calves. J Dairy Sci. 2012;95(1):286–93. doi: 10.3168/jds.2011-4405. [DOI] [PubMed] [Google Scholar]
- 44.Phillips C. The effects of forage provision and group size on the behavior of calves. J Dairy Sci. 2004;87(5):1380–8. doi: 10.3168/jds.S0022-0302(04)73287-7. [DOI] [PubMed] [Google Scholar]
- 45.Jami E, Israel A, Kotser A, Mizrahi I. Exploring the bovine rumen bacterial community from birth to adulthood. The ISME journal. 2013;7(6):1069–79. doi: 10.1038/ismej.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li RW, Connor EE, Li C, Baldwin VIRL, Sparks ME. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ Microbiol. 2012;14(1):129–39. doi: 10.1111/j.1462-2920.2011.02543.x. [DOI] [PubMed] [Google Scholar]
- 47.Jami E, Mizrahi I. Composition and similarity of bovine rumen microbiota across individual animals. PLoS One. 2012;7(3):e33306. doi: 10.1371/journal.pone.0033306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y, Penner GB, Li M, Oba M, Guan LL. Rumen bacterial community transition during adaptation to high-grain diet. Anaerobe. 2000;6(5):273–84. [Google Scholar]
- 49.Tajima K, Aminov RI, Nagamine T, Matsui H, Nakamura M, Benno Y. Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl Environ Microbiol. 2001;67(6):2766–74. doi: 10.1128/AEM.67.6.2766-2774.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horn GW, Beck PA, Andrae JG, Paisley SI. Designing supplements for stocker cattle grazing wheat pasture. J Anim Sci. 2005;83(13 sul):E69–E78. [Google Scholar]
- 51.Fernando SC, Purvis HT, 2nd, Najar FZ, et al. Rumen microbial population dynamics during adaptation to a high-grain diet. Appl Environ Microbiol. 2010;76(22):7482–90. doi: 10.1128/AEM.00388-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Russell JB, Cotta MA, Dombrowski DB. Rumen bacterial competition in continuous culture: Streptococcus bovis versus Megasphaera elsdenii. Appl Environ Microbiol. 1981;41(6):1394–9. doi: 10.1128/aem.41.6.1394-1399.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Owens FN, Secrist DS, Hill WJ, Gill DR. Acidosis in cattle: a review. J Anim Sci. 1998;76(1):275–86. doi: 10.2527/1998.761275x. [DOI] [PubMed] [Google Scholar]
- 54.Slyter LL. Influence of acidosis on rumen function. J Anim Sci. 1976;43(4):910–29. doi: 10.2527/jas1976.434910x. [DOI] [PubMed] [Google Scholar]
- 55.Tajima K, Nagamine T, Matsui H, Nakamura M, Aminov RI. Phylogenetic analysis of archaeal 16S rRNA libraries from the rumen suggests the existence of a novel group of archaea not associated with known methanogens. FEMS Microbiol Lett. 2001;200(1):67–72. doi: 10.1111/j.1574-6968.2001.tb10694.x. [DOI] [PubMed] [Google Scholar]
- 56.Russell JB, Baldwin RL. Substrate preferences in rumen bacteria: evidence of catabolite regulatory mechanisms. Appl Environ Microbiol. 1978;36(2):319–329. doi: 10.1128/aem.36.2.319-329.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petri RM, Schwaiger T, Penner GB, et al. Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis. Appl Environ Microbiol. 2013;79(12):3744–55. doi: 10.1128/AEM.03983-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finster K, Kjeldsen K. Desulfovibrio oceani subsp. oceani sp. nov., subsp. nov. and Desulfovibrio oceani subsp. galateae subsp. nov., novel sulfate-reducing bacteria isolated from the oxygen minimum zone off the coast of Peru. Antonie Van Leeuwenhoek. 2010;97(3):221–9. doi: 10.1007/s10482-009-9403-y. [DOI] [PubMed] [Google Scholar]
- 59.Hamdi O, Ben Hania W, Postec A, et al. Isolation and characterization of Desulfocurvus thunnarius sp. nov., a sulfate-reducing bacterium isolated from an anaerobic sequencing batch reactor treating cooking wastewater. Int J Syst Evol Microbiol. 2013;63(pt 11):4237–42. doi: 10.1099/ijs.0.051664-0. [DOI] [PubMed] [Google Scholar]
- 60.Sadet S, Martin C, Meunier B, Morgavi D. PCR-DGGE analysis reveals a distinct diversity in the bacterial population attached to the rumen epithelium. Animal. 2007;1(07):939–44. doi: 10.1017/S1751731107000304. [DOI] [PubMed] [Google Scholar]
- 61.Drackley JK. Biology of dairy cows during the transition period: the final frontier? J Dairy Sci. 1999;82(11):2259–73. doi: 10.3168/jds.s0022-0302(99)75474-3. [DOI] [PubMed] [Google Scholar]
- 62.Wang X, Li X, Zhao C, et al. Correlation between composition of the bacterial community and concentration of volatile fatty acids in the rumen during the transition period and ketosis in dairy cows. Appl Environ Microbiol. 2012;78(7):2386–92. doi: 10.1128/AEM.07545-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohammed R, Stevenson D, Weimer P, Penner G, Beauchemin K. Individual animal variability in ruminal bacterial communities and ruminal acidosis in primiparous Holstein cows during the periparturient period. J Dairy Sci. 2012;95(11):6716–30. doi: 10.3168/jds.2012-5772. [DOI] [PubMed] [Google Scholar]
- 64.Herd R, Arthur P. Physiological basis for residual feed intake. J Anim Sci. 2009;87(14 suppl):E64–E71. doi: 10.2527/jas.2008-1345. [DOI] [PubMed] [Google Scholar]
- 65.Guan LL, Nkrumah JD, Basarab JA, Moore SS. Linkage of microbial ecology to phenotype: correlation of rumen microbial ecology to cattle’s feed efficiency. FEMS Microbiol Lett. 2008;288(1):85–91. doi: 10.1111/j.1574-6968.2008.01343.x. [DOI] [PubMed] [Google Scholar]
- 66.Hernandez-Sanabria E, Goonewardene LA, Wang Z, Durunna ON, Moore SS, Guan LL. Impact of feed efficiency and diet on adaptive variations in the bacterial community in the rumen fluid of cattle. Appl Environ Microbiol. 2012;78(4):1203–14. doi: 10.1128/AEM.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Carberry CA, Kenny DA, Han S, McCabe MS, Waters SM. Effect of phenotypic residual feed intake and dietary forage content on the rumen microbial community of beef cattle. Appl Environ Microbiol. 2012;78(14):4949–58. doi: 10.1128/AEM.07759-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCann JC, Wiley LM, Forbes TD, Rouquette FM, Jr, Tedeschi LO. Relationship between the rumen microbiome and residual feed intake-efficiency of Brahman bulls stocked on bermudagrass pastures. PLoS One. 2014;9(3):e91864. doi: 10.1371/journal.pone.0091864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rius AG, Kittelmann S, Macdonald KA, Waghorn GC, Janssen PH, Sikkema E. Nitrogen metabolism and rumen microbial enumeration in lactating cows with divergent residual feed intake fed high-digestibility pasture. J Dairy Sci. 2012;95(9):5024–34. doi: 10.3168/jds.2012-5392. [DOI] [PubMed] [Google Scholar]
- 70.Hernandez-Sanabria E, Guan LL, Goonewardene LA, et al. Correlation of Particular Bacterial PCR-Denaturing Gradient Gel Electrophoresis Patterns with Bovine Ruminal Fermentation Parameters and Feed Efficiency Traits. Appl Environ Microbiol. 2010;76(19):6338–50. doi: 10.1128/AEM.01052-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lawrence P, Kenny D, Earley B, Crews D, McGee M. Grass silage intake, rumen and blood variables, ultrasonic and body measurements, feeding behavior, and activity in pregnant beef heifers differing in phenotypic residual feed intake. J Anim Sci. 2011;89(10):3248–61. doi: 10.2527/jas.2010-3774. [DOI] [PubMed] [Google Scholar]
- 72.Callaway TR, Dowd SE, Edrington TS, et al. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J Anim Sci. 2010;88(12):3977–83. doi: 10.2527/jas.2010-2900. [DOI] [PubMed] [Google Scholar]
- 73.Attwood GT, Reilly K. Identification of proteolytic rumen bacteria isolated from New Zealand cattle. J Appl Microbiol. 1995;79(1):22–9. doi: 10.1111/j.1365-2672.1995.tb03119.x. [DOI] [PubMed] [Google Scholar]
- 74.Reilly K, Carruthers VR, Attwood GT. Design and use of 16S ribosomal DNA-directed primers in competitive PCRs to enumerate proteolytic bacteria in the rumen. Microb Ecol. 2002;43(2):259–70. doi: 10.1007/s00248-001-1052-2. [DOI] [PubMed] [Google Scholar]
- 75.Mullins CR, Grigsby KN, Anderson DE, Titgemeyer EC, Bradford BJ. Effects of feeding increasing levels of wet corn gluten feed on production and ruminal fermentation in lactating dairy cows. J Dairy Sci. 2010;93(11):5329–37. doi: 10.3168/jds.2010-3310. [DOI] [PubMed] [Google Scholar]
- 76.de Menezes AB, Lewis E, O’Donovan M, O’Neill BF, Clipson N, Doyle EM. Microbiome analysis of dairy cows fed pasture or total mixed ration diets. FEMS Microbiol Ecol. 2011;78(2):256–65. doi: 10.1111/j.1574-6941.2011.01151.x. [DOI] [PubMed] [Google Scholar]
- 77.Strobel HJ. Vitamin B12-dependent propionate production by the ruminal bacterium Prevotella ruminicola 23. Appl Environ Microbiol. 1992;58(7):2331–3. doi: 10.1128/aem.58.7.2331-2333.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O’Neill BF, Deighton MH, O’Loughlin BM, et al. Effects of a perennial ryegrass diet or total mixed ration diet offered to spring-calving Holstein-Friesian dairy cows on methane emissions, dry matter intake, and milk production. J Dairy Sci. 2011;94(4):1941–51. doi: 10.3168/jds.2010-3361. [DOI] [PubMed] [Google Scholar]
- 79.Kim M, Yu Z. Quantitative comparisons of select cultured and uncultured microbial populations in the rumen of cattle fed different diets. J Anim Sci Biotechnol. 2012;3(1):28. doi: 10.1186/2049-1891-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang R, Zhu W, Zhu W, Liu J, Mao S. Effect of dietary forage sources on rumen microbiota, rumen fermentation and biogenic amines in dairy cows. J Sci Food Agric. 2013 doi: 10.1002/jsfa.6508. [DOI] [PubMed] [Google Scholar]
- 81.Zhu W, Fu Y, Wang B, et al. Effects of dietary forage sources on rumen microbial protein synthesis and milk performance in early lactating dairy cows. J Dairy Sci. 2013;96(3):1727–34. doi: 10.3168/jds.2012-5756. [DOI] [PubMed] [Google Scholar]
- 82.Thoetkiattikul H, Mhuantong W, Laothanachareon T, et al. Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16S rRNA gene pyrosequencing. Curr Microbiol. 2013;67(2):1–8. doi: 10.1007/s00284-013-0336-3. [DOI] [PubMed] [Google Scholar]
- 83.Jenkins TC, Palmquist DL. Effect of fatty acids or calcium soaps on rumen and total nutrient digestibility of dairy rations. J Dairy Sci. 1984;67(5):978–86. doi: 10.3168/jds.S0022-0302(84)81396-X. [DOI] [PubMed] [Google Scholar]
- 84.Shingfield KJ, Kairenius P, Arölä A, et al. Dietary fish oil supplements modify ruminal biohydrogenation, alter the flow of fatty acids at the omasum, and induce changes in the Ruminal Butyrivibrio population in lactating cows. J Nutr. 2012;142(8):1437–48. doi: 10.3945/jn.112.158576. [DOI] [PubMed] [Google Scholar]
- 85.Maia MRG, Chaudhary LC, Figueres L, Wallace RJ. Metabolism of polyunsaturated fatty acids and their toxicity to the microflora of the rumen. Antonie Van Leeuwenhoek. 2007;91(4):303–14. doi: 10.1007/s10482-006-9118-2. [DOI] [PubMed] [Google Scholar]
- 86.Zened A, Combes S, Cauquil L, et al. Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiol Ecol. 2013;83(2):504–14. doi: 10.1111/1574-6941.12011. [DOI] [PubMed] [Google Scholar]
- 87.Huws SA, Kim EJ, Lee MR, et al. As yet uncultured bacteria phylogenetically classified as Prevotella, Lachnospiraceae incertae sedis and unclassified Bacteroidales, Clostridiales and Ruminococcaceae may play a predominant role in ruminal biohydrogenation. Environ Microbiol. 2011;13(6):1500–12. doi: 10.1111/j.1462-2920.2011.02452.x. [DOI] [PubMed] [Google Scholar]
- 88.Köster HH, Cochran RC, Titgemeyer EC, Vanzant ES, Abdelgadir I, St-Jean G. Effect of increasing degradable intake protein on intake and digestion of low-quality, tallgrass-prairie forage by beef cows. J Anim Sci. 1996;74(10):2473–81. doi: 10.2527/1996.74102473x. [DOI] [PubMed] [Google Scholar]
- 89.Mathis CP, Cochran RC, Stokka GL, Heldt JS, Woods BC, Olson KC. Impacts of increasing amounts of supplemental soybean meal on intake and digestion by beef steers and performance by beef cows consuming low-quality tallgrass-prairie forage. J Anim Sci. 1999;77(12):3156–62. doi: 10.2527/1999.77123156x. [DOI] [PubMed] [Google Scholar]
- 90.Winterholler SJ, McMurphy CP, Mourer GL, Krehbiel CR, Horn GW, Lalman DL. Supplementation of dried distillers grains with solubles to beef cows consuming low-quality forage during late gestation and early lactation. J Anim Sci. 2012;90(6):2014–25. doi: 10.2527/jas.2011-4152. [DOI] [PubMed] [Google Scholar]
- 91.Bohnert DW, Schauer CS, Bauer ML, DelCurto T. Influence of rumen protein degradability and supplementation frequency on steers consuming low-quality forage: I. Site of digestion and microbial efficiency. J Anim Sci. 2002;80(11):2967–77. doi: 10.2527/2002.80112967x. [DOI] [PubMed] [Google Scholar]
- 92.Farmer CG, Cochran RC, Nagaraja TG, Titgemeyer EC, Johnson DE, Wickersham TA. Ruminal and host adaptations to changes in frequency of protein supplementation. J Anim Sci. 2004;82(3):895–903. doi: 10.2527/2004.823895x. [DOI] [PubMed] [Google Scholar]
- 93.Nguyen V, Wanapat M, Khejornsart P, Kongmun P. Nutrient digestibility and ruminal fermentation characteristic in swamp buffaloes fed on chemically treated rice straw and urea. Trop Anim Health Prod. 2012;44(3):629–36. doi: 10.1007/s11250-011-9946-6. [DOI] [PubMed] [Google Scholar]
- 94.Drewery ML. Post-extraction Algal Residue as a Protein Source for Cattle Consuming Forage [MS thesis] College Station, TX: Texas A&M University; 2012. [Google Scholar]
- 95.Whitford MF, Forster RJ, Beard CE, Gong J, Teather RM. Phylogenetic analysis of rumen bacteria by comparative sequence analysis of cloned 16S rRNA genes. Anaerobe. 1998;4(3):153–63. doi: 10.1006/anae.1998.0155. [DOI] [PubMed] [Google Scholar]
- 96.Bryant MP, Small N, Bouma C, Chu H. Bacteroides ruminicola n. sp. and Succinimonas amylolytica; the new genus and species; species of succinic acid-producing anaerobic bacteria of the bovine rumen. J Bacteriol. 1958;76(1):15–23. doi: 10.1128/jb.76.1.15-23.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bekele AZ, Koike S, Kobayashi Y. Genetic diversity and diet specificity of ruminal Prevotella revealed by 16S rRNA gene-based analysis. FEMS Microbiol Lett. 2010;305(1):49–57. doi: 10.1111/j.1574-6968.2010.01911.x. [DOI] [PubMed] [Google Scholar]
- 98.Avgustin G, Wallace RJ, Flint HJ. Phenotypic diversity among ruminal isolates of Prevotella ruminicola: proposal of Prevotella brevis sp. nov., Prevotella bryantii sp. nov., and Prevotella albensis sp. nov. and redefinition of Prevotella ruminicola. Int J Syst Bacteriol. 1997;47(2):284–8. doi: 10.1099/00207713-47-2-284. [DOI] [PubMed] [Google Scholar]
- 99.Ramsak A, Peterka M, Tajima K, et al. Unravelling the genetic diversity of ruminal bacteria belonging to the CFB phylum. FEMS Microbiol Ecol. 2000;33(1):69–79. doi: 10.1111/j.1574-6941.2000.tb00728.x. [DOI] [PubMed] [Google Scholar]
- 100.Matsui H, Ogata K, Tajima K, et al. Phenotypic characterization of polysaccharidases produced by four Prevotella type strains. Curr Microbiol. 2000;41(1):45–9. doi: 10.1007/s002840010089. [DOI] [PubMed] [Google Scholar]
- 101.Purushe J, Fouts DE, Morrison M, et al. Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb Ecol. 2010;60(4):721–9. doi: 10.1007/s00248-010-9692-8. [DOI] [PubMed] [Google Scholar]
- 102.Fondevila M, Dehority BA. Interactions between Fibrobacter succinogenes, Prevotella ruminicola, and Ruminococcus flavefaciens in the digestion of cellulose from forages. J Anim Sci. 1996;74(3):678–84. doi: 10.2527/1996.743678x. [DOI] [PubMed] [Google Scholar]
- 103.Dehority BA. Effects of microbial synergism on fibre digestion in the rumen. Proc Nutr Soc. 1991;50(02):149–59. doi: 10.1079/pns19910026. [DOI] [PubMed] [Google Scholar]
- 104.Minic Z, Jouanin L. Plant glycoside hydrolases involved in cell wall polysaccharide degradation. Plant Physiol Biochem. 2006;44:7–9. 435–49. doi: 10.1016/j.plaphy.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 105.Ross EM, Moate PJ, Bath CR, et al. High throughput whole rumen metagenome profiling using untargeted massively parallel sequencing. BMC Genet. 2012;13(1):53. doi: 10.1186/1471-2156-13-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee HJ, Jung JY, Oh YK, Lee S-S, Madsen EL, Jeon CO. Comparative survey of rumen microbial communities and metabolites across one caprine and three bovine groups, using bar-coded pyrosequencing and 1H nuclear magnetic resonance spectroscopy. Appl Environ Microbiol. 2012;78(17):5983–93. doi: 10.1128/AEM.00104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li RW, Wu S, Baldwin RLVI, Li W, Li C. Perturbation dynamics of the rumen microbiota in response to exogenous butyrate. PLoS One. 2012;7(1):e29392. doi: 10.1371/journal.pone.0029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Berg MillerME, Yeoman CJ, Chia N, et al. Phage–bacteria relationships and CRISPR elements revealed by a metagenomic survey of the rumen microbiome. Environ Microbiol. 2012;14(1):207–27. doi: 10.1111/j.1462-2920.2011.02593.x. [DOI] [PubMed] [Google Scholar]
- 109.Wu S, Baldwin Vi RL, Li W, Li C, Connor EE, Li RW. The bacterial community composition of the bovine rumen detected using pyrosequencing of 16S rRNA genes. Metagenomics. 2012:1–11. [Google Scholar]
- 110.Ramirez HR, Nestor K, Tedeschi L, et al. The effect of brown midrib corn silage and dried distillers’ grains with solubles on milk production, nitrogen utilization and microbial community structure in dairy cows. Can J Anim Sci. 2012;92(3):365–80. [Google Scholar]
- 111.Broadway P, Callaway T, Carroll J, et al. Evaluation of the ruminal bacterial diversity of cattle fed diets containing citrus pulp pellets. Agric Food Anal Bacteriol. 2012;2:297–308. [Google Scholar]
- 112.Ross E, Moate P, Marett L, Cocks B, Hayes B. Investigating the effect of two methane-mitigating diets on the rumen microbiome using massively parallel sequencing. J Dairy Sci. 2013;96(9):6030–46. doi: 10.3168/jds.2013-6766. [DOI] [PubMed] [Google Scholar]
- 113.Mao SY, Zhang RY, Wang DS, Zhu WY. Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe. 2013;24:12–9. doi: 10.1016/j.anaerobe.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 114.de Oliveira MN, Jewell KA, Freitas FS, et al. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Vet Microbiol. 2013;164:3–4. 307–14. doi: 10.1016/j.vetmic.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 115.Pinloche E, McEwan N, Marden J-P, Bayourthe C, Auclair E, Newbold CJ. The effects of a probiotic yeast on the bacterial diversity and population structure in the rumen of cattle. PLoS One. 2013;8(7):e67824. doi: 10.1371/journal.pone.0067824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sandri M, Manfrin C, Pallavicini A, Stefanon B. Microbial biodiversity of the liquid fraction of rumen content from lactating cows. Animal. 2014;8(04):572–9. doi: 10.1017/S1751731114000056. [DOI] [PubMed] [Google Scholar]
- 117.Rey M, Enjalbert F, Combes S, Cauquil L, Bouchez O, Monteils V. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J Appl Microbiol. 2014;116(2):245–57. doi: 10.1111/jam.12405. [DOI] [PubMed] [Google Scholar]
- 118.Jami E, White BA, Mizrahi I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS One. 2014;9(1):e85423. doi: 10.1371/journal.pone.0085423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Castillo-Lopez E, Ramirez Ramirez HA, Klopfenstein TJ, et al. Effect of feeding dried distillers grains with solubles on ruminal biohydrogenation, intestinal fatty acid profile, and gut microbial diversity evaluated through DNA pyro-sequencing. J Anim Sci. 2014;92(2):733–43. doi: 10.2527/jas.2013-7223. [DOI] [PubMed] [Google Scholar]
- 120.Malmuthuge N, Griebel PJ, Guan LL. Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Appl Environ Microbiol. 2014;80(6):2021–8. doi: 10.1128/AEM.03864-13. [DOI] [PMC free article] [PubMed] [Google Scholar]