

Abstract
Background
Phytochemical-mediated alterations in P-glycoprotein (P-gp) activity may result in herb-drug interactions by altering drug pharmacokinetics. Shengmai-san, a traditional Chinese herbal medicine composed by Panax Ginseng, Ophiopogon Japonicus, and Schisandra Chinensis, is routinely being used for treating various coronary heart diseases. In our previous studies, Schisandra Lignans Extract (SLE) was proved as a strong P-gp inhibitor, and herein, the compatibility of Shengmai-san was studied by investigating the influence of SLE on the pharmacokinetics of the ginsenosides from the perspective of P-gp.
Methodology
Pharmacokinetic experiments were firstly performed based on in vitro uptake, efflux and transport experiments in Caco-2, LLC-PK1 wild-type and MDR1-overexpressing L-MDR1 cells. During the whole experiment, digoxin, a classical P-gp substrate, was used as a positive control drug to verify the cells used are the valid models. Meanwhile, the effects of SLE on the pharmacokinetics of ginsenosides were further investigated in rats after single-dose and multi-dose of SLE.
Results and Conclusions
The efflux ratios of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 were found more than 3.5 in L-MDR1 cells and can be decreased significantly by verapamil (a classical P-gp inhibitor). Contrarily, the efflux ratios of other ginsenosides (Rh1, F1, Re, and Rg1) were lower than 2.0 and not affected by verapamil. Then, the effects of SLE on the uptake and transport of ginsenosides were investigated, and SLE was found can significantly enhance the uptake and inhibit the efflux ratio of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 in Caco-2 and L-MDR1 cells. Besides, In vivo experiments showed that single-dose and multi-dose of SLE at 500 mg/kg could increase the area under the plasma concentration time curve of Rb2, Rc and Rd significantly without affecting terminal elimination half-time. In conclusion, SLE could enhance the exposure of ginsenosides Rb2, Rc, Rg2, Rg3, Rd and Rb1 significantly.
Introduction
Traditional Chinese medicine (TCM) has been utilized in the Chinese culture to treat the complex of symptoms for thousands of years [1], [2]. As the comprehensive effect of medical system, TCM advocates combinatory therapeutic strategies by prescriptions called ‘formulae’ which can hit multiple targets and produce a synergistic therapeutic effect based on their multi-targeting, multi-ingredient preparations, in contrast to modern pharmacology and drug development that often focus on a single chemical entity [3], [4]. In TCM theory, compatibility refers to the combined use of two or more herbal medicines based on the clinical efficacy and the properties of each herb, and the principles of herbal medicine in TCM can be divided into “monarch”, “minister”, “assistant” and “messenger” [5], [6]. Among them, the “monarch” drug directly aims at the main disease or principal syndrome, and “minister” drug has auxiliary therapeutic effect on the main or accompanying syndrome, as well as to aim at the accompanying diseases or symptoms. Besides, “assistant” drug is used to promote “minister” drug treat the chief syndromes / disease, or to aim at minor symptoms, and the “messenger” drug mainly used to modulate the interactions among the herbs [7], [8]. TCM compatibility has shown its great significance in long-term clinical practices, and revealing the compatibility mechanism of TCM is a key issue to its modernization, while the research has faced many obstacles due to the unimaginable complexity of multi-herb formulae [9], [10]. In fact, pharmacokinetic interactions among the bioactive constituents of herbal formulae can supply important clues to the compatibility research for TCM. In recently years, there was an increasing focus on the compatibility study for TCM based on pharmacokinetic interaction among herbs [11]–[13].
Shengmai-san, composed by Panax Ginseng, Ophiopogon Japonicus, and Schisandra Chinensis, has been applied for cardiovascular diseases routinely and prophylactically for thousands of years in China as a well-known traditional Chinese herbal prescription [14]–[16]. Among them, Panax Ginseng was monarch drug, Ophiopogon Japonicus was minister drug, and Schisandra Chinensis acted as the role of assistant and messenger drug. According to previous reports, several kinds of Schisandra lignans were found can alter pharmacokinetics of substrates of P-gp [17]–[19]. In recent years, several publications have already discussed the relationship between ginsenosides and P-gp [20]–[25], and most of the studies are carried based on the single component of Ginsenosides Extract. However, the curative effects of traditional Chinese medicines are principally based on the synergic effect of their multi-targeting, multi-ingredient preparations, in contrast to modern pharmacology and drug development that often focus on a single chemical entity. P-gp, a member of the ATP binding cassette, plays an important role in the exposure and disposition for many xenobiotics through its action as an efflux pump [26]. Thus, phytochemical-mediated alterations in P-gp activity may result in herb-drug interaction by altering drug pharmacokinetics [27]. In 2013, SLE was proved as a strong P-gp inhibitor, and could significantly down-regulate of the P-gp expression and inhibit P-gp activity by our research group [28]. Schisandra Chinensis, an assistant and messenger drug in Shengmai-san, was used to improve the pharmacological activity of monarch drug (Panax Ginseng) and ministerial drug (Ophiopogon Japonicus). However, the mechanism of improving the pharmacological activity produced by Schisandra Chinensis in Shengmai-san was not clear until now. Herein, the compatibility of Shengmai-san was studied by investigating the influence of Schisandra lignans on the pharmacokinetics of the ginsenosides from the perspective of P-gp.
In this study, several kinds of ginsenosides were systematically proved as P-gp substrates through investigating the effects of P-gp inhibitor (verapamil) on ginsenosides transport and uptake on in Caco-2, LLC-PK1 wild-type and MDR1-overexpressing L-MDR1 cells. Digoxin, a classical P-gp substrate, was used as a positive control drug to verify the cells used are the valid models. Firstly, ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 were confirmed as P-gp substrates. Then, the effects of SLE on the pharmacokinetics of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 were thoroughly investigated based on in vitro and in vivo models, and SLE was found can greatly enhance the exposure of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 in cells and rats. According to the results above, the profound mystery of compatibility principles for Shengmai-san may have been revealed to some extent.
Materials and Methods
Animals
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the China Pharmaceutical University. All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering. Female healthy Sprague-Dawley rats (200±20 g) were purchased from the Laboratory Animal Center of Peking University Health Science Center (Beijing, PR China) and kept in an environmentally controlled breeding room for at least 3 days before experimentation. The rats were fed with standard laboratory food and water and fasted overnight but with free access to water before the test.
Materials
SLE was purchased from Nanjing Qingze Medical Technological Development Co. Ltd (Nanjing, China). Ginsenosides Extract (GE) was purchased from Jilin University (Jilin, China). Digoxin and verapamil were purchased from Sigma-Aldrich (St. Louis, MO). Ginsenoside Rh1, F1, Rb2, Rc, Rg2, Rg3, Re, Rd, Rb1 and Rg1 were purchased from Jilin University (Jilin, China), and their structures are shown in Figure S1. HPLC-grade acetonitrile and methanol were purchased from Merck (Merck, Germany). Deionized water was prepared by the Milli-Q system (Millipore Corporation, Billerica, MA) and was used throughout. Other reagents and solvents were commercially available and of analytical grade.
Methods
LLC-PK1 and L-MDR1 cell culture
LLC-PK1 cells, a cell line lacking P-gp, and the derived cell line L-MDR1, which over-express human MDR1-encoded P-gp [29], were grown in minimum essential medium with supplements at 37°C, 5% CO2, and 95% humidity. LLC-PK1 and L-MDR1 cells were seeded at a density of 12×106 cells per 150-cm2 flask and grown for 7 days in medium 199 supplemented with 2 mM glutamine, 10% fetal bovine serum, 10 mg/500 mL of streptomycin, and 10,000 IU/500 mL of penicillin. To ensure a constant expression level of transport protein, L-MDR1 cells were grown in the presence of 640 nM vincristine. Cells were grown as monolayer on polycarbonate membrane filters (Transwell; Costar Corporation, Cambridge, MA) as outlined previously [30]. Transepithelial resistance was measured in each well using a Millicell ERS ohmmeter (Millipore, Bedford, MA); wells registering a resistance of 200 ohms or greater, after correcting for the resistance obtained in control blank wells, were used in the transport experiments.
Inhibiting transport of ginsenosides by verapamil and SLE in cultured LLC-PK1 and L-MDR1 cells
The LLC-PK1 and L-MDR1 cells were washed with warm Hank's balanced salt solution HBSS (pH 7.4) twice before transport experiment. HBSS containing verapamil (10 µM), SLE (2.0 and 10.0 µg/mL) or blank HBSS (control) was then loaded into both apical and basolateral chambers. After incubation at 37°C for 1.5 h, GE (100 µg/mL) was added to either the apical or basolateral side to evaluate the transport in absorptive and secretive directions, respectively. The cell monolayer was then incubated for another 2 h. Finally, the concentrations of ginsenosides were determined by LC/MS. All experiments were conducted in triplicate.
Enhancing uptake of ginsenosides by verapamil and SLE in cultured LLC-PK1 and L-MDR1 cells
LLC-PK1 and L-MDR1 cells were incubated with SLE (2.0 and 10.0 µg/mL) or verapamil (10 µM) for 1.0 h, and then were washed to discard SLE or verapamil before adding 100 µg/mL of GE. The accumulation of GE lasted for 2 h, and the intracellular concentrations of ginsenosides were measured by LC/MS. All experiments were conducted in triplicate.
Caco-2 cell culture
Caco-2 cells were purchased from American Type Culture Collection (Manassas, VA), and were routinely cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% nonessential amino acids, 1 mM sodium pyruvate, and 100 U/mL penicillin and streptomycin (Invitrogen, Carlsbad, CA). The cells were grown in an atmosphere of 5% CO2 at 37°C, and cell medium was changed every other day. Cells were seeded at a density of 4×104 cells per well in 24-well microporous polycarbonate insert filter plates (0.4-µm pore size) (Corning Costar plates; Corning Life Sciences, Lowell, MA) and grown for 15 days in Caco-2 growth medium. The medium was replaced every 3 to 4 days. Before running the assay, the culture medium was replaced by HBSS buffer supplemented with 10 mM Hepes and 20 mM glucose.
Inhibiting transport of ginsenosides by verapamil and SLE in Caco-2 cell monolayer
Caco-2 cells with a density of 1.2×105 cells/insert were seeded on a permeable polycarbonate insert (Millicell cell culture inserts, 0.4 µm pore size, 12 mm diameter; Millipore Corporation) in 24-well tissue culture plates and were used for the experiment 18 to 21 days after seeding. [14C]Mannitol permeability and transepithelial electrical resistance measurements (Millicell-ERS epithelial volt-ohm meter; Millipore Corporation) were used to evaluate the integrity of Caco-2 cell monolayer. The monolayer used in transport studies had transepithelial electrical resistance values exceeding 600 Ω·cm2, and the leakage of mannitol was less than 0.3% of the dose/h. Before transport initiation studies, the cell monolayer was first washed with warm HBSS (pH 7.4) twice. HBSS containing SLE (2.0 and 10.0 µg/mL) or 1% ethanol (control) was then loaded into both apical and basolateral chambers. After incubation at 37°C for 1.5 h, 100 µg/mL of GE was added to either the apical or basolateral side to evaluate the transport in absorptive and secretory directions, and the cell monolayer was incubated for another 2 h. 10 µM verapamil was used as a positive control inhibitor. At the designated time point, samples were taken from the receiving chamber for analysis. Ginsenosides were determined by LC-MS. All experiments were conducted in triplicate.
Enhancing uptake of ginsenosides by verapamil and SLE in Caco-2 cell
Caco-2 cells were incubated with SLE (2.0 and 10.0 µg/mL) or verapamil (10 µM) for 1.0 h. Then the cells were washed with Hank's to discard SLE or verapamil before the addition of GE (100 µg/mL). The accumulation of ginsenosides lasted for 2 h, and the intracellular concentrations of ginsenosides were measured by LC/MS. All experiments were conducted in triplicate.
In vivo pharmacokinetic studies
To evaluate the effect of single treatment of SLE on the pharmacokinetics of ginsenosides, 24 rats were divided into four groups (group I ∼ group IV) averagely. In group I: the rats were administrated a single dose of SLE at 500 mg/kg by gavage. 2 h later, digoxin (0.5 mg/kg), a P-gp substrate, was given to the rats by intragastric administration. In group II (control group): the rats received a single dose of vehicle (0.5% CMC-Na), and digoxin (0.5 mg/kg) was also given to the rats by intragastric administration. In group III: the rats received a single dose of SLE at 500 mg/kg suspended in 0.5% CMC-Na. 2 h later, GE was administrated to the rats at a dose of 120 mg/kg by gavage. In group IV (control group): the rats received a single dose of vehicle (0.5% CMC-Na), and GE (120 mg/kg) was also given to the rats by gavage.
To evaluate the effect of long-term treatment with SLE on the pharmacokinetics of ginsenosides, 24 rats were also divided into four groups (group I∼ group IV) with 6 animals each. In the group I and group III, SLE was administrated intragastrically to rats at a dose of 500 mg/kg once a day for 10 continuous days, whereas the other two groups (group II and group IV) received the vehicle (0.5% CMC-Na), serving as the control groups. On the 11th day, digoxin was administered intragastrically to rats (group I and group II) at a single dose of 0.5 mg/kg, GE was administered intragastrically to rats (group III and group IV) at a single dose of 120 mg/kg.
In the experiments above, 0.2 mL blood samples were collected at 5, 10, 20, 40, 60, 120, 240, 360, 480 and 720 min after digoxin dosing. After intragastric administration of GE, 0.2 mL blood samples were collected at 5, 10, 20, 40, 60 min, 2, 4, 6, 8, 12, 24, 48, 72 and 96 h. Plasma was obtained by centrifugation at 5000 g for 10 min and stored at −20°C before analysis.
Analysis of ginsenosides in cell Lysates, buffer Samples and rat plasma
The primary stock solutions of 10 ginsenosides were prepared by dissolving 10.0 mg of corresponding standards in 10 mL acetonitrile, and were stored at 4°C until analysis. Working solutions of the analytes were prepared by appropriate dilution of the primary stock solutions in acetonitrile. Then the stock solutions were diluted with blank rat plasma to make the calibration standards at the concentrations of 0.001, 0.002, 0.005, 0.01, 0.02, 0.05, 0.10, 0.20, 0.50, 1.00, 2.00, 5.00 and 10.00 µg/mL for all the ginsenosides.
The plasma / cell samples were extracted using a liquid–liquid extraction technique. To each tube containing 100 µL plasma, 10 µL internal standard (digoxin, 1.0 µg/mL) and 1.0 mL of n-butanol were added. The mixture was then vortex-extracted for 2 min, and centrifuged for 10 min at 10,000 g. The n-butanol extract was evaporated to dryness in a rotary evaporator and the residue was reconstituted in 200 µL acetonitrile. 5 µL aliquot was analyzed by LC/MS.
LC experiments were conducted on a Shimadzu (Kyoto, Japan) HPLC system. Chromatographic separation was achieved on a Phenomenex C18 column (150 mm×2.0 mm, 5 µm, Lunar) at 40°C. Water mobile phase (solvent A) was H2O containing 0.1 mM ammonium chloride; and the organic phase (solvent B) was acetonitrile. A 30-minute binary gradient elution (delivered at 0.2 mL/min) was performed for the separation: an isocratic elution of 30 % solvent B for the initial 1.0 min, followed by a linear gradient elution of 30–75% solvent B from 1.0 to 23 min; after holding the composition of 75 % solvent B for the next 2 min, the column was returned to its starting conditions till the end of the gradient program at 30 minute for column equilibration.
The ESI source was used in negative ionization mode. The [M+CL]− ions of ginsenosides (Re, Rg1, Rb1, Rb2, Rg2, Rc, Rh1, F1, Rd, and Rg3 ) and internal standard (digoxin, m/z 815.55) were used for SIM detection, the chromatography of ginsenosides are shown in Figure S2. Peak areas were used for quantification. The optimum MS operating conditions were as follows: drying gas 2.5 L min−1, curved desolvent line temperature 250°C, block temperature 200°C, detector voltage 1.65 kV, peak width 0.7 FWHM, interval time 0.2 s.
Data Analysis
For the transport assay, the apparent permeability (P
app, cm/s) in each direction (A→B and B→A) was calculated as follows, where A is the surface area of the membrane inserts (0.0804 cm2), C
0 is the initial concentration of the compound applied in the donor compartment (2 µM), and △Q is the amount (micromoles) of compound transported over time △t (5 h = 18,000 s) [31]: 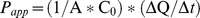
The efflux ratio (ER) was a dimensionless number calculated as the ratio of the apparent permeability in the B→A direction divided by the apparent permeability in the A→B direction.
Calculation of pharmacokinetic parameters
The plasma concentration-time curves of ginsenosides in rats were obtained by plotting the mean plasma concentrations versus time. Pharmacokinetic parameters were calculated using the WinNonlin 6.3 program (Pharsight Inc., Mountain View, CA). The elimination half-life (t 1/2) value was calculated as 0.693/β, where β is the elimination rate constant calculated from the terminal linear portion of the log plasma concentration-time curve. The total areas under the plasma concentration- time curve from time 0 to the last quantifiable time point (AUC0-t) and from time 0 to infinity (AUC0-∞) were calculated using the log trapezoidal rule.
The maximum plasma concentrations (C max) for ginsenosides were obtained by visual inspection of the plasma concentration-time curve, whereas the initial drug concentration (the extrapolated concentration at zero time) of the drug after intravenous injection was calculated by back-extrapolation of the plasma concentration-time curve to the y-axis.
Systematic workflow
The systematic workflow is schematically depicted in Figure 1. In step 1, ginsenosides were identified as P-gp substrates through comparing the effects of P-gp inhibitor on ginsenosides transport and uptake in LLC-PK1 wild-type and MDR1-overexpressing L-MDR1 cells. Besides, the effects of P-gp inhibitor on transport and uptake in Caco-2 cell were also investigated to further confirm several kinds of ginsenosides were P-gp substrates. In step 2, the effects of SLE on the transport and uptake of ginsenosides were inspected in Caco-2, LLC-PK1 and L-MDR1 cells since SLE were reported as a strong P-gp inhibitor, and in step 3, the effects of SLE on the pharmacokinetics of ginsenosides were investigated in vivo through comparing the pharmacokinetics of ginsenosides in rats after single/ multiple dose of SLE with that in the control rats which received the vehicle except GE.
Figure 1. The systematic workflow of this study.

Results
The effects of P-gp inhibitor on ginsenosides transport and uptake
In order to identify whether ginsenosides are P-gp substrates or not, we compared the effect of a classical P-gp inhibitor (verapamil) on the transport / uptake abilities of ginsenosides in LLC-PK1 cells and the derived cell line L-MDR1. Besides, the transport and uptake of ginsenosides were also investigated on Caco-2 cell in the absence and presence of verapamil to further confirm which ginsenosides were P-gp subtrates. In order to verify the reliability of the cell models, digoxin was used as a positive control drug during the whole course of the experiment.
The effects of P-gp inhibitor (verapamil) on Ginsenosides transport in cultured LLC-PK1 and L-MDR1 cells
The effect of verapamil (a classical P-gp inhibitor) on the transport ability of ginsenosides has been investigated in LLC-PK1 cells and the derived cell line L-MDR1. Figure 2(A) illustrates the ERs of digoxin (positive control drug), ginsenoside Rh1, F1, Rb2, Rc, Rg2, Rg3, Re, Rd, Rb1 and Rg1 on LLC-PK1 cells (lacking P-gp), in the absence and presence of 10 µM verapamil as a P-gp inhibitor. No transport difference was observed when verapamil was added, and the efflux level of digoxin and ginsenosides across LLC-PK1 cell monolayer in the absorptive (AP→BL) was similar to the secretive (BL→AP) directions. It was mean that the ER of ginsenosides across LLC-PK1 cell monolayer and the corresponding P app values were not affected by verapamil. On the other hand, we also compared the transport of digoxin, ginsenoside Rh1, F1, Rb2, Rc, Rg2, Rg3, Re, Rd, Rb1 and Rg1 on L-MDR1 cells (LLC-PK1 cells stably transfected with the human MDR1 gene), in the absence and presence of 10 µM verapamil as P-gp inhibitor (shown in Figure 2(B)). Obviously, the ERs of digoxin, ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 on L-MDR1 cells were 26.23, 5.14, 4.12, 4.20, 4.71, 8.22 and 6.69, respectively (far greater than 2.0). Moreover, the addition of verapamil caused a significantly decrease of ER through increasing the transport of digoxin, ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 in the AP→BL direction, and decreasing their transport across L-MDR1 cell monolayers in the BL→AP direction. For the other ginsenosides (Rh1, F1, Re and Rg1), the ERs across L-MDR1 cells were lower than 2.0, and the corresponding P app values were almost not affected by verapamil (P>0.05).
Figure 2. The effect of verapamil on the efflux ratio of digoxin, ginsenoside Rh1, F1, Rb2, Rc, Rg2, Rg3, Re, Rd, Rb1 and Rg1 on LLC-PK1 and L-MDR1 cell.

(A) The efflux ratio of digoxin and ginsenosides on LLC-PK1 cells in the absence and presence of verapamil. (B) The efflux ratio of digoxin and ginsenosides on L-MDR1cells in the absence and presence of verapamil.
The effects of verapamil on the uptake of ginsenosides in cultured LLC-PK1 and L-MDR1 cells
Herein, we examined the effect of verapamil on the uptake of ginsenosides on LLC-PK1 cells and the derived cell line L-MDR1. As shown in Figure 3(A), intracellular accumulation of digoxin, ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 on LLC-PK1 cells had no significant difference in the absence and presence of verapamil. In contrast, the intracellular concentrations of digoxin, ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 on L-MDR1 cells were greatly increased in the presence of P-gp inhibitor verapamil (Figure 3(B)), and the uptake levels of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 were enhanced by 3.2-, 1.7-, 1.5-, 1.6-, 1.5-, 1.8 and 2.6- fold in the presence of 10 µM verapamil, respectively (P<0.05).
Figure 3. The effect of verapamil on the uptake of ginsenosides in LLC-PK1 and L-MDR1 cell and the effect of verapamil on the uptake and transport of digoxin and ginsenosides in Caco-2 cell.

(A) The concentration of digoxin and ginsenosides in LLC-PK1 cell in the absence and presence of verapamil. (B) The concentration of digoxin and ginsenosides in L-MDR1 cell in the absence and presence of verapamil. (C) The concentration of digoxin and ginsenosides in Caco-2 cell in the absence and presence of verapamil. (D) The efflux ratio of digoxin and ginsenosides on Caco-2 cells in the absence and presence of verapamil.
The effects of verapamil on the transport and uptake of ginsenosides in Caco-2 cell
Caco-2 cells, a human colon adenocarcinoma cell line, had demonstrated numerous morphological and biochemical characteristics of enterocytes with a well-developed brush border and associated enzymes and transporters [32]. Herein, we studied the effect of verapamil on the transport ability of ginsenosides in Caco-2 cell. Figure 3(C) illustrates the ERs of digoxin, ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1in Caco-2 cells, in the absence and presence of 10 µM verapamil as P-gp inhibitor. Ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 exhibited highly polarized transport across Caco-2 cell monolayers with marked efflux ratio value at 3.23, 4.42, 4.36, 4.26, 3.85, 4.47 and 3.77, respectively, which were far greater than 2.0, and their high efflux ratios were decreased significantly in the presence of 10 µM verapamil. In addition, the effect of verapamil on the uptake of ginsenosides had been investigated, and the results are shown in Figure 3(D). The uptake of digoxin, ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 was found can be significantly enhanced by verapamil.
In summary, the results above indicated that the ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 were moderate P-gp substrates, and ginsenosides Rh1, F1, Re, and Rg1 almost could not be transported by P-gp.
The effects of SLE on ginsenosides transport and uptake
According to our previous study [22], SLE was a strong P-gp inhibitor, and could significantly decrease the ERs of P-gp substrates (digoxin and vincrisine) in L-MDR1 and Caco-2 cells. Herein, the effects of SLE on the transport and uptake of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 would be investigated since these ginsenosides were been proven as moderate P-gp substrates based on the experiments above.
Inhibition of ginsenosides transport by SLE in cultured LLC-PK1 and L-MDR1 cells
Herein, we investigated the effect of SLE on the transport of ginsenosides to explore the compatibility rule between ginsenosides and SLE in Shengmai-san based on P-gp. Figure 4(A) illustrates the absorptive (AP→BL) and secretory (BL→AP) transport of ginsenoside Rb2, Rc, Rg2, Rg3, Rd, Rb1 and digoxin on LLC-PK1 cells (lacking P-gp), in the absence and presence of SLE (2.0 and 10.0 µg/mL) as P-gp inhibitor. No transport difference was observed when SLE was added. It was mean that the efflux of ginsenoside Rb2, Rc, Rg2, Rg3, Rd, Rb1 and digoxin across LLC-PK1 cell monolayer in the absorptive (AP→BL) and secretory (BL→AP) directions and the corresponding P app values were not affected by SLE. On the other hand, we also compared the transport of ginsenoside Rb2, Rc, Rg2, Rg3, Rd, Rb1 and digoxin on L-MDR1 cells, in the absence and presence of SLE (2.0 and 10.0 µg/mL) as P-gp inhibitor (shown in Figure 4(B)). Obviously, the ERs of ginsenoside Rb2, Rc, Rg2, Rg3, Rd, Rb1 and digoxin could be significantly decreased by SLE.
Figure 4. The effect of SLE on the uptake and transport of digoxin and ginsenosides in LLC-PK1 and L-MDR1 cell.

(A) The transport of digoxin and ginsenosides on LLC-PK1 cell in the absence and presence of SLE at 2.0 µg/mL (L) and 10.0 µg/mL (H). (B) The transport of digoxin and ginsenosides on L-MDR1 cell in the absence and presence of SLE at 2.0 µg/mL (L) and 10.0 µg/mL (H). (C) The concentration of digoxin and ginsenosides in LLC-PK1 cell in the absence and presence of SLE at 2.0 µg/mL (L) and 10.0 µg/mL (H). (D) The concentration of digoxin and ginsenosides in L-MDR1 cell in the absence and presence of SLE at 2.0 µg/mL (L) and 10.0 µg/mL (H).
The Effect of SLE on the uptake of ginsenosides in cultured LLC-PK1 and L-MDR1 cells
Herein, we examined the effect of SLE on the uptake of ginsenosides on LLC-PK1 cells and the derived cell line L-MDR1. As shown in Figure 4(C), intracellular accumulation of ginsenoside Rb2, Rc, Rg2, Rg3, Rd, Rb1 and digoxin on LLC-PK1 cells had no significant difference in the absence and presence of SLE. In contrast, SLE concentration dependently increased intracellular concentrations of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 in L-MDR1 cells. When 2.0 µg/mL SLE was added, the intracellular concentrations of ginsenoside Rb2, Rc, Rg2, Rg3, Rd, Rb1 and digoxin in L-MDR1 cells were increased by 2.0-, 1.9-, 1.7-, 2.5-, 2.9, 2.9 and 2.7- fold. The intracellular concentrations of ginsenoside Rb2, Rc, Rg2, Rg3, Rd, Rb1 and digoxin in L-MDR1 cells were increased by 3.7-, 3.5-, 2.1-, 2.9-, 4.3, 5.1-, and 3.6- fold with the presence of 10.0 µg/mL SLE (Figure 4(D)).
Inhibition of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 transport across Caco-2 monolayer by SLE
The ability of SLE to affect the transport of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 was further confirmed in Caco-2 cells. Ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 exhibited highly polarized transport across Caco-2 cell monolayer with marked efflux ratio values at 4.42, 4.36, 4.26, 3.85, 4.47 and 3.77, respectively, which were far greater than 2.0, and the presence of SLE could concentration dependently decrease the transport capability of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 across Caco-2 monolayer in the BL-AP direction, which correspondingly led to the decrease in efflux ratios (As shown in Figure 5(A)).
Figure 5. The effect of SLE on the transport and uptake of digoxin and ginsenosides in Caco-2 cell.

(A) The transport of digoxin and ginsenosides on Caco-2 cell in the absence and presence of SLE at 2.0 µg/mL (L) and 10.0 µg/mL (H). (B) The uptake of digoxin and ginsenosides in Caco-2 cell in the absence and presence of SLE at 2.0 µg/mL (L) and 10.0 µg/mL (H).
Enhancing uptake of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 by SLE in Caco-2 cell
The effect of SLE on the uptake of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 had been investigated and the results are shown in Figure 5(B)). SLE could concentration dependently increase intracellular concentrations of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 in Caco-2 cells when 2.0 and 10.0 µg/mL of SLE were loaded (P<0.01).
Single-dose SLE increased plasma concentrations of ginsenosides in rats
In this process, the effect of single-dose SLE on the plasma pharmacokinetics of ginsenosides in rats was investigated. The plasma concentration of ginsenosides after intragastric administration of GE at the dose of 150 mg/kg or in combination with SLE was determined by LC/MS. As a result, ginsenoside Rb1, Rb2, Rc and Rd were the main components in rat plasma, and the exposure of ginsenoside Rb1, Rb2, Rc and Rd was much higher than other components. The plasma concentration-time profiles of ginsenoside Rb1, Rb2, Rc and Rd are shown in Figure 6 and their pharmacokinetic parameters are shown in Table 1. Obviously, coadministration of SLE could evidently increase the AUC0-∞ of ginsenoside Rb2 for 2.18-fold, from 10551.63±1728.81 ng.h/mL in the vehicle treated group to 23198.61±5566.23 ng.h/mL in the SLE-treated group (P<0.01). For ginsenoside Rc and Rd, similarly, coadministration of SLE could increase the AUC for 1.49-fold and 1.86-fold (P<0.05). For ginsenoside Rb1, coadministration of SLE could increase the AUC for 1.29-fold, but no statistical difference was found between the two groups after carrying t test (P>0.05). Besides, maximum plasma concentrations (C max) for Rb1 and Rb2 were also significantly enhanced by SLE treatment (P<0.05), and C max of ginsenoside Rc and Rd was not dramatically changed after SLE treatment. At the same time, the time of maximum plasma concentration (T max) and half-life time (t1/2) were not altered after coadministration of SLE. As a positive control, the effect of single-dose SLE on the pharmacokinetics of digoxin in rats was investigated. The plasma concentration-time profiles when digoxin was single administered alone or in combination with SLE at 500 mg/kg are shown in Fig. 6 and the pharmacokinetic parameters are list in Table 1. Obviously, coadministration of SLE could significantly increase the AUC0-τ of digoxin 1.25-fold, from 61.80±13.37 ng.h/mL in the vehicle treated group to 84.38±7.25 ng.h/mL in the SLE-treated group.
Figure 6. The plasma concentration-time profiles of digoxin and ginsenoside Rb1, Rb2, Rc and Rd after single-dose of vehicle / SLE and GE.

Table 1. The pharmacokinetic parameters of digoxin and ginsenoside Rb1, Rb2, Rc and Rd after single ig SLE and 150/kg GE in rats (n = 5).
| Ginsenosides | t1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUCall (ng.h/mL) | AUCINF_obs (ng.h/mL) | Cl_F_obs (L/h) | MRTINF_obs (h) | |
| Control | Digoxin | 3.08±1.23 | 1.28±0.36 | 15.04±5.07 | 61.80±13.37 | 67.51±13.43 | 7.66±1.53 | 5.15±1.09 |
| Rb1 | 13.34±2.43 | 7.50±1.00 | 491.40±61.38 | 14256±2378 | 14394±2469 | 0.007±0.001 | 23.10±4.31 | |
| Rb2 | 19.42±2.87 | 7.00±2.00 | 330.46±70.86 | 10224±1706 | 10551±1728 | 0.012±0.003 | 27.91±4.20 | |
| Rc | 16.90±2.14 | 9.00±2.00 | 429.99±16.66 | 14523±879 | 14877±876 | 0.007±0.000 | 28.30±1.70 | |
| Rd | 75.42±42.74 | 7.50±1.00 | 236.51±79.05 | 5347±1405 | 7409±2177 | 0.014±0.003 | 21.63±48.98 | |
| SLE | Digoxin | 2.98±0.36 | 1.58±0.38 | 17.46±2.26 | 84.38±7.25** | 90.98±8.39** | 5.53±0.48** | 5.06±0.42 |
| Rb1 | 11.34±2.74 | 11.00±2.00 | 650.56±126.56 | 18457±5779 | 18511±5733 | 0.006±0.002 | 23.53±2.68 | |
| Rb2 | 19.03±3.84 | 9.50±3.00 | 540.94±45.03 | 22310±5273** | 23198±5566** | 0.004±0.001 | 25.17±3.63 | |
| Rc | 17.34±4.54 | 9.50±3.00 | 514.89±142.09 | 21642±4411* | 24127±3845* | 0.004±0.001 | 23.61±8.09 | |
| Rd | 21.83±6.78 | 7.50±1.00 | 227.47±57.82 | 7962±2002* | 9045±2203* | 0.011±0.003 | 25.09±7.45 |
In control group: the rats were intragastrically administrated a single dose of vehicle (0.5% sodium CMC), 2 h later, digoxin (0.5 mg/kg) / GE (120 mg/kg) was given to the rats by intragastric administration. In SLE group: the rats were administrated a single dose of SLE intragastrically at 500 mg/kg suspended in 0.5% sodium CMC. 2 h later, digoxin (0.5 mg/kg)/GE (120 mg/kg) was given to the rats by intragastric administration.
*:P<0.05 & **:P<0.01.
Long-term treatment with SLE increased plasma concentrations of ginsenosides in rats
In order to investigate the effect of long-term treatment with SLE on the pharmacokinetics of ginsenosides, 5 rats were given a dose of SLE intragastrically at 500 mg/kg suspended in 0.5% sodium CMC once a day for 10 continuous days, whereas the other 5 rats received the vehicle (0.5% sodium CMC), serving as the control groups. On the 11th day, GE was administered to rats at a single dose of 150 mg/kg by gavage. As shown in Figure 7, 10 continuous days administration of SLE could increase the AUC0-∞ of ginsenoside Rb2 for 1.33-fold, from 22557.12±6121.56 ng.h/mL in the vehicle treated group to 16600.84±3511.26 ng.h/mL in the SLE-treated group, but no statistical difference was found between the two groups after carrying t test (P>0.05). For ginsenoside Rc and Rd, similarly, coadministration of SLE could increase the AUC for 1.16-fold and 1.60-fold. Besides, 10 continuous days administration of SLE could increase the AUC0-∞ of ginsenoside Rb1 1.82-fold, from 14324.65±5178.69 ng.h/mL in the vehicle treated group to 27017.32±2381.53 ng.h/mL in the SLE-treated group (P<0.01). The pharmacokinetic parameters of of ginsenoside Rb1, Rb2, Rc and Rd in rats are shown in Table 2. Besides, the effect of multi-dose SLE on the pharmacokinetics of digoxin (a positive drug) in rats was investigated. 10 continuous days administration of SLE could increase the AUC0-τ of digoxin about 2-fold, from 68.30±25.96 ng.h/mL in the vehicle treated group to 123.24±52.78 ng.h/mL in the SLE-treated group (P<0.05).
Figure 7. The plasma concentration-time profiles of digoxin and ginsenoside Rb1, Rb2, Rc and Rd after multi-dose of vehicle / SLE and GE.

Table 2. The pharmacokinetic parameters of digoxin and ginsenoside Rb1, Rb2, Rc and Rd after multiple ig SLE and 150/kg GE in rats (n = 5).
| Ginsenosides | t1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUCall (ng.h/mL) | AUCINF_obs (ng.h/mL) | Cl_F_obs (L/h) | MRTINF_obs (h) | |
| Control | Digoxin | 2.99±2.32 | 2.33±1.03 | 11.75±4.22 | 68.30±25.96 | 76.14±30.04 | 7.48±3.09 | 5.65±2.85 |
| Rb1 | 17.24±2.16 | 10.00±4.47 | 430.32±135.11 | 14066±5067 | 14324±5178 | 0.0079±0.0035 | 28.09±4.81 | |
| Rb2 | 20.15±3.66 | 12.00±0.00 | 512.33±77.75 | 15937±3310 | 16600±3511 | 0.0062±0.0013 | 29.50±2.61 | |
| Rc | 20.81±9.64 | 11.20±1.79 | 475.56±64.04 | 12569 ±1821 | 14462 ±2343 | 0.0143±0.0035 | 22.35±9.98 | |
| Rd | 20.81±9.64 | 8.00±2.45 | 261.64±103.87 | 6569±1821 | 7462±2343 | 0.0143±0.0035 | 22.35±9.98 | |
| SLE | Digoxin | 2.19±0.87 | 3.28±1.73 | 22.94±13.67* | 123.24±52.78* | 126.79±52.07* | 4.49±1.75* | 4.78±0.91 |
| Rb1 | 19.31±8.29 | 9.60±2.19 | 735.89±142.95 | 25644±1305** | 27017±2381** | 0.0037±0.0003 | 33.50±7.91 | |
| Rb2 | 24.28±8.42 | 10.00±2.83 | 585.87±95.24 | 21261±6522** | 22557±6121** | 0.0047±0.0011 | 26.51±6.58 | |
| Rc | 26.75±3.08 | 11.20±1.79 | 422.52±86.49 | 14641±2687 | 16729±8560 | 0.0028±0.0002 | 40.03±4.04 | |
| Rd | 17.97±75.93 | 8.00±2.45 | 264.68±49.43 | 7641±2687 | 11729±8560* | 0.0125±0.0084 | 26.32±90.58 |
In control group: rats were given the vehicle (0.5% sodium CMC) once a day for 10 continuous days. On the 11th day, digoxin (0.5 mg/kg) / GE (120 mg/kg) was administered intragastrically to rats. In SLE+GE group: rats were given a dose of SLE intragastrically at 500 mg/kg suspended in 0.5% sodium CMC once a day for 10 continuous days. On the 11th day, digoxin (0.5 mg/kg) / GE (120 mg/kg) was administered intragastrically to rats.
*:P<0.05 & **:P<0.01.
Discussion and Conclusions
Over 1,000 Chinese herb formulae have been used in China since hundreds of years and their modern preparations are also widely applied in clinic [7]. In the tide of the modernization of Chinese medicine, combined application of TCM can achieve a synergistic interaction capable of yielding more distinct effect at lower doses than that produced by single Chinese herbal medicine [33]. Shengmai-san has protective effects against oxidative damages in the cells or tissues of the cardiovascular and nervous systems, and play an important role on treatment of symptoms related to cardiovascular diseases such as heart failure, stroke, shock, etc [34]. Schisandra Chinensis in Shengmai-san formulae, a strong P-gp inhibitor, was used as an adjuvant drug to improve the pharmacological activity of monarch drug (Panax Ginseng). In order to investigate the mechanism of enhancing the pharmacological activity produced by Schisandra Chinensis, the influence of SLE on the pharmacokinetics of the ginsenosides was systematically investigated on in vitro and in vivo models based on P-glycoprotein.
Firstly, the effect of P-gp inhibitor (verapamil) on the transport of ginsenosides in LLC-PK1 cells and the derived cell line L-MDR1 were compared to investigate which ginsenosides were P-gp substrates. As a result, ERs of ginsenosides across LLC-PK1 cell monolayers and the corresponding P app values were not affected by verapamil, while the use of verapamil caused a significantly decrease on ERs of ginsenosides (Rb2, Rc, Rg2, Rg3, Rd and Rb1) in L-MDR1 cells. Then, the uptake of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 in L-MDR1 cells was found be greatly increased by verapamil, while the uptake of ginsenoside Rh1, F1, Re, and Rg1 in LLC-PK1 cell was found almost not affected by verapamil. According to the results above, the ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 were identified as P-gp substrates. In 2006, Caco-2 cell monolayer was used as an in vitro model to reveal the transport mechanism of Rb1 across the intestinal mucosa [21]. Ginsenoside Rb1 was found not be transported by P-gp, which was contradictory to our research. The main possible reason was that the expression and activity of P-gp on Caco-2 cell were not high enough to confirm whether Rb1 was a moderate P-gp substrate or not. Besides, existence of other transporters and metabolic enzymes on Caco-2 cell may be another reason.
Based on the research above, it seems that an important breakthrough on the compatibility of Ginseng and Schisandra Chinensis in Shengmai-san was found, and SLE may enhance the exposure of ginsenosides through inhibiting the activity and expression of P-gp. In order to confirm the views above, the effect of SLE on the pharmacokinetics of ginsenosides was systematically investigated based on both in vitro and in vivo models. As we infer, the efflux ratios of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 in L-MDR1 cells could be significantly decreased by SLE, and the transport of ginsenoside Rh1, F1, Re, and Rg1 was found almost not be affected by SLE. Besides, the intracellular concentration of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 on L-MDR1 cells was greatly increased with the presence of SLE in L-MDR1 cells. In contrast, the transport and uptake could not be altered by SLE in the LLC-PK1 cell. Likewise, the effects of SLE on the transport and uptake of ginsenosides were also investigated in Caco-2 cell which was always used to assess bioavailability for several years. As a result, SLE could concentration dependently decrease the efflux ratios and increase the uptake of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 in Caco-2 cell. In the in vivo model, single administration of SLE was found can increase the AUC of ginsenosides (Rb1, Rb2, Rc and Rd), and the exposure of ginsenoside Rb1, Rb2, Rc and Rd could also be significantly enhanced by multiple administration (for 10 continuous days) of SLE.
Overall, our results suggested that the exposure of ginsenoside Rb2, Rc, Rg2, Rg3, Rd and Rb1 could be enhanced by SLE, and the compatibility law of Ginseng and Schisandra Chinensis in Shengmai-san could be explained based on P-gp regulation to some extent.
Supporting Information
The structures of ginsenoside Rh1, F1, Rb2, Rc, Rg2, Rg3, Re, Rd, Rb1, Rg1 and digoxin (internal standard).
(TIF)
The chromatography of ginsenoside Rh1, F1, Rb2, Rc, Rg2, Rg3, Re, Rd, Rb1, Rg1 and digoxin (internal standard).
(TIF)
Funding Statement
This study was supported by the National Nature Science Foundation (81102881, 81273589, 81374054), Natural Science Foundation of Jiangsu Province (Grants BK20131311). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hoenders HJ, Appelo MT, Milders CF (2006) Complementary and alternative medicine and psychiatry: opinions of patients and psychiatrists. Tijdschr Psychiatr 48: 733–737. [PubMed] [Google Scholar]
- 2. Cao H, Han M, Ng EH, Wu X, Flower A, et al. (2013) Can Chinese herbal medicine improve outcomes of in vitro fertilization? A systematic review and meta-analysis of randomized controlled trials. PLoS One 8: e81650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang L, Zhou GB, Liu P, Song JH, Liang Y, et al. (2008) Dissection of mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia. Proc Natl Acad Sci USA 105: 4826–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang X, Zhang A, Wang P, Sun H, Wu G, et al. (2013) Metabolomics coupled with proteomics advancing drug discovery toward more agile development of targeted combination therapies. Mol Cell Proteomics 12: 1226–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bao H, Gao J, Huang T, Zhou ZM, Zhang B, et al. (2010) Relationship between traditional Chinese medicine syndrome differentiation and imaging characterization to the radiosensitivity of nasopharyngeal carcinoma. Chin J Cancer 29: 937–945. [DOI] [PubMed] [Google Scholar]
- 6. Wang S, Hu Y, Tan W, Wu X, Chen R, et al. (2012) Compatibility art of traditional Chinese medicine: from the perspective of herb pairs. J Ethnopharmacol 143: 412–423. [DOI] [PubMed] [Google Scholar]
- 7. Lin T, Li Y (2009) Discussion on the Three Levels of the Prescription-Composition Principle of TCM. Deutsche Zeitschrift für Akupunktur 52: 27–30. [Google Scholar]
- 8. Feng Y, Wu Z, Zhou X, Zhou Z, Fan W (2006) Knowledge discovery in traditional Chinese medicine: state of the art and perspectives. Artif Intell Med 38: 219–236. [DOI] [PubMed] [Google Scholar]
- 9. Zhang JM, Liao W, He YX, He Y, Yan D, et al. (2013) Study on intestinal absorption and pharmacokinetic characterization of diester diterpenoid alkaloids in precipitation derived from fuzi-gancao herb-pair decoction for its potential interaction mechanism investigation. J Ethnopharmacol 147: 128–135. [DOI] [PubMed] [Google Scholar]
- 10. Zhang J, Gao W, Hu X, Liu Z, Liu C (2012) The influence of compatibility of traditional Chinese medicine on the pharmacokinetic of main components in Fructus aurantii. J Ethnopharmacol 144: 277–283. [DOI] [PubMed] [Google Scholar]
- 11. Yu CP, Tsai SY, Kao LJ, Chao PD, Hou YC (2013) A Chinese herb formula decreases the monocarboxylate transporter-mediated absorption of valproic acid in rats. Phytomedicine 20: 648–653. [DOI] [PubMed] [Google Scholar]
- 12. Seo TB, Baek K, Kwon KB, Lee SI, Lim JS, et al. (2009) Shengmai –san –mediated enhancement of regenerative responses of spinal cord axons after injury in rats. J Pharmacol Sci 110: 483–492. [DOI] [PubMed] [Google Scholar]
- 13. Wang J, Yang X, Chu F, Chen J, He Q, et al. (2013) The effects of xuefu zhuyu and shengmai on the evolution of syndromes and inflammatory markers in patients with unstable angina pectoris after percutaneous coronary intervention: a randomised controlled clinical trial. Evid Based Complement Alternat Med 2013: 896467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen CY, Lu LY, Chen P, Ji KT, Lin JF, et al. (2013) Shengmai injection, a traditional chinese patent medicine, for intradialytic hypotension: a systematic review and meta-analysis. Evid Based Complement Alternat Med 2013: 703815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yao HT, Chang YW, Chen CT, Chiang MT, Chang L, et al. (2008) Shengmai San reduces hepatic lipids and lipid peroxidation in rats fed on a high-cholesterol diet. J Ethnopharmacol 116: 49–57. [DOI] [PubMed] [Google Scholar]
- 16. Li LH, Wang JS, Kong LY (2013) Protective effects of shengmai san and its three fractions on cerebral ischemia-reperfusion injury. Chin J Nat Med 11: 222–230. [DOI] [PubMed] [Google Scholar]
- 17. Fan L, Mao XQ, Tao GY, Wang G, Jiang F, et al. (2009) Effect of Schisandra chinensis extract and Ginkgo biloba extract on the pharmacokinetics of talinolol in healthy volunteers. Xenobiotica 39: 249–254. [DOI] [PubMed] [Google Scholar]
- 18. Xue XP, Qin XL, Xu C, Zhong GP, Wang Y, et al. (2013) Effect of Wuzhi Tablet (Schisandra sphenanthera extract) on the Pharmacokinetics of Cyclosporin A in Rats. Phytother Res 27: 1255–1259. [DOI] [PubMed] [Google Scholar]
- 19. Yoo HH, Lee M, Lee MW, Lim SY, Shin J, et al. (2007) Effects of Schisandra lignans on P-glycoprotein-mediated drug efflux in human intestinal Caco-2. Planta Med 73: 444–450. [DOI] [PubMed] [Google Scholar]
- 20. Yang Z, Wang JR, Niu T, Gao S, Yin T, et al. (2012) Inhibition of P-glycoprotein leads to improved oral bioavailability of compound K, an anticancer metabolite of red ginseng extract produced by gut microflora. Drug Metab Dispos 40: 1538–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Han M, Sha X, Wu Y, Fang X (2006) Oral absorption of ginsenoside Rb1 using in vitro and in vivo models. Planta Med 72: 398–404. [DOI] [PubMed] [Google Scholar]
- 22. Liu H, Yang J, Du F, Gao X, Ma X, et al. (2009) Absorption and disposition of ginsenosides after oral administration of Panax notoginseng extract to rats. Drug Metab Dispos 37: 2290–2298. [DOI] [PubMed] [Google Scholar]
- 23. Zhang J, Zhou F, Niu F, Lu M, Wu X, et al. (2012) Stereoselective regulations of P-glycoprotein by ginsenoside Rh2 epimers and the potential mechanisms from the view of pharmacokinetics. PLoS One 7: e35768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meng Z1, Zhang H, Zhao Y, Lan J, Du L (2007) Transport behavior and efflux of Rg1 in rat pulmonary epithelial cells. Biomed Chromatogr 21: 635–641. [DOI] [PubMed] [Google Scholar]
- 25. Kim SW, Kwon HY, Chi DW, Shim JH, Park JD, et al. (2003) Reversal of P-glycoprotein-mediated multidrug resistance by ginsenoside Rg(3). Biochem Pharmacol 65: 75–82. [DOI] [PubMed] [Google Scholar]
- 26. Hashiguchi Y, Hamada A, Shinohara T, Tsuchiya K, Jono H, et al. (2013) Role of P-glycoprotein in the efflux of raltegravir from human intestinal cells and CD4+T-cells as an interaction target for anti-HIV agents. Biochem Biophys Res Commun 439: 221–227. [DOI] [PubMed] [Google Scholar]
- 27. Mills E, Wu P, Johnston BC, Galliacano K (2005) Natural health product-drug interactions: a systematic review of clinical trials. Ther. Drug Monit 27: 549–557. [DOI] [PubMed] [Google Scholar]
- 28. Liang Y, Zhou Y, Zhang J, Liu Y, Guan T, et al. (2013) In vitro to in vivo evidence of the inhibitor characteristics of Schisandra lignans toward P-glycoprotein. Phytomedicine 20: 1030–1038. [DOI] [PubMed] [Google Scholar]
- 29. Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P (1995) Absence of the mdr1a P-glycoprotein in mice affects tissue distribution and pharmacokineticsof dexamethasone, digoxin, and cyclosporin A. J Clin Invest 96: 1698–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gnoth MJ, Buetehorn U, Muenster U, Schwarz T, Sandmann S (2011) In vitro and in vivo P-glycoprotein transport characteristics of rivaroxaban. J Pharmacol Exp Ther 338: 372–80. [DOI] [PubMed] [Google Scholar]
- 31. Zhou S, Feng X, Kestell P, Paxton JW, Baguley BC, et al. (2005) Transport of the investigational anti-cancer drug 5,6-dimethylxanthenone-4-acetic acid and its acyl glucuronide by human intestinal Caco-2 cells. Eur J Pharm Sci 24: 513–524. [DOI] [PubMed] [Google Scholar]
- 32. Angela PA, Manju B (2000) Reddy Caco-2 Cells Can Be Used to Assess Human Iron Bioavailability from a Semipurified Meal. J Nutr 130: 1329–1334. [DOI] [PubMed] [Google Scholar]
- 33. Zhang J, Gao W, Hu X, Liu Z, Liu C (2012) The influence of compatibility of traditional Chinese medicine on the pharmacokinetic of main components in Fructus aurantii. J Ethnopharmacol 144: 277–83. [DOI] [PubMed] [Google Scholar]
- 34. Seo TB, Baek K, Kwon KB, Lee SI, Lim JS, et al. (2009) Shengmai –san -mediated enhancement of regenerative responses of spinal cord axons after injury in rats. J Pharmacol Sci 110: 483–492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The structures of ginsenoside Rh1, F1, Rb2, Rc, Rg2, Rg3, Re, Rd, Rb1, Rg1 and digoxin (internal standard).
(TIF)
The chromatography of ginsenoside Rh1, F1, Rb2, Rc, Rg2, Rg3, Re, Rd, Rb1, Rg1 and digoxin (internal standard).
(TIF)