

Abstract
The objective of this study was to investigate the effect of equine arteritis virus (EAV) on type I interferon (IFN) production. Equine endothelial cells (EECs) were infected with the virulent Bucyrus strain (VBS) of EAV and expression of IFN-β was measured at mRNA and protein levels by quantitative real-time RT-PCR and IFN bioassay using vesicular stomatitis virus expressing the green fluorescence protein (VSV-GFP), respectively. Quantitative RT-PCR results showed that IFN-β mRNA levels in EECs infected with EAV VBS were not increased compared to those in mock-infected cells. Consistent with quantitative RT-PCR, Sendai virus- (SeV-) induced type I IFN production was inhibited by EAV infection. Using an IFN-β promoter-luciferase reporter assay, we subsequently demonstrated that EAV nsps 1, 2, and 11 had the capability to inhibit type I IFN activation. Of these three nsps, nsp1 exhibited the strongest inhibitory effect. Taken together, these data demonstrate that EAV has the ability to suppress the type I IFN production in EECs and nsp1 may play a critical role to subvert the equine innate immune response.
1. Introduction
Equine arteritis virus (EAV) is the causative agent of equine viral arteritis, a respiratory and reproductive disease of horses [1, 2]. EAV is a small enveloped virus with a positive-sense, single-stranded RNA genome of ~12.7 kb. It belongs to the family Arteriviridae (genus Arterivirus, order Nidovirales), which also includes porcine reproductive and respiratory syndrome virus (PRRSV), simian hemorrhagic fever virus (SHFV), and lactate dehydrogenase-elevating virus (LDV) of mice [3–5]. The EAV genome includes at least ten known functional open reading frames (ORFs 1a, 1b, 2a, 2b, 3, 4, 5a, 5b, 6, and 7) [5–7]. ORFs 1a and 1b are located at the 5′-proximal three-quarters of the genome and are translated to produce replicase polyproteins pp1a and pp1ab (1,727 and 3,175 amino acids, resp.). Translation of ORF1b depends on a −1 ribosomal frameshift located just before termination of ORF1a translation [8]. The two replicase precursor polyproteins are cleaved by three ORF1a-encoded proteases that reside in nsp1, nsp2, and nsp4, yielding at least 13 end-products, namely, nonstructural proteins (nsps) 1 to 12, including a recently described nsp7α and 7β [5, 9, 10]. The remaining eight ORFs (2a, 2b, and 3, 4, 5a, 5b, and 6-7) are located in the 3′ quarter of the genome and encode the structural proteins (E, GP2, GP3, GP4, ORF5a protein, GP5, M, and N, resp.) of the virus [5, 6, 11–13].
Type I interferon (IFN-α/β) is a key component of the host innate immune response to viral infection [14]. Recognition of pathogen-associated molecular patterns (PAMPs) in double-stranded RNA (dsRNA) by intracellular receptors, such as retinoic acid inducible gene I (RIG-I) and melanoma differentiation-associated antigen 5 (MDA-5) [15], activates protein signaling cascades that result in the activation of transcription factors, including interferon regulatory factor-3 (IRF-3) and nuclear factor-κB (NF-κB) [14]. The IFN-β promoter contains positive regulatory domains (PRDs), including the binding sites for different transcription factors, IRF-3 (PRDs I and III) and NF-κB (PRD II). Activation of these transcription factors triggers the formation of enhanceosomes in the cell nucleus and induces the expression of IFN-α/β [14, 16]. Both IRF-3 and NF-κB activation are mediated by mitochondrial antiviral signaling (MAVS) protein, which functions downstream of RIG-I and MDA-5 and upstream of the IκB kinase (IKK) complex and TANK-binding kinase-1 (TBK1) [17–20]. Among the various factors involved in type I IFN production, IRF-3 plays a critical role. IRF-3 is expressed in most cell types and resides in the cytoplasm in an inactive form. When stimulated, IRF-3 becomes phosphorylated and undergoes conformational changes, resulting in dimerization with the exposure of nuclear localization signal. In the nucleus, IRF-3 recruits coactivator CBP/p300 and forms a complex to bind IRF-3 responsive elements (PRDs I and III) of the IFN-β promoter [14]. In addition to IRF-3, NF-κB is also a critical regulator of host innate and adaptive immunity. It plays an important role in the regulation of cell proliferation as well as cell survival. Many viruses have evolved strategies to counteract key elements of the IFN response and prevent development of an antiviral response in the host [14]. Through evolution, viruses can either activate or inhibit the NF-κB pathway in order to replicate in host cells. Viruses such as African swine fever virus and influenza A virus block NF-κB activation to counteract the host innate immune response [21, 22]. In contrast, viruses such as hepatitis C virus, reovirus, and herpes simplex virus have developed mechanisms to directly activate NF-κB to support production of progeny viruses and intracellular spreading [23–25].
Until now, the innate immune response to EAV infection was poorly characterized and the information pertaining to type I IFN production was largely derived from studies of PRRSV and other nidoviruses [26–32]. Recently, van Kasteren et al. [33] reported that the EAV PLP2 has de-ubiquitylation function which suppresses RIG-I to control innate immune signaling in EAV-infected cells [34, 35]. In all these studies, the investigators have used recombinant proteins (e.g., nsp2 protein of EAV) and a specific immune suppression mechanism. However, the effect of the whole virus and involvement of other EAV proteins in the suppression of host cell immune responses are largely unknown. To elucidate the molecular mechanism of EAV involved in host immune suppression, the objective of this study was to investigate the effect of EAV on type I IFN production and to further identify the specific viral proteins responsible for the suppression of IFN-β activity.
2. Materials and Methods
2.1. Virus and Cells
Equine pulmonary artery endothelial cells (EECs [36]), baby hamster kidney-21 (BHK-21 [ATCC CCL-10], Manassas, VA), and HEK293T (ATCC CRL-11228) cells were maintained in Dulbecco's modified essential medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (FBS; HyClone Laboratories, Inc., Logan, UT), 100 U/μg per mL penicillin-streptomycin, and 200 mM L-glutamine (Gibco, Carlsbad, CA) in a humidified incubator with 5% CO2 at 37°C. HeLa cells (NIH AIDS Research and Reference Reagent Program, Germantown, MD) were propagated in minimum essential medium (MEM) supplemented with 10% heat-inactivated FBS in a humidified incubator with 5% CO2 at 37°C. MDBK (ATCC CCL-22) cells were grown in Eagle's minimum essential medium with 10% ferritin-supplemented bovine calf serum (HyClone Laboratories, Inc., Logan, UT) and 100 U/μg per mL penicillin-streptomycin (Gibco, Carlsbad, CA). The virulent Bucyrus strain of EAV (EAV VBS, horse passage 15 pleural fluid; ATCC VR-796, Manassas, VA) was passaged once in EECs to obtain high-titered working stocks for the present study using the method described previously [37]. Sendai virus, the Cantell strain (SeV; ATCC VR-907, Manassas, VA), was propagated in embryonated chicken eggs. The virus titer was determined by hemagglutination inhibition (HI) assay using chicken red blood cells as described previously [38]. Vesicular stomatitis virus expressing green fluorescent protein (VSV-GFP [39]) was kindly provided by Dr. Adolfo Garcia-Sastre (Mt. Sinai School of Medicine, New York, NY).
2.2. Plasmids
Plasmids for expression of recombinant EAV nsp1 to nsp12 in mammalian cells were constructed as previously described [40]. Briefly, the coding regions of each of the twelve nsps were PCR-amplified from the EAV rVBS full-length infectious cDNA clone [41] and cloned into the eukaryotic expression vector pCAGGS [33]. The nsp5, nsp6, nsp10, and nsp12 were expressed as C-terminal FLAG-tagged fusion proteins. To express the recombinant EAV nsps, BHK-21 cells were transfected with each individual plasmid containing an nsp coding region. Transfection was performed using FuGENE HD (Promega, Madison, WI) according to manufacturer's instructions. The specificity of each recombinant protein was confirmed by immunofluorescence and Western blot analysis. Reporter plasmids expressing the firefly luciferase under the control of either the IFN-β promoter (p125-Luc) or an artificial promoter containing three IRF-3 binding sites (p55-CIB-Luc) were kindly provided by Yoneyama et al. [42]. The pNF-κB-Luc reporter plasmid (Stratagene, La Jolla, CA) expresses the firefly luciferase under the control of a promoter with NF-κB-response element. The pRL-SV40 plasmid (Promega, Madison, WI) expresses a Renilla luciferase under the control of a simian virus (SV40) promoter. The pEFneo-RIG-I, pEFneo-MDA-5, and pEFneo-IKKε were kindly provided by Komatsu et al. [43]. The pcDNA3-TRIF and pCMV2-IKK2-WT were purchased from Addgene (Cambridge, MA). Construction of the pCAGGS-IRF-3 and pCAGGS-NS1 plasmids was described previously [44].
2.3. Antibodies
To detect EAV antigens in infected cells, monoclonal antibodies (MAbs) against EAV nsp1 (MAb 12A4) and N protein (MAb 3E2) were used [45, 46]. Specific polyclonal rabbit antisera recognizing EAV nsp2 [47], nsp3 [48], nsp4 [47], nsp7-8 [47], and nsp10 [49] have been described previously. In addition, antisera against nsp9 and nsp11 were raised by immunizing rabbits with purified full-length recombinant proteins expressed in E. coli (J.C. Zevenhoven, D. D. Nedialkova, and E. J. Snijder, unpublished data). Anti-FLAG MAb (F3165) purchased from Sigma (St. Louis, MO) was used to detect FLAG-tagged EAV fusion proteins in immunofluorescence assay. Rabbit polyclonal antibodies for human IRF-3 (sc-9082) and NF-κB p65 (sc-7151) were purchased from Santa Cruz Biotechnologies Inc. (Santa Cruz, CA). Alexa Fluor 488-conjugated and Alexa Fluor 594-conjugated secondary antibodies were purchased from Invitrogen (Carlsbad, CA).
2.4. RNA Extraction and Quantitative Real-Time RT-PCR (qRT-PCR)
Total RNA was extracted using MagMAX-96 Total RNA Isolation kit (Ambion, Austin, TX) in a MagMAX Express-96 magnetic particle processor (Applied Biosystems, Foster City, CA). RNA from each culture was treated with DNase to remove any contaminating genomic DNA (gDNA). The RNA concentration was assessed at OD260 nm and purity was verified by the OD260/OD280 ratio using NanoDrop (Thermo Scientific, Wilmington, DE). The reverse transcription reaction was performed with 1 μg of total RNA using RT random primers and a MultiScribe reverse transcriptase (High Capacity cDNA Reverse Transcription kit with RNase inhibitor (Applied Biosystems, Foster City, CA)) according to the manufacturer's instructions. The following IFN-β primers and probe set were used for PCR amplification with an Applied Biosystems 7500 Fast Real-Time PCR System: EqIL-IFN-βF: 5′-AATGGCCCTCCTGCTGTGT-3′, EqIL-IFN-βR: 5′-CCGAAGCAAGTCATAGTTCACAGA-3′, and EqIL-IFN-β: probe 5′-FAM-CTCCACCACGGCTC-NFQ-3′. For each sample, cDNA corresponding to the β-glucuronidase (β-GUS) gene was amplified and used as an endogenous control. All PCR efficiency values were determined using LinReg [50]. The relative concentration of target gene mRNA was equal to 2−ΔΔCt where ΔΔCt = [(Avg. gene of interest Ct − Avg. β-GUS Ct)sample − (Avg. gene of interest Ct − Avg. β-GUS Ct)calibrator]. The calibrator was calculated from the mean ΔCt of mock-infected samples for each individual gene.
2.5. Interferon Bioassay
The interferon bioassay was performed using a recombinant vesicular stomatitis virus (VSV) that expresses green fluorescent protein (VSV-GFP) as previously described [31, 39, 51]. Briefly, EECs were either infected with EAV or Sendai virus (SeV) alone or dually infected with both EAV and SeV at an m.o.i. of 1 and incubated for 24 h at 37°C. Culture supernatants were collected and virus in supernatant was inactivated by ultraviolet (UV) irradiation for 30 min. Two-fold dilutions of supernatants were made in DMEM and used in IFN bioassays. MDBK cells were grown in 96-well plates to 70% confluency and incubated with two-fold dilutions of each of the supernatants. After 24 h incubation at 37°C, cells were infected with VSV-GFP at an m.o.i. of 0.1 and further incubated for 18 h. Cells were fixed with 4% paraformaldehyde and expression of green fluorescence protein was examined under an inverted fluorescence microscope.
2.6. Cytotoxicity Test of EAV nsp1 on HEK293T Cells
HEK293T cells in 96-well plates were transfected with increased amount of plasmid expressing EAV nsp1 (0, 0.05, 0.1, 0.2, or 0.4 μg/well) using FuGENE HD (Promega, Madison, WI) transfection reagent (0.8 μL/well). At 24 h after transfection, cytotoxicity was determined by using a cell proliferation assay (CellTiter 96 Aqueous One Solution, Life Technologies, Grand Island, NY).
2.7. Luciferase Reporter Assay
HEK293T cells were seeded in 24-well plates and transfected with various combinations of plasmid DNAs: the pEFneo-RIG-I, pEFneo-MDA-5, pEFneo-IKKε, pcDNA3-TRIF, or pCAGGS-IRF-3 was mixed with a plasmid expressing an EAV protein (or empty pCAGGS vector), a luciferase reporter plasmid, and the pRL-SV40 plasmid. Transfection was performed using FuGENE HD (Promega, Madison, WI) transfection reagent following the manufacturer's instruction. For the SeV or IFN stimulation, HEK293T cells were transfected with a plasmid expressing an EAV protein (or empty pCAGGS vector), a reporter plasmid, and the pRL-SV40 plasmid. The plasmid pCAGGS-NS1 expressing swine influenza virus NS1 was used as positive control. At 20 h after transfection, cells were infected with SeV at 5000 HA unit/0.5 mL/well for 12–16 h or induced by treatment with 2000 IU/0.5 mL/well of IFN-α or IFN-β for 16 h. Cells were harvested at the indicated time points. Cell lysates were subjected to reporter gene assay using the dual luciferase reporter system (Promega, Madison, WI) according to manufacturer's instruction. Firefly and Renilla luciferase activities were measured in a luminometer (Berthold Technologies, Oak Ridge, TN). Values for each sample were normalized using the Renilla luciferase values. Relative luciferase (RLU) activity is defined as the ratio of firefly luciferase reporter activity to Renilla luciferase activity.
2.8. Immunofluorescence Assay
Cells were seeded on coverslips and grown to 80% confluency. DNA transfection was performed in HeLa cells using Lipofectamine 2000 (Invitrogen; Carlsbad, CA) according to the manufacturer's instructions. A transfection mix containing DNA and Lipofectamine 2000 in OPTI-MEM I (Invitrogen; Carlsbad, CA) was incubated at room temperature for 20 min and added to each well. After incubation, the transfection mix was replaced with fresh medium, and cells were incubated for 12 h to allow gene expression. For IRF-3 staining, cells were stimulated with 1 μg/mL of polyinosinic:polycytidylic [(poly(I:C)]; Sigma, St. Louis, MO) or untreated for 8 h. For NF-κB p65 staining, HeLa cells were treated with 20 ng/mL of TNF-α for 45 min or untreated. After incubation, cells were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 10 min at room temperature (RT), and then permeabilized using 0.1% Triton X-100 for 10 min at RT. After blocking with 1% bovine serum albumin (BSA) in PBS for 30 min, cells were incubated with primary antibody in PBS containing 1% BSA for 2 h followed by incubation with Alexa Fluor 488- and/or Alexa Fluor 594-conjugated secondary antibody for 1 h. Nuclear staining was performed with DAPI (4′,6-diamidino-2-phenylindole; Sigma, St. Louis, MO) for 3 min at RT. After washing with PBS, coverslips were mounted onto microscope slides using Fluoromount-G mounting medium (Southern Biotech, Birmingham, AL) and examined under the fluorescence microscope (Leitz Laborlux 12).
3. Results
3.1. Suppression of Type I Interferon Production by EAV
To investigate the effect of EAV on type I IFN production, IFN-β mRNA expression in infected cells was analyzed using qRT-PCR. Equine pulmonary endothelial cells (EECs) were infected with EAV VBS at an m.o.i. of 5 for 8 h, followed by infection with SeV for 3 or 6 hpi. SeV-alone-infected cells were used as positive control for type I IFN induction and mock-infected cells were used as negative control. As shown in Figure 1(a), IFN-β mRNA was barely detected in cells infected with EAV alone, whereas SeV infection induced a strong expression, approximately 190–200-fold increase of IFN-β mRNA expression level in comparison to that of mock-infected cells at 3 or 6 hpi. In contrast, SeV-induced IFN-β mRNA expression, at both 3 and 6 hpi, was significantly suppressed in cells previously infected with EAV. In parallel with the quantitative measurement of IFN-β mRNA, EAV-induced suppression of IFN production was confirmed by IFN bioassay using VSV-GFP, since VSV is IFN sensitive and presence of IFN-α/β blocks VSV replication. EECs were either infected with EAV or SeV alone or infected with both EAV and SeV. Mock-infected cells were used as negative control. Subsequently, MDBK cells were incubated with two-fold serial dilutions of virus-inactivated cell culture supernatant and then infected with VSV-GFP. The VSV infectivity was determined by monitoring the level of GFP expression. As shown in Figure 1(b), VSV-GFP replicated well in cell culture supernatant from mock-infected cells, while VSV replication was effectively inhibited in those MDBK cells that were preincubated with supernatant from SeV-infected EECs. No VSV-GFP replication was observed up to a dilution of 1 : 32 of the culture supernatant from SeV-infected cells. In contrast, VSV-GFP replication was not inhibited in MDBK cells that were preincubated with cell culture supernatant from EECs infected with EAV alone. Consistent with quantitative RT-PCR results, SeV-induced type I IFN production was significantly inhibited by EAV exposure since VSV-GFP replication was detected at a lower level compared to that from EECs only infected with SeV. Taken together, the data suggest that EAV has an ability to suppress the induction of IFN in infected cells.
Figure 1.
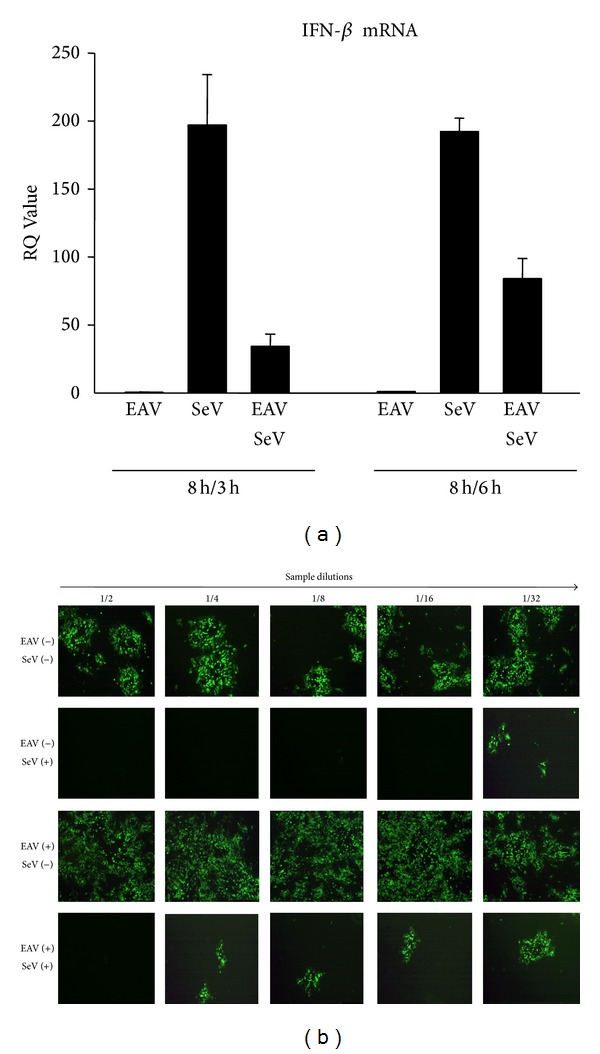
Inhibition of type I IFN production after EAV infection. (a) Expression levels of IFN-β mRNA in EAV infected cells. EECs were mock-infected or infected with EAV VBS at an m.o.i. of 5 for 8 h. Subsequently, cells were infected with Sendai virus (SeV; 100 HAU/mL) for 3 h or 6 h. Total RNA was isolated and real-time RT-PCR was performed for the detection of equine IFN-β. Bar graph showing relative quantitation (RQ) values of IFN-β mRNA expression from three independent experiments are shown. (b) VSV bioassay for IFN production. EECs were mock-infected or infected with EAV VBS at an m.o.i. of 1 for 24 h. SeV was used as an IFN stimulator. Cell culture supernatants were collected and UV-irradiated for 30 min prior to use in the assay. MDBK cells were grown in 96-well plates and incubated with 2-fold dilution series of the supernatant up to 1/32. After 24 h incubation, cells were infected with VSV-GFP at an m.o.i. of 0.1, and 18 h after infection GFP expression was assessed by fluorescence microscopy. Each dilution was tested in duplicate.
3.2. EAV nsp1, nsp2, and nsp11 Exhibited Strong Inhibition on IFN-β Promoter Activation
To investigate the role of EAV proteins as potential IFN antagonists, we focused on the nonstructural proteins of EAV. Each of the 12 nsp-encoding regions from the genome of EAV VBS was cloned individually into a mammalian expression vector, pCAGGS [40]. The expression of recombinant proteins in the plasmid DNA transfected cells was verified by immunofluorescence assay (Figure 2(a)). To determine whether these nsps have an effect on IFN-β activation, we used an IFN-β promoter-luciferase reporter assay. HEK293T cells were cotransfected with individual nsp-expressing plasmid and the luciferase reporter plasmid (p125-Luc) along with a Renilla luciferase expression plasmid (pRL-SV40) for normalizing purpose of sample expression levels. As a positive control, pCAGGS-NS1 plasmid expressing swine influenza virus NS1 (sw-ns1) gene was used to cotransfect the cells with the reporter plasmid, since sw-ns1 is a known IFN antagonist [52]. At 24 h after transfection, cells were infected with SeV to induce luciferase production. The IFN-β promoter-luciferase reporter assay result is presented in Figure 2(b). As we expected, the expression of sw-ns1 significantly inhibited IFN-β promoter-driven luciferase expression. In contrast, a strong reporter signal was observed in cells transfected with empty pCAGGS plasmid after infection with SeV. IFN-β promoter activation by SeV infection was suppressed to various degrees by expression of several EAV nsps, among which nsp1, nsp2, and nsp11 showed strong inhibition of IFN-β promoter-driven luciferase expression. In particular, nsp1 exhibited the strongest inhibitory effect, followed by nsp11 and nsp2. The results suggest that several EAV nsps are capable of suppressing IFN-β promoter activation.
Figure 2.
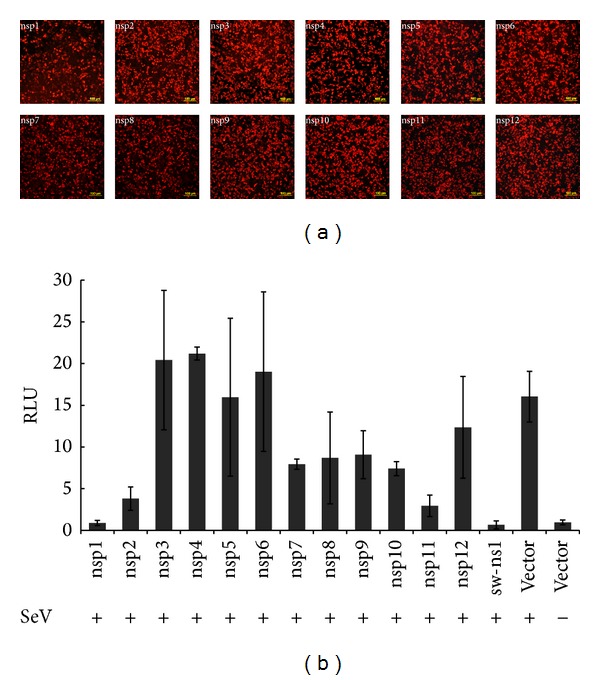
EAV nonstructural proteins (nsps) involved in suppression of IFN-promoter activation. (a) Expression of nsps in transfected cells detected by indirect immunofluorescence assay. HEK293T cells were transfected with each EAV nsp and fixed at 24 h after transfection. Cells were stained with EAV nsp1 MAb, EAV nonstructural protein-specific rabbit antisera (nsp2, nsp3, nsp4, nsp7-8, nsp9, nsp10, and nsp11), or anti-FLAG monoclonal antibody (nsp5, nsp6, and nsp12). DyLight 549-conjugated goat anti-rabbit antibody was used as secondary antibody. (b) HEK293T cells were cotransfected with p125-Luc, pRL-SV40, and pCAGGS expressing nsps or pCAGGS empty vector for 24 h. Cells were harvested and measured for firefly and Renilla luciferase activities. Relative luciferase (RLU) activity is defined as a ratio of firefly luciferase reporter activity to Renilla luciferase activity. Each data point shown represents a mean value ± standard error of the mean (SEM) from three experiments.
3.3. EAV nsp1 Interferes with IRF-3- and NF-κB-Mediated Signaling Pathways for IFN-β Production
Since the nsp1 of EAV showed the strongest inhibitory effect on the IFN-β promoter activation, we further determined the specific IFN production signaling pathway(s) associated with nsp1 expression. Toward this end, we tested nsp1 in IRF-3- and NF-κB-promoter-driven luciferase reporter systems. Cells were cotransfected with control plasmids or with plasmids expressing the EAV nsp1 protein, the plasmid pRL-SV40, and a luciferase reporter plasmid.
As shown in Figure 3(a), upon SeV stimulation, the level of IRF-3-dependent luciferase expression was significantly reduced in cells expressing EAV nsp1 and sw-ns1 compared to that in cells transfected with control plasmid (empty pCAGGS vector). Similarly, NF-κB promoter-dependent luciferase expression was suppressed in cells expressing EAV nsp1 (Figure 3(b)). These results suggest that EAV nsp1 suppresses IFN-β production by interfering with the IRF-3 and NF-κB signaling pathways. Furthermore, the viability of the HEK293T cells expressing EAV nsp1 24 h after transfection was compared to that of untransfected control cells. As shown in Figure 3(c), the cell viability appeared to be not affected by the nsp1 protein expression. These data further suggest that the decreased type I IFN production is not due to cellular cytotoxicity, which further confirmed the important role of nsp1 in the interfering with IRF-3 and NF-κB signaling pathways.
Figure 3.
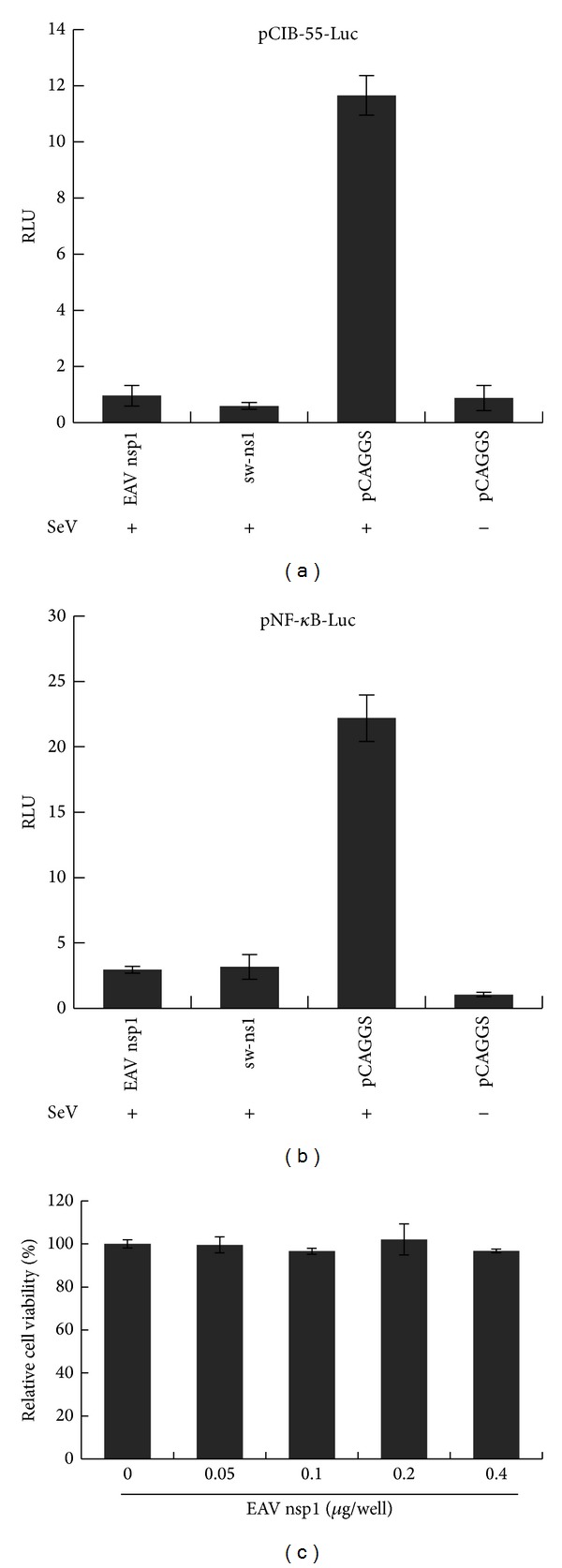
EAV nsp1 inhibits both IRF-3 and NF-κB pathways. HEK293T cells were cotransfected with reporter plasmid of (a) p55-CIB-Luc and (b) NF-κB-Luc and a plasmid that constitutively expresses Renilla luciferase and plasmids expressing EAV nsps or the indicated control plasmids. Cells were infected with Sendai virus (SeV) at 24 hours post transfection. Cells were harvested and analyzed for firefly and Renilla luciferase. Data were normalized using the Renilla luciferase values. Data are averages ± standard deviation for three experiments. (c) Cytotoxicity test of EAV nsp1 on HEK293T cells. Graph shows the results (mean and SD) of a representative quadruplicate experiment. Relative luciferase (RLU) activity is defined as a ratio of firefly luciferase reporter activity to Renilla luciferase activity.
3.4. Effect of EAV nsp1 on the IRF-3-Dependent Signaling Pathway
We further investigated specific steps in the IRF-3 signaling pathway that EAV nsp1 could possibly block. We tested each step in the signaling pathway of IRF-3 activation. First, we investigated whether the nsp1 was interfering with the mitochondrial antiviral signaling (MAVS) complex activity. Since MDA-5 or RIG-I is associated with the MAVS complex, cells were cotransfected with a plasmid expressing RIG-I, MDA-5, or MAVS protein, a plasmid expressing EAV nsp1, the plasmid pRL-SV40, and the p55-CIB-Luc reporter plasmid. As shown in Figures 4(a)–4(c), IRF-3 promoter-dependent luciferase expression was suppressed in the presence of EAV nsp1. These results suggested that EAV nsp1 might inhibit the MAVS-mediated IFN-β induction or downstream portion of the signaling pathway. Therefore, we further tested the effect of EAV nsp1 on TRIF- and IKKε-mediated IFN-β induction. The results showed that nsp1 had the ability to suppress TBK1- and IKKε-mediated reporter gene expression (Figures 4(d) and 4(e)). Similarly, overexpression of IRF-3 itself did not activate the transcription of the luciferase reporter gene either (Figure 4(f)). These results suggest that EAV nsp1 might block the signaling process downstream IRF-3 activation, possibly in the nucleus.
Figure 4.
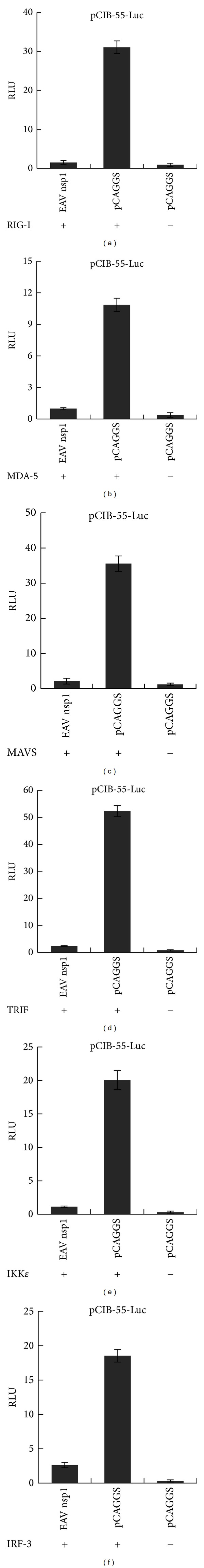
EAV nsp1 inhibits the IRF-3 signaling pathway. Suppression of (a) RIG-I, (b) MDA-5, (c) MAVS, (d) TRIF, (e) IKKε, and (f) IRF-3-induced IRF-3 dependent reporter gene expression by EAV nsp1 in HEK293T cells. (a)–(f) Cells cultured in 24-well plates were cotransfected with plasmid pEFneo-RIG-I, pEFneo-MDA-5, pEGFP-N1-MAVS, pcDNA3-TRIF, pEFneo-IKKε, or pCAGGS-IRF-3 along with a pCAGGS expressing nsp1 protein or pCAGGS empty vector, pRL-SV40, and a luciferase reporter plasmid p55-CIB-Luc for 24 h. Cells were harvested and measured for firefly and Renilla luciferase activities. Relative luciferase (RLU) activity is defined as a ratio of firefly luciferase reporter activity to Renilla luciferase activity. Mean value ± SEM from three experiments is shown.
3.5. Effect of EAV nsp1 on the NF-κB -Dependent Signaling Pathway
Since EAV nsp1 also inhibited the activation of the NF-κB-dependent signaling pathway (Figure 3), the mechanism by which EAV nsp1 can inhibit the NF-κB signaling pathway was analyzed in detail. As shown in Figure 5(a), in the presence of EAV nsp1, NF-κB-dependent reporter gene expression was strongly inhibited with the stimulation of the TNF-α, a potent inducer for the activation of NF-κB signaling pathway. Subsequently, the effect of EAV nsp1 on the MAVS complex and Toll/interleukin-1 receptor domain-containing adaptor protein (TRIF-) and IkB kinase beta (IKKβ-) mediated NF-κB activation was evaluated. Overexpression of any of these proteins induced activation of NF-κB-dependent reporter gene expression. However, when cells coexpressed the EAV nsp1 with one of these signaling molecules, the expression level of luciferase reporter was significantly reduced (Figures 5(b)–5(d)). Similarly, overexpression of p65, a subunit of the NF-κB complex, had no effect on the activation of the transcription of NF-κB-driven reporter gene, but EAV nsp1 significantly reduced the expression level of luciferase reporter signal (Figure 5(e)).
Figure 5.
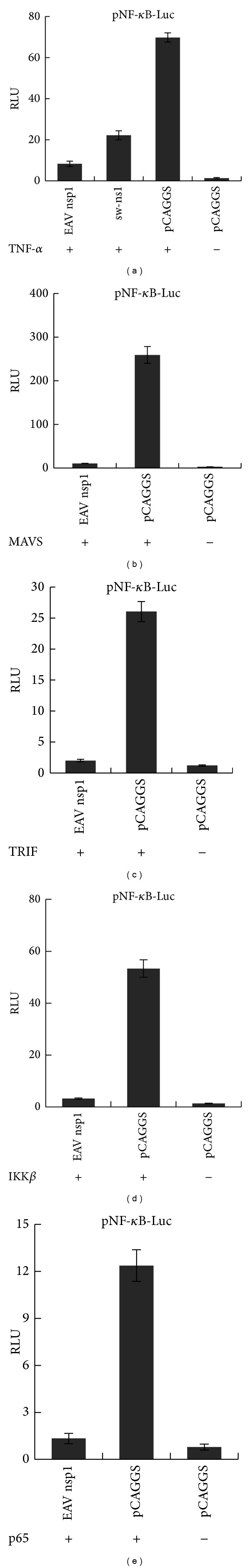
EAV nsp1 inhibits the NF-κB signaling pathway. Suppression of (a) TNF-α, (b) MAVS, (c) TRIF, (d) IKKβ, and (e) p65 subunit of NF-κB-induced NF-κB-dependent reporter gene expression by EAV nsp1 in HEK293T cells. (a) Cells cultured in 24-well plates were cotransfected with pNF-κB-Luc, pRL-SV40, and pCAGGS expressing nsp1 or pCAGGS empty vector for 24 h and subsequently stimulated with TNF-α (20 ng/mL) for 6 h. (b)–(e) Cells were cotransfected with plasmid pEGFP-N1-MAVS, pcDNA3-TRIF, pCMV2-IKK2-WT, or pEGFP-N1-p65 along with a pCAGGS expressing nsp1 protein or pCAGGS empty vector, pRL-SV40, and a luciferase reporter plasmid pNF-κB-Luc for 24 h. Cells were harvested and measured for firefly and Renilla luciferase activities. Relative luciferase (RLU) activity is defined as a ratio of firefly luciferase reporter activity to Renilla luciferase activity. Mean value ± SEM from three experiments is shown.
3.6. Effect of EAV nsp1 Expression on Nuclear Translocation of IRF-3 and NF-κB
The mechanism of EAV nsp1 effect on IRF-3- and NF-κB-dependent gene expression was further examined by observing the nuclear localization of IRF-3 and NF-κB, respectively. EAV nsp1 transfected HeLa cells were stimulated with poly(I:C), and subsequently expression of IRF-3 was stained with anti-IRF-3 antibody. In the absence of stimulation, IRF-3 was homogeneously distributed throughout the cell, whereas upon poly(I:C) stimulation IRF-3 was mainly translocated into the nucleus. As shown in Figure 6(a), expression of EAV nsp1 did not block the nuclear translocation of IRF-3. The result suggested that nsp1-mediated suppression of IFN production occurs downstream of the IRF-3 nuclear translocation. Similarly, the nuclear translocation of NF-κB p65 subunit was determined in cells expressing EAV nsp1 by immunofluorescence assay (Figure 6(b)). NF-κB remained largely not only in the cytoplasm but also in the nucleus to some extent in unstimulated cells, whereas TNF-α stimulation induced the nuclear translocation of p65. In cells expressing EAV nsp1, TNF-α stimulation did not notably change the p65 nuclear translocation, and p65 normally remained in the nucleus, indicating that EAV nsp1 did not block p65 nuclear translocation. These results suggest that, in the NF-κβ-dependent signaling pathway for IFN-β production, the activation of IFN-β transcription would be blocked by nsp1 somewhere downstream in the nucleus after NF-κB nuclear translocation occurred.
Figure 6.
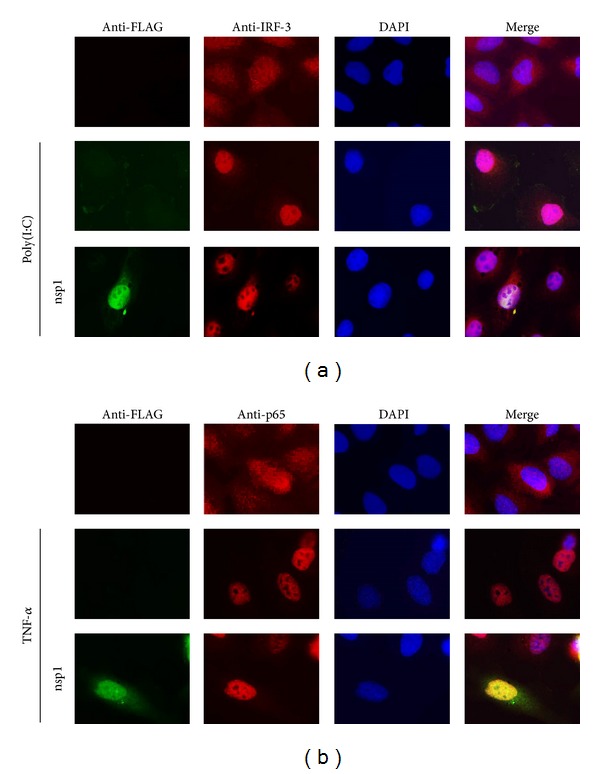
Effect of EAV nsp1 in IRF-3 and NF-κB nuclear translocation. (a) HeLa cells were transfected with FLAG-tagged EAV nsp1 for 12 h and cells were stimulated with poly(I:C) (1 μg/mL) for 8 h. Cells were fixed and stained with rabbit anti-IRF-3 Ab for endogenous IRF-3 and mouse anti-FLAG Ab for FLAG-tagged EAV nsp1, followed by incubation with Alexa Fluor 594-conjugated (red) and 488-conjugated (green) secondary antibodies, respectively, along with DAPI for nucleus staining (blue). (b) HeLa cells were transfected with FLAG-tagged EAV nsp1 for 12 h and subsequently treated with 20 ng/mL of TNF-α for 45 min or untreated. Cells were then costained with mouse anti-FLAG MAb for FLAG-tagged EAV nsp1 and rabbit anti p65 polyclonal antibody for endogenous NF-κB p65 subunit followed by Alexa Fluor 488-labeled (green) anti-mouse Ab and Alexa Fluor 594-labeled (red) anti-rabbit Ab, respectively, along with DAPI for nucleus staining (blue).
4. Discussion
Synthesis and secretion of type I IFNs, such as IFN-α and IFN-β, are critical aspects of the antiviral immune response [14, 53, 54]. Viruses use different mechanisms to inhibit interferon response in order to evade the host innate immune response. Many viruses encode more than one protein capable of inhibiting the interferon response, which act synergistically to ensure complete blocking of interferon activity. For example, Ebola, Nipah, and SARS-CoV encode multiple viral proteins capable of inhibiting interferon activity, suggesting important roles for these proteins in pathogenesis and disease outcome [55–58]. In this study, we investigated whether EAV has the ability to interfere with the host innate immune response, in particular, type I IFN production. Our results demonstrated that EAV infection in EECs significantly inhibited type I IFN production at both mRNA and protein levels, whereas infection with SeV stimulated a high level of type I IFN production. Furthermore, EAV infection significantly inhibited SeV-induced type I IFN production as well. Based on IFN-β promoter-luciferase reporter assay results, three EAV nonstructural proteins, nsp1, nsp2, and nsp11, were identified as potential IFN antagonists. Previous studies reported that EAV nsp2-encoded papain-like proteinase (PLP2) is capable of inhibiting Ub- and ISG15-dependent innate immune responses [34, 35, 59]. The EAV nsp11 encodes NendoU endoribonuclease [60], which is highly cytotoxic upon its in vitro expression. In PRRSV, the homologous nsp11 was proposed to be an IFN antagonist [26, 31, 61]. However, whether this effect is due to the cytosolic version of the enzyme targeting on the overall RNA population of the cell or the specific suppression of IFN production needs to be determined in the future. It could be possible that the three potential immune antagonists of EAV, nsp1, nsp2, and nsp11, target different parts of the host cellular immune system and their synergistic effect during the course of infection could be able to shut down the host cell innate immune response completely. This may explain why the induction of interferon and some immunomodulatory cytokines are inhibited during in vivo EAV infection [62].
Among the three potential IFN antagonists of EAV, expression of nsp1 had the strongest inhibitory effect on IFN-β promoter activation. In comparison to our findings, previous studies on PRRSV also demonstrated that the EAV nsp1 homologous, PRRSV nsp1α/β has the strongest ability to inhibit type I IFN synthesis [26, 31, 51, 63–67]. Further analysis revealed that EAV nsp1 inhibited two key signaling pathways for IFN-β activation, the IRF-3- and NF-κB-dependent signaling pathways. In our study, EAV nsp1 blocked each signaling step upstream of IRF-3 or NF-κB activation, suggesting that EAV nsp1 acts downstream of all those tested steps in both signaling pathways. Immunofluorescence microscopy analysis further showed that nsp1 did not have much effect on the nuclear accumulation of IRF-3 and NF-κB. Therefore, we postulated that EAV nsp1 might have an effect on the IFN-β promoter inside the nucleus. Previous studies have shown that PRRSV nsp1α/β modulates type I IFN response by blocking dsRNA-induced IRF-3 activation and IFN promoter activities, but IRF-3 phosphorylation and its nuclear translocation occur normally in the presence of the nsp1α/β [51]. Recent study reported that PRRSV nsp1α blocks the IRF-3 activation by degrading the CREB (cyclic AMP response element binding)-binding protein (CBP) and subsequently inhibiting formation of enhanceosomes in the nucleus [51, 63]. EAV nsp1 was reported to be largely nuclear located, similar to PRRSV nsp1β [68], suggesting that it may have an effect on the formation of the transcription enhanceosome on the IFN-β promoter inside the nucleus. However, PRRSV nsp1β does not degrade CBP or interrupt the formation of enhanceosome [68]. Thus, EAV nsp1 may have a unique function in modulating the IFN production in the nucleus. The EAV nsp1 does not contain the traditional nuclear localization signal [68], which suggests that nsp1 might be bound with cellular protein(s) and shuttled into the nucleus. Further studies are required to map the exact point(s) on the IFN induction pathway at which EAV nsp1 acts. It will be interesting to determine the requirement for nuclear localization of the EAV nsp1 that relates to its interferon antagonist function.
In summary, this study is the first report on molecular mechanisms of EAV involved in IFN antagonistic activity in its natural host cells. Our data indicate that several EAV replicase proteins, including nsp1, nsp2, and nsp11, possess IFN antagonistic activity and may have potential roles in the regulation of host innate immune responses. Among these proteins, nsp1 may play a key role as IFN antagonist. Further studies are needed to elucidate the detailed mechanisms that EAV proteins are involved in counteracting the host innate immune response.
Acknowledgments
This work was supported by the Agriculture and Food Research Initiative Competitive Grant nos. 2013-68004-20360, 2012-67015-21823, and 2013-767015-21243 (to Udeni Balasuriya, Ying Fang, and Dongwan Yoo, resp.) from the USDA-National Institute of Food and Agriculture. Yun Young Go was supported by a Geoffrey C. Hughes Foundation graduate fellowship from the Maxwell H. Gluck Equine Research Center during her graduate studies at the University of Kentucky.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Yun Young Go and Yanhua Li contributed equally to this work.
References
- 1.Balasuriya UB, Go YY, Maclachlan NJ. Equine arteritis virus. Veterinary Microbiology. 2013;167(1-2):93–122. doi: 10.1016/j.vetmic.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Timoney PJ, McCollum WH. Equine viral arteritis. The Veterinary Clinics of North America. 1993;9(2):295–309. doi: 10.1016/S0749-0739(17)30397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Archives of Virology. 1997;142:629–633. [PubMed] [Google Scholar]
- 4.Snijder EJ, Kikkert M, Fang Y. Arterivirus molecular biology and pathogenesis. Journal of General Virology. 2013;94:2141–2163. doi: 10.1099/vir.0.056341-0. [DOI] [PubMed] [Google Scholar]
- 5.Snijder EJ, Meulenberg JJM. The molecular biology of arteriviruses. Journal of General Virology. 1998;79(5):961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- 6.Firth AE, Zevenhoven-Dobbe JC, Wills NM, et al. Discovery of a small arterivirus gene that overlaps the gp5 coding sequence and is important for virus production. Journal of General Virology. 2011;92(5):1097–1106. doi: 10.1099/vir.0.029264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snijder EJ. Arteriviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, Pa, USA: Lippincott Williams and Wilkins; 2001. pp. 1205–1220. [Google Scholar]
- 8.den Boon JA, Snijder EJ, Chirnside ED, de Vries AAF, Horzinek MC, Spaan WJM. Equine arteritis virus is not a togavirus but belongs to the coronaviruslike superfamily. Journal of Virology. 1991;65(6):2910–2920. doi: 10.1128/jvi.65.6.2910-2920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Aken D, Zevenhoven-Dobbe J, Gorbalenya AE, Snijder EJ. Proteolytic maturation of replicase polyprotein pp1a by the nsp4 main proteinase is essential for equine arteritis virus replication and includes internal cleavage of nsp7. Journal of General Virology. 2006;87(12):3473–3482. doi: 10.1099/vir.0.82269-0. [DOI] [PubMed] [Google Scholar]
- 10.Ziebuhr J, Snijder EJ, Gorbalenya AE. Virus-encoded proteinases and proteolytic processing in the Nidovirales. Journal of General Virology. 2000;81(4):853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 11.de Vries AAF, Chirnside ED, Horzinek MC, Rottier PJM. Structural proteins of equine arteritis virus. Journal of Virology. 1992;66(11):6294–6303. doi: 10.1128/jvi.66.11.6294-6303.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snijder EJ, van Tol H, Pedersen KW, Raamsman MJB, de Vries AAF. Identification of a novel structural protein of arteriviruses. Journal of Virology. 1999;73(8):6335–6345. doi: 10.1128/jvi.73.8.6335-6345.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wieringa R, de Vries AAF, Raamsman MJB, Rottier PJM. Characterization of two new structural glycoproteins, GP3 and GP4, of equine arteritis virus. Journal of Virology. 2002;76(21):10829–10840. doi: 10.1128/JVI.76.21.10829-10840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. Journal of General Virology. 2008;89(1):1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 15.Luo R, Xiao S, Jiang Y, et al. Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-β production by interfering with the RIG-I signaling pathway. Molecular Immunology. 2008;45(10):2839–2846. doi: 10.1016/j.molimm.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thanos D, Maniatis T. Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell. 1995;83(7):1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 17.Buddaert W, van Reeth K, Pensaert M. In vivo and in vitro interferon (IFN) studies with the porcine reproductive and respiratory syndrome virus (PRRSV) Advances in Experimental Medicine and Biology. 1998;440:461–467. doi: 10.1007/978-1-4615-5331-1_59. [DOI] [PubMed] [Google Scholar]
- 18.Choi C, Cho W-S, Kim B, Chae C. Expression of interferon-gamma and tumour necrosis factor-alpha in pigs experimentally infected with porcine reproductive and respiratory syndrome virus (PRRSV) Journal of Comparative Pathology. 2002;127(2-3):106–113. doi: 10.1053/jcpa.2002.0566. [DOI] [PubMed] [Google Scholar]
- 19.Rowland RRR, Robinson B, Stefanick J, et al. Inhibition of porcine reproductive and respiratory syndrome virus by interferon-gamma and recovery of virus replication with 2-aminopurine. Archives of Virology. 2001;146(3):539–555. doi: 10.1007/s007050170161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan J, Wu W, Xia C. Expression of porcine interferon-gamma gene in Pichia pastoris and its effect of inhibiting porcine reproductive and respiratory syndrome virus. Sheng Wu Gong Cheng Xue Bao. 2002;18(6):683–686. [PubMed] [Google Scholar]
- 21.Chang H, Jeng C, Liu JJ, et al. Reduction of porcine reproductive and respiratory syndrome virus (PRRSV) infection in swine alveolar macrophages by porcine circovirus 2 (PCV2)-induced interferon-alpha. Veterinary Microbiology. 2005;108(3-4):167–177. doi: 10.1016/j.vetmic.2005.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wesley RD, Lager KM, Kehrli ME., Jr. Infection with Porcine reproductive and respiratory syndrome virus stimulates an early gamma interferon response in the serum of pigs. Canadian Journal of Veterinary Research. 2006;70(3):176–182. [PMC free article] [PubMed] [Google Scholar]
- 23.Chung H-K, Lee J-H, Kim S-H, Chae C. Expression of interferon-α and Mx1 protein in pigs acutely infected with porcine reproductive and respiratory syndrome virus (PRRSV) Journal of Comparative Pathology. 2004;130(4):299–305. doi: 10.1016/j.jcpa.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Meier WA, Galeota J, Osorio FA, Husmann RJ, Schnitzlein WM, Zuckermann FA. Gradual development of the interferon-γ response of swine to porcine reproductive and respiratory syndrome virus infection or vaccination. Virology. 2003;309(1):18–31. doi: 10.1016/s0042-6822(03)00009-6. [DOI] [PubMed] [Google Scholar]
- 25.Miller LC, Laegreid WW, Bono JL, Chitko-McKown CG, Fox JM. Interferon type I response in porcine reproductive and respiratory syndrome virus-infected MARC-145 cells. Archives of Virology. 2004;149(12):2453–2463. doi: 10.1007/s00705-004-0377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beura LK, Sarkar SN, Kwon B, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 1β modulates host innate immune response by antagonizing IRF3 activation. Journal of Virology. 2010;84(3):1574–1584. doi: 10.1128/JVI.01326-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beura LK, Subramaniam S, Vu HL, Kwon B, Pattnaik AK, Osorio FA. Identification of amino acid residues important for anti-IFN activity of porcine reproductive and respiratory syndrome virus non-structural protein 1. Virology. 2012;433:431–439. doi: 10.1016/j.virol.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang Y, Fang L, Wang Y, et al. Porcine reproductive and respiratory syndrome virus nonstructural protein 2 contributes to NF-kappaB activation. Virology Journal. 2012;9(article 83) doi: 10.1186/1743-422X-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung K, Renukaradhya GJ, Alekseev KP, Fang Y, Tang Y, Saif LJ. Porcine reproductive and respiratory syndrome virus modifies innate immunity and alters disease outcome in pigs subsequently infected with porcine respiratory coronavirus: implications for respiratory viral co-infections. Journal of General Virology. 2009;90(11):2713–2723. doi: 10.1099/vir.0.014001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Zhu L, Lawson SR, Fang Y. Targeted mutations in a highly conserved motif of the nsp1beta protein impair the interferon antagonizing activity of porcine reproductive and respiratory syndrome virus. Journal of General Virology. 2013;94:1972–1983. doi: 10.1099/vir.0.051748-0. [DOI] [PubMed] [Google Scholar]
- 31.Subramaniam S, Kwon B, Beura LK, Kuszynski CA, Pattnaik AK, Osorio FA. Porcine reproductive and respiratory syndrome virus non-structural protein 1 suppresses tumor necrosis factor-alpha promoter activation by inhibiting NF-κB and Sp1. Virology. 2010;406(2):270–279. doi: 10.1016/j.virol.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 32.Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. Journal of Virology. 2012;86:3839–3850. doi: 10.1128/JVI.06466-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 34.van Kasteren PB, Bailey-Elkin BA, James TW, et al. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E838–E847. doi: 10.1073/pnas.1218464110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Kasteren PB, Beugeling C, Ninaber DK, et al. Arterivirus and nairovirus ovarian tumor domain-containing deubiquitinases target activated RIG-I to control innate immune signaling. Journal of Virology. 2012;86(2):773–785. doi: 10.1128/JVI.06277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hedges JF, Demaula CD, Moore BD, Mclaughlin BE, Simon SI, James Maclachlan N. Characterization of equine E-selectin. Immunology. 2001;103(4):498–504. doi: 10.1046/j.1365-2567.2001.01262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore BD, Balasuriya UBR, Hedges JF, MacLachlan NJ. Growth characteristics of a highly virulent, a moderately virulent, and an avirulent strain of equine arteritis virus in primary equine endothelial cells are predictive of their virulence to horses. Virology. 2002;298(1):39–44. doi: 10.1006/viro.2002.1466. [DOI] [PubMed] [Google Scholar]
- 38.Sever JL. Application of a microtechnique to viral serological investigations. The Journal of Immunology. 1962;88:320–329. [PubMed] [Google Scholar]
- 39.Dalton KP, Rose JK. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology. 2001;279(2):414–421. doi: 10.1006/viro.2000.0736. [DOI] [PubMed] [Google Scholar]
- 40.Go YY, Snijder EJ, Timoney PJ, Balasuriya UBR. Characterization of equine humoral antibody response to the nonstructural proteins of equine arteritis virus. Clinical and Vaccine Immunology. 2011;18(2):268–279. doi: 10.1128/CVI.00444-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balasuriya UBR, Snijder EJ, Heidner HW, et al. Development and characterization of an infectious cDNA clone of the virulent Bucyrus strain of Equine arteritis virus. Journal of General Virology. 2007;88(3):918–924. doi: 10.1099/vir.0.82415-0. [DOI] [PubMed] [Google Scholar]
- 42.Yoneyama M, Suhara W, Fukuhara Y, Sato M, Ozato K, Fujita T. Autocrine amplification of type I interferon gene expression mediated by interferon stimulated gene factor 3 (ISGF3) The Journal of Biochemistry. 1996;120:160–169. doi: 10.1093/oxfordjournals.jbchem.a021379. [DOI] [PubMed] [Google Scholar]
- 43.Komatsu T, Takeuchi K, Gotoh B. Bovine parainfluenza virus type 3 accessory proteins that suppress beta interferon production. Microbes and Infection. 2007;9(8):954–962. doi: 10.1016/j.micinf.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 44.Overend C, Mitchell R, He D, Rompato G, Grubman MJ, Garmendia AE. Recombinant swine beta interferon protects swine alveolar macrophages and MARC-145 cells from infection with Porcine reproductive and respiratory syndrome virus. Journal of General Virology. 2007;88(3):925–931. doi: 10.1099/vir.0.82585-0. [DOI] [PubMed] [Google Scholar]
- 45.MacLachlan NJ, Balasuriya UB, Rossitto PV, Hullinger PA, Patton JF, Wilson WD. Fatal experimental equine arteritis virus infection of a pregnant mare: immunohistochemical staining of viral antigens. Journal of Veterinary Diagnostic Investigation. 1996;8:367–374. doi: 10.1177/104063879600800316. [DOI] [PubMed] [Google Scholar]
- 46.Wagner HM, Balasuriya UBR, MacLachlan NJ. The serologic response of horses to equine arteritis virus as determined by competitive enzyme-linked immunosorbent assays (c-ELISAs) to structural and non-structural viral proteins. Comparative Immunology, Microbiology and Infectious Diseases. 2003;26(4):251–260. doi: 10.1016/S0147-9571(02)00054-1. [DOI] [PubMed] [Google Scholar]
- 47.Snijder EJ, Wassenaar ALM, Spaan WJM. Proteolytic processing of the replicase ORF1a protein of equine arteritis virus. Journal of Virology. 1994;68(9):5755–5764. doi: 10.1128/jvi.68.9.5755-5764.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pedersen KW, van der Meer Y, Roos N, Snijder EJ. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. Journal of Virology. 1999;73(3):2016–2026. doi: 10.1128/jvi.73.3.2016-2026.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Dinten LC, Wassenaar AL, Gorbalenya AE, Spaan WJ, Snijder EJ. Processing of the equine arteritis virus replicase ORF1b protein: identification of cleavage products containing the putative viral polymerase and helicase domains. Journal of Virology. 1996;70:6625–6633. doi: 10.1128/jvi.70.10.6625-6633.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramakers C, Ruijter JM, Deprez RH, Moorman AFM. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neuroscience Letters. 2003;339(1):62–66. doi: 10.1016/s0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- 51.Kim O, Sun Y, Lai FW, Song C, Yoo D. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by non-structural protein 1 in MARC-145 and HeLa cells. Virology. 2010;402(2):315–326. doi: 10.1016/j.virol.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solórzano A, Webby RJ, Lager KM, Janke BH, García-Sastre A, Richt JA. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. Journal of Virology. 2005;79(12):7535–7543. doi: 10.1128/JVI.79.12.7535-7543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haller O, Weber F. Pathogenic viruses: smart manipulators of the interferon system. Current Topics in Microbiology and Immunology. 2007;316:315–334. doi: 10.1007/978-3-540-71329-6_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samuel CE. Antiviral actions of interferons. Clinical Microbiology Reviews. 2001;14(4):778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramanan P, Shabman RS, Brown CS, Amarasinghe GK, Basler CF, Leung DW. Filoviral immune evasion mechanisms. Viruses. 2011;3(9):1634–1649. doi: 10.3390/v3091634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Albina E, Carrat C, Charley B. Interferon-α response to swine arterivirus (PoAV), the porcine reproductive and respiratory syndrome virus. Journal of Interferon and Cytokine Research. 1998;18(7):485–490. doi: 10.1089/jir.1998.18.485. [DOI] [PubMed] [Google Scholar]
- 57.Basler CF. Nipah and hendra virus interactions with the innate immune system. Current Topics in Microbiology and Immunology. 2012;359:123–152. doi: 10.1007/82_2012_209. [DOI] [PubMed] [Google Scholar]
- 58.Totura AL, Baric RS. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Current Opinion in Virology. 2012;2:264–275. doi: 10.1016/j.coviro.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frias-Staheli N, Giannakopoulos NV, Kikkert M, et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host and Microbe. 2007;2(6):404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nedialkova DD, Ulferts R, van den Born E, et al. Biochemical characterization of arterivirus nonstructural protein 11 reveals the nidovirus-wide conservation of a replicative endoribonuclease. Journal of Virology. 2009;83(11):5671–5682. doi: 10.1128/JVI.00261-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shi X, Wang L, Li X, et al. Endoribonuclease activities of porcine reproductive and respiratory syndrome virus nsp11 was essential for nsp11 to inhibit IFN-β induction. Molecular Immunology. 2011;48(12-13):1568–1572. doi: 10.1016/j.molimm.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Go YY, Cook RF, Fulgêncio JQ, et al. Assessment of correlation between in vitro CD3+ T cell susceptibility to EAV infection and clinical outcome following experimental infection. Veterinary Microbiology. 2012;157(1-2):220–225. doi: 10.1016/j.vetmic.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 63.Han M, Du Y, Song C, Yoo D. Degradation of CREB-binding protein and modulation of type I interferon induction by the zinc finger motif of the porcine reproductive and respiratory syndrome virus nsp1alpha subunit. Virus Research. 2013;172:54–65. doi: 10.1016/j.virusres.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 64.Yoo D, Song C, Sun Y, Du Y, Kim O, Liu H. Modulation of host cell responses and evasion strategies for porcine reproductive and respiratory syndrome virus. Virus Research. 2010;154(1-2):48–60. doi: 10.1016/j.virusres.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shi X, Wang L, Zhi Y, et al. Porcine reproductive and respiratory syndrome virus (PRRSV) could be sensed by professional beta interferon-producing system and had mechanisms to inhibit this action in MARC-145 cells. Virus Research. 2010;153(1):151–156. doi: 10.1016/j.virusres.2010.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shi X, Zhang G, Wang L, et al. The nonstructural protein 1 papain-like cysteine protease was necessary for porcine reproductive and respiratory syndrome virus nonstructural protein 1 to inhibit interferon-β induction. DNA and Cell Biology. 2011;30(6):355–362. doi: 10.1089/dna.2010.1188. [DOI] [PubMed] [Google Scholar]
- 67.Song C, Krell P, Yoo D. Nonstructural protein 1α subunit-based inhibition of NF-κB activation and suppression of interferon-β production by porcine reproductive and respiratory syndrome virus. Virology. 2010;407(2):268–280. doi: 10.1016/j.virol.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 68.Tijms MA, van der Meer Y, Snijder EJ. Nuclear localization of non-structural protein 1 and nucleocapsid protein of equine arteritis virus. Journal of General Virology. 2002;83(4):795–800. doi: 10.1099/0022-1317-83-4-795. [DOI] [PubMed] [Google Scholar]