

Key Points
The fludarabine and bendamustine combination is cytotoxic to CLL cells even in the presence of a protective microenvironment.
H2AX activation was maximum with the combination, and unscheduled DNA synthesis induced by bendamustine was blocked by fludarabine.
Abstract
The fludarabine and cyclophosphamide couplet has become the backbone of the chronic lymphocytic leukemia (CLL) standard of care. Although this is an effective treatment, it results in untoward toxicity. Bendamustine is a newly approved and better-tolerated alkylating agent. We hypothesized that similar to cyclophosphamide, bendamustine-induced DNA damage will be inhibited by fludarabine, resulting in increased cytotoxicity. To test this hypothesis and the role of the stromal microenvironment in this process, we treated CLL lymphocytes in vitro with each drug alone and in combination. Simultaneous or prior addition of fludarabine to bendamustine resulted in maximum cytotoxicity assayed by 3,3′-dihexyloxacarbocyanine iodine negativity, annexin positivity, and poly (adenosine 5′-diphosphate-ribose) polymerase cleavage. Cytotoxicity elicited by combination of both agents was similar in these malignant B cells cultured either in suspension or on marrow stroma cells. Cell death was associated with DNA damage response, which was determined by phosphorylation of H2AX and unscheduled DNA synthesis. H2AX activation was maximum with the drug combination, and unscheduled DNA synthesis induced by bendamustine was blocked by fludarabine. In parallel, ATM, Chk2, and p53 were phosphorylated and PUMA was induced. Cell death was caspase independent; however, caspases did decrease levels of Mcl-1 survival protein. These data provide a rationale for combining fludarabine with bendamustine for patients with CLL.
Introduction
The most efficacious therapies in chronic lymphocytic leukemia (CLL) include alkylating agents and the combination of these DNA-damaging drugs with purine nucleoside analogs. In fact, the combination of cyclophosphamide and fludarabine or pentostatin has long been the standard of care for CLL.
Bendamustine is a newly approved alkylating agent. Chemically, bendamustine is 4-{5-[bis(2-chloroethyl)amino]-1-methyl-2-bezimidazolyl} butyric acid hydrochloride.1 Structurally, it is an alkylating agent with a benzimidazole ring and a butyric acid side chain, which improves water solubility.2 The nitrogen mustard group of bendamustine resembles a similar group on chlorambucil and cyclophosphamide, the 2 most commonly used alkylating agents for CLL.
When chlorambucil, which is among the oldest drugs used for the treatment of CLL,3 was compared with bendamustine for efficacy and toxicity profiles, the overall response rate to bendamustine was 68%, which was more than double the observed rate with chlorambucil.4 Based on these results, bendamustine was approved by the US Food and Drug Administration for the treatment of CLL.5 In another randomized clinical study of untreated CLL, fludarabine resulted in higher response rates and a longer duration of remission and progression-free survival than did single-agent chlorambucil.6 Collectively, these data illustrate the benefits of single-agent bendamustine or fludarabine for the treatment of CLL.
The most-used alkylating agent for CLL, although not alone but rather in combination with fludarabine, is cyclophosphamide. When compared head to head, the fludarabine and cyclophosphamide regimen was preferred to fludarabine or chlorambucil.7 With these clinical investigations, the fludarabine plus cyclophosphamide couplet with or without monoclonal antibodies (mAbs) has become standard of care for patients with CLL.8-11
The choice of this combination of a purine nucleoside analog (fludarabine) with an alkylating agent (cyclophosphamide) was based on the mechanism of action. Cyclophosphamide-mediated DNA damage results in monoadducts, biadducts, and intra- and interstrand crosslinks. This DNA damage initiates a repair response, and in most cases cells repair the damage with no or minimal biological response, especially in cells such as CLL lymphocytes that are characterized by an increased DNA repair ability.12,13 This biological property provides a rationale for combining alkylating agents with chemotherapeutic drugs such as fludarabine that inhibit DNA synthesis. Such rationales have led to clinical investigations of these 2 agents in combination.14 These preclinical data and the above-mentioned clinical results with bendamustine underscore the potential importance of combining fludarabine with bendamustine.
To test such an approach, we combined bendamustine with fludarabine in primary CLL cells. We identified the optimal schedule and determined the mechanistic basis for the effectiveness of this combination by quantitating DNA damage, maintenance of damage response, effect on DNA/RNA synthesis, and effect on proteins impacted by DNA damage and repair response. Furthermore, we evaluated the biological consequences of the single agents and their combination in primary CLL cells. Finally, we compared the presence of a stroma-influenced DNA damage response and repair and the biological consequences in CLL lymphocytes treated with one or the other or both drugs.
Materials and methods
Drugs and chemicals
Bendamustine hydrochloride was obtained from Cephalon (Frazer, PA), and fludarabine was a gift from Berlex Laboratories (Alameda, CA). Fludarabine was dephosphorylated,15 and free nucleoside 2-fluoro-arabinosyladenine (F-ara-A) was used. Z-VAD was purchased from MP Biomedicals (Solon, OH).
Patient samples
The in vitro studies were carried out in freshly isolated lymphocytes obtained from peripheral blood of 38 patients with CLL. Samples were obtained twice from patients 9 and 13 (total samples = 40; see supplemental Table 1 available on the Blood Web site). All patients signed a written informed consent to participate in this laboratory protocol, which was approved by the institutional review board of the University of Texas MD Anderson Cancer Center. This study was conducted in accordance with the Declaration of Helsinki.
Isolation of peripheral blood MNCs, and suspension and stromal cocultures
MNCs were isolated from heparinized peripheral blood by Ficoll gradient separation (Ficoll-Hypaque specific gravity, 1.086; Life Technologies, Grand Island, NY) and were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated human serum, 5% antibiotics (streptomycin, penicillin) (Invitrogen, Carlsbad, CA), and 5% l-glutamate (Mediatech, Homodon, VA). A Coulter Channelyzer (Coulter Electronics, Hialeah, FL) was used to determine cell number and the mean cell volume.
For CLL-stromal cocultures, lymphocytes were cultured on confluent layers of human stromal cells (NK-Tert; RIKEN cell bank, Tsukuba, Japan) at a ratio of 100 CLL cells to 1 stromal cell. Stromal cells were plated first in medium at 1 × 105 cells per well, and after 8 to 24 hours, the medium was removed and 1 × 107 CLL cells per well were added.
CB CD34-positive cells
Cord blood (CB) units were provided under protocols approved by The University of Texas MD Anderson Cancer Center institutional review board. CB mononuclear cells (MNCs) were prepared by centrifugation on Ficoll layers (Histopaque 1077, Sigma) at 400g for 20 minutes before collecting the interface cells. Washed CB MNCs were incubated in Clinimacs buffer (phosphate-buffered saline [PBS] EDTA buffer, Miltenyi Biotec) containing CD34 microbeads (CD34 reagent, Miltenyi Biotec), 0.5% human serum albumin (Baxter Healthcare), and an Fc blocker (Gammagard, Baxter Healthcare) for 30 minutes on ice. After incubation, CB CD34-positive cells were selected by magnetic-activated cell sorting (LS Column, Miltenyi Biotec) according to the manufacturer’s instructions.
Cell viability assays
DiOC6 staining.
Cell viability was determined using 3,3′-dihexyloxacarbocyanine iodine (DiOC6) dye to test the mitochondrial membrane potential and propidium iodide (PI) to test cell membrane permeability using an FACScalibur (Becton Dickinson).
Annexin V/PI staining.
Cells were incubated with annexin V/fluorescein isothiocyanate (5 µL) (BD Pharmingen, San Diego, CA), and to the labeled cells 10 µL PI (50 µg/mL) was added. The cells were analyzed immediately with an FACScalibur. At least 10 000 events per sample were recorded and processed using Cell Quest software (Becton Dickinson).
Flow cytometry detection of phosphorylated-H2AX
CLL cells were fixed by adding 4% paraformaldehyde (BD Biosciences, San Jose, CA) and after washing were resuspended in 75% ice-cold ethanol. The fixed CLL cells were permeabilized, washed, and incubated for 1 hour with 3% donkey serum in PBS to suppress nonspecific antibodies. Cells were then resuspended in 100 µL of 3% donkey serum containing anti-phosphohistone H2AX (Ser139) mAb (Millipore; 1:125 titer). The cells were then washed and resuspended in 3% donkey serum containing fluorescein isothiocyanate–conjugated mAb (Millipore, 1:100). The labeled cells were resuspended in PBS containing the counterstain PI (15 µg/mL) and RNase (Roche) (2.5 µg/mL) and analyzed using FACScalibur.
Uridine and thymidine incorporation
For RNA and DNA synthesis assays, radioactivity incorporation was measured using [3H]uridine and [3H]thymidine (specific activity, 48 and 68.5 Ci/mmol; Moravek Biochemicals, Brea, CA), respectively. For this, 10 μCi/mL [3H]uridine or [3H]thymidine were added to the cell cultures and left for half an hour, and aliquots were collected in triplicate. The radioactivity was measured by liquid scintillation counter (Millipore, Bedford, MA) and expressed as percentage of control (untreated) cells.
Immunoblot analysis
CLL cell pellets were lysed at 4°C in radioimmunoprecipitation assay buffer supplemented with 1 Mini cOmplete protease inhibitor tablet (Roche). Aliquots (30-50 µg) of total protein were loaded onto 4% to 12% sodium dodecyl sulfate polyacrylamide gels and transferred to nitrocellulose membranes (GE Osmonics Labstore, Minnetonka, MN).16 The membranes were blocked and incubated with primary antibodies against the following: total ATM (Abcam, Cambridge, MA), phospho-ATM (Millipore, Billerica, MA), Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam), Mcl-1 (Santa Cruz Biotechnology), PUMA (ProSci, Poway, CA), total p53 (EMD Chemicals, Gibbstown, NJ), phospho-p53 (Cell Signaling Technology, Danvers, MA), and poly (adenosine 5′-diphosphate-ribose) polymerase (PARP) (BD Pharmingen). After being washed, membranes were incubated with infrared-labeled secondary antibodies and then visualized using a LI-COR Odyssey infrared imager.
Statistical analyses
For 2-group comparisons, we used Student t tests. For multigroup comparisons, we used 1-way and 2-way analysis of variance (ANOVA) models. Generalized linear regression mixed models were used to study the effects of bendamustine, fludarabine, and their combination on responses measured over time such as cell apoptosis and percent γ-H2AX (Figure 4). An unstructured covariance structure is used to account for interpatient variability over time, and responses in different treatment groups and different media (eg, stroma/suspension) were assumed to be independent. Comparison of treatment effects by groups was done using the CONTRAST statement in PROC MIXED. SAS version 9.1 and S-Plus version 8.0 were used to carry out the computations for all analyses. Adjustments for multiple comparisons were made using the Bonferroni procedure.
Figure 4.
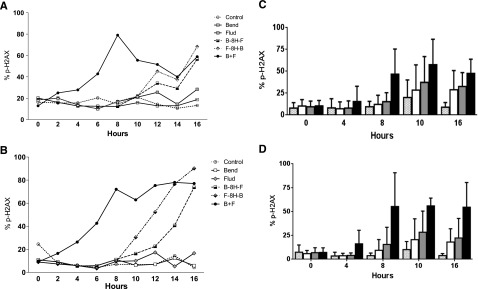
Phosphorylation of H2AX in CLL cells after treatment with bendamustine, fludarabine, or their combination. CLL cells were either cultured in suspension (A,C) or cocultured with bone marrow stroma cells (B,D). Panels A and B show samples from 1 patient that were either untreated (control) or incubated with 30 µM bendamustine, 10 µM fludarabine, their simultaneous combination, or their sequential combination (bendamustine followed by fludarabine or fludarabine followed by bendamustine) for the indicated times. Phosphorylation of H2AX was measured using flow cytometry. Panels C and D show results from 10 patient samples, and cells were either untreated (speckled bars) or treated with bendamustine alone (white bar), fludarabine alone (gray bar), or the simultaneous combination of fludarabine and bendamustine (black bar). Phosphorylation of H2AX was measured using flow cytometry, and data are mean ± SD. 8H, 8 hours; B, bendamustine; F, fludarabine.
Results
Induction of apoptosis in CLL cells by bendamustine, fludarabine, and their combination
For all cell death assays, a time-matched control (untreated cells) was used, and this background value (generally 5% to 20%) was subtracted from the drug-treated value. Data were similar with both assays (DiOC6 negativity, Figure 1; annexin V/PI positivity, supplemental Figure 1). There was a dose- and time-dependent increase in cell death from the same 3 samples (1, 2, and 3) when incubated with either bendamustine or fludarabine for 24, 48, and 72 hours. We selected 3, 10, and 30 µM concentrations of these drugs, because during therapy, the fludarabine peak plasma level is 3 µM17 and the bendamustine level is 30 µM.18 Maximum cell death was generally observed with 30 µM bendamustine and 10 µM fludarabine.
Figure 1.
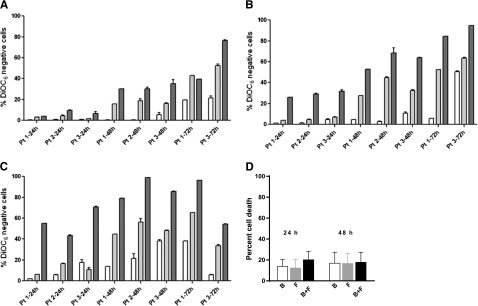
Induction of apoptosis by bendamustine, fludarabine, or their combination in CLL or CD34-positive cells. (A-C) CLL cells from 3 patients (1, 2, and 3) were incubated with bendamustine, fludarabine, or their combination for 24, 48, and 72 hours. Induction of apoptosis by bendamustine (white bars), fludarabine (gray bars), or their combination (black bars) in CLL (A-C) or CD34-positive (D) cells is shown. Cell death was measured by DiOC6 assay as described in “Materials and methods.” Concentrations of the drugs were 3 µM (A), 10 µM (B), or 30 µM (C), and data are from 3 patient samples. Measurements for patients 2 and 3 were done in triplicate. (D) Purified CD34-positive cells from CB were incubated with 30 µM bendamustine, 10 µM fludarabine, or their combination for 24 or 48 hours. All data are presented as mean ± standard deviation (SD). Results are corrected to a time-matched control (untreated sample). B, bendamustine; F, fludarabine.
In simultaneous incubations, compared with bendamustine alone, the combination of bendamustine and fludarabine was significantly higher at lower concentrations (3 and 10 µM) and at 24 and 48 hours (Figure 1A: P = .013 [24 hours], P = .001 [48 hours]; Figure 1B: P = .0006 [24 hours]; supplemental Figure 1A: P = .04 [24 hours], P = .002 [48 hours]; and supplemental Figure 1B: P = .0005 [24 hours]). Longer incubations (72 hours) and higher concentrations (30 µM) did not produce significantly different results (Figure 1C). Similarly, compared with fludarabine alone, there was a small but consistent increase with the combination with significant differences at lower concentrations and at 24 and 48 hours.
Induction of apoptosis in CD34-positive cells or stroma cells
Cell death at 24 or 48 hours was minimum (<15%) in CD34-positive cells, and this was not significantly increased with the combination (Figure 1D).
Similarly, for NK-Tert stroma cells, there was only 5% to 10% increase in apoptosis with the combination of both drugs (data not shown).
Sequence-specific effect of bendamustine and fludarabine
To determine if the sequence of drug affected sensitivity, CLL lymphocytes were incubated with fludarabine and 8 hours later with bendamustine or with bendamustine followed 8 hours later by fludarabine or both drugs together. In all samples, in both suspension and stroma culture conditions, at 24 hours, compared with single-agent treatment (bendamustine or fludarabine), there was a significantly higher cell death when the drugs were combined simultaneously or fludarabine was administered first, followed by bendamustine (compare P values in Figure 2A-B). At 48 hours, the trend was similar (Figure 2C-D). In contrast, when bendamustine was followed by fludarabine, the results were not different from either bendamustine alone or fludarabine alone (Figure 2). These data suggest that the presence of fludarabine with bendamustine was necessary for maximal apoptosis.
Figure 2.
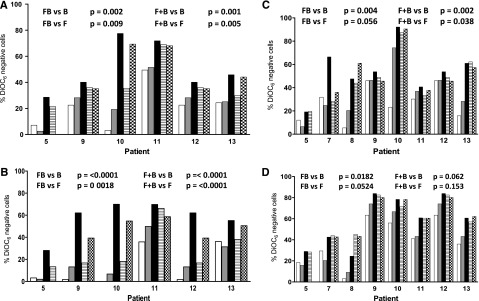
Effect of bone marrow stroma coculturing on the induction of apoptosis in CLL cells by bendamustine, fludarabine, or their combination. CLL cells were either cultured in suspension (A,C) or cocultured with bone marrow stroma cells (B,D) and incubated with 30 µM bendamustine (white bars), 10 µM fludarabine (gray bars), their simultaneous combination (black bars), or their sequential combination (bendamustine followed by fludarabine, line bar; or fludarabine flowed by bendamustine, check bar) for 24 (A-B) or 48 (C-D) hours. Cell death was measured using a DiOC6 assay as described in “Materials and methods.” Data are from individual patient samples, and results are corrected to a time-matched control (untreated sample) for each patient sample. B, bendamustine; F, fludarabine; FB, fludarabine followed by bendamustine; F+B, fludarabine plus bendamustine.
Effect of bone marrow stroma coculturing on induction of apoptosis in CLL cells
For coculturing, we used a standardized method19 in which CLL lymphocytes were cultured with NK-Tert MSCs or in suspension. There was a 10% to 30% survival advantage when CLL cells were cultured on stroma (data not shown). Also, in most samples, 24-hour treatment with either fludarabine or bendamustine resulted in a decrease in apoptosis when cells were cocultured with stroma (compare Figure 2A with Figure 2B, white and gray bars). However, when the drugs were combined, the difference in apoptosis was minor or nonexistent between suspension and stroma cultures, suggesting that the combination overcame stroma-induced resistance. At 24 hours, the percent apoptosis above background values was similar and ranged from 20% to 80% (P = .23 DiOC6 assay, Figure 3A; 0.88 annexin V/PI assay, supplemental Figure 2A). Similarly for 48 hours, data were not significantly different between suspension and stroma coculturing (P = .25 DiOC6, Figure 3B; 0.62 annexin V/PI, supplemental Figure 2B).
Figure 3.

Effect of bone marrow stroma coculturing on the induction of apoptosis in CLL cells by the simultaneous combination of bendamustine and fludarabine. CLL cells were cultured in suspension (white bar) or cocultured with bone marrow stroma cells (gray bar) and incubated with 30 µM bendamustine and 10 µM fludarabine simultaneously for 24 (A) or 48 hours (B). Cell death was measured using a DiOC6 assay as described in “Materials and methods.” Data are from individual patient samples, and results are corrected to a time-matched control (untreated sample) for each patient sample.
Phosphorylation of H2AX in CLL cells
Previous studies14 have established that alkylating agents induce DNA damage and that readout of this DNA damage response and repair is the phosphorylation of H2AX. Compared with untreated cells, CLL cells showed only a 5% to 10% increase in the H2AX phosphorylation (γ-H2AX) signal after treatment with fludarabine or bendamustine for up to 20 hours. This increase was similar in both culture conditions. Treatment with either fludarabine or bendamustine for 8 hours followed by a second drug increased the signal in both culture systems. Between the 2 treatment sequences, fludarabine followed by bendamustine resulted in the higher DNA damage response. The simultaneous addition of both drugs increased the signal 2 hours after the start of incubation and reached 60% or higher in both model systems (Figure 4A-B). The slopes were similar for H2AX as soon as both drugs were combined.
To determine if simultaneous addition of the drugs consistently increased the H2AX phosphorylation signal, similar experiments were performed with the lymphocytes of 10 patients cultured either in suspension (Figure 4C) or on stroma (Figure 4D). Compared with the single agents, the signal was higher when the drugs were combined (P values between .014 and .039 for suspension culture, Figure 4C; P < .001 for stroma cocultures, Figure 4D). Direct comparison of H2AX phosphorylation in stroma vs suspension culture in all 10 patients suggested that although there was heterogeneity among samples, the damage response appears to be similar in both culture systems (P = .12-.50, supplemental Figure 3).
Changes in RNA and DNA synthesis
There was no significant change in uridine incorporation between untreated and bendamustine-treated cells. Fludarabine alone decreased total RNA synthesis in CLL cells cultured either in suspension or on stroma. The combination of bendamustine and fludarabine produced the maximum inhibition, which was similar under both culture conditions (Figure 5A-B).
Figure 5.

RNA and DNA synthesis after treatment with bendamustine, fludarabine, or their combination in CLL cells. RNA and DNA synthesis after treatment with bendamustine alone (▼), fludarabine alone (▲), or their combination (▪) in CLL cells in suspension culture (A,C) and in CLL cells cocultured on human stromal cells (B,D). Cells were obtained from 3 patients (24, 25, and 26). After incubation for 0, 5, and 10 hours, RNA and DNA synthesis activity was measured by [3H]uridine incorporation assay (A-B) and [3H]thymidine incorporation assay (C-D) as described in “Materials and methods.” Values are mean ± SD from 3 patient samples, and each one was done in triplicate. Data are presented as percent of control, where control is the 0-hour untreated sample.
CLL cells from peripheral blood cultured in vitro are in G0 to G1 phase and replicationally quiescent. However, DNA damage response results in an increase in thymidine incorporation due to the initiation of unscheduled DNA synthesis. Fludarabine-treated cells did not show any significant change. In contrast, bendamustine produced a 2-fold and significant increase in DNA synthesis in CLL cells (Figure 5C-D) that was abrogated when fludarabine was added.
Changes in DNA damage signaling molecules
Total ATM was unchanged, but there was an increase in phospho-ATM, which was most prominent with fludarabine treatment. With both drugs alone and in combination, there was a stabilization of p53 protein, which was also evident with phosphorylation of p53 at ser15. At 8 hours, only combination treatment produced phosphorylation of ser15-p53; however, at 24 hours, single agents and the combination of drugs resulted in phosphorylation of p53. Phosphorylation of p53 is a response to DNA damage and repair stress. With the combination of bendamustine and fludarabine, the damage was higher at an earlier time (as reflected in the H2AX data) and hence a stress response was observed at 8 hours. However, with single agents, this response took a longer time (ie, 24 hours). Phosphorylation of p53 at ser20 was also measured, which was induced, but the bands were very faint (not shown). Consistent with the p53 phosphorylation data, there was induction of PUMA in these samples (Figure 6A). The increase in p53 and PUMA was further confirmed in 4 additional patient samples (Figure 6B).
Figure 6.

Immunoblot analysis of DNA damage response. (A) CLL lymphocytes from patient 25 were cultured in suspension or on stroma. These were untreated or treated with bendamustine, fludarabine, or their combination for 8 or 24 hours and then harvested and lysed. Immunoblot analysis of the total and phosphoprotein levels of ATM, total and ser-15 phospho-p53, and total PUMA and Mcl-1 were analyzed. GAPDH was used as a loading control. (B) CLL lymphocytes from patients (35-38) were cultured in suspension and were untreated or treated with bendamustine, fludarabine, or their combination for 24 hours and then harvested and lysed. Immunoblot analysis of the total p53, PUMA, and GAPDH levels were analyzed. (C-D) CLL lymphocytes from patients (31-38) were cultured in suspension and were untreated or treated with bendamustine, fludarabine, or their combination for 24 hours and then harvested and lysed. Immunoblot analysis of the total and phospho thr-68 Chk2 (C) and total Chk1 (D) levels was done. GAPDH was used as a loading control. B, bendamustine; C, control (untreated); F, fludarabine.
Chk1 and Chk2 have been established as mediators of the DNA damage response and phosphorylate p5320-22 and are activated by ATM and ATR kinases.23,24 Although Chk1 was expressed at low levels and phosphorylation was below the limit of detection (Figure 6C), Chk2 was present at high levels and was phosphorylated after treatment with bendamustine, fludarabine, or their combination (Figure 6D). H2AX phosphorylation measured by flow cytometry (Figure 4) was confirmed by immunoblot assays (supplemental Figure 4).
Except for the combination treatment at 24 hours, Mcl-1 protein was unchanged in all samples. At 24 hours, when treated with both agents, there was a slight decrease in Mcl-1 protein that was consistent with the cell-death induction data. Data were similar in both types of culture conditions (Figure 6A).
Impact of a pan-caspase inhibitor in DNA damage signaling molecules and cell death
To evaluate role of caspase activation and to block caspase-mediated protein degradation, cultures were also pretreated with Z-VAD, a pan-specific caspase inhibitor (Figure 7A). Compared with control untreated cells, there was an increase in p53 protein due to stabilization of the protein with either fludarabine or bendamustine. This occurred in concert with increases in phospho-p53 protein. However, at this time point (48 hours) in bendamustine- and fludarabine-treated cells, there was a decrease in p-p53 protein, possibly due to increased cell death. Expression of PUMA protein, a proapoptotic molecule, paralleled the phospho-p53 increase. Mcl-1 protein levels were stable and similar in all conditions after treatment with a single compound but were dramatically decreased with both drugs. This decrease was abrogated when cells were pretreated with Z-VAD, indicating that the dissipation of Mcl-1 protein was due to caspase-mediated cleavage of this antiapoptotic protein. In contrast to Mcl-1, Bcl-2 protein levels remained unchanged with these treatments (Figure 7A).
Figure 7.

Impact of Z-VAD, a pan-caspase inhibitor on cell death. Impact of Z-VAD on cell-death–associated proteins (A) and cell death (B-E). (A) CLL lymphocytes from patient 29 were cultured in suspension or on stroma. These were pretreated with or without Z-VAD and then either untreated or treated with bendamustine, fludarabine, or their combination for 48 hours, harvested, and lysed. Immunoblot analysis of the total and phosphoprotein levels of p53 and total PUMA, Bcl-2, Mcl-1, and cleaved and uncleaved PARP was performed. GAPDH was used as a protein loading control. (B-E) CLL cells from patients (n = 3) cultured in suspension (B-C) or on stroma (D-E) were either untreated (−) or pretreated (+) with Z-VAD followed by a 24- or 48-hour treatment with bendamustine and fludarabine together and assayed for DiOC6 negativity (B,D) or annexin V/PI positivity (C,E). Data are presented as mean with SD from samples from 3 patients, and results are corrected to a time-matched control (untreated sample) for each patient sample. B, bendamustine; C, control (untreated); F, fludarabine.
Because Z-VAD pretreatment mitigated the decline in Mcl-1 protein levels, we evaluated the effect of caspase inhibition on apoptosis of CLL cells. As shown for 2 representative patients, PARP cleavage was similar in CLL cells irrespective of the culture conditions (suspension vs stroma). Furthermore, Z-VAD pretreatment did not affect PARP cleavage. These data suggest that PARP cleavage was caspase independent. Similar data were obtained in a total of 6 patient samples for stroma and suspension culture comparison and in 3 patient samples in the presence and absence of Z-VAD.
To further confirm and extend these investigations, CLL cells from patients (n = 3) cultured in suspension (Figure 7B-C) or on stroma (Figure 7D-E) were either untreated or pretreated with Z-VAD followed by treatment with both drugs. At both 24 and 48 hours, there were similar amounts of cell death in both culture conditions (P = .82 and .84 at 24 and 48 hours in the DiOC6 assay and P = .93 and .93 at 24 and 48 hours in the annexin V/PI assay with Z-VAD; P = .57 and .82 at 24 and 48 hours in the DiOC6 assay and P = .68 and .60 at 24 and 48 hours in the annexin V/PI assay without Z-VAD). Furthermore, cell death was not affected by the presence of Z-VAD (P = .86 and .96 at 24 and 48 hours in the DiOC6 assay, P = .97 and .88 at 24 and 48 hours in the annexin V/PI assay in suspension culture; P = .37 and .60 at 24 and 48 hours in the DiOC6 assay and P = .78 and .47 at 24 and 48 hours in the annexin V/PI assay in stromal cocultures).
Discussion
Bendamustine is a newly approved alkylating agent that showed effectiveness during a clinical trial in patients with lymphoma and CLL.4 This drug appears to be well tolerated and milder than cyclophosphamide. Hence, it is desirable to test bendamustine with a purine nucleoside analog. Previous studies have provided some utility of the bendamustine and fludarabine combination, but the role of survival factors from the microenvironment and mechanistic investigations were missing.
Bendamustine alone showed dose- and time- dependent CLL cell death (Figure 1). Although a distinct pattern of cytotoxicity and mechanisms were observed for bendamustine,25 the primary action was the activation of a DNA damage stress response and apoptosis.26 Although mitotic checkpoint and catastrophe have also been reported specifically in cell lines,26,27 those responses were irrelevant in our system because primary CLL lymphocytes are replicationally quiescent. The cytotoxic response to bendamustine has been shown to be caspase and p53 independent28 and resulted in Puma and Noxa induction28 without impacting the levels of Bcl-2 and Bax,29 and prior treatment did not affect the outcome.29 In the current work, both in suspension and stroma cocultures, we demonstrate phosphorylation of Chk2, phosphorylation and stabilization of p53, and induction of Puma but not Noxa (not shown).
The addition of fludarabine to bendamustine significantly increased cell death29 (Figure 2). Combination index determinations with isobolograms demonstrated that 48-hour incubations of CLL cells with both drugs were synergistic.29 Furthermore, the best combination was simultaneous incubations. The addition of fludarabine followed by bendamustine was also effective, but bendamustine followed by fludarabine was the least potent among these 3 combination strategies (Figure 2). These results underscore the presence of intracellular fludarabine triphosphate at the time of bendamustine treatment, consistent with previous results with the cyclophosphamide and fludarabine couplet.14
Because the primary action of bendamustine is the induction of a DNA damage response and the initiation of DNA repair, we reasoned that fludarabine will block DNA repair and induce higher levels of cytotoxicity. Fludarabine triphosphate inhibits DNA repair by several mechanisms. First, fludarabine triphosphate gets incorporated opposite T’s in the nascent DNA chain during replication or repair patch synthesis.15 Fludarabine incorporation into DNA may be enhanced in replicationally quiescent CLL lymphocytes by the addition of DNA-damaging agents, thus presumably enhancing the cytotoxic activity of fludarabine.14,30 Second, because the fludarabine moiety incorporated at the 3′ end of DNA is resistant to proofreading exonucleases, DNA synthesis terminates.31 Third, fludarabine triphosphate inhibits the DNA primase needed to synthesize primer to initiate DNA synthesis.32,33 Fourth, DNA ligase, required to fill the nick after DNA repair patch synthesis, is inhibited by fludarabine.34 Fifth, fludarabine triphosphate incorporation during messenger RNA synthesis35 may play a role in the inhibition of transcript synthesis, which may include the transcription of genes needed for DNA repair. Finally, fludarabine triphosphate inhibits ribonucleotide reductase, an enzyme responsible for the maintenance of optimal levels of 2′-deoxynucleoside 5′-triphosphate pools for DNA synthesis.36,37
The level and activity of ribonucleotide reductase are low in quiescent CLL lymphocytes; however, DNA damage response and repair induce this enzyme38-43 in an ATM- and p53-dependent pathway to synthesize deoxynucleotides.44 Thus, fludarabine triphosphate–mediated inhibition may block the synthesis of deoxynucleotides required for DNA repair. Collectively, these multifaceted actions would impact DNA damage repair elicited by alkylating agent45; our data on H2AX phosphorylation, which is an indicator of DNA damage, repair, and its inhibition, were in concert with this notion. Once again, an increase in the γ-H2AX signal was observed with the simultaneous combination or increased dramatically as soon as fludarabine was added in the culture of CLL cells being incubated with bendamustine (Figure 4A-B). In parallel with H2AX response data, a 5-hour bendamustine treatment induced a significant increase in DNA synthesis in CLL cells. This was blocked in cells treated with the bendamustine and fludarabine couplet (Figure 5C-D). Although direct fludarabine incorporation in the DNA was not investigated in our system, these data indirectly provide evidence for the fludarabine-mediated inhibition of a bendamustine-induced DNA repair response.
Previous investigations have demonstrated that bone marrow stroma cells (including the NK-Tert human stroma cell line) result in the development of resistance to an alkylating agent19 or purine nucleoside analog.46 Interestingly, our data clearly suggest that the combination of these agents abrogated resistance; cell death effects were similar in CLL samples whether kept in suspension or on stroma (Figure 3).
Although several B-cell receptor pathway inhibitors, such as ibrutinib,47,48 idelalisib,49,50 and IPI-145,51,52 are showing unprecedented clinical responses and prolonged overall survival, complete remissions and cures are rare. These data further underscore the need for cytotoxic therapeutics, because they lead to remissions. Our present data provide the rationale to combine fludarabine and bendamustine for CLL treatment. A preliminary report suggested a benefit of this combination strategy in patients with CLL and mantle cell lymphoma.53 A phase 1/2 investigation at MD Anderson is testing bendamustine and fludarabine with a single dose of rituximab.54 Clinical response data in 35 CLL patients with relapsed/refractory disease suggest that the regimen is well tolerated and results in 71% objective clinical remissions. Furthermore, DNA damage biomarker studies validate our current in vitro data and demonstrate an increase in DNA damage with the combination.
Acknowledgments
The authors thank Yuling Chen for her assistance in obtaining blood samples, Susan Lerner and Susan Smith for providing information on patient characteristics and clinical laboratory observations, and Heather Lin for assisting Dr Baladandayuthapani with the statistical analyses. The authors thank Dr Jan A. Burger for providing the NK-Tert cell line and Dr Walter Pagel for editing the manuscript.
This work was supported in part by grant Lymphoma SPORE CA136411 and CLL PO1 CA81534 from the National Cancer Institute at the National Institutes of Health, Department of Health and Human Services, a CLL Global Research Foundation Alliance grant, and a sponsored research agreement from Cephalon. W.G.W. and V.G. are members of the CLL Research Consortium.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.A.E.-M. designed research, performed experiments, and analyzed data; M.L.A. performed immunoblot analyses and assisted in performing experiments; V.B. analyzed all data statistically and wrote statistical section; E.J.S. identified donors for CB and directed isolation and culture of CD34-positive cells; M.J.K. and W.G.W. identified patients for blood samples, provided clinical and patient related input, and reviewed the manuscript; and V.G. conceptualized and supervised the research, obtained funding, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: V.G. received research funding from Cephalon (now Teva). The remaining authors declare no competing financial interests.
Correspondence: Varsha Gandhi, Department of Experimental Therapeutics, Unit 1950, The University of Texas MD Anderson Cancer Center, Houston, TX 77230-1429; e-mail: vgandhi@mdanderson.org.
References
- 1.Gandhi V. Metabolism and mechanisms of action of bendamustine: rationales for combination therapies. Semin Oncol. 2002;29(4 Suppl 13):4–11. doi: 10.1053/sonc.2002.34872. [DOI] [PubMed] [Google Scholar]
- 2.Gandhi V, Burger JA. Bendamustine in B-Cell Malignancies: The New 46-Year-Old Kid on the Block. Clin Cancer Res. 2009;15(24):7456–7461. doi: 10.1158/1078-0432.CCR-08-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foon KA, Hallek MJ. Changing paradigms in the treatment of chronic lymphocytic leukemia. Leukemia. 2010;24(3):500–511. doi: 10.1038/leu.2009.266. [DOI] [PubMed] [Google Scholar]
- 4.Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol. 2009;27(26):4378–4384. doi: 10.1200/JCO.2008.20.8389. [DOI] [PubMed] [Google Scholar]
- 5.Cheson BD, Rummel MJ. Bendamustine: rebirth of an old drug. J Clin Oncol. 2009;27(9):1492–1501. doi: 10.1200/JCO.2008.18.7252. [DOI] [PubMed] [Google Scholar]
- 6.Rai KR, Peterson BL, Appelbaum FR, et al. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N Engl J Med. 2000;343(24):1750–1757. doi: 10.1056/NEJM200012143432402. [DOI] [PubMed] [Google Scholar]
- 7.Catovsky D, Richards S, Matutes E, et al. UK National Cancer Research Institute (NCRI) Haematological Oncology Clinical Studies Group; NCRI Chronic Lymphocytic Leukaemia Working Group. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet. 2007;370(9583):230–239. doi: 10.1016/S0140-6736(07)61125-8. [DOI] [PubMed] [Google Scholar]
- 8.Flinn IW, Neuberg DS, Grever MR, et al. Phase III trial of fludarabine plus cyclophosphamide compared with fludarabine for patients with previously untreated chronic lymphocytic leukemia: US Intergroup Trial E2997. J Clin Oncol. 2007;25(7):793–798. doi: 10.1200/JCO.2006.08.0762. [DOI] [PubMed] [Google Scholar]
- 9.Keating MJ, O’Brien S, Albitar M, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol. 2005;23(18):4079–4088. doi: 10.1200/JCO.2005.12.051. [DOI] [PubMed] [Google Scholar]
- 10.Tam CS, O’Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112(4):975–980. doi: 10.1182/blood-2008-02-140582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wierda W, O’Brien S, Wen S, et al. Chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab for relapsed and refractory chronic lymphocytic leukemia. J Clin Oncol. 2005;23(18):4070–4078. doi: 10.1200/JCO.2005.12.516. [DOI] [PubMed] [Google Scholar]
- 12.Buschfort C, Muller MR, Seeber S, Rajewsky MF, Thomale J. DNA excision repair profiles of normal and leukemic human lymphocytes: functional analysis at the single-cell level. Cancer Res. 1997;57(4):651–658. [PubMed] [Google Scholar]
- 13.Geleziunas R, McQuillan A, Malapetsa A, et al. Increased DNA synthesis and repair-enzyme expression in lymphocytes from patients with chronic lymphocytic leukemia resistant to nitrogen mustards. J Natl Cancer Inst. 1991;83(8):557–564. doi: 10.1093/jnci/83.8.557. [DOI] [PubMed] [Google Scholar]
- 14.Yamauchi T, Nowak BJ, Keating MJ, Plunkett W. DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4-hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin Cancer Res. 2001;7(11):3580–3589. [PubMed] [Google Scholar]
- 15.Huang P, Chubb S, Plunkett W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J Biol Chem. 1990;265(27):16617–16625. [PubMed] [Google Scholar]
- 16.Balakrishnan K, Wierda WG, Keating MJ, Gandhi V. Mechanisms of cell death of chronic lymphocytic leukemia lymphocytes by RNA-directed agent, 8-NH2-adenosine. Clin Cancer Res. 2005;11(18):6745–6752. doi: 10.1158/1078-0432.CCR-05-0553. [DOI] [PubMed] [Google Scholar]
- 17.Gandhi V, Plunkett W. Cellular and clinical pharmacology of fludarabine. Clin Pharmacokinet. 2002;41(2):93–103. doi: 10.2165/00003088-200241020-00002. [DOI] [PubMed] [Google Scholar]
- 18.Ogura M, Uchida T, Taniwaki M, et al. Japanese Bendamustine Lymphoma Study Group. Phase I and pharmacokinetic study of bendamustine hydrochloride in relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. Cancer Sci. 2010;101(9):2054–2058. doi: 10.1111/j.1349-7006.2010.01633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurtova AV, Balakrishnan K, Chen R, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114(20):4441–4450. doi: 10.1182/blood-2009-07-233718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caspari T. How to activate p53. Curr Biol. 2000;10(8):R315–R317. doi: 10.1016/s0960-9822(00)00439-5. [DOI] [PubMed] [Google Scholar]
- 21.Hirao A, Kong YY, Matsuoka S, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287(5459):1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 22.Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14(3):289–300. [PMC free article] [PubMed] [Google Scholar]
- 23.Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11(1):71–77. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- 24.Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31(7):402–410. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Leoni LM, Hartley JA. Mechanism of action: the unique pattern of bendamustine-induced cytotoxicity. Semin Hematol. 2011;48(suppl 1):S12–S23. doi: 10.1053/j.seminhematol.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Leoni LM, Bailey B, Reifert J, et al. Bendamustine (Treanda) displays a distinct pattern of cytotoxicity and unique mechanistic features compared with other alkylating agents. Clin Cancer Res. 2008;14(1):309–317. doi: 10.1158/1078-0432.CCR-07-1061. [DOI] [PubMed] [Google Scholar]
- 27.Gaul L, Mandl-Weber S, Baumann P, Emmerich B, Schmidmaier R. Bendamustine induces G2 cell cycle arrest and apoptosis in myeloma cells: the role of ATM-Chk2-Cdc25A and ATM-p53-p21-pathways. J Cancer Res Clin Oncol. 2008;134(2):245–253. doi: 10.1007/s00432-007-0278-x. [DOI] [PubMed] [Google Scholar]
- 28.Roué G, López-Guerra M, Milpied P, et al. Bendamustine is effective in p53-deficient B-cell neoplasms and requires oxidative stress and caspase-independent signaling. Clin Cancer Res. 2008;14(21):6907–6915. doi: 10.1158/1078-0432.CCR-08-0388. [DOI] [PubMed] [Google Scholar]
- 29.Schwänen C, Hecker T, Hübinger G, et al. In vitro evaluation of bendamustine induced apoptosis in B-chronic lymphocytic leukemia. Leukemia. 2002;16(10):2096–2105. doi: 10.1038/sj.leu.2402651. [DOI] [PubMed] [Google Scholar]
- 30.Yang LY, Li L, Keating MJ, Plunkett W. Arabinosyl-2-fluoroadenine augments cisplatin cytotoxicity and inhibits cisplatin-DNA cross-link repair. Mol Pharmacol. 1995;47(5):1072–1079. [PubMed] [Google Scholar]
- 31.Kamiya K, Huang P, Plunkett W. Inhibition of the 3′ —> 5′ exonuclease of human DNA polymerase epsilon by fludarabine-terminated DNA. J Biol Chem. 1996;271(32):19428–19435. doi: 10.1074/jbc.271.32.19428. [DOI] [PubMed] [Google Scholar]
- 32.Catapano CV, Chandler KB, Fernandes DJ. Inhibition of primer RNA formation in CCRF-CEM leukemia cells by fludarabine triphosphate. Cancer Res. 1991;51(7):1829–1835. [PubMed] [Google Scholar]
- 33.Catapano CV, Perrino FW, Fernandes DJ. Primer RNA chain termination induced by 9-beta-D-arabinofuranosyl-2-fluoroadenine 5′-triphosphate. A mechanism of DNA synthesis inhibition. J Biol Chem. 1993;268(10):7179–7185. [PubMed] [Google Scholar]
- 34.Yang SW, Huang P, Plunkett W, Becker FF, Chan JY. Dual mode of inhibition of purified DNA ligase I from human cells by 9-beta-D-arabinofuranosyl-2-fluoroadenine triphosphate. J Biol Chem. 1992;267(4):2345–2349. [PubMed] [Google Scholar]
- 35.Huang P, Sandoval A, Van Den Neste E, Keating MJ, Plunkett W. Inhibition of RNA transcription: a biochemical mechanism of action against chronic lymphocytic leukemia cells by fludarabine. Leukemia. 2000;14(8):1405–1413. doi: 10.1038/sj.leu.2401845. [DOI] [PubMed] [Google Scholar]
- 36.Parker WB, Bapat AR, Shen JX, Townsend AJ, Cheng YC. Interaction of 2-halogenated dATP analogs (F, Cl, and Br) with human DNA polymerases, DNA primase, and ribonucleotide reductase. Mol Pharmacol. 1988;34(4):485–491. [PubMed] [Google Scholar]
- 37.Tseng WC, Derse D, Cheng YC, Brockman RW, Bennett LL., Jr In vitro biological activity of 9-beta-D-arabinofuranosyl-2-fluoroadenine and the biochemical actions of its triphosphate on DNA polymerases and ribonucleotide reductase from HeLa cells. Mol Pharmacol. 1982;21(2):474–477. [PubMed] [Google Scholar]
- 38.Elledge SJ, Davis RW. DNA damage induction of ribonucleotide reductase. Mol Cell Biol. 1989;9(11):4932–4940. doi: 10.1128/mcb.9.11.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elledge SJ, Zhou Z, Allen JB, Navas TA. DNA damage and cell cycle regulation of ribonucleotide reductase. Bioessays. 1993;15(5):333–339. doi: 10.1002/bies.950150507. [DOI] [PubMed] [Google Scholar]
- 40.Hurta RA, Wright JA. Alterations in the activity and regulation of mammalian ribonucleotide reductase by chlorambucil, a DNA damaging agent. J Biol Chem. 1992;267(10):7066–7071. [PubMed] [Google Scholar]
- 41.Kuo ML, Kinsella TJ. Expression of ribonucleotide reductase after ionizing radiation in human cervical carcinoma cells. Cancer Res. 1998;58(10):2245–2252. [PubMed] [Google Scholar]
- 42.Lee YD, Wang J, Stubbe J, Elledge SJ. Dif1 is a DNA-damage-regulated facilitator of nuclear import for ribonucleotide reductase. Mol Cell. 2008;32(1):70–80. doi: 10.1016/j.molcel.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sandoval MR. Alternations in the expression of ribonucleotide reductase induced by DNA damage in human chronic lymphocytic leukemia cells. Houston, TX: Graduate School of Biomedical Sciences, The University of Texas Health Science Center at Houston; 1999:109. [Google Scholar]
- 44.Tanaka H, Arakawa H, Yamaguchi T, et al. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404(6773):42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 45.Moufarij MA, Sampath D, Keating MJ, Plunkett W. Fludarabine increases oxaliplatin cytotoxicity in normal and chronic lymphocytic leukemia lymphocytes by suppressing interstrand DNA crosslink removal. Blood. 2006;108(13):4187–4193. doi: 10.1182/blood-2006-05-023259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–2663. [PubMed] [Google Scholar]
- 47.Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603–3612. doi: 10.1182/blood-2011-05-352492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winkler DG, Faia KL, DiNitto JP, et al. PI3K-δ and PI3K-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol. 2013;20(11):1364–1374. doi: 10.1016/j.chembiol.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 52.Flinn I, Patel M, Kahl BS, et al. Phosphoinositide-3-kinase-δ,γ, in patients with chronic lymphocytic leukemia. Paper presented at the Annual Meeting of the American Society of Hematology. December 8-11, 2013. New Orleans, LA. [Google Scholar]
- 53.Koenigsmann M, Knauf W, Herold M, et al. Fludarabine and bendamustine in refractory and relapsed indolent lymphoma—a multicenter phase I/II Trial of the east german society of hematology and oncology (OSHO). Leuk Lymphoma. 2004;45(9):1821–1827. doi: 10.1080/1042819042000223822. [DOI] [PubMed] [Google Scholar]
- 54.Wierda WG, Balakrishnan K, Ferrajoli A, et al. Fludarabine, bendamustine, and rituximab (FBR) chemoimmunotherapy is a safe and active regimen for relapsed/refractory CLL with in vivo mechanism of action for combination chemotherapy [abstract]. Blood. 2012:120(21):437. [Google Scholar]