

Abstract
The IκB kinase complex induces nuclear factor kappa B activation and has recently been recognized as a key player of autoimmunity in the central nervous system. Notably, IκB kinase/nuclear factor kappa B signalling regulates peripheral myelin formation by Schwann cells, however, its role in myelin formation in the central nervous system during health and disease is largely unknown. Surprisingly, we found that brain-specific IκB kinase 2 expression is dispensable for proper myelin assembly and repair in the central nervous system, but instead plays a fundamental role for the loss of myelin in the cuprizone model. During toxic demyelination, inhibition of nuclear factor kappa B activation by conditional ablation of IκB kinase 2 resulted in strong preservation of central nervous system myelin, reduced expression of proinflammatory mediators and a significantly attenuated glial response. Importantly, IκB kinase 2 depletion in astrocytes, but not in oligodendrocytes, was sufficient to protect mice from myelin loss. Our results reveal a crucial role of glial cell-specific IκB kinase 2/nuclear factor kappa B signalling for oligodendrocyte damage during toxic demyelination. Thus, therapies targeting IκB kinase 2 function in non-neuronal cells may represent a promising strategy for the treatment of distinct demyelinating central nervous system diseases.
Keywords: oligodendrocyte, demyelination, remyelination, NF-κB, glia, cuprizone, multiple sclerosis
Introduction
The rapid conduction of action potentials in the mammalian nervous system depends on myelin, which forms an insulating multi-lamellar structure around axons. This specialized organelle is produced by Schwann cells in the PNS and by oligodendrocytes in the CNS. The unique association between axons and glia is also a key regulator of nerve regeneration after injury, promoting axonal regrowth in the periphery while preventing it in the CNS. During development of the CNS, oligodendroglial cells ensheath axonal processes, assemble compact myelin and form the white matter. Brain myelin formation during development is initiated by, as yet, poorly identified signals from axons to oligodendrocytes. There are interactions between axonal ligands and glial receptors, e.g. neuregulins and ErbB receptor tyrosine kinases, which integrate cell surface signals and modulate myelin thickness (Falls 2003; Michailov et al., 2004; Brinkmann et al., 2008).
Among the signalling pathways that orchestrate axonal ensheathment in the PNS via Schwann cell differentiation to a myelinating phenotype, nuclear factor kappa B (NF-κB) was shown to be crucial for insulating axons (Nickols et al., 2003). NF-κB was defined as one essential differentiation signal in the PNS; it was highly upregulated in premyelinating Schwann cells and its blockage markedly attenuated myelination (Nickols et al., 2003). Oct-6 and Krox-20, two additional transcription factors, known to be induced after axonal contact and essential for the proper temporal expression of myelin-specific genes in Schwann cells, are both regulated via NF-κB (Topilko et al., 1994; Jaegle et al., 1996; Nickols et al., 2003).
It is largely unknown whether NF-κB exerts similar functions in oligodendrocytes of the CNS. NF-κB could also modulate the remyelination process by oligodendrocytes, especially since tumour necrosis factor (TNF), a prototypical inducer of NF-κB, was shown to be required for both remyelination and proliferation of oligodendrocyte progenitor cells (Arnett et al., 2001). Activation of the canonical NF-κB pathway is induced by the IκB kinase (IKK) complex, composed of two catalytic subunits, IKK1/α and IKK2/β, and a regulatory subunit, NF-κB essential modulator (NEMO)/IKKγ (Hayden and Ghosh 2008). An alternative NF-κB activation pathway has been described that involves NF-κB-inducing kinase and IKK1 and mediates processing of the p100 precursor protein resulting in the release of p52-containing dimers.
The present study addressed the role of NF-κB signalling in oligodendrocyte differentiation and in the complex process of CNS myelination throughout development and during de- and remyelination using the demyelinating toxin cuprizone (bis-cyclohexanone oxaldihydrazone). By applying CNS cell-restricted inhibition of IKK2-mediated activation of the canonical NF-κB pathway, we show that IKK/NF-κB signalling in glial cells is critical for driving oligodendrocyte destruction and demyelination in vivo. In contrast, IKK2 is dispensable for insulating axons during development and remyelination. These data reveal a new function for IKK2-mediated NF-κB activity during toxic demyelination. The demonstration that NF-κB inhibition is protective during non-inflammatory demyelination suggests that pharmacological targeting of the IKK/NF-κB pathway, specifically in glial cells, might have therapeutic effects in other demyelinating pathologies such as multiple sclerosis.
Materials and methods
Mice
For IKK2 inactivation in all cells of the CNS or in oligodendrocytes only, mice carrying loxP-IKK2 alleles (Pasparakis et al., 2002) were crossed with transgenic mice expressing Cre recombinase under the control of the nestin (Tronche et al., 1999) or the MOG promoter (Hovelmeyer et al., 2005). Glial fibrillary acidic protein (GFAP)-IκBα-dn animals were described previously (Brambilla et al., 2005). All mice used in this study were backcrossed to the C57Bl/6 genetic background for at least seven generations. Mice were bred in-house under pathogen-free conditions.
Cuprizone and lysocithin treatment
Cuprizone experiments were performed on 6–8-week-old female mice. The animals were housed under standard laboratory conditions with food and water ad libitum. Mice were fed for 5 or 10 weeks with 0.2% (wt/wt) cuprizone (Sigma, St Louis, MO, USA) in the ground breeder chow. The control group received breeder chow without cuprizone admixture. We conducted two experimental series: in the first experimental setup, analysis of light and ultrastructural level and cytokine production was performed after 5 weeks of cuprizone treatment; in the second setup, the cuprizone diet was discontinued after 5 weeks and animals were maintained for a further 5 weeks under normal diet to allow spontaneous remyelination. For the remyelination experiments, lysolecithin was stereotactically injected into the spinal cord of 10–12-week-old mice, as described previously (Ousman and David, 2000). Mice were anaesthetized with ketamine:xylazine (100 mg/kg:20 mg/kg) and a dorsal laminectomy was performed at spinal cord segment Th10. One microlitre of lysolecithin (Sigma), at a concentration of 2 µg/µl, was injected into the ventral part of the spinal cord, lateral to the central artery. Animals were sacrificed after either four or 14 days and perfused with 2% paraformaldehyde/3% glutaraldehyde for electron microscopy.
Histology and electron microscopy
Histology was performed as described recently (Prinz et al., 2006, 2008). Brains were removed and fixed in 4% buffered formalin. Tissue was paraffin embedded before staining with haematoxylin and eosin, luxol fast blue to assess the degree of demyelination, MAC-3 (BD Pharmingen) for microglia, GFAP for astrocytes (Dako, Hamburg, Germany), degraded myelin basic protein (Chemicon, Billerica, USA) for myelin destruction, amyloid precursor protein for axonal damage (Chemicon), Nogo-A (mAb11c7; Oertle et al., 2003; kindly provided by M. Schwab, University of Zurich), proteolipid protein, 2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNP), and myelin basic protein (Biozol, Eching) for mature oligodendrocytes and Olig 2 for oligodendrocyte precursors (Immuno-Biological Laboratories, USA). Serial coronal sections were examined between levels −0.7 and −1.7 mm bregma according to the mouse brain atlas of Franklin and Paxinos (1997). Demyelination was scored as described, ranging from 0 (no demyelination) to 3 (complete demyelination) (Hiremath et al., 1998). The total number of MAC-3+ microglia and GFAP+ astrocytes in the lateral and medial part of the corpus callosum was counted using ×400 magnification and cellularity was calculated as cells per mm2. For assessment of oligodendrocyte precursors, rabbit anti-NG2 antibody (Chemicon) was used 1:200 on floating cryosections. For immunohistochemical detection of nuclear translocated p65, sections were stained with antibodies to NF-κB p65 (Santa Cruz). Terminal deoxynucleotidyl transferase dUTP nick end labelling staining was performed according to the manufacturer's protocol (TUNEL, Oncor).
For electron microscopy, sections from Epon®-embedded, glutaraldehyde-fixed corpus callosum were cut and stained with toluidine blue. The tissue was then trimmed and reoriented so that ultrathin cross sections of the midline corpus callosum could be cut and treated with uranyl acetate and lead citrate. Electron micrographs were analysed for fibre diameter, axon diameter and myelin thickness using the analySIS Docu System (Soft Imaging System GmbH, Germany). G ratios were defined as diameter of the axon divided by fibre diameter (axon plus myelin). We calculated a G ratio for each fibre and subsequently averaged all G ratios from one brain. In addition, the number of myelinated fibres in relation to all fibres was expressed as percentage of axons with a diameter of >250 nm to all visible axons.
Cell cultures
Primary astrocytes and microglia were prepared from newborn mice as described previously (Prinz and Hanisch, 1999; Mildner et al., 2007). Purified astrocytes and microglia were seeded at a density of 2 × 105 cells/6 wells and murine recombinant TNFα (10 ng/ml), IL-1β (10 ng/ml) or IFNγ (10 ng/ml) was added to the cell culture medium for 30 min to 1 h. For cell stimulation, cuprizone was added to primary glial cells at 25 µM for 90 min. For p65/RelA nuclear translocation cells were analysed by immunofluorescence using antibodies against GFAP (Chemicon) and p65/RelA (Santa Cruz). Five random optical fields were taken, with 100 cells counted per optical field; the results are presented as mean ± SEM. Oligodendrocyte enriched glial cell cultures were prepared according to a modified protocol for rats as described earlier (Prinz et al., 2004).
Western blot
Immunoblots were performed as described previously (van Loo et al., 2006). In brief, total cell or brain extracts were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis (10%) and transferred to polyvinylidene fluoride membranes (Bio-Rad, München, Germany). Membranes were probed with mouse anti-IKK2 (Imgenex, San Diego, USA) and mouse anti-actin (Chemicon) antibodies and detected by chemiluminescent peroxidase substrate (Sigma, Steinheim, Germany). For detection of myelin proteins, brains were homogenized in Laemmli buffer and protein lysates were separated by sodium dodecyl sulphate polyacrylamide gel electrophoresis. Upon blotting, nitrocellulose membranes were probed with antibodies recognizing CNPase (Sigma 11-5B) and myelin basic protein (Millipore AB980) as well as anti β-actin to monitor loading.
Laser microdissection
Microdissection of 150 astrocytes and microglia per mouse was performed using a Leica AS LMD system (DM 6000B, Leica Microsystems) as described before, with modifications (Burbach et al., 2004). Fast immunochemistry of serial brain sections from cuprizone-treated and untreated mice was performed for astrocytes with GFAP (DAKO, Hamburg, Germany) and for microglia with CD11b antibodies (BMA Biomedicals, Augst, Switzerland). Immunostained sections were counterstained with Hoechst 33342 (Sigma-Aldrich) to facilitate the identification of individual cells. RNA was isolated with the RNeasy® Micro Plus Kit (Qiagen, Hilden, Germany) and reverse transcription, pre-amplification and real-time polymerase chain reaction were performed using Applied Biosystems (Darmstadt, Germany) reagents according to the manufacturer's recommendations.
Real-time polymerase chain reaction
RNA was extracted from diseased mice challenged with cuprizone. The tissue was flushed with ice cold Hank's buffered salt solution and RNA was isolated using RNeasy® Mini kits (Qiagen), following the manufacturer's instructions, and the polymerase chain reaction was performed as described recently (Prinz et al., 2006). The following primer probe pairs were used: CCL2 (sense TCTGGGCCTGCTGTTCACC, antisense TTGGGATCATCTTGCTGGTG), CXCL10 (sense TGCTGGGTCTGAGTGGGACT, antisense CCCTATGGCCCTCATTCTCAC), CCL3 (sense CACCACTGCCCTTGCTGTT, antisense AGGAGAAGCAGCAGGCAGTC), TNFα (sense CATCTTCTCAAAATTCGA GTGACAA, antisense TGGGAGTAGACAAGGTACAACCC), IL-1β (sense ACAAGAGCTTCAGGCAGGCAGTA, antisense ATATGGGTCCGACAGCACGAG), CNP (sense TGGTGTCCGCTGATGCTTAC, antisense CCGCTCGTGGTTGGTATCAT), myelin-associated glycoprotein (sense GGTGTTGAGGGAGGCAGTTG, antisense CGTTCTCTGCTAGGCAAGCA), Olig1 (sense CGACGCCAAAGAGGAACAG, antisense GCCAAGTTCAGGTCCTGCAT) and SOX2 (sense AACTTTTGTCCGAGACCGAGAA, antisense CCTCCGGGAAGCGTGTACT).
Statistical analysis
Statistical differences of clinical scores were evaluated using a non-paired Student's t-test. Differences were considered significant when P < 0.05.
Results
Formation of myelin in the central nervous system in the absence of the canonical nuclear factor kappa B pathway activator IκB kinase 2
As NF-κB activation has been shown to be an essential signal for the progression of axon-associated Schwann cells into a myelinating phenotype in the PNS (Nickols et al., 2003), we hypothesized that NF-κB might also be required for the formation of myelin in the brain. We therefore generated mice with CNS-restricted knockout of IKK2 (IKK2CNS-KO) by crossing mice carrying loxP-flanked IKK2 alleles (Pasparakis et al., 2002) with Nestin-Cre transgenic mice (Tronche et al., 1999). Immunoblot analysis of brain region extracts as well as cell lysates from neuroectodermal astrocytes confirmed the efficient ablation of IKK2 in IKK2CNS-KO mice (Fig. 1A). Mesodermal-derived microglia were not affected.
Figure 1.

The canonical NF-κB pathway activator IKK2 is dispensable for formation of myelin in the CNS. (A) Immunoblot analysis of lysates demonstrating brain and neuroectoderm-specific deletion of IKK2. Data are representative of four independent experiments. Lysates from total brain, cortex and subcortex were generated from mice at the age of 6–8 weeks. Lysates from astrocytes and microglia derived from newborn mice primary cell cultures. (B) Proteolipid protein expression reveals proper myelination and the absence of any gross abnormalities within the CNS in the absence of IKK2 (left panel) at the age of 6–8 weeks. GFAP-expressing astrocytes are normally distributed within the white matter tract of the corpus callosum (right). Analysis was performed with mice at the age of 6–8 weeks. Scale bars = 500 µm (left panel) and 50 µm (right panel). (C) Ultrastructural micrographs showing myelination in the corpus callosum at different time points of myelination. Scale bar = 400 nm (left). Measurements of axon diameter, myelin thickness, G ratio and percentage of myelin fibres in the corpus callosum of three and 25-week old IKK2CNS-KO (filled squares) or wild-type (WT) mice (open squares, right). There are no differences between the mean group values for any parameter in either genotype or at either developmental stage. Each point represents the mean value from at least 100 individual axonal measurements. (D) The number of oligodendroglial progenitor cells and mature oligodendrocytes was quantitatively examined on histological sections of corpus callosum and cortex in 2–3-week old IKK2CNS-KO and IKK2CNS-wild-type mice. Olig2 immunohistochemistry (left) and quantification thereof (right). Data are expressed as mean ± SEM of at least three mice per group. (E) Unaltered oligodendrocyte differentiation and morphology in IKK2CNS-KO mice. Primary oligodendrocytes were cultured and kept for different maturation stages. Double immunofluorescence with DAPI for nuclear staining (blue) and NG2 (red), myelin basic protein (red) or CNP (green), respectively (left panel). Scale bar = 10 µm. Quantification of differentially expressed maturation marker for oligodendrocytes after three days in culture (right). Data indicate the mean of cells expressing the differentiation marker. Data are representative of two independent experiments. MBP = myelin basic protein.
We next performed a detailed histological examination of the CNS of adult IKK2CNS-KO mice for any signs of dys- or demyelination. Adult IKK2CNS-KO brains showed normal myelin distribution and no signs of altered myelination at the light microscopy level (Fig. 1B). Frontal sections of the brain revealed normal myelin fibre tracts on proteolipid protein immunohistochemistry (Fig. 1B, left) in the corpus callosum. Accordingly, immunohistochemistry for the astrocyte-specific marker GFAP did not show any gliotic changes and exhibited morphologically unaltered astrocytes in IKK2-deficient brains (Fig. 1B, right). Further electron microscopy examinations showed comparable numbers of myelinated axons with structurally intact myelin sheath and regular thickness between IKK2CNS-KO and IKK2CNS-wild-type mice (Fig. 1C). In order to examine whether myelin composition is normal in the absence of NF-κB signalling, we performed quantitative real-time polymerase chain reaction and immunoblot analysis of the expression of myelin proteins CNP, myelin-associated glycoprotein, oligodendrocyte transcription factor 1 (Olig1), myelin basic protein and the neural stem cell marker SOX2 from brain lysate (Supplementary Fig. 1A and B). Expression of all myelin proteins tested was similar in mutant animals and wild-type controls. Quantification of Olig2+ oligodendroglial progenitor cells and mature oligodendrocytes revealed normal numbers in both corpus callosum and cortex in the absence of IKK2 (Fig. 1D). Furthermore, normal numbers of oligodendrocyte precursor cells (NG2+) were obtained from newborn IKK2CNS-KO mice, with no obvious differentiation defects or morphological abnormalities in primary cultures (Fig. 1E). Cells from both wild-type and mutant mice expressed CNP and myelin basic protein at a mature developmental stage and displayed similar numbers of well-developed large processes, extensive lacy arborisations and membrane sheets. In summary, these data show that IKK2-mediated NF-κB activation is dispensable for oligodendrocyte maturation in vitro and in vivo, and subsequent insulation of axons in the CNS. These results are in sharp contrast to the previously reported crucial role of NF-κB signalling for myelination in the PNS (Nickols et al., 2003), suggesting that NF-κB has different functions in peripheral and central nervous system myelination. We therefore examined NF-κB activation during myelination in the CNS in the white and grey matter and in the PNS by immunostaining for the p65/RelA NF-κB subunit during the second post-natal week (Supplementary Fig. 2). Consistent with the observed phenotypes, robust nuclear p65 signal, indicating NF-κB activation, could be found in the Schwann cells of the peripheral nerves, whereas white matter oligodendrocytes and cortical neurons did not show nuclear p65 at the same age.
Pivotal role of the classical NF-κB pathway for toxic demyelination
To assess the role of CNS-specific IKK2-mediated NF-κB activation for CNS myelin damage in vivo, IKK2CNS-KO mice were tested in the cuprizone model, a model of toxic demyelination (Mildner et al., 2007). Treatment with cuprizone leads to substantial demyelination in the corpus callosum after 4–5 weeks of administration. Spontaneous remyelination can be observed as early as four days after withdrawal of the agent, making cuprizone an excellent tool to study the mechanisms involved in de- and remyelination (Matsushima and Morell, 2001). Furthermore, cuprizone-induced demyelination shows similarities to lesions seen in experimental autoimmune encephalomyelitis and multiple sclerosis (Gold et al., 2006). We first determined in wild-type mice in which cells NF-κB is activated during toxic demyelination (Fig. 2A). We readily detected nuclear translocation of p65/RelA, the main transactivating NF-κB subunit, in the demyelinating corpus callosum after 5 weeks of cuprizone exposure. Increased nuclear p65 immunoreactivity was mainly visible in GFAP+ astrocytes (192.0 ± 44.3 cells/mm2) and MAC-3+ microglia (814.9 ± 142.9 cells/mm2) and only occasionally in Nogo-A+ oligodendrocytes at the lesion border.
Figure 2.
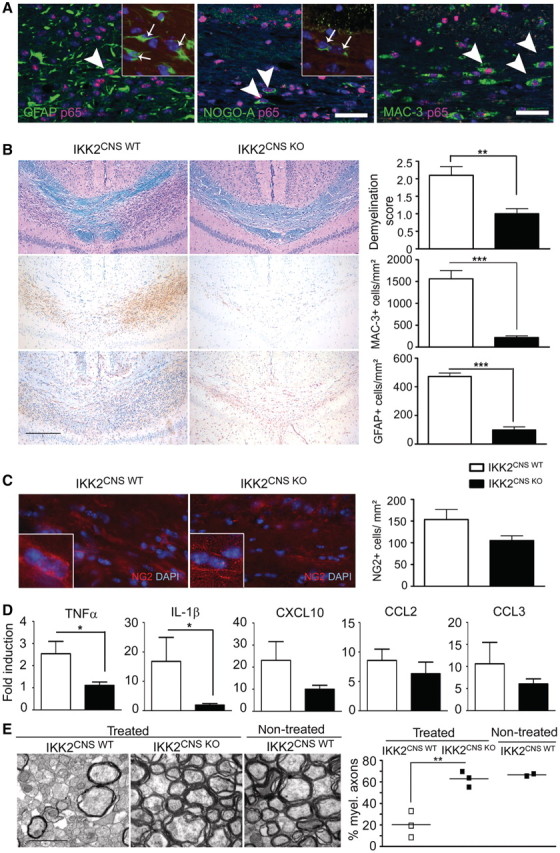
Inhibition of NF-κB signalling in the brain ameliorates demyelination and reduces induction of toxic cytokines in the CNS. (A) NF-κB p65 translocation is induced in neuroectodermal and myeloid cells of the brain during acute demyelination. Immunofluorescence of p65 depicts strong nuclear signal in numerous GFAP+ astrocytes (arrow, left panel), Nogo-A+ oligodendrocytes (arrows, middle panel) and MAC-3+ microglia (arrows, right panel). Inserts illustrate the absence of nuclear p65 translocation in neuroectodermal glia cells of IKK2CNS-KO mice. Scale bar = 25 µm. (B) Histopathological analysis of the corpus callosum from IKK2CNS-wild-type and IKK2CNS-KO mice 5 weeks after cuprizone treatment. Sections were examined for demyelination by luxol fast blue (LFB), for microglia by MAC-3 and for astrogliosis by GFAP immunohistochemistry. Representative sections are shown left and quantification thereof is depicted on the right. (C) The presence of NG2+ cells in the corpus callosum of mice treated with cuprizone is shown by immunofluorescence (red). Nuclei are stained in blue (DAPI). Inserts depict higher magnification. Scale bars = 30 µm and 7 µm (inserts). The number of NG2+ cells per mm2 is shown on the right (mean ± SEM). (D) Quantification of cytokine and chemokine messenger RNA expression in the corpus callosum of IKK2CNS-wild-type and IKK2CNS-KO mice 5 weeks after cuprizone treatment. (E) Electron micrographs depicting highly preserved myelin sheaths in mice lacking IKK2 in all neuroectodermal cells of the brain. The number of myelinated axons is greatly reduced in the presence of canonical NF-κB signalling as quantified in at least 100 axons of a diameter ≥250 nm after acute corpus callosum demyelination. Scale bar = 400 nm. *P < 0.05; **P < 0.01, ***P < 0.001.
To investigate the possible effects of NF-κB inhibition for oligodendrocyte damage, we examined histologically the corpus callosum of IKK2CNS-KO mice fed with cuprizone for 5 weeks (Fig. 2B). In control mice, luxol fast blue staining revealed extensive loss of myelin accompanied by the accumulation of numerous activated MAC-3+ microglia and strong astrogliosis (GFAP). In contrast, loss of myelin and gliosis were strongly reduced in the brain of IKK2CNS-KO mice. Accordingly, the number of NG2+ oligodendrocyte precursor cells in the corpus callosum tended to be lower in protected IKK2CNS-KO mice (Fig. 2C). We then determined the expression of the NF-κB-dependent cytokines and chemokines IL-1β, TNFα, CCL2, CCL3 and CXCL10 that potentially contribute to oligodendrocyte toxicity in this model (Fig. 2D). In IKK2CNS-KO mice, the induction of IL-1β and TNFα was significantly diminished (P < 0.05). Furthermore, the expression of CCL2, CCL3 and CXCL10 was reduced in IKK2CNS-KO mice compared with wild-type controls.
To estimate the degree of demyelination on ultrastructural level, we counted the number of myelinated axons in Epon®-embedded corpora callosa (Fig. 2E). In wild-type mice, only 20 ± 12% of all axons remained myelinated after cuprizone treatment. In contrast, 63 ± 7% (P < 0.05) of all axons were enwrapped with myelin sheaths in IKK2CNS-KO mice.
In order to examine demyelination kinetics in brain-specific IKK2CNS-KO animals, we investigated the degree of demyelination at further time points during prolonged exposure to cuprizone for up to 10 weeks (Fig. 3). The number of Nogo-A+ oligodendrocytes was dramatically reduced in wild-type animals already at 3 weeks (124.7 ± 7.8 cells/mm2 in treated versus 412.1 ± 23.4 cells/mm2 in untreated mice) and further declined until 5 weeks (28.9 ± 2.1 cells/mm2; Fig. 3A). In sharp contrast, IKK2CNS-KO animals exhibited only a subtle oligodendrocyte loss after 3 weeks (378.4 ± 14.5 cells/mm2 in treated versus 414.7 ± 18.8 cells/mm2 in untreated mice) that reached its maximum at 5 weeks (237.2 ± 16.5 cells/mm2) and remained stable during the chronic disease phase. Accordingly, demyelination measured by luxol fast blue staining was less prominent in brain-specific IKK2-deficient animals (Fig. 3A and C). The increase in demyelination was paralleled by a decrease in myelin proteins evident at 3 weeks (Fig. 3B). Consistent with the histologically evident protection from demyelination, IKK2CNS-KO animals showed only mildly decreased myelin protein gene expression. Interestingly, the expression of CNP, Olig1 and myelin-associated glycoprotein spontaneously increased until 5 and 10 weeks. This transcriptional response associated with repair was further characterized by the subsequent increase of the neuronal stem cell factor SOX2. Despite significant myelin preservation in brains lacking NF-κB signalling, the glial response was vigorous after 10 weeks of cuprizone with abundant microglia and astrocytes that were found in similar numbers in the mutant mice (1168.0 ± 89.9 MAC-3+ cells/mm2 compared with 1174.1 ± 321.5 cells/mm2 in wild-type mice and 408.5 ± 22.1 GFAP+ cells/mm2 versus 373.7 ± 44.3 cells/mm2 in wild-type, Fig. 3C). Taken together, these findings show that inhibition of IKK2 mediated NF-κB signalling by deletion of IKK2 in all CNS-resident neuroectodermal cells is associated with decreased expression of key proinflammatory mediators involved in toxic damage of oligodendrocytes, resulting in protection from myelin loss.
Figure 3.

Gene expression pattern is temporally altered during chronic demyelination in the absence of IKK2. (A) Time course of demyelination (upper panel) and the number Nogo-A+ oligodendrocytes (lower panel) in IKK2CNS-wild-type (open squares) and IKK2CNS-KO (filled squares) mice. Data show mean ± SEM from at least three mice per group. (B) Levels of myelin transcripts in the corpus callosum of IKK2CNS-wild-type (open squares) and IKK2CNS-KO (filled squares) mice treated with cuprizone over time, measured by quantitative real-time polymerase chain reaction, normalized to GAPDH and expressed in relation to untreated mice (n ≥ 3). Mean ± SEM from at least three mice per group is depicted. (C) Attenuated demyelination is found in the corpus callosum of chronically treated animals after 10 weeks in the absence of canonical NF-κB activation. Representative histological sections are shown left and the quantification is given right. Data show mean ± SEM from one representative experiment out of two with at least five mice per group. *P < 0.05. Scale bar = 300 µm. LFB = luxol fast blue; MAG (myelin associated glycoprotein) = MAC-3 for microglia and GFAP for astrocytes.
Oligodendrocyte-restricted IκB kinase 2 is dispensable for demyelination and remyelination in vivo
As the Nestin-Cre transgene is highly expressed in all neuroectodermal cells, which results in efficient gene inactivation in neurons, astrocytes and oligodendrocytes, we wondered whether the resistance of IKK2CNS-KO mice to demyelination is due to a cell autonomous protective function of IKK2 inhibition in oligodendrocytes. Cell specificity of Cre recombination in MOGi-Cre transgenic mice has been demonstrated (Hovelmeyer et al., 2005). We therefore generated mice lacking IKK2 only in oligodendrocytes (IKK2Oligo-KO) by crossing Ikk2FL mice with MOGi-Cre knock-in mice, which express Cre specifically in mature oligodendrocytes. Mice with oligodendrocyte-restricted ablation of IKK2 developed normally without any clinical signs of disease. Upon histological examination the number of mature Nogo-A+ oligodendrocytes was found to be normal in all brain regions tested (Supplementary Fig. 3A). Furthermore, CNS myelination was unchanged and oligodendrocyte morphology and differentiation in vitro were normal (Supplementary Fig. 3B).
However, upon toxin-induced (cuprizone, Fig. 4) or autoimmune-mediated (MOG35-55 specific T cells in experimental autoimmune encephalomyelitis; data not shown) demyelination, IKK2Oligo-KO mice showed extensive myelin loss and pronounced gliosis similar to wild-type mice. Accordingly, equal numbers of NG2+ oligodendrocyte precursor cells were found in the corpus callosum of mutant and control mice after 5 weeks of cuprizone treatment (Fig. 4C). Disease incidence, onset and clinical course of EAE were identical in IKK2Oligo-KO and IKK2Oligo-wild-type mice (data not shown). Thus, oligodendrocyte-restricted IKK2-deficiency does not affect oligodendrocyte damage in either chemically induced or autoimmune demyelination.
Figure 4.

NF-κB signalling in mature oligodendrocytes is not pathogenic during demyelination. (A) Analysis of paraffin sections indicate the same pattern (left) and extend (right) of demyelination (luxol fast blue, LFB), infiltrating microglia (MAC-3) as well as astrogliosis (GFAP) after cuprizone treatment for 5 weeks. Data show mean ± SEM from one representative experiment out of three. Scale bar = 300 µm. (B) Similar number of NG2+ progenitor cells in IKK2Oligo-wild-type and IKK2Oligo-KO mice 5 weeks after cuprizone treatment. Representative mean ± SEM of two similar experiments is shown. Scale bar = 20 µm.
IκB kinase 2 is not required for remyelination in the central nervous system
Remyelination is a complex repair process ensuring cellular integrity after neuronal damage, as well as after primary loss of mature oligodendrocytes. The cuprizone model is a suitable tool to study both the kinetics and the underlying molecular mechanisms of remyelination in vivo. Since IKK2Oligo-KO mice showed similar cuprizone-induced demyelination compared with control animals, we used them to examine the role of IKK2-mediated NF-κB activation in oligodendrocytes during remyelination. IKK2Oligo-KO mice were treated with cuprizone for 5 weeks followed by 5 weeks feeding with normal chow without cuprizone to allow the generation of new oligodendrocytes. Semiquantitative assessment of luxol fast blue stainings and ultrastructural morphometric analysis (data not shown) revealed no obvious differences between both genotypes. Thus, IKK2/NF-κB signalling in oligodendrocytes in the cuprizone demyelination model is not essential for remyelination in the adult CNS.
We therefore wanted to address the role of IKK2 in other cells of the CNS during remyelination using the IKK2CNS-KO animals. Since IKK2CNS-KO mice were protected from cuprizone-induced demyelination, we explored alternative models and chose lysolecithin as a potential alternative to induce demyelination in the CNS (Arnett et al., 2004). First, lysolecithin was stereotactically injected into the spinal cord of wild-type mice and subsequent demyelination was assessed. Comparative histopathological analysis of cuprizone- and lysolecithin-induced changes in the CNS revealed striking differences in the lesion pattern as well as in the inflammatory make up of the infiltrates (Fig. 5). Lysolecithin evoked at peak of destruction fulminant axonal damage as revealed by amyloid precursor protein accumulation in axonal spheroids accompanied by strong accumulation of infiltrating macrophages/microglia (MAC-3), whereas the astrocytic response was only scarce. In contrast, at a comparable disease stage, cuprizone provoked a strong astrogliosis and no obvious axonal damage.
Figure 5.

Diverse lesion pattern and different inflammatory make up in two distinct demyelination models. (A) Immunohistochemical examination of inflammation and myelin and axonal damage during acute toxic demyelination in the lysolecithin model at peak of disease (4 days after injection) compared with the maximal response after cuprizone challenge (5 weeks of treatment). MAC-3 staining revealed a plethora of macrophages/microglia in both lesion models, whereas significantly more GFAP+ astrocytes were found in cuprizone-induced lesions during acute myelin loss. Lymphocytes (CD3 for T cells, B220 for B cells) were scarce in both models. Notably, amyloid precursor protein (APP) deposits representing axonal damage were clearly detectable only on the lysolecithin model. (B) Histopathology of infiltration and axonal damage during remyelination after lysolecithin (14 days after injection) and the cuprizone (5 weeks treatment and 5 weeks recovery) model. Data show mean ± SEM from one representative experiment out of two with at least four or five mice per group. Scale bar = 400 μm low magnification; scale bar = 40 μm high magnification. *P < 0.05, **P < 0.01, LFB-periodic acid-Schiff = luxol fast blue-periodic acid Schiff.
Interestingly, in contrast to our findings in the cuprizone model, lysolecithin-induced demyelination was found to be almost complete even in IKK2-deficient brains similarly to wild-type mice (Fig. 6A). The complete demyelination in the lysolecithin model in contrast to cuprizone exposure might be due to differences in the underlying pathophysiology and their diverse consequences on the CNS in terms of subsequent damage and demyelination. IKK2CNS-KO and control mice were injected with lysolecithin and were analysed 14 days later for remyelination. Ultrastructural morphometric analysis of remyelinated spinal cords at Day 14 after lysolecithin injection revealed no significant differences in the G ratios (G ratio 0.93 ± 0.01 for both genotypes), axon diameter, myelin thickness or the percentage of myelinated axons between IKK2CNS-KO and control mice (48.0 ± 5.6% for IKK2CNS-KO and 41.0 ± 3.3 for wild-type P > 0.05) (Fig. 6B). Taken together, these data demonstrate that the process of myelin repair in the lysolecithin model of demyelination is independent of CNS cell-specific IKK2 expression.
Figure 6.

Remyelination in the CNS is independent from canonical NF-κB activation by IKK2. (A) Transmission electron microscopy revealed a similar extent of demyelination induced by lysolecithin four days after treatment (left). Percentage of myelinated fibres is shown for IKK2CNS-wild-type (open squares) and IKK2CNS-KO mice (filled squares). Each symbol represents the mean value from at least 20 individual measurements in each mouse. (B) Morphologically compatible structures of myelin in the remyelination phase 14 days after lysolecithin exposure in both genotypes (left). Measurements of the percentages of myelinated axons, myelin thickness, G ratio and axon diameter in the spinal cord of IKK2CNS-wild-type (open squares) and IKK2CNS-KO mice (filled squares). Scale bar = 400 nm. Each point represents the mean value from at least 50 individual axonal measurements.
Inhibition of neuroectodermal nuclear factor kappa B activation by IκB kinase 2 results in reduced myelin loss
The experiments in IKK2Oligo-KO mice showed that the protection of IKK2CNS-KO mice from cuprizone-induced demyelination is not caused by oligodendrocyte-intrinsic IKK2-deficiency.
Astrocytes are one of the major cell types responding to acute as well as chronic damage of the CNS. Following a lesion, they rapidly proliferate and secrete a variety of immunological factors such as proinflammatory cytokines, as well as extracellular matrix molecules (Dong and Benveniste, 2001). Upregulation of GFAP is one hallmark of astrocytic response in the damaged brain.
To determine whether astrocytes are cellular sources of potential detrimental cytokines during demyelination, messenger RNA levels for different NF-κB-dependent cytokines and chemokines were measured in conditions of acute demyelination. For microdissection of astrocytes and microglia, GFAP and CD11b immunohistochemistry on brain sections containing the corpus callosum was performed (Fig. 7A). Immunostained sections were counterstained with Hoechst 33342 to facilitate the identification of individual cells. These immunostained serial sections were then used to dissect GFAP+ astrocytes (upper row) and CD11b+ microglia (lower row) from the lesion site by a pulsed UV laser beam. Cytokine messenger RNA expression was subsequently analysed by quantitative real-time polymerase chain reaction (Fig. 7B). We found a 9.1 ± 2.4-fold increase of CXCL10 messenger RNA in astrocytes from wild-type mice subjected to cuprizone compared with cells from untreated mice. Inhibition of neuroectodermal NF-κB resulted in strongly reduced expression of CXCL10 in IKK2CNS-KO mice (3.6 ± 0.1-fold) whereas microglial CXCL10 levels were only slightly reduced (8.4 ± 2.9-fold in IKK2CNS-wild-type and 6.6 ± 1.8-fold in IKK2CNS-KO). Similarly, CCL2 transcripts were upregulated in wild-type astrocytes in diseased brains (6.1 ± 2.7-fold) and robustly decreased in IKK2 lacking astrocytes (0.7 ± 0.5-fold) whereas microglial CCL2 was not influenced by the absence of the NF-κB activator IKK2 in neuroectodermal cells (1.3 ± 0.4-fold in IKK2CNS-wild-type and 0.9 ± 0.5-fold in IKK2CNS-KO). Expression of CCL3 was only slightly modulated in astrocytes and microglia IKK2CNS-KO mice. Inactivation of IKK2-mediated NF-κB activation in IKK2CNS-KO mice decreased TNFα transcripts from 6.5 ± 3.0-fold in wild-type astrocytes to 1.2 ± 0.4-fold. Notably, TNFα expression in microglia was not influenced in IKK2CNS-wild-type and IKK2CNS-KO cuprizone-treated mice (26.1 ± 8.6-fold compared with 27.3 ± 4.3-fold).
Figure 7.

Selective downregulation of NF-κB activation in neuroectodermal significantly inhibits myelin loss. (A) Laser microdissection of GFAP+ astrocytes (upper row) and CD11b+ microglia (lower row) from the lesion site in the corpus callosum after 5 weeks of cuprizone treatment. Location of cells before (left) and after (right) microdissection. Scale bar = 50 µm. (B) Quantification of cytokine and chemokine messenger RNA in microdissected astrocytes and microglia reveals IKK2-dependent suppression of gene induction in astrocytes. Messenger RNA levels were normalized to β-actin and compared with untreated controls. Mean ± SEM from at least three mice per group are exhibited. *P < 0.05. (C) Histopathological analysis of myelin loss and accompanying gliosis in GFAP-IκBα-dn and wild-type (WT) mice 5 weeks after cuprizone treatment. Sections were assessed for myelin damage by luxol fast blue (LFB) and for gliosis (immunostaining for astrocytes by GFAP and for microglia by MAC-3, respectively). Neuropathological changes were quantified (below) and statistically significant changes were marked with *P < 0.05. (D) Expression of demyelination-associated genes in the corpus callosum of GFAP-IκBα-dn (black bars) and wild-type (white bars) animals during toxic demyelination. Data are expressed as mean ± SEM of at least four animals per group and significant changes are indicated *P < 0.05.
Furthermore, we addressed the contribution of astroglial NF-κB myelin loss in vivo by using a transgenic mouse strain in which NF-κB was selectively inhibited in astrocytes by overexpression of a dominant negative form of the NF-κB inhibitor IκBα (IκBα-dn) under the control of the astrocyte-specific GFAP promoter (Brambilla et al., 2005). Histological analysis of the corpus callosum after 5 weeks of cuprizone-mediated demyelination showed clearly more preserved myelin in GFAP-IkBα-dn mice compared with control wild-type littermate mice (Fig. 7C). In addition, reduced GFAP and MAC-3 immunoreactivity was detected in mutant animals indicating a diminished gliosis. To investigate the molecular mechanisms behind the improved myelin preservation in GFAP-IκBα-dn mice, we evaluated the expression of NF-κB-dependent genes in the corpus callosum (Fig. 7D). Corpus callosum messenger RNA levels for multiple cytokines and chemokines were statistically significant different between wild-type and transgenic animals showing a clear reduction in TNFα, IL-1β and CCL2 in GFAP-IκBα-dn. On the contrary, levels of CCL3 and CXCL10 were not statistically different. These results show that inhibiting NF-κB in astrocytes effectively reduces the inflammatory response that is associated with myelin damage in the cuprizone model.
To further investigate the mechanisms that underlie myelin protection in IKK2CNS-KO mice on the cellular level, we prepared astrocytes and microglia from IKK2CNS-KO and wild-type mice. We measured NF-κB nuclear localization in cultured astrocytes upon stimulation with lipopolysaccharide. Astrocytes from wild-type mice had a robust NF-κB activation, as demonstrated by nuclear translocation of p65 (Supplementary Fig. 4A). In contrast, mutant astrocytes displayed impaired p65 nuclear translocation, showing that IKK2 deficiency inhibits NF-κB activation in IKK2CNS-KO astrocytes. We then tested the capacity of IKK2-deficient astrocytes to express NF-κB target genes upon stimulation. IKK2-ablated astrocytes failed to mount a strong expression of IL-1β, CCL2 or CXCL10 messenger RNAs, demonstrating that IKK2-dependent NF-κB activation is crucial for efficient induction of proinflammatory mediators by astrocytes. In contrast to astrocytes, microglia derive from the mesoderm (Priller et al., 2001; Mildner et al., 2007) and should therefore not be targeted by the Nestin-Cre transgene. Indeed, microglia from wild-type and mutant mice showed similar NF-κB activation and induction of proinflammatory genes in response to lipopolysaccharide, demonstrating that microglia in IKK2CNS-KO mice do not have impaired NF-κB signalling (Supplementary Fig. 4B). Cuprizone, however, could not induce nuclear p65 translocation in astrocytes in vitro, not alone or in combination with lipopolysaccharide or TNFα, nor could it induce the induction of TNFα, IL-1β, IP-10 messenger RNAs after stimulation (data not shown). Indicating that, cuprizone does not seem to induce an inflammatory response leading to demyelination by directly acting on astrocytes. It seems more likely that astrocyte activation is an indirect effect of an initial insult induced in oligodendrocytes by the cuprizone treatment. With ongoing degradation of oligodendrocytes and their ensheathing membranes, glial cells are recruited to the site of damage where they are activated and release a plethora of potentially harmful factors contributing to and amplifying tissue damage in vivo.
Discussion
Here we investigated the role of NF-κB activator in mediating oligodendrocyte maturation, myelination, de- and remyelination in the CNS using different experimental paradigms. The NF-κB pathway can be activated by diverse stimuli, including inflammatory cytokines such as TNF, neurotrophic factors like nerve growth factor, neurotransmitters, cell adhesion molecules and various types of cell stress (Hayden and Ghosh, 2008). NF-κB was also shown to play an important role in regulating cell survival and synaptic plasticity in the CNS (Mattson and Camandola, 2001). Only recently, activation of the classical NF-κB pathway has been shown to be crucial in CNS-related pathologies such as in ischaemic stroke and autoimmune inflammation (Herrmann et al., 2005; van Loo et al., 2006; Brambilla et al., 2009). In general, the reported roles of IKK2 vary considerably depending on the context of investigation and the model systems that were applied. The described functions of IKK2 range from the promotion to the inhibition of cancer, tissue development, tissue regeneration, inflammation, as well as the induction or inhibition of apoptosis (Gerondakis et al., 2006; Schmid and Birbach, 2008).
Although previous elegant studies have established the cellular dynamics of myelination (Bunge et al., 1989), it remains a mystery how signals are communicated to myelinating cells from axons and adjacent myelinating cells. Some cues from axons stimulate a genetic programme in Schwann cells and oligodendrocytes that involves the production of specialized membranes with very high content of cholesterol, sphingolipids and myelin-specific proteins (Michailov et al., 2004; Kassmann et al., 2007).
Previously, Nickols and colleagues (2003) provided compelling evidence that NF-κB orchestrates the process of myelination in Schwann cells of the PNS. They found that NF-κB activity in the sciatic nerve of developing rodents was at its maximum during the period when Schwann cells were actively myelinating axons. Most importantly, an essential role for NF-κB in Schwann cell differentiation and myelination in vitro was established by blocking NF-κB activity with a peptide inhibitor or by overexpression of a mutant (dominant repressor) form of IκBα. Moreover, the production of myelin was drastically reduced in dorsal root ganglion neurons cultured from mice lacking the p65 subunit of NF-κB. We show here that in contrast to the PNS, NF-κB activation by IKK2 in the CNS is dispensable for the differentiation of oligodendrocytes and the process of myelination during development. These results may come as a surprise, since inflammatory cytokines, such as those produced by glial cells during demyelination and remyelination, are potent inducers of NF-κB in a variety of cell types, including oligodendrocytes and Schwann cells (Arnett et al., 2001; Nicholas et al., 2001; Huang et al., 2002; Hamanoue et al., 2004). Nevertheless, we could not establish an essential role for the IKK2-mediated activation of the NF-κB pathway during developmental myelination and remyelination using cell-specific inhibition of NF-κB signalling in neuroectodermal cells of the brain. Consistent with these results, we found that NF-κB was activated in Schwann cells in the PNS but not in oligodendrocytes in the CNS during myelination, similar to previous reports (Bakalkin et al., 1993; Schmidt-Ullrich et al., 1996; Nickols et al., 2003). As the nestin promoter, driving Cre expression in IKK2CNS-KO mice as used in our study, is already active during early embryogenesis, oligodendrocytes having their maximal activity during post-natal myelination should be targeted. We therefore conclude that in contrast to Schwann cells in the PNS, oligodendrocytes do not require IKK2 mediated NF-κB activation for orchestrating axon myelination in the CNS. Furthermore, our data also argue against a role for IKK2-mediated NF-κB activation in regulating the remyelination process by oligodendrocytes. This is an important issue as TNF was shown to be required for both remyelination and proliferation of oligodendrocyte progenitor cells (Arnett et al., 2001). NF-κB activation is further thought to be important in response to stress and injury (Vollgraf et al., 1999). Other in vitro studies have shown that NF-κB exhibits antagonistic properties in oligodendrocytes; it has a pro-survival role and promotes maturation of oligodendrocyte progenitor cells (Nicholas et al., 2004), a finding that was not apparent by our in vivo study as we observed a similar amount of terminal deoxynucleotidyl transferase dUTP nick end labelling positive cells in the developing IKK2-deficient brain (not shown) and a normal number of mature oligodendrocytes in the adult corpus callosum. Immunoreactivity for p65 NF-κB in oligodendrocytes located at the edge of active lesions and in macrophages/microglia throughout plaques has been reported in multiple sclerosis, suggesting oligodendrocyte involvement in the repair process (Bonetti et al., 1999). Despite the fact that our data do not indicate an active role for oligodendrocyte-specific IKK2 during remyelination, we can not exclude that the use of a different oligodendrocyte-specific promoter driving the Cre (such as CNP Cre) might depict a role for NF-κB activation in immature oligodendrocyte progenitor cells. Future studies will address the role of NF-κB signalling in non-myelinating oligodendrocyte progenitor cells.
Most importantly, we demonstrate an essential pathogenic role for brain-specific IKK2-dependent NF-κB signalling during toxin-induced demyelination in vivo. The amelioration of demyelination in mice with brain-restricted NF-κB inhibition correlated with impaired induction of inflammatory cytokines, which are potentially toxic for oligodendrocytes. Despite the fact that IKK2-mediated canonical NF-κB signalling is known to be involved in the regulation of a plethora of effects, these findings suggest that the mechanism by which inhibition of canonical NF-κB signalling in the CNS mediates its myelin-protecting effect is by preventing the expression of proinflammatory mediators by CNS-resident cells. However, since IKK2-mediated canonical activation of NF-κB was described to be involved in the regulation of numerous factors like chemokines, cell-adhesion molecules and other transcription factors (www.NF-kB.org) (Perkins, 2007; Vallabhapurapu and Karin, 2009), additional paradigms of the role of canonical NF-κB signalling in the context of myelin-protection in the CNS might be considered as well. Moreover, despite the fact that the most prominent function of IKKβ is allocated to the canonical activation of NF-κB (by phosphorylation of inhibitory molecules) (Hayden and Ghosh, 2008), recent studies indicate that IKK2 is also able to target/phosphorylate proteins independent of NF-κB (Chariot, 2009). The growing list of target molecules includes signalling molecules such as 14-3-3β, (Gringhuis et al., 2005), as well as transcription factors such as FOXO3a (Hu et al., 2004). Hence, a potential influence of IKK2 dependent but NF-κB independent functions in the context of de- and remyelination in the CNS, cannot be completely excluded, and therefore require further investigation.
Which are the cells mediating oligodendrocyte damage during demyelinating events? The plethora of brain endogenous cytokines and chemokines is produced by microglia and astrocytes (Hanisch, 2002). However, microglial cells were not targeted by the CNS-specific approach we used and we have demonstrated unaltered NF-κB activation in microglia. Using GFAP-IκBα-dn mice, we provide evidence that inactivation of NF-κB in astrocytes protects from myelin loss, reduces the expression of proinflammatory mediators and suppresses gliosis during cuprizone-induced demyelination. These NF-κB-dependent processes initiated in astrocytes are potentially responsible for subsequent activation of other glial cells, especially microglia that could enhance myelin loss. This is also supported by the fact that dissected microglia from IKK2CNS-KO animals also showed decreased gene induction of CCL3 and CXCL10 during toxic demyelination. NF-κB activation in astrocytes induces both detrimental and protective effects on CNS recovery. For the beneficial effects, reactive astrocytes have a protective role in brain ischaemia that is linked to changes in glutamate transport important for neuronal restoration and the control of glial gap junctions (Li et al., 2008). NF-κB activation in astrocytes further leads to the synthesis of neurotrophins such as nerve growth factor and brain derived neurotrophic factor that are essential for neuronal survival (Zaheer et al., 2001). As for the detrimental functions, astroglial NF-κB activation increases excitotoxic brain injury, resulting in decreased neuroprotection (Acarin et al., 2001). Further, NF-κB stimulation promotes the transition of astrocytes to a substrate non-permissive to neurite outgrowth (de Freitas et al., 2002). After spinal cord injury, astrocyte-specific suppression of NF-κB signalling improves functional recovery by modulation of proteoglycan expression and modulation of the inflammatory response (Brambilla et al., 2005). Finally, NF-κB-dependent expression of vascular cell adhesion protein-1 by astrocytes participates in autoimmune demyelination, which is completely abolished in CNS-specific IKK-deficient mice (van Loo et al., 2006). Collectively, these data point to an important role for NF-κB in astroglial cells controlling brain damage. Importantly, the majority of our data were obtained in specific mouse models of toxic demyelination using either the copper chelator cuprizone or lysolecithin. The value of these models for studying demyelination is the high reproducibility and robustness of myelin damage allowing accurate description of oligodendrocyte features during de-and remyelination. Our experimental approach, however, does not allow us to assess the specific contribution of antigen-specific T lymphocytes to oligodendrocyte damage as found in the brains of patients with multiple sclerosis or in its mouse model experimental autoimmune encephalomyelitis. Although it is possible from our own previous report that showed a pathogenic role of NF-κB in neuroectodermal cells during experimental autoimmune encephalomyelitis (van Loo et al., 2006), these findings have to be confirmed for their relevance in multiple sclerosis. Similarly, whether oligodendrocyte loss is also IKK2-mediated in other demyelinating diseases such as leukodystrophies has to be validated in the respective models such as the twitcher murine model of Krabbe disease and others.
In conclusion, our study highlights IKK2-dependent NF-κB signalling as a key pathogenic pathway during toxic CNS demyelination, which could potentially be targeted to suppress demyelination-associated diseases as well.
Funding
The Gemeinnützige Hertie-Stiftung (GHST); the German Research Council [Deutsche Forschungsgemeinschaft, PR 577/5-1 (to M.P.), Sonderforschungsbereich TRR43/A7 (to J.P. and M.P.), TRR43/B3 (to D.M.), DE 551/8-1 to (T.D.)]; BMBF funded competence network of Multiple Sclerosis (KKNMS to M.P.); Deutsche Forschungsgemeinschaft funded research unit (FOR to M.P.) 1336 ‘From monocytes to brain macrophages-conditions influencing the date of myeloid cells in the brain’; Gertrud Reemtsma foundation, fellowship (to A.M.).
Supplementary material
Supplementary material is available at Brain online.
Acknowledgements
We would like to thank Olga Kowatsch, Mariann Schedensack, Jessica Weiland, Heike Korff and Mandy Paul for excellent technical assistance, Rüdiger Klein for providing the Nestin-Cre mice and Florian Kirchheim for technical support. We are grateful to Klaus-Armin Nave, Göttingen, and Rudolf Martini, Würzburg, for critical reading the article and their constructive comments.
Glossary
Abbreviations
- CNP
2′, 3′-cyclic nucleotide 3′-phosphodiesterase
- GFAP
glial fibrillary acidic protein
- IKK
IκB kinase
- NF-κB
nuclear factor kappa B
- TNF
tumour necrosis factor
References
- Acarin L, Gonzalez B, Castellano B. Triflusal posttreatment inhibits glial nuclear factor-kappaB, downregulates the glial response, and is neuroprotective in an excitotoxic injury model in postnatal brain. Stroke. 2001;32:2394–402. doi: 10.1161/hs1001.097243. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Fancy SP, Alberta JA, Zhao C, Plant SR, Kaing S, et al. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;306:2111–5. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–22. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Bakalkin GY, Yakovleva T, Terenius L. NF-kappa B-like factors in the murine brain. Developmentally-regulated and tissue-specific expression. Brain Res Mol Brain Res. 1993;20:137–46. doi: 10.1016/0169-328x(93)90119-a. [DOI] [PubMed] [Google Scholar]
- Bonetti B, Stegagno C, Cannella B, Rizzuto N, Moretto G, Raine CS. Activation of NF-kappaB and c-jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am J Pathol. 1999;155:1433–8. doi: 10.1016/s0002-9440(10)65456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, et al. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–56. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, et al. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol. 2009;182:2628–40. doi: 10.4049/jimmunol.0802954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann BG, Agarwal A, Sereda MW, Garratt AN, Müller T, Wende H, et al. Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron. 2008;59:581–95. doi: 10.1016/j.neuron.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunge RP, Bunge MB, Bates M. Movements of the Schwann cell nucleus implicate progression of the inner (axon-related) Schwann cell process during myelination. J Cell Biol. 1989;109:273–84. doi: 10.1083/jcb.109.1.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach GJ, Dehn D, Del Turco D, Staufenbiel M, Deller T. Laser microdissection reveals regional and cellular differences in GFAP mRNA upregulation following brain injury, axonal denervation, and amyloid plaque deposition. Glia. 2004;48:76–84. doi: 10.1002/glia.20057. [DOI] [PubMed] [Google Scholar]
- Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009;19:404–13. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- de Freitas MS, Spohr TC, Benedito AB, Caetano MS, Margulis B, Lopes UG, et al. Neurite outgrowth is impaired on HSP70-positive astrocytes through a mechanism that requires NF-kappaB activation. Brain Res. 2002;958:359–70. doi: 10.1016/s0006-8993(02)03682-x. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–90. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- Gerondakis S, Grumont R, Gugasyan R, Wong L, Isomura I, Ho W, et al. Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene. 2006;25:6781–99. doi: 10.1038/sj.onc.1209944. [DOI] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–71. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Gringhuis SI, Garcia-Vallejo JJ, Van Het HB, Van Dijk W. Convergent actions of I kappa B kinase beta and protein kinase C delta modulate mRNA stability through phosphorylation of 14-3-3 beta complexed with tristetraprolin. Mol Cell Biol. 2005;25:6454–63. doi: 10.1128/MCB.25.15.6454-6463.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanoue M, Yoshioka A, Ohashi T, Eto Y, Takamatsu K. NF-kappaB prevents TNF-alpha-induced apoptosis in an oligodendrocyte cell line. Neurochem Res. 2004;29:1571–6. doi: 10.1023/b:nere.0000029571.39497.56. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–55. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, et al. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–9. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- Hiremath MM, Saito Y, Knapp GW, Ting JP, Suzuki K, Matsushima GK. Microglial/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J Neuroimmunol. 1998;92:38–49. doi: 10.1016/s0165-5728(98)00168-4. [DOI] [PubMed] [Google Scholar]
- Hovelmeyer N, Hao Z, Kranidioti K, Kassiotis G, Buch T, Frommer F, et al. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:5875–84. doi: 10.4049/jimmunol.175.9.5875. [DOI] [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–37. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Huang CJ, Nazarian R, Lee J, Zhao PM, Espinosa-Jeffrey A, de VJ. Tumor necrosis factor modulates transcription of myelin basic protein gene through nuclear factor kappa B in a human oligodendroglioma cell line. Int J Dev Neurosci. 2002;20:289–96. doi: 10.1016/s0736-5748(02)00022-9. [DOI] [PubMed] [Google Scholar]
- Jaegle M, Mandemakers W, Broos L, Zwart R, Karis A, Visser P, et al. The POU factor Oct-6 and Schwann cell differentiation. Science. 1996;273:507–10. doi: 10.1126/science.273.5274.507. [DOI] [PubMed] [Google Scholar]
- Kassmann CM, Lappe-Siefke C, Baes M, Brügger B, Mildner A, Werner HB, et al. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet. 2007;39:969–76. doi: 10.1038/ng2070. [DOI] [PubMed] [Google Scholar]
- Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, et al. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab. 2008;28:468–81. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–16. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–54. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, et al. Axonal neuregulin-1 regulates myelin sheath thickness. Science. 2004;304:700–3. doi: 10.1126/science.1095862. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, et al. Microglia in the adult brain arise from Ly-6C(hi)CCR2(+) monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–53. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Nicholas RS, Wing MG, Compston A. Nonactivated microglia promote oligodendrocyte precursor survival and maturation through the transcription factor NF-kappa B. Eur J Neurosci. 2001;13:959–67. doi: 10.1046/j.0953-816x.2001.01470.x. [DOI] [PubMed] [Google Scholar]
- Nickols JC, Valentine W, Kanwal S, Carter BD. Activation of the transcription factor NF-kappaB in Schwann cells is required for peripheral myelin formation. Nat Neurosci. 2003;6:161–7. doi: 10.1038/nn995. [DOI] [PubMed] [Google Scholar]
- Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, et al. Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J Neurosci. 2003;23:5393–406. doi: 10.1523/JNEUROSCI.23-13-05393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousman SS, David S. Lysophosphatidylcholine induces rapid recruitment and activation of macrophages in the adult mouse spinal cord. Glia. 2000;30:92–104. [PubMed] [Google Scholar]
- Pasparakis M, Courtois G, Hafner M, Schmidt-Supprian M, Nenci A, Toksoy A, et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature. 2002;417:861–6. doi: 10.1038/nature00820. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Priller J, Flugel A, Wehner T, Boentert M, Haas CA, Prinz M, et al. Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat Med. 2001;7:1356–61. doi: 10.1038/nm1201-1356. [DOI] [PubMed] [Google Scholar]
- Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, et al. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006;116:456–64. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Hanisch UK. Murine microglial cells produce and respond to interleukin-18. J Neurochem. 1999;72:2215–8. doi: 10.1046/j.1471-4159.1999.0722215.x. [DOI] [PubMed] [Google Scholar]
- Prinz M, Montrasio F, Furukawa H, van der Haar ME, Schwarz P, Rülicke T, et al. Intrinsic resistance of oligodendrocytes to prion infection. J Neurosci. 2004;24:5974–81. doi: 10.1523/JNEUROSCI.0122-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. 2008;28:675–86. doi: 10.1016/j.immuni.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)–a key molecule in signaling to the transcription factor NF-kappaB. Cytokine Growth Factor Rev. 2008;19:157–65. doi: 10.1016/j.cytogfr.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Schmidt-Ullrich R, Memet S, Lilienbaum A, Feuillard J, Raphael M, Israel A. NF-kappaB activity in transgenic mice: developmental regulation and tissue specificity. Development. 1996;122:2117–28. doi: 10.1242/dev.122.7.2117. [DOI] [PubMed] [Google Scholar]
- Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, et al. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–9. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- van Loo G, De LR, Schmidt H, Huth M, Mildner A, Schmidt-Supprian M, et al. Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol. 2006;7:954–61. doi: 10.1038/ni1372. [DOI] [PubMed] [Google Scholar]
- Vollgraf U, Wegner M, Richter-Landsberg C. Activation of AP-1 and nuclear factor-kappaB transcription factors is involved in hydrogen peroxide-induced apoptotic cell death of oligodendrocytes. J Neurochem. 1999;73:2501–9. doi: 10.1046/j.1471-4159.1999.0732501.x. [DOI] [PubMed] [Google Scholar]
- Zaheer A, Yorek MA, Lim R. Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion. Neurochem Res. 2001;26:1293–9. doi: 10.1023/a:1014241300179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.